

Abstract
Francisella tularensis subsp. tularensis is the etiologic agent of tularemia and has been designated a category A biothreat agent by the CDC. Tularemia is characterized by replication and dissemination within host phagocytes. Intramacrophage growth is dependent upon the regulation of Francisella pathogenicity island (FPI) virulence genes, which is poorly understood. Two-component regulatory systems (TCS) are widely employed by Gram-negative bacteria to monitor and respond to environmental signals. Virulent strains of F. tularensis subsp. tularensis are devoid of classical, tandemly arranged TCS genes, but orphaned members, such as that encoding the response regulator PmrA, have been identified. In the F. novicida model system, previous work has shown that a pmrA mutant shows decreased expression of FPI genes, is deficient for intramacrophage growth, and is avirulent in the mouse model. Here, we determine that phosphorylation aids PmrA binding to regulated promoters pmrA and the FPI-encoded pdpD, and KdpD is the histidine kinase primarily responsible for phosphorylation of PmrA at the aspartic acid at position 51 (D51). A strain expressing PmrA D51A retains some DNA binding but exhibits reduced expression of the PmrA regulon, is deficient for intramacrophage replication, and is attenuated in the mouse model. With regard to virulence gene induction, PmrA coprecipitates with the FPI transcription factors MglA and SspA, which bind RNA polymerase. Together, these data suggest a model of Francisella gene regulation that includes a TCS consisting of KdpD and PmrA. Once phosphorylated, PmrA binds to regulated gene promoters recruiting free or RNA polymerase-bound MglA and SspA to initiate FPI gene transcription.
Francisella tularensis subsp. tularensis is a Gram-negative nonmotile facultative intracellular pathogen and the causative agent of tularemia. F. tularensis has been extensively researched as a biological weapon and has been designated a category A biothreat agent by the Centers for Disease Control and Prevention (CDC). There is a low frequency of tularemia in the United States, but in those occurring cases, misdiagnosis can lead to a poor prognosis (22). F. tularensis can be acquired from the bite of an infected arthropod, contact with an infected animal, or ingestion of contaminated food, water, or air (23). Different routes of entry can lead to several forms of the disease, with pneumonic tularemia being the most serious (22). There are three recognized subspecies of F. tularensis (subspecies tularensis, holarctica, and mediasiatica), and Francisella novicida has been proposed to be reclassified as F. tularensis subsp. novicida. Type A (F. tularensis subsp. tularensis) strains, recovered primarily from North America, are the most virulent. Less virulent type B (F. tularensis subsp. holarctica) strains are found in Europe. F. novicida is closely related to type A F. tularensis (>96% DNA homology) (13) and causes a tularemia-like disease in mice but does not cause disease in immunocompetent humans.
Infections with F. tularensis are characterized by the invasion of and replication within host phagocytes. In fact, mutagenesis experiments have identified very few mutations that are attenuated and yet replicate within macrophages (17, 29). Upon entry into macrophages, Francisella modifies the endocytic pathway, preventing phagolysosomal fusion. Once the phagosomal maturation is halted, the bacteria escape to the cytosol, where they replicate (7). It is clear that in vivo expression of the Francisella pathogenicity island (FPI) is critical for the ability of this pathogen to cause disease (14, 25). Mutations in the FPI result in attenuation, inability to escape the phagosome, and deficient replication within macrophages (21). MglA, SspA, FevR (also called PigR [6]), MigR, Hfq, and PmrA have been shown to be necessary for Francisella virulence and transcription of the FPI, and MglA and SspA have been shown to bind to RNA polymerase (4, 6, 15, 19, 20). How these proteins coordinate regulation of the FPI is not understood.
Two-component regulatory systems (TCS) play a critical role in the regulation of virulence determinants for many bacterial pathogens. TCS are composed of a sensor kinase and a response regulator. Typically, response regulators are phosphorylated at a conserved aspartate residue by the sensor kinase that has autophosphorylated at a conserved histidine residue. Autophosphorylation occurs in response to an environmental signal that is detected by the membrane-bound sensor kinase. The phosphorylated response regulator then causes changes in transcription by binding to gene promoters. Traditional TCS have tandemly arranged genes in an operon transcribed from a single promoter (27). There are no tandemly arranged TCS genes in virulent Francisella, but orphaned members, including pmrA, are present (20). The pmrA gene was named as such because of the similarity of its product to the PmrA protein of Salmonella spp.; however, PmrA also demonstrates similarity to other response regulators, including PhoP and QseB.
Our previous report showed that an F. novicida pmrA null mutant is defective for intramacrophage survival and is attenuated in mice. Also, microarray and genetic analyses indicate that PmrA positively regulates its own transcription and that of the FPI (20). Here, we demonstrate that PmrA binds to regulated gene promoters and that, though lacking a linked kinase, DNA binding is enhanced by phosphorylation. The primary kinase phosphorylating PmrA is KdpD, and this occurs at D51. In addition, data indicate that PmrA may physically interact with MglA and SspA, suggesting a model in which PmrA promoter binding recruits MglA and SspA to initiate FPI transcription. This work represents the first detailed characterization of a regulated promoter and the first demonstration of DNA binding by a transcriptional activator in Francisella.
MATERIALS AND METHODS
Protein purification.
The entire mglA gene was amplified by PCR from F. novicida genomic DNA. The primers (JG1071, 5′-CGGGATCCGAGGATACAATCTTGCTTTTATACAC-3′, and JG1072, 5′-AACTGCAGTTAAGCTCCTTTTGCTTTGATAGT-3′) were engineered to include a BamHI restriction site on the forward primer and a PstI restriction site on the reverse primer (Table 1). The PCR products were digested and cloned into the pQE30 His-tagged expression vector (Qiagen, Valencia, CA). The presence of mglA was confirmed by DNA sequencing. His-tagged proteins were purified using immobilized metal affinity chromatography (IMAC) native affinity and desalting columns with the Profinia protein purification system (Bio-Rad, Hercules, CA). The concentration of the purified protein was determined using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). The purified protein was also analyzed by SDS-PAGE separation and staining with GelCode Blue (Pierce Biotechnology Inc., Rockford, IL).
