

Abstract
The upstream protein kinases responsible for thousands of phosphorylation events in the phosphoproteome remain to be discovered. We developed a three-component chemical reaction which converts the transient non-covalent substrate–kinase complex into a covalently cross-linked product by utilizing a dialdehyde based cross-linker, 1. Unfortunately the reaction of 1 with a lysine in the kinase active site and an engineered cysteine on the substrate to form an isoindole cross-linked product could not be performed in the presence of competing cellular proteinsdue to non-specific side reactions. In order to more selectively target the cross-linker to protein kinases in cell lysates we replaced the weak, kinase-binding adenosine moiety of 1 with a potent protein kinase inhibitor scaffold. In addition, we replaced the o-phthaldialdehyde moiety in 1 with a less reactive thiophene-2,3-dicarboxaldehyde moiety. The combination of these two structural modifications provides for cross-linking of a cysteine containing substrate to its corresponding kinase in the presence of competing cellular proteins.
1. Introduction
Protein phosphorylation represents one of the most dominant and evolutionarily conserved posttranslational modifications for information transfer in cells and organisms.1 The human genome encodes 518 eukaryotic protein kinases and thousands of phosphorylation sites are present in the mammalian proteome.2 In order to develop a global view of eukaryotic phosphorylation networks, it will be necessary to map the connectivity between kinases and phosphoproteins. To this end, we and others have developed methods that enable identification of kinase substrates.3 The inverse problem, that of identifying upstream kinases responsible for specific phosphorylation events has proven much more challenging.4 Examples of phosphorylation sites with unknown upstream kinases include: Ser-170 of the protein BAD which enhances its anti-apoptotic activity,5 Ser-497 of the natiuretic peptide receptor, which is responsible for desensitization of the receptor's anti-hypertensive activity,6 and Ser-325 of the tumor suppressor LKB1, which is phosphorylated in response to the biguanide anti-diabetic agent metformin.7 These examples, as well as the thousands of new “orphan” phosphorylation sites that have been identified in the mammalian phosphoproteome,8 highlight the need to develop methods for reverse mapping of phosphorylation networks as a complement to methods for kinase substrate identification.
Several years ago we reported a three-component chemical reaction designed to facilitate identification of the kinase responsible for a given phosphorylation event by covalently cross-linking a protein substrate of interest to its upstream kinase (Figure 1).9 This is accomplished by replacing a phosphorylated serine or threonine residue of interest with a cysteine. This substitution provides a nucleophilic residue that is in close proximity to the site of phosphotransfer. To covalently link this cysteine to a lysine in the upstream kinase, a bi-functional, ATP-binding site directed cross-linker (1, Figure 1) was developed. This bi-functional reagent contains an o-phthaldialdehyde, which can chemoselectively cross-link a cysteine of the substrate to a lysine in the kinase. The irreversible nature of the cross-linking reaction provides the means to trap weak and transient kinase–substrate binding interactions.
Figure 1.

Kinase – substrate cross-linking strategy. (A) Phosphorylation of the protein substrate by an upstream kinase. (B) The serine of the kinase is replaced with a reactive cysteine moiety that facilitates kinase – substrate cross-link by crosslinker 1.
Identification of kinase-substrate pairs is ideally carried out in a cellular environment which contains a full complement of cellular proteins and kinases, including scaffolding proteins which enhance kinase-substrate specificity.10 Successful execution of cross-linking reactions in complex mixtures requires low background labeling of highly abundant cellular proteins that are not relevant to the phosphorylation event being studied. Unfortunately, we have found that cross-linker 1 reacts with many proteins in mammalian cell lysates and is not suitable for use in complex proteomes.
In theory, selectivity can by improved by specifically targeting the cross-linking agent to proteins of interest and by modulating the reactivity of the dialdehyde cross-linker. This strategy is similar to the design of highly selective irreversible enzyme inhibitors,11 with the distinction that the iso-indole cross-linking reaction is a three-component reaction rather than a bi-molecular one. Yao and coworkers recently reported a modified version of cross-linker 1 that is based on 2,3-naphthalenedicarboxaldehyde rather than on o-phthaldialdehyde. This substitution improves the efficiency of the cross-linking reaction in cell lysates.12 In this report, we systematically explore two of the structural elements that diminish the utility of cross-linker 1 in cell lysates and describe a new series of cross-linkers with greatly enhanced selectivity for the desired kinase substrate cross-linking reaction. In order to more selectively target protein kinases in cell lysates we have replaced the weak, kinase-binding adenosine moiety of 1 with a general protein kinase inhibitor. In addition, we have replaced the o-phthaldialdehyde cross-linking moiety in 1 with a less reactive five-membered heterocyclic dialdehyde. The combination of these two structural modifications has led to a compound capable of cross-linking a cysteine containing substrate to its corresponding kinase in the presence of a vast excess of competing cellular proteins without producing significant background cross-linking.