TABLE 1.
Primers and strains used in this study
| Primer or strain | Sequence or genotype/phenotype | Description or reference/source |
|---|---|---|
| Primers | ||
| JG1071 | CGGGATCCGAGGATACAATCTTGCTTTTATACAC | mglA forward primer BamHI |
| JG1072 | AACTGCAGTTAAGCTCCTTTTGCTTTGATAGT | mglA reverse primer PstI |
| JG1289 | ATGATTATGAGTGAGATGATAACAAG | iglC qRT-PCR forward primer |
| JG1290 | CTATGCAGCTGCAATATATCC | iglC qRT-PCR reverse primer |
| JG1412 | GTATGATATAGTCGTCTTAGCCATTGGTATGCCAATAAAAAC | PmrA site-directed mutant Asp51 to Ala51 from GAT to GCC forward primer |
| JG1413 | TAAGACGACTATATCATACAATCCAGATTCTATAAAAGTTTGCGC | PmrA site-directed mutant D51 to A51 from GAT to GCC reverse primer |
| JG1462 | ATATCTTATGGGCTTGGGCGAT | pmrA primer extension PCR forward primer |
| JG1463 | ATAAAAGTTTGCGCTGCCTCACCA | pmrA primer extension/gel shift reverse primer |
| JG1508 | AAAAGTTTGATGTAACTTTAGAAAACATTTTCA | pmrA gel shift forward primer |
| JG1510 | GCAACCGGAGCAAAAAGTAG | pdpD gel shift forward primer |
| JG1511 | GAGGTCATCAGTATCATATAATAAATCGTT | pdpD gel shift reverse primer |
| JG1514 | FAM-GCCTTCACCCAAATGAAGATC | pmrA primer extension FAM primer |
| JG1515 | FAM-TAGCCATGACATCCATCGTTT | pdpD primer extension FAM primer |
| JG2079 | CGGGATCCCGGAGCTTTTACACTTCTAAAAGAACGAG | kdpD C-terminal fragment forward primer |
| JG2080 | GGGGTACCCCGTGATCACTTGATAGCTAATTTGTAC | kdpD C-terminal fragment reverse primer |
| Strains | ||
| JSG1819 | F. novicida U112 | ATCC |
| JSG2250 | F. novicida mglA::Erm | 15 |
| JSG2845 | JSG1819 with ΔpmrA::Kan | 20 |
| JSG2846 | E. coli DH5α with Francisella pmrA carried on pkk214pgroEL | 20 |
| JSG2847 | JSG2845 complemented with pmrA carried on pKK214pgroEL | 20 |
| JSG2894 | F. novicida transposon T20 insertion at FTN1715 (kdpD) | 10 |
| JSG3033 | JSG2845 complemented with pmrA D51A carried on pKK214pgroEL | This work |
| JSG2970 | sspA tagged with His and inserted in the chromosome of F. novicida (single copy expression) | 3 |
| JSG2890 | F. novicida transposon T20 insertion at FTN1453 | 10 |
| JSG2892 | F. novicida transposon T20 insertion at FTN1617 (qseC) | 10 |
| JSG2894 | F. novicida transposon T20 insertion at FTN1715 (kdpD) | 10 |
| JSG2895 | F. novicida transposon T20 insertion at FTN1714 (kdpE) | 10 |
| E. coli JM109 | endA1 recA1 gyrA96 thi hsdR17 (rK− mK+) relA1 supE44 Δ(lac-proAB) [F′ traD36 proAB laqIqZΔM15] | Promega |
| JSG3031 | E. coli JM109(pQE30[mglA]) | This work |
| JSG3032 | E. coli XL-1 Blue MRF(pQE30XA[pmrA]) | 20 |
| FT4 | Francisella tularensis Schu4 | BEI Resources via Rick Lyons |
Site-directed mutagenesis.
The site-directed mutant PmrA(D51A) was generated by using a procedure with overlapping PCR primers to create a single base pair substitution that resulted in the desired amino acid conversion (Table 1). The pKK214pgroEL plasmid carrying pmrA was purified from JSG2846. Purified plasmid was methylated with CpG methyltransferase (New England Biolabs, Ipswich, MA). The methylated plasmid DNA was used as a template for PCR with primers JG1412 and JG1413. The resulting PCR products were transformed into F. novicida, where the methylated template was degraded, leaving the PCR-produced plasmid DNA with the mutated sequence. The constructed pmrA(D51A) gene was digested from the pKK214 plasmid and cloned into a pQE30 His expression vector (Qiagen, Valencia, CA), using the restriction sites BamHI and PstI. The pQE30[pmrA D51A] plasmid was maintained in Escherichia coli JM109. The codon change in pmrA(D51A) was confirmed by DNA sequencing.
Primer extension.
These studies were performed essentially as described previously (16). 6-Carboxyfluorescein (FAM)-labeled primers JG1514 and JG1515 were designed to bind downstream of the ATG of the gene of interest (Table 1). The primer was bound to a PCR product that contained the fluorescent probe binding site and the predicted location of the promoter. The fluorescently labeled fragment was then sequenced. One nanomole of the fluorescently labeled primer was also bound to 50 μg of RNA and used as a template to generate a single-stranded DNA (ssDNA) by using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). Denatured ssDNAs were analyzed in an ABI 3770 capillary electrophoresis sequencer. The length of the fluorescently labeled ssDNA fragment is equal to the distance from the primer to the start of transcription. The fluorescent signals from the sequence reaction and the ssDNA reaction were aligned to determine the exact base at which transcription was initiated.
Electrophoretic mobility shift assays (EMSA).
Promoter regions for pmrA and pdpD were amplified and labeled by PCR from F. novicida wild-type genomic DNA. Standard PCR conditions were used, and the reaction mixtures were spiked with [γ-32P]dATP. Primer pair JSG1508 and JSG1463 was used to amplify the pmrA promoter region. Primers JG1510 and JG1511 were used to amplify the pdpD promoter region (Table 1). Nonradioactive control reactions were used to estimate the concentration and purity of the PCR products by NanoDrop spectrophotometry and gel electrophoresis, followed by staining with ethidium bromide. Purified His-tagged proteins were added to 12 ng of labeled DNA and incubated for 30 to 40 min at room temperature in binding buffer (0.1 mM dithiothreitol [DTT], 2 mM MgCl2, 1% glycerol, 0.2 mM EDTA, 20 mM KCl, 2 mM Tris-HCl [pH 7.5], 1 μg poly[d{I-C}]). Agarose loading buffer was added to each sample and then electrophoresed on prerun 5% acrylamide Tris-borate-EDTA (TBE) gels at 20 milliamps. Gels were dried, and the DNA was detected by autoradiography. Purified protein was phosphorylated by incubating with acetyl phosphate (40 mM acetyl phosphate, 50 mM Tris-HCl, 20 mM MgCl2, and 0.1 mM DTT) for 30 to 40 min at 37°C prior to adding to the DNA.
Phosphotransfer with CheA.
The conditions used for autophosphorylation and phosphotransfer from CheA have been previously described (30). Autophosphorylation of CheA was performed with 50 μCi of [γ-32P]ATP at room temperature in 10 μl of phosphorylation buffer (100 mM Tris-HCl [pH 7.4], 5 mM MgCl2, 50 mM KCl) for 1 h. Phosphotransfer was performed with 1 μg of purified His-PmrA or His-PmrA(D51A) with 100 ng of autophosphorylated CheA in phosphorylation buffer at 37°C for 1 h. The reaction was stopped with equal volumes of Laemmli sample buffer (Bio-Rad, Hercules, CA) plus 0.5% β-mercaptoethanol (β-ME). Radiolabeled products were separated on a 12.5% SDS-PAGE gel and detected by autoradiography. Duplicate reactions with nonradioactive ATP were electrophoresed through a 12.5% SDS-PAGE gel and proteins were detected by staining with GelCode Blue (Pierce, Rockford, IL). Phosphotransfer experiments with the C-terminal cytoplasmic phosphotransfer domains of His-KdpD and His-PmrA were performed in an identical manner. The KdpD fragment was amplified with primers JG2079 and JG2080, cloned into pTRC-Topo-2, and purified as described above.
Phosphotransfer from bacterial membrane fractions.
Phosphotransfer from membrane fractions was performed as described by Gunn et al. (11a). F. novicida wild-type (JSG1819), F. novicida ΔFTN1453 (JSG2890), F. novicida ΔFTN1617 (JSG2892 qseC), F. novicida ΔFTN1715 (JSG2894 kdpD), F. novicida ΔFTN1714 (JSG2893 kdpE), and F. tularensis Schu4 strains were grown in 100 ml of Trypticase soy broth (TSB) plus 0.1% cysteine to an optical density at 600 nm (OD600) of 1.0. Bacteria were harvested by centrifugation at ∼9,000 × g for 15 min at 4°C. Pellets were resuspended in 5 ml of Bugbuster protein extraction buffer (Novagen, Madison, WI) and sonicated for 2 min in 10-s bursts at an output of 50%, using a Sonics Vibra-Cell processor (Sonics and Materials, Inc., Newtown, CT). Lysates were centrifuged at 100,000 × g for 1 h at 4°C. The pellet was resuspended in 10% glycerol. Total protein was measured using the BCA protein assay kit (Pierce, Rockford, IL) and the NanoDrop spectrophotometer. Membranes (5 μg), His-PmrA or His-PmrA(D51A) (1 μg), and [γ-32P]ATP (3 pmol) were incubated at 37°C for 1 h. The reaction was stopped with equal volumes of Laemmli sample buffer (Bio-Rad, Hercules, CA) plus 0.5% β-ME. Radiolabeled proteins were separated on a 12.5% SDS-PAGE gel and detected by autoradiography or exposed to a phosphor screen and imaged with a Typhoon variable mode imager (GE Healthcare, Pittsburgh, PA). Duplicate control reaction mixtures with equal amounts of nonradioactive ATP were analyzed by Western blotting. The autoradiograph from the Western blot was overlaid with the phosphotransfer assay to confirm the location of His-PmrA. Growth and lysis of type A Schu S4 strain were performed in CDC-approved BSL3/ABSL3 suites with Biosafety Committee-approved protocols at The Ohio State University.
Immunoprecipitation.
Coprecipitation experiments were performed essentially as previously described (2). F. novicida ΔpmrA (JSG2845), F. novicida ΔmglA (JSG2250), and F. novicida SspA-His (JSB2970) strains were grown in TSB plus 0.1% cysteine at 37°C to an OD600 of 1.0. The bacteria were harvested by centrifugation at ∼9,000 × g for 15 min at 4°C. Pellets were resuspended in 5 ml of Bugbuster protein extraction buffer (Novagen, Madison, WI) and sonicated for 2 min in 10-s bursts at an output of 50%, using a Sonics Vibra-Cell processor (Newtown, CT). Lysates were centrifuged at 100,000 × g for 1 h at 4°C. The supernatant was recovered for immunoprecipitation. Purified His-PmrA, His-MglA, or His-PhoP was added to 1 ml of the appropriate soluble fractions and incubated for 1 h at 4°C. His-tagged and associated proteins were precipitated by incubating with 75 μl of His-bind resin (Novagen, Madison, WI) overnight at 4°C with purification per the manufacturer's instructions. Equal volumes of Laemmli sample buffer (Bio-Rad, Hercules, CA) plus 0.5% β-ME was added to the samples, and they were stored at −70°C. Proteins were detected by Western blotting.
Western blotting.
Proteins were separated on SDS-PAGE gels. The gels were transferred to nitrocellulose by using the Trans-Blot SD semidry transfer cell (Bio-Rad, Hercules, CA) for 1 h at 12 V. Immunoblots were blocked with 1% casein (Novagen, Madison, WI). PmrA was detected using anti-PmrA-His rabbit antisera. MglA was detected using anti-MglA-His rabbit antisera. Antisera were produced by Alpha Diagnostic Intl., Inc. (San Antonio, TX). His-tagged proteins were detected using a His-tagged monoclonal antibody (Novagen, Madison, WI).
Quantitative real-time PCR (qRT-PCR).