Results and Discussion
In this report, we utilized the serine/threonine kinase Akt as a model kinase for our studies.13 Akt is a central node in growth factor signaling, and is hypothesized to be responsible for over a hundred substrate phosphorylation events.14 Although, a substrate consensus motif has been identified for Akt (R-X-R-X-X-S/T-B, where X represents any amino acid and B represents bulky hydrophobic residue),15 25% of reported Akt substrates do not contain this sequence13 making unambiguous determination of Akt-substrate pairs a major challenge. Additionally, phosphorylation of the kinase substrates in vitro may not properly recapitulate phosphorylation events in cells. For example, although Akt was first shown to phosphorylate mTOR on S2448 in vitro,16 further studies revealed that S6K1, itself a substrate of mTOR, reciprocally phosphorylates mTOR at S2448 in HEK293 cells.17
To optimize Akt-substrate cross-linking we chose a model peptide substrate sequence (RPRTSSF) derived from GSK3β.18 Our previous studies demonstrated that a fluorescein-labeled Akt consensus motif peptide (Fluorescein-ZZRPRTSCF, 2, Z = aminohexanoic acid), in which the phosphorylated serine residue is replaced with a cysteine, can be cross-linked to Akt1 in the presence of 1 (Figure 2, lane 2). However, the fluorescently labeled Akt1-peptide complex cannot be detected when the cross-linking reaction is conducted in the presence of 7 μg of HeLa cell lysate, and the observed non-specific fluorescent products are not discernable from those produced with HeLa cell lysate alone (Figure 2, lane 3-4). We attribute the absence of Akt1 cross-linked product and abundance of background protein labeling, with proteins with molecular weights much smaller than known kinases, to lysine containing proteins which cross-link to fluorescently labeled peptide, 2.
Figure 2.

Cross-linking of Akt1 with cross-linker 1 and Fluorescein-ZZRPRTSCF-OH 2, in the presence of HeLa cell lysate proteins. Fluorescent peptide 2 (1 μM), cross-linker 1 (20 μM), Akt1 (22 nM) and HeLa cell lysate (7 μg) were incubated for 20 min at r.t., followed by SDS PAGE and in gel fluorescent scanning.
In order to reduce the high amount of background labeling, we initially chose to focus on increasing the affinity of the adenosine portion of 1 for protein kinases. Adenosine is a weak, micromolar ligand for most protein kinases (Figure 3A) and is capable of binding to many other ATP utilizing proteins outside of the protein kinase family. We reasoned that replacing the adenosine fragment in cross-linker 1 with an alternative molecular scaffold, that has higher specificity and binding affinity for kinase ATP-binding sites would reduce the amount of background labeling. A key requirement for such a molecular scaffold is to maintain promiscuous kinase binding to ensure the resulting cross-linkers would be general and could be used to covalently cross-link any kinase with the substrate in question.
Figure 3.
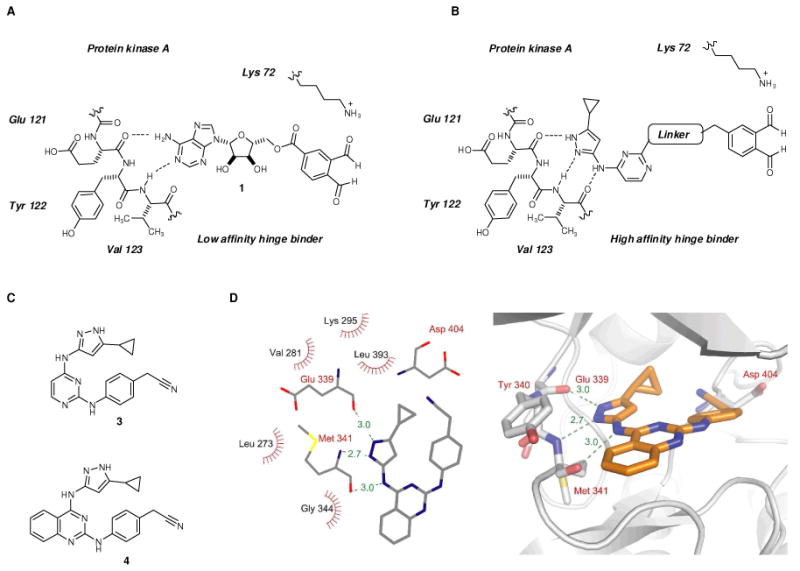
(A) Schematic representation of cross-linker 1 interacting with the hinge region of PKA. (B) Schematic representation of the aminopyrazole scaffold based cross-linker interacting with the hinge region of PKA. (C) Chemical structures of promiscuous kinase inhibitors 3 and 4. (D) Crystal structure of kinase inhibitor 4 bound to c-Src at 2.35 Å resolution. Key interactions and hydrogen bond distances are highlighted.
An analysis of non-selective kinase inhibitors revealed a common structural motif that is responsible for promiscuous kinase-binding behavior.19 Aminopyrazole 3 (Figure 3C), contains all the necessary structural properties for promiscuous inhibition, and therefore, appeared to be a promising molecular replacement for the adenosine fragment in cross-linker 1 (Figure 3B). In order to evaluate ability of aminopyrazole 3 to serve as a general scaffold for an o-phthaldialdehyde based cross-linker, we first determined if it is capable of binding to a wide assortment of protein kinases.20 The affinity of aminopyrazole 3 for the ATP-binding site of 402 diverse kinases was assessed with a phage-encoded kinase library based method as described previously.21 Compound 3 was screened against a panel of 402 kinases (including 43 clinical mutant kinases) at a single concentration (10 μM) to identify the number of kinases addressable by 3. The screen revealed that compound 3 binds 337 out of 359 wild type kinases tested (supplementary Figure 4 and supplementary Table 1). A quantitative dissociation constant Kd was then determined for 40 representative kinases that displayed binding interactions with the kinase inhibitor 3.22 CDK2, CDK5 and JNK3 kinases were among the most potently inhibited kinases with Kds of 4, 27 and 26 nM respectively. Inhibitor 3 has low affinity for several kinases, including Akt1, making cross-linking to this kinase a particularly stringent test of the broad utility of the aminopyrazole based cross-linker.
Having established that compound 3 is a promiscuous kinase binder, we turned to identifying the appropriate linkage to the o-phthaldialdehyde moiety. We attempted to co-crystallize 3 bound to the tyrosine kinase c-Src23 to facilitate proper positioning of the o-phthaldialdehyde moiety with respect to the kinase active site. Although we solved the crystal structure of the c-Src-3 co-complex to 2.0 Å resolution, it was not possible to unambiguously determine the binding orientation of inhibitor 3 in the active site. Thus, we prepared a close structural analog of 3, with a quinazoline rather than a pyrimidine core, 4 (Figure 3C) in hopes that an extra phenyl ring would aid in modeling the bound inhibitor 4 into the electron density map, without disrupting binding to the kinase active site.
Kinase inhibitor 4 was prepared starting from 2,4-dichloroquinazoline utilizing the synthetic route used for 3.24 Under the previously optimized crystallization conditions, we were able to obtain a 2.35 Å resolution crystal structure of compound 4 bound to c-Src and unambiguously visualize its binding mode. The complex of Src with compound 4 crystallized under very similar conditions as the Src•imatinib complex25 and the unit cell dimensions of the two crystals are also closely related. Because the four Src complexes (which we designate chain A, B, C and D) with compound 4 in the crystallographic asymmetric unit are highly similar, we will only discuss one of them (chain A).
Compound 4 forms a total of three hydrogen bonds with c-Src. The aminopyrazole forms three hydrogen bonds with the backbone of Glu 339 and Met 341, which mimic the hydrogen bonding pattern of adenine in ATP binding to the kinase. The quinazoline core in compound 4 forms hydrophobic contacts with the sidechain of Leu 273 (Figure 3D). Two additional recently deposited structures of compound 4, co-crystallized with human Ste20-like kinase and human calcium calmodulin dependent protein kinase IIδ, show similar binding modes26 which suggests that this class of inhibitors bind to divergent kinases in a consistent manner. In addition, quinazoline based kinase inhibitor 4, similar to 3, displayed promiscuous kinase binding properties and bound 317 out of 359 wild type kinases tested (Supplementary Table 1). Similar kinase binding profiles for compounds 3 and 4 indicated that the replacement of pyrimidine scaffold in 3 with quinazoline for crystallographic purposes was appropriate and did not significantly impair promiscuous kinase binding properties of the resulting inhibitor 4. Taken together, these results demonstrate that compound 3 is a promiscuous kinase binder and may serve as alternative molecular scaffold to replace the adenosine fragment in the initially discovered cross-linker 1.