Expression analysis was performed as described previously (20). RNA was extracted from mid-log-phase (OD600, 0.4 to 0.5) F. novicida (JSG1819), F. novicida ΔpmrA (JSG2845), F. novicida ΔpmrA pKK214pgroEL(pmrA) (JSG2847), and F. novicida ΔpmrA pKK14pgroEL(pmrA D51A) (JSG3033) bacteria by using the RNeasy Kit (Qiagen, Valencia, CA). The RNA quality and quantity were determined with the Experion automated electrophoresis system (Bio-Rad, Hercules, CA) and NanoDrop spectrophotometry (NanoDrop Products, Wilmington, DE). One microgram of total RNA was reverse transcribed to cDNA with Superscript II (RNase H−) and reverse transcriptase (Invitrogen, Carlsbad, CA) and normalized according to the concentration. Two nanograms of the converted cDNA was used for quantitative PCR with the SYBR green PCR master mixture in the Bio-Rad iCycler apparatus (Bio-Rad, Hercules, CA). Relative quantification was used to evaluate the expression of the chosen genes. All primers were designed to give 200- to 220-nucleotide amplicons, have a G+C content range of 30 of 50%, and a melting temperature of 58 to 60°C. Relative copy numbers and expression ratios of the selected genes were normalized to the expression of two housekeeping genes (the 16S rRNA gene and dnaK) and calculated as described by Gavrilin et al. (11).
Intramacrophage survival assay.
Gentamicin protection assays were performed as previously described (20). F. novicida (JSG1819), F. novicida ΔpmrA (JSG2845), F. novicida ΔpmrA pKK214pgroEL(pmrA) (JSG2847), and F. novicida ΔpmrA pKK 214pgroEL(pmrA D51A) (JSG3033) strains were used to infect phorbol myristate acetate (PMA)-induced (10 ng/ml) THP-1 cells, a human macrophage-like cell line, at a multiplicity of infection of 50:1. Wells were seeded with 2 × 105 macrophages, and 1.0 × 107 bacteria were added to each well. After 2 h of incubation at 37°C and 5% CO2, gentamicin (50 μg/ml) was added to the medium to eliminate extracellular organisms. After 30 min, wells were washed twice with phosphate-buffered saline (PBS) and incubated with their respective media supplemented with 10 μg/ml gentamicin. The macrophages were lysed with 0.1% sodium dodecyl sulfate (SDS) at 2 h, 12 h, and 24 h postinfection, and the lysates were serially diluted in PBS and plated onto chocolate II agar plates (BD, Franklin Lakes, NJ) for determination of viable counts.
Mouse survival studies.
Virulence experiments were performed as described by Mohapatra et al. (20). Francisella sp. strains were given intranasally to groups of five anesthetized female 6- to 8-week-old BALB/c mice (Harlan Sprague, Indianapolis, IN) at a dose of 1 × 103 to 1 × 106 CFU/20 μl PBS. Actual bacterial counts delivered were determined by plate counts from the inoculum. Mice were monitored for 5 weeks postinfection.
RESULTS
PmrA binds to the promoter regions of regulated genes, and binding is increased upon treatment with a phosphate donor.
In order to perform DNA binding studies with PmrA, it was necessary to identify the promoter regions of regulated genes. The transcriptional initiation sites for pmrA and pdpD were determined by primer extension. Fluorescently labeled primers were designed to bind to the RNA 50 to 100 bp downstream of the start codon of the gene of interest (Table 1). These primers were annealed to F. novicida wild-type RNA and used as a template for a reverse transcription reaction. The genes pmrA and pdpD were chosen for initial analysis, as pmrA is positively autoregulated and unaffected by MglA/SspA, while pdpD transcription required MglA, SspA, and PmrA (2, 6, 20). The start of transcription of pmrA was determined to be 40 bp upstream of the ATG and that of pdpD 32 bp upstream of its ATG (see Fig. S1 and S2 in the supplemental material). This confirmed the existence of promoters upstream for both of these genes, indicating that they are the first genes of their respective operons (17, 24).
The primer extension data were used to design PCR primers that would amplify a region of DNA flanking the start of transcription. The pmrA promoter region PCR product is approximately 350 bp, extending 190 bp upstream of the start of transcription. The pdpD probe is 200 bp in length, extending approximately 128 bp upstream of the start of transcription. PCRs incorporating radiolabeled nucleotides generated probes that were incubated with purified His-PmrA in EMSA. The results show that purified His-PmrA binds to the promoter regions of pmrA and pdpD in a dose-dependent manner (Fig. 1A). Binding of His-PmrA was interrupted when unlabeled DNA was added as a specific competitor; however, similar amounts of a nonspecific competitor (an internal region of the iglC open reading frame) did not affect binding (Fig. 1B and C). Purified His-tagged MglA was unable to bind to these same promoter regions. When His-MglA and His-PmrA were added to the labeled promoter DNA, no additional shift (supershift) was observed, suggesting that these two proteins do not physically associate under these conditions (Fig. 2).
FIG. 1.