Structural analysis of 4 bound to the three kinases (c-Src, Ste20-like kinase and human calcium calmodulin dependent protein kinase IIδ) reveals the nitrile group in 4 is in the position corresponding to the 5′-hydroxyl group of ATP. This position is the site of attachment for the o-phthaldialdehyde in 1. We thus replaced the nitrile group of 3 with the o-phthaldialdehyde to produce candidate cross-linker 10. The synthetic route for 10 is illustrated in Figure 4A. Treatment of the previously synthesized intermediate 527 with the commercially available p-phenylenediamine in the presence of catalytic amount of HCl in BuOH at 100°C furnished amine 7. The final installation of the dialdehyde moiety was conducted by the treatment of amine 7 with aldehyde 8 under reductive amination conditions followed by acidic hydrolysis to yield cross-linker 10.
Figure 4.
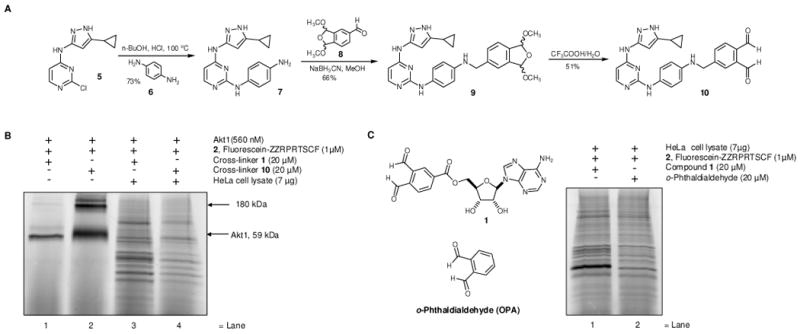
(A) Synthesis of cross-linker 10. (B) Cross-linking Akt1 and fluorescent substrate peptide 2 with cross-linkers 1 and 10 in the presence or absence of HeLa cell lysate proteins. Akt1, fluorescent peptide 2 with or without HeLa cell lysates, were treated with either cross-linker 1 or 10 for 20 min, followed by SDS-PAGE and in gel fluorescence scanning. (C) Cross-linking of endogenous HeLa cell lysate proteins with substrate peptide 2, in the presence of cross-linker 1 or o-phthaldialdehyde. HeLa cell lysates and substrate peptide 2 were treated with cross-linker 1 or o-phthaldialdehyde for 20 min at r.t., followed by SDS-PAGE and in gel fluorescence scanning.
The aminopyrazole based cross-linker 10 efficiently cross-linked Akt1 kinase to cysteine containing peptide 2 with a two-fold increase in intensity of the cross-linked product band when compared with the adenosine based cross-linker 1 (Figure 4B, lanes 1-2). In addition the crosslinking reaction was accompanied by the formation of higher molecular weight (∼180 kDa) fluorescent species (Figure 4B, lane 2). We attribute these products to further oligomerization of the Akt1-peptide crosslinked product induced by the dialdehyde 10 reacting with lysines on the Akt1 kinase surface. The observation that the cross-linking reaction tolerates significant changes in chemical structure of the initially developed dialdehyde 1 points to the robustness of the core iso-indole forming reaction.
We asked if cross-linking of Akt1 with its substrate peptide 2 could be observed in the presence of HeLa cell extract. To determine if the newly designed cross-linker was more selective in the presence of cellular proteins, a mixture of recombinant Akt1 (560 nM), substrate peptide 2 (1 μM) and HeLa cell lysate (7 μg) were treated with either cross-linker 1 or 10. Unfortunately, even the newly designed aminopyrazole based cross-linker suffered a significant decrease in the efficiency of cross-linking of Akt1 with its substrate peptide 2 (Figure 4B, lanes 3-4) when compared with experiments without cell lysate present (Figure 4B, lanes 1-2). The fluorescently labeled reaction products formed a complex pattern of fluorescently labeled bands which spanned the entire molecular weight range of the cell lysate proteome which was similar to that produced with 1 (Figure 4B, lanes 3-4, Figure 2, lanes 3-4, respectively). The intensity of these background bands from the HeLa cell lysate was somewhat mitigated when cross-linker 10 was employed rather than 1 (Figure 4B lane 3-4). Our interpretation is that the kinase binding portion of the cross-linker reagent is partially responsible for the non-specific background reactions yet other elements of cross-linker 10 are responsible for the majority of the undesired reactivity of the cross-linker. Therefore, we set out to identify and modify the other remaining elements of 10 in order to obtain a kinase-substrate cross-linker suitable for whole cell lysate studies.