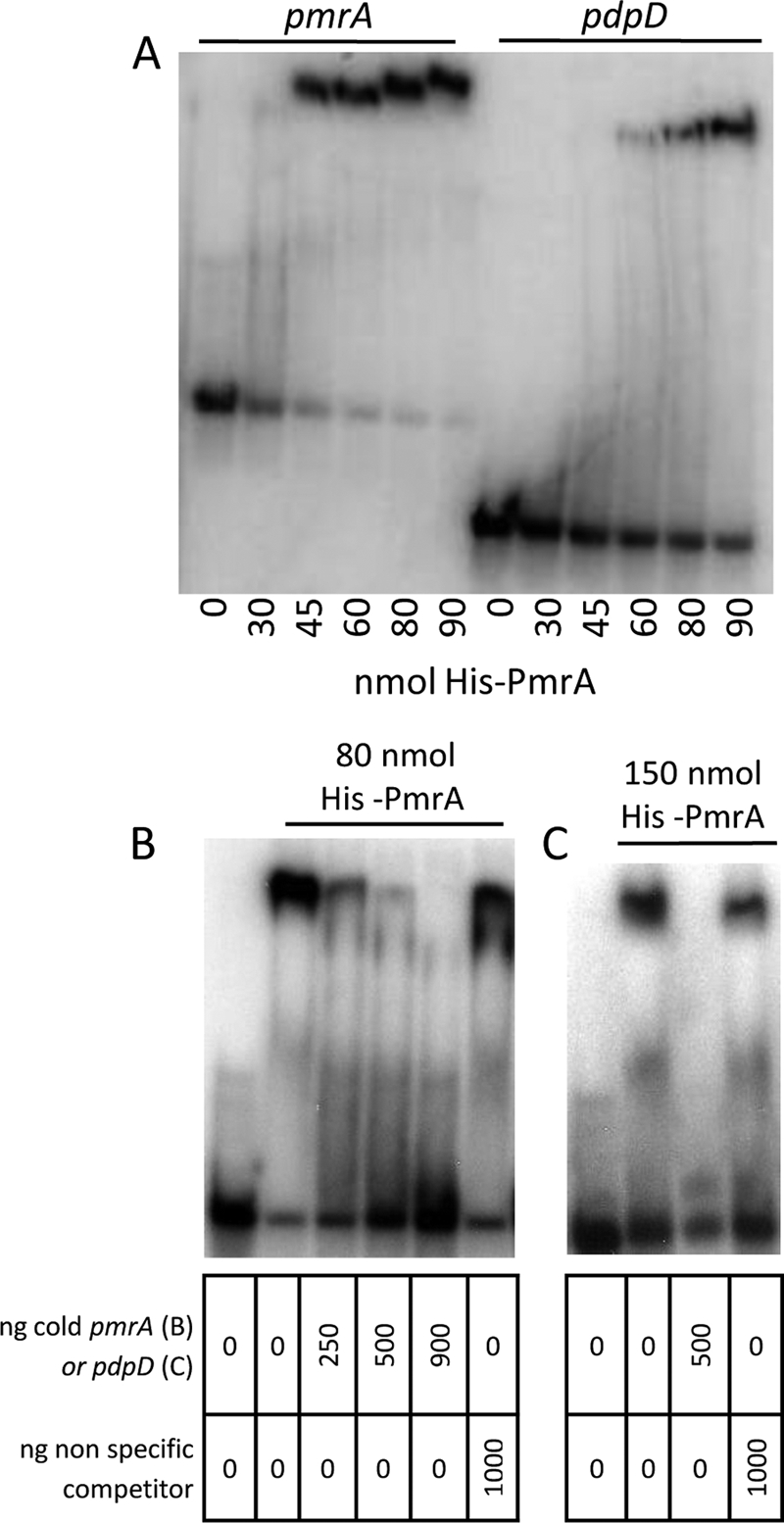
EMSA. PmrA binding. Mobility shift assays were used to determine if purified proteins bound to regulated gene promoters. (A) His-PmrA binds to the pmrA (12 ng; lanes 1 to 6) and pdpD (12 ng; lanes 7 to 12) promoters in a dose-dependent manner. (B) His-PmrA binding to the pmrA promoter is affected by specific competitor DNA but not nonspecific competitor DNA. Each lane contains 12 ng of labeled pmrA promoter. (C) His-PmrA binding to the pdpD promoter is affected by specific but not nonspecific competitor DNA. Each lane contains 12 ng of labeled pdpD promoter.
FIG. 2.
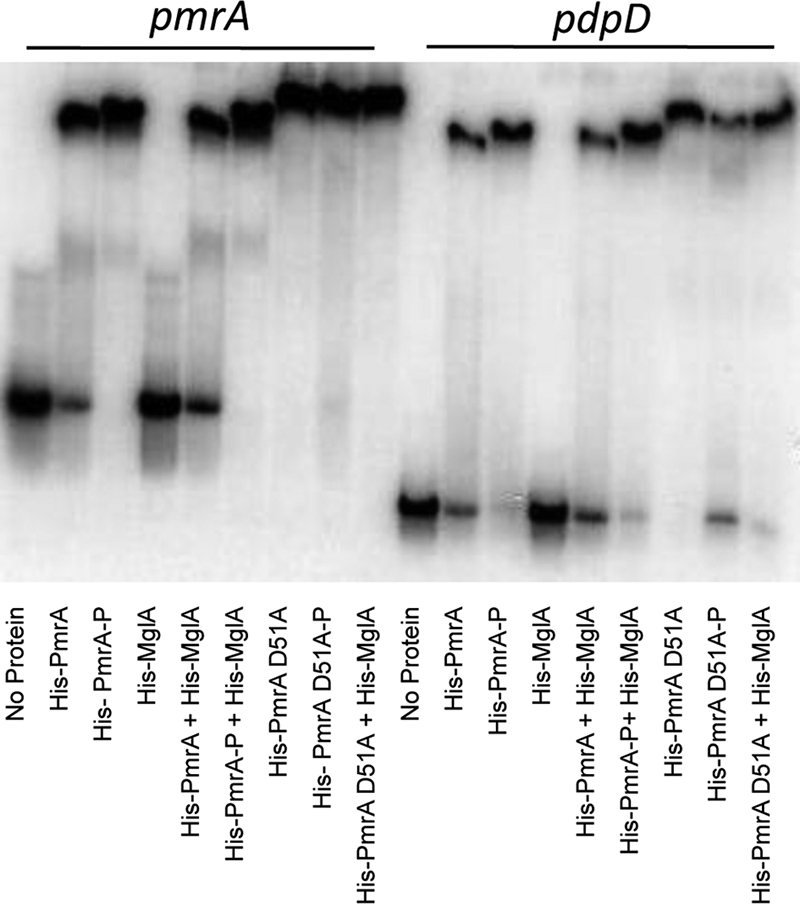
EMSA. PmrA phosphorylation. Phosphorylating His-PmrA increases binding to regulated promoters. His-MglA does not bind to either the pdpD or the pmrA promoter, the combined addition of His-PmrA and His-MglA to promoters does not result in a supershift, and His-PmrA(D51A) retains its ability to bind regulated promoters. Lanes 1 to 9, 12 ng of labeled pmrA promoter and 1 μg of each indicated protein. Lanes 10 to 18, 12 ng of labeled pdpD promoter and 150 nanomoles of each indicated protein. -P indicates that the protein was treated with acetyl phosphate.