We focused on the o-phthaldialdehyde moiety since its reactivity is a key determinant of cross-linking reaction. We asked if o-phthaldialdehyde alone, without any kinase directed adenosine or aminopyrazole element, is capable of cross-linking cysteine containing peptide 2 to cellular proteins. o-Phthaldialdehyde has been used to intramolecularly label enzymes that possess proximal lysines and cysteines, such as creatine kinase28 and hexokinase,29 and thus is itself capable of mediating isoindole formation in a cellular context. We asked if o-phthaldialdehyde would efficiently carry out intermolecular cross-linking of peptide 2 to cellular proteins. HeLa cell lysates were treated with fluorescently labeled peptide substrate 2, followed by the treatment with cross-linker 1 or with o-phthaldialdehyde. Surprisingly, o-phthaldialdehyde and 1 produced similar patterns of cross-linked fluorescent bands, with only modest differences in the fluorescent intensity (Figure 4C). Since o-phthaldialdehyde contains no particular protein targeting moieties (e.g. adenosine), yet exhibits similar cross-linking to a diverse set of proteins we conclude the broad spectrum of cross-linked products produced by 1 and 10 are a result of the high reactivity of o-phthaldialdehyde itself. Therefore we aimed to reduce the reactivity of o-phthaldialdehyde towards non-kinase proteins in the cell lysates.
The observation that o-phthaldialdehyde efficiently cross-links background proteins in cell lysates led us to use this assay as a screen for a second generation of dialdehyde species with reduced reactivity. Consideration of the isoindole forming reaction mechanism and previous structure activity studies of the reaction,30 led us to investigate a ketone functionality as a replacement for one aldehyde group in o-phthaldialdehyde, to produce o-acetylbenzaldehyde 11 (Figure 5A). This analog is predicted to serve as a less reactive substitute to o-phthaldialdehyde, since it contains a sterically hindered and less electron deficient acetyl group, while still reacting with thiols and amines to produce isoindoles.31 Naphtalene-2,3-dialdehyde 12 was also included in the screen since Yao and coworkers reported an improved version of cross-linker 1, containing naphthalene-2,3-dialdehyde.12 Replacement of the acetyl group in o-acetylbenzaldehyde 11 with a cyanogroup led to o-cyanobenzaldehyde 13 as another possible candidate with reduced reactivity, this cyanoaldehyde forms a thiazolidine based product rather than isoindole.32 We also asked if replacing the six membered aromatic ring in o-phthaldialdehyde with a five membered aromatic heterocycle, such as thiophene, could eliminate non-specific cross-linking in cell lysates, due to additional steric strain of the resulting thieno[2,3,c]pyrrole based cross-linked product when compared with isoindole. Indeed, commercially available thiophene-2,3-dialdehyde 14 (Figure 5A) has been shown to react with primary thiols and amines to produce thieno[2,3-c]pyrroles,33 which are isoelectronic to isoindoles.
Figure 5.
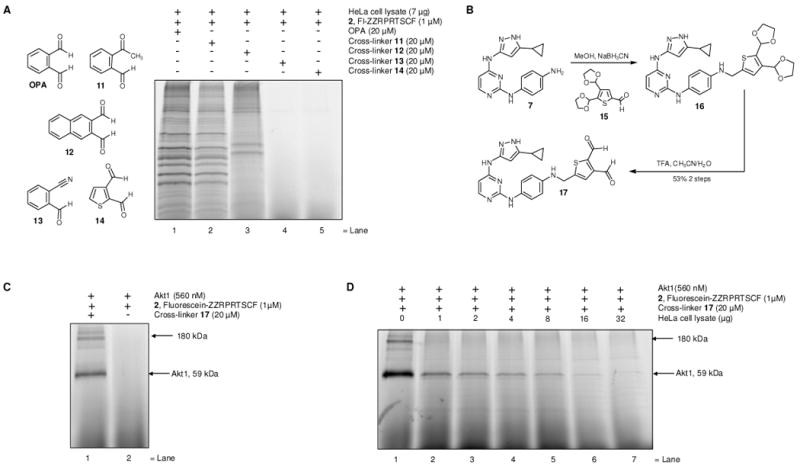
(A) Cross-linking of endogenous HeLa cell lysate proteins with fluorescent Akt1 substrate peptide 2 in the presence of different cross-linkers OPA or aldehydes 11-14. HeLa cell lysates and fluorescent Akt1 substrate peptide 2 were treated with cross-linkers OPA or 11-14 for 20 min at r.t., followed by SDS-PAGE and in gel scanning fluorescence. (B) Synthesis of the thiophene-2,3-dialdehyde based cross-linker 17. (C) Cross-linking of Akt1 and fluorescent substrate peptide 2 with cross-linker 17. Akt1 and fluorescent peptide 2 were treated with cross-linker 17, for 20 min at r.t., followed by SDS-PAGE and in gel scanning fluorescence. (D) Cross-linking of Akt1 and fluorescent peptide substrate 2 with cross-linker 17 in the presence of increasing amounts of HeLa cell lysate proteins. Akt1 and substrate peptide 2 were treated with cross-linker 17 for 20 min at r.t., followed by SDS-PAGE and in gel scanning fluorescence.