Phosphorylation of response regulators typically results in an increase in DNA binding, providing a mechanism for regulation by phosphotransfer. We treated His-PmrA with acetyl phosphate, which has been shown to phosphorylate response regulators in vitro (18). The protein was then added to radiolabeled DNA in EMSA. Treatment of His-PmrA with acetyl phosphate resulted in a 15 to 20% increase in binding (as assessed by densitometry) versus untreated His-PmrA to the promoter regions of pmrA and pdpD (Fig. 2).
A mutation in the predicted site of PmrA phosphorylation (aspartate at position 51) was constructed, and the mutant protein was purified. The site-directed mutant protein His-PmrA(D51A) was also evaluated by EMSA for its ability to bind DNA promoters. Purified His-PmrA(D51A) retains the ability to bind to both the pmrA and the pdpD promoters. In fact, binding appears to be even more robust for His-PmrA(D51A) than that for the same amount of His-PmrA (Fig. 2). Using lower concentrations of His-PmrA(D51A) that did not result in a 100% shift of the pmrA promoter fragment, the addition of acetyl phosphate did not enhance promoter binding (data not shown).
PmrA is likely phosphorylated at aspartate 51.
To determine if D51 is the site of phosphorylation, we again utilized the site-directed mutant, His-PmrA(D51A). The enteric histidine kinase CheA has been used to phosphorylate other response regulators in vitro (30). The ability of CheA to phosphorylate His-PmrA and its ability to phosphorylate His-PmrA(D51A) were compared. CheA was autophosphorylated by incubation with [γ-32P]dATP. CheA was able to phosphorylate His-PmrA but not His-PmrA(D51A) (Fig. 3A). A duplicate SDS-PAGE gel stained for total protein showed equal amounts of His-PmrA and His-PmrA(D51A), ruling out the possibility that PmrA(D51A) was degraded during the reaction (Fig. 3B). It is important to note that His-PmrA(D51A) runs slightly faster on SDS-PAGE than does His-PmrA, even though the gene sequence is identical to that of pmrA except for the codon 51. In addition, the His tag is still present as the protein reacts with an anti-His antibody on Western blots (data not shown).
FIG. 3.

Phosphotransfer from CheA. (A) PmrA but not PmrA(D51A) is phosphorylated by the enteric histidine kinase CheA. Each lane consists of 0.1 ng phosphorylated CheA and 1 μg His-PmrA or His-PmrA(D51A). (B) Duplicate reaction mixtures were stained for total protein to demonstrate the presence of His-PmrA and His-PmrA(D51A) throughout the reaction.
To determine if PmrA is phosphorylated by Francisella kinases, we prepared a membrane fraction of F. novicida wild-type bacteria. This membrane fraction was incubated with His-PmrA or His-PmrA(D51A) in the presence of [γ-32P]dATP. Phosphorylation was detected after separation by SDS-PAGE. His-PmrA was phosphorylated, while the mutated D51A protein was not (Fig. 4A, lanes 1 and 2). Duplicate reactions were run using unlabeled ATP and probed for PmrA by Western blotting to confirm that neither His-PmrA nor His-PmrA(D51A) was lost during the reaction (Fig. 4B, lanes 3 and 4). These data, combined with the results from the CheA phosphorylation experiment, strongly suggest that the site of PmrA phosphorylation is aspartate 51.
FIG. 4.
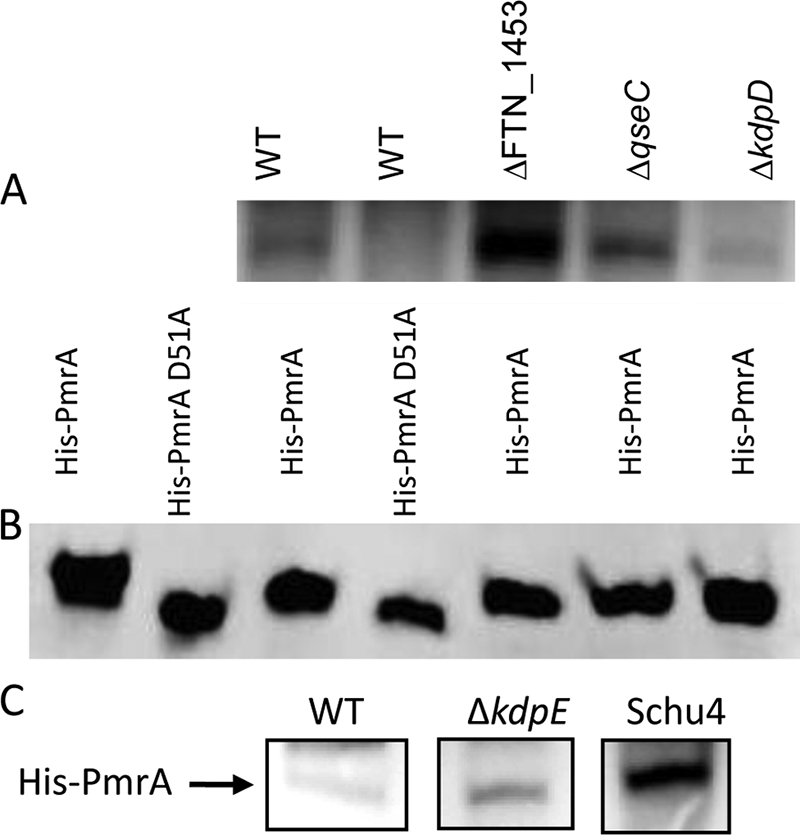
Phosphotransfer from membrane fractions. (A) Francisella membrane fractions phosphorylate His-PmrA but not His-PmrA(D51A), and the putative histidine kinase KdpD is primarily responsible for phosphorylating His-PmrA. Each lane consists of 5 μg of membrane fractions (lane designations above panel A) and 1 μg His-PmrA or His-PmrA(D51A) (lane designations directly above panel B). (B) Western blot analysis detecting PmrA in duplicate phosphotransfer reaction mixtures. (C) His-PmrA shows enhanced phosphorylation by the ΔkdpE mutant and F. tularensis Schu4 membrane fractions. WT, wild type.
PmrA D51 is important for gene regulation.
If phosphorylation of PmrA increases binding to regulated promoters, it should have a positive effect on the expression of regulated genes. Similarly, if the site of phosphorylation (D51) is removed, then the expression of regulated gene promoters should decrease. To test if phosphorylation of PmrA is important for expression of regulated genes, we performed qRT-PCR for iglC in F. novicida wild-type strains, F. novicida ΔpmrA (JSG2845) strains, F. novicida ΔpmrA strains complemented with pmrA carried on pKK214pgroEL (JSG2847), and F. novicida ΔpmrA strains complemented with pmrA(D51A) on pKK214pgroEL (JSG3033). Complemented strains produce amounts of PmrA similar to the amounts produced by the wild-type strain (data not shown). As expected, expression of iglC was almost undetectable in the ΔpmrA (JSG2845) strain compared to the wild type, and transcript levels were restored in the ΔpmrA complemented strain (JSG2847). The ΔpmrA strain complemented with PmrA(D51A) (JSG3033) had decreased expression of iglC compared to the wild type, and the strain complemented with native PmrA (JSG2847) (Fig. 5). These data indicate that expression of PmrA-regulated genes is dependent upon the aspartate at position 51, and therefore, phosphorylation of PmrA is important for gene regulation.
FIG. 5.

RT-PCR analysis of iglC in an F. novicida ΔpmrA strain complemented with PmrA or PmrA(D51A). Expression of iglC, which is within the PmrA-regulated FPI, is significantly reduced in an F. novicida ΔpmrA strain compared to wild-type (WT) bacteria. Complementation of an F. novicida ΔpmrA strain with PmrA resulted in restored expression of iglC, while complementation with PmrA(D51A) failed to restore iglC expression. *, P < 0.05.
PmrA D51 is required for intramacrophage growth.
Host macrophages have been widely described as a site of replication for Francisella bacteria (7, 8, 9, 26). Previous data demonstrated that the F. novicida ΔpmrA (JSG2845) strain is defective for replication within macrophages (20). To determine the importance of phosphorylation of PmrA on Francisella replication within macrophages, we infected PMA-induced THP-1 cells with the F. novicida wild-type and F. novicida ΔpmrA (JSG2845) strains, the F. novicida ΔpmrA strain complemented with native PmrA (JSG2847) and the F. novicida ΔpmrA strain complemented with pmrA(D51A) (JSG3033) (Table 1). As expected, the wild-type and native complement strains were capable of replication within THP-1 macrophages, increasing in number by more than 2 logs during 24 h of infection. Conversely the ΔpmrA strain and the strain complemented with the D51A mutant were largely defective for replication within macrophages. Some replication was observed for the pmrA(D51A) complemented strain, but at 24 h postinfection, the increase in CFU was less than 1/2 log (Fig. 6A). These data indicate that PmrA aspartate 51 and likely phosphorylation of PmrA are important for the replication of F. novicida within macrophages.
FIG. 6.
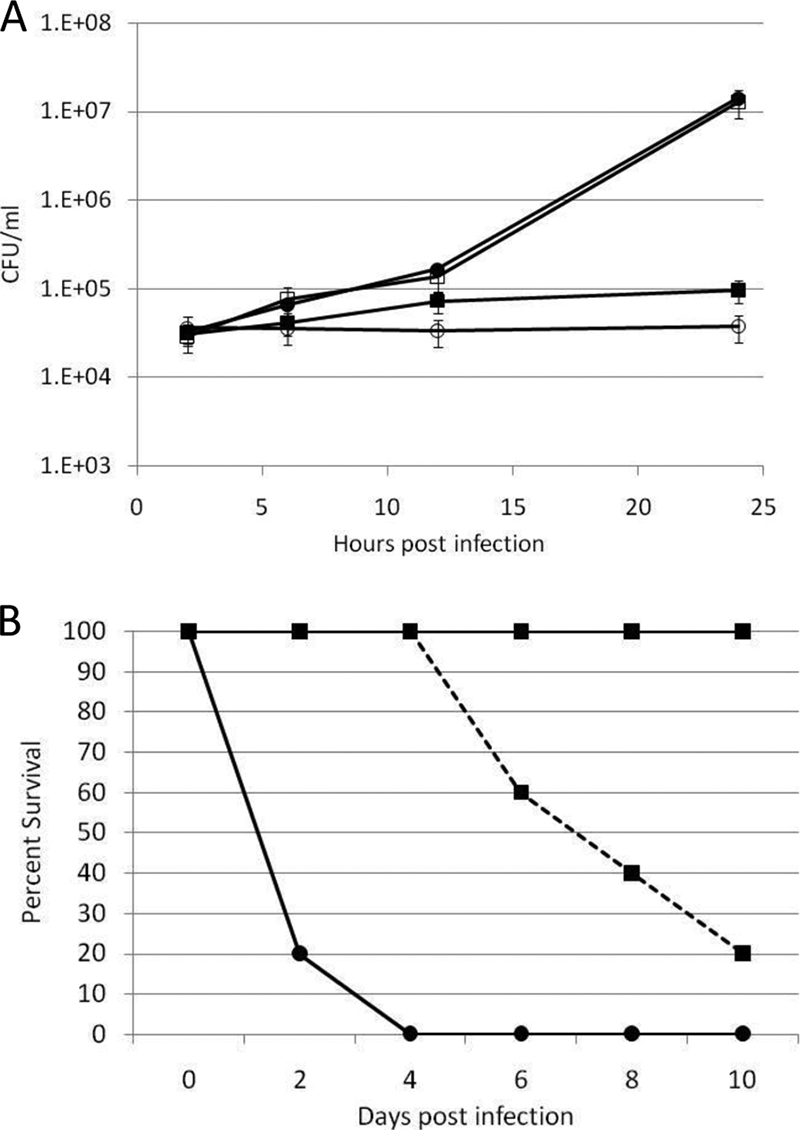
PmrA(D51A) is important for intramacrophage replication and mouse virulence. (A) PmrA D51 is required for replication within PMA-induced (10 ng/ml) THP-1 cells at a multiplicity of infection of 50:1. Closed circles, F. novicida wild type; open circles, F. novicida ΔpmrA mutant; open squares, F. novicida ΔpmrA pKK214pgroEL [pmrA]; closed squares, F. novicida ΔpmrA pKK214pgroEL [pmrA D51A]; closed triangles, F. novicida ΔkdpD strain. (B) PmrA D51 is required for maximal virulence of F. novicida in mice. Bacteria were given intranasally to groups of five anesthetized female 6- to 8-week-old BALB/c mice at a dose of 1 × 103 or 1 × 106 CFU. Closed circles, 1 × 103 F. novicida ΔpmrA pKK214pgroEL [pmrA]; closed squares with solid line, 1 × 103 F. novicida ΔpmrA pKK214pgroEL [pmrA D51A]; closed squares with dashed line, 1 × 106 F. novicida ΔpmrA pKK214pgroEL [pmrA D51A].