We compared the cross-linking of fluorescently labeled Akt1 substrate peptide 2 to HeLa cell lysate proteins in the presence of each of the unsubstituted cross-linkers, o-phthaldialdehyde or 11-14 (Figure 5A). o-Acetylbenzaldehyde 11 produced a less intense, but similar pattern of fluorescently labeled bands compared to o-phthaldialdehyde (Figure 5A, lane 2). Naphthalene-2,3-dialdehyde 12 produced fewer but did not completely eliminate non-specifically labeled fluorescent cross-linked products when compared with o-phthaldialdehyde or o-acetylbenzaldehyde 11. However both o-cyanobenzaldehyde 13 and thiophene-2,3-dialdehyde 14 showed dramatic reductions in the non-specific labeling of cell lysate proteins (Figure 5A, lane 3-4).
Based on the dramatic reduction of the background reactions of cellular proteins with the thiophene dialdehyde 14, and its ability to form thieno[2,3-c]pyrrole based products upon reaction with amines and thiols,33 we asked if we could direct thiophene-2,3-dialdehyde to the kinase ATP-binding site and produce a kinase-substrate cross-link. Since aminopyrazole based cross-linker 10 appeared to produce less background labeling when compared to adenosine based cross-linker 1, we chose to append the thiophene dialdehyde moiety directly to aminopyrazole 7. The required dialdehyde 15 was prepared in two steps from commercially available 2-3-thiophenedialdehyde 14 (Figure 5B).34 Reaction of the aldehyde 15 with the previously synthesized amine 7, under reductive amination conditions, furnished compound 16 which was subsequently hydrolyzed under mild acidic conditions to afford cross-linker 17.
First, we tested if cross-linker 17 was capable of cross-linking recombinant Akt1 kinase to the fluorescent peptide substrate 2. Treatment of Akt1 and substrate peptide 2 with cross-linker 17, produced an intensely fluorescent band of labeled Akt1 kinase, confirming that the newly prepared cross-linker 17 is capable of mediating the desired kinase-substrate covalent cross-link (Figure 5C). However, as in the case of cross-linker 10 (Figure 4B, lane 2), the crosslinking reaction was accompanied by the formation of higher molecular weight (∼180 kDa) species (Figure 5C, lane 1). We next optimized conditions for kinase substrate cross-linking employing the newly synthesized cross-linker 17. Studies on the pH dependence of the cross-linking reaction indicate that pH 5 is optimal for the cross-linking reaction (Supplemental Figure 1A). The cross-linking reaction is also resistant to the presence of competing thiol nucleophiles, tolerating up to 1000-fold excess of β-mercaptoethanol relative to cysteine containing peptide substrate 2 (Supplememental Figure 1B), thus illustrating remarkable chemoselectivity toward the thiol containing peptide. Covalently cross-linked kinase-substrate complex was readily cleaved upon heating as no detectable cross-linked band was observed when samples were boiled prior to SDS-PAGE analysis (Supplemental Figure 1C). The reaction showed linear dose dependence, with respect to both cross-linker 17 and fluorescent peptide 2 concentrations (Supplemental Figures 2 and 3). Saturation of the crosslinked signal was observed when the concentration of crosslinker 17 and peptide 2 reached approximately 40 μM and 60 μM, respectively (Supplemental Figure 2 and 3).