PmrA D51 is required for mouse virulence.
Since PmrA D51 is required for intramacrophage survival and replication within macrophages is closely tied to virulence, this residue may also be important for virulence. The lethality of the F. novicida ΔpmrA strain complemented with native PmrA (JSG2847) and the F. novicida ΔpmrA strain complemented with pmrA(D51A) (JSG3033) was compared. Groups of five BALB/c mice were infected via the intranasal route. The F. novicida ΔpmrA strain complemented with pmrA (dose of 1,000 CFU) resulted in 100% lethality within 4 days, while no mice died after receiving 1,000 CFU of the pmrA mutant complemented with pmrA(D51A). Another group of mice was infected with 106 pmrA(D51A) complemented bacteria. Eighty percent of these mice died within 10 days of infection (Fig. 6B). Our previous report showed that the F. novicida ΔpmrA strain was completely attenuated in mice at a dose of 108 CFU delivered (20). Therefore, the pmrA(D51A) complemented strain is attenuated but not to the extent of a strain that is completely lacking PmrA. These data indicate that PmrA D51 and likely the phosphorylation of PmrA are important for F. novicida virulence.
PmrA is a primary target of the histidine kinase KdpD.
The aspartate at position 51 of PmrA is important for survival within macrophages and virulence of F. novicida. Though the data suggest that this amino acid was the site of phosphotransfer from CheA and F. novicida membrane extracts, the native Francisella kinase responsible for phosphorylating PmrA at D51 has not been identified. Transposon mutants of the three putative histidine kinase genes identified (http://go.francisella.org/cgi-bin/frangb/genomelist.cgi) in the F. novicida genome, FTN1453 (JSG2890), FTN1617 (JSG2892 qseC), and FTN1715 (JSG2894 kdpD), were used to attempt to identify which kinase is responsible for phosphorylation of PmrA (10). Membrane fractions from these mutants were incubated with [γ-32P]dATP and either His-PmrA or His-PmrA(D51A). Phosphorylated proteins were separated on an SDS-PAGE gel and detected by autoradiography. Duplicate reactions were run without His-PmrA or His-PmrA(D51A) as a control to identify phosphorylated proteins unrelated to PmrA. His-PmrA was phosphorylated to some degree by each of the membrane fractions. Densitometry readings indicate that the FTN1453 mutant membrane fraction phosphorylated His-PmrA four times as much as F. novicida wild-type membranes, while the qseC mutant phosphorylated His-PmrA in a manner similar to that of the wild type. The kdpD mutant membrane preparations phosphorylated His-PmrA only 20% of the level of wild-type membranes. This suggested that the kinase primarily responsible for phosphorylation of PmrA was the histidine kinase KdpD (Fig. 4A, lanes 3 to 5). Duplicate reactions were run with unlabeled ATP and analyzed by Western blotting with anti-PmrA antisera to confirm that neither His-PmrA nor His-PmrA(D51A) was lost during the reaction and that equal amounts of target proteins were present (Fig. 4B, lanes 5 to 7). In addition, we purified the predicted C-terminal cytoplasmic portion (residues 499 to 893) of KdpD as a His-tagged protein and examined its ability to mediate phosphotransfer with PmrA. Phosphotransfer was observed, demonstrating that KdpD can phosphorylate PmrA (data not shown). The F. novicida strain containing a transposon mutation in kdpD is also deficient for intramacrophage survival (Fig. 6), thus demonstrating the expected similarity to the pmrA null mutation strain.
Further phosphorylation studies were conducted to determine the relative amounts of PmrA phosphorylation in different Francisella strains. Although there are no typical tandemly arranged genes encoding TCS in human virulent Francisella bacteria, there is one in F. novicida comprised of KdpD and KdpE. We hypothesized that in the absence of KdpE, phosphorylation of PmrA by KdpD would be increased. Indeed, using a membrane fraction from a KpdE (FTN_1714) transposon mutant (10), His-PmrA was phosphorylated 14 times more than by the F. novicida wild type (Fig. 4C). The fully virulent F. tularensis Schu4 strain does not have homologs to either kdpE or FTN1453; therefore, we hypothesized that phosphorylation of PmrA would be higher in this strain. When His-PmrA is incubated with an F. tularensis Schu4 membrane preparation in the presence of radiolabeled ATP, it was phosphorylated approximately 50 times more than that observed with F. novicida wild-type membranes (Fig. 4C).
PmrA coprecipitates with MglA and SspA.
PmrA, MglA, SspA, MigR, and FevR are required for Francisella intramacrophage survival and regulate the FPI (1, 2, 4, 6, 15). FevR has poor homology to DNA binding proteins and appears to act in a manner not involving direct promoter interaction (2). Recently, FevR (PigR) has also been demonstrated to interact with MglA and SspA, though there are conflicting data in the literature regarding this (2, 5). One hypothesis to explain the involvement of PmrA, MglA, and SspA is that PmrA binds to regulated promoters and interacts with MglA and/or SspA, which in turn recruits RNA polymerase to the site (or interacts with MglA and/or SspA bound to RNA polymerase), initiating transcription of regulated genes. For our model to be correct, this would require PmrA to physically interact