Next, we tested if cross-linker 17 could specifically cross-link Akt1 kinase to its substrate peptide 2 in a complex cell lysate. We carried out the cross-linking reaction with Akt1 and its substrate peptide 2 (1 μM or 49 ng of peptide 2 per reaction) alone (Figure 5D, lane 1), and in the presence of increasing amounts of cell lysate from 1μg–lane 2 up to 32μg–lane 7 (Figure 5D). Remarkably, fluorescein-containing cross-linked product is readily apparent in the presence of up to 8 μg of HeLa cell lysate (Figure 5D, lane 6). Thus, thiophene based cross-linker 17 represents a significant improvement over cross-linkers 1 or 10 which produced abundant non-kinase cross-linked products in the presence of cell lysates, (Figure 4B, lanes 3-4).
In conclusion, a three component chemical reaction leading to the formation of the covalently cross-linked kinase-substrate complex in vitro, has been developed to cross-link recombinant Akt kinase with its cysteine containing peptide substrate in the presence of cell lysates. This was achieved through extensive structural modifications to the adenosine and dialdehyde fragments in cross-linker 1. The adenosine fragment in cross-linker 1, was replaced with a promiscuous kinase inhibitor that has a nanomolar binding affinity toward a wide range of kinases, to enhance targeting to protein kinase active sites. We determined that o-phthaldialdehyde itself is largely responsible for the non-specific labeling of cell lysate proteins. We have developed an alternative dialdehyde, thiophene-2,3-dicarboxaldehyde 14, that shows a remarkable decrease in the non-specific background labeling of cell lysate proteins. Replacing the o-phthaldialdehyde derived fragment in our newly designed cross-linker 10 with the thiophene-2,3-dicarboxaldehyde moiety led to cross-linker 17 that allowed selective cross-linking of recombinant Akt1 kinase with its cysteine containing peptide substrate in the presence of competing cellular proteins.
Our findings represent a significant step towards a solution to the challenge of identifying upstream kinases for the given substrate of interest, although further challenges remain. The next step is to perform the cross-linking reaction between the kinase and its full length substrate protein. The replacement of the substrate peptide with the full length protein may further increase the specificity of the cross-linking reaction in the presence of cell lysates, due to additional recognition elements presented on the protein substrate surface, that are distal from the local phosphorylation site. For example p38 kinase phosphorylates the protein substrate MAPKAP2 150-fold more efficiently than the corresponding 14-residue substrate peptide resembling local phosphorylation site.35 Once conditions for the cross-linking reaction between full length kinase and substrate protein are optimized, the next challenge is to perform the cross-linking of the protein substrate with its endogenous kinases in cell lysates. Since many endogenous kinases are present at very low abundance in cells, the detection limits of the cross-linking reaction must still be addressed. Additional structural modifications to 15 such as optimization of the linker which connects the kinase binding scaffold with the dialdehyde moiety may further increase the efficiency of the reaction in the presence of cellular proteins. The ultimate challenge is to perform the cross-linking reaction in intact cells, as the intact cellular environment provides all the elements necessary encoding kinase substrate specificity via scaffolding proteins, specific protein-protein interaction domains, and subcellular co-localization of kinases and their substrates.
Supplementary Material
Acknowledgments
MAS was supported by a Johnson & Johnson fellowship of the Life Science Research Foundation, Baltimore and NIH grant K99 GM080097. KMS thanks NIH RO1-EB001987 for funding as well as a grant from Eli Lilly.
Footnotes
Supporting Information Available: Complete experimental procedures, kinase binding data for compounds 3 and 4, X-ray crystallographic data, compound characterization. Complete ref. 3(c), 21(a) and 21(b). This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.(a) Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]; (b) Johnson LN, Lewis RJ. Chem Rev. 2001;101:2209–2242. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- 2.Villén J, Beausoleil SA, Gerber SA, Gygi SP. PNAS. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Manning BD, Cantley LC. Sci STKE. 2002;2002:pe49. doi: 10.1126/stke.2002.162.pe49. [DOI] [PubMed] [Google Scholar]; (b) Ptacek J, Snyder M. Trends Genet. 2006;22:545–554. doi: 10.1016/j.tig.2006.08.005. [DOI] [PubMed] [Google Scholar]; (c) Ptacek J, et al. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]; (d) Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. Curr Biol. 1994;4:973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]; (e) Obenauer JC, Cantley LC, Yaffe MB. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Berwick DC, Tavare JM. Trends Biochem Sci. 29:227–232. doi: 10.1016/j.tibs.2004.03.004. [DOI] [PubMed] [Google Scholar]; (g) Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hübner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, Katayama CD, Hedrick SM, Shokat KM. Nat Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Blethrow JD, Glavy JS, Morgan DO, Shokat KM. PNAS. 2008;105:1442–1447. doi: 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Parang K, Kohn JA, Saldanha SA, Cole PA. FEBS Lett. 2002;520:156–160. doi: 10.1016/s0014-5793(02)02778-3. [DOI] [PubMed] [Google Scholar]; (b) Shen K, Cole PA. J Am Chem Soc. 2003;125:16172–16173. doi: 10.1021/ja0380401. [DOI] [PubMed] [Google Scholar]
- 5.Dramsi S, Scheid MP, Maiti A, Hojabrpour P, Chen X, Schubert K, Goodlett DR, Aebersold R, Duronio V. J Biol Chem. 2002;277:6399–6405. doi: 10.1074/jbc.M109990200. [DOI] [PubMed] [Google Scholar]
- 6.Potter LR, Hunter T. Mol Cell Biol. 1998;18:2164–2172. doi: 10.1128/mcb.18.4.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Forcet C, Billaud M. Sci STKE. 2007;404:pe51. doi: 10.1126/stke.4042007pe51. [DOI] [PubMed] [Google Scholar]; (b) Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Biochem J. 2002;362:481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villén J, Li J, Cohn MA, Cantley LC, Gygi SP. Proc Natl Acad Sci. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maly DJ, Allen JA, Shokat KM. J Am Chem Soc. 2004;126:9160–1. doi: 10.1021/ja048659i. [DOI] [PubMed] [Google Scholar]
- 10.Pawson T, Nash P. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 11.Walsh CT. Annu Rev Biochem. 1984;53:493–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- 12.Liu K, Kalesh KA, Bing Ong L, Yao SQ. Chembiochem. 2008 doi: 10.1002/cbic.200800212. ahead of print. [DOI] [PubMed] [Google Scholar]
- 13.Manning BD, Cantley LC. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 15.Alessi DR, Caudwell FB, Andjelkovic Mb, Hernmings BA, Cohen P. FEBS letters. 1996;399:333. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 16.(a) Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC. PNAS. 1998;95:7772–7777. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- 17.(a) Chiang GG, Abraham RT. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]; (b) Holz MK, Blenis J. J Biol Chem. 2005;280:26089–26093. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- 18.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 19.Aronov AM, Murcko MA. J Med Chem. 2004;47:5616–5619. doi: 10.1021/jm049793g. [DOI] [PubMed] [Google Scholar]
- 20.See supplementary material for the synthesis of aminopyrazole 3.
- 21.(a) Fabian MA, et al. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]; (b) Karaman MW, et al. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 22.See supplementary material.
- 23.Benati D, Baldari CT. Curr Med Chem. 2008;15:1154–65. doi: 10.2174/092986708784310404. [DOI] [PubMed] [Google Scholar]
- 24.See supplementary material.
- 25.Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. Structure. 2007;15:299–311. doi: 10.1016/j.str.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 26.(a) Pike AC, Rellos P, Niesen FH, Turnbull A, Oliver AW, Parker SA, Turk BE, Pearl LH, Knapp S. EMBO J. 2008;27:704–714. doi: 10.1038/emboj.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roos AK, Rellos P, Salah E, Pike ACW, Fedorov O, Pilka ES, Von Delft F, Arrowsmith CH, Weigelt J, Edwards A, Bountra C, Knapp S. PDB deposited (PDB 2VN9) [Google Scholar]
- 27.See supplementary material.
- 28.Sheikh S, Mukunda K, Katiyar SS. Biochim Biophys Acta. 1993;1203:276–81. doi: 10.1016/0167-4838(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 29.Puri RN, Bhatnagar D, Roskoski R., Jr Biochim Biophys Acta. 1988;957:34–46. doi: 10.1016/0167-4838(88)90154-9. [DOI] [PubMed] [Google Scholar]
- 30.Zuman P. Chem Rev. 2004;104:3217–3238. doi: 10.1021/cr0304424. [DOI] [PubMed] [Google Scholar]
- 31.Sternson LA, Stobaugh JF, Repta AJ. Anal Biochem. 1985:233–246. doi: 10.1016/0003-2697(85)90111-3. [DOI] [PubMed] [Google Scholar]
- 32.Oliver GL, Dann JR, Gates JW. J Am Chem Soc. 1958;80:702–706. [Google Scholar]
- 33.El-borai MA, Aly EA, Gerges SS. Phosphorus, Sulfur Silicon Relat Elem. 1995;101:9–15. [Google Scholar]
- 34.See supplementary material.
- 35.Hawkins J, Zheng S, Frantz B, LoGrasso P. Arch Biochem Biophys. 2000;382:310. doi: 10.1006/abbi.2000.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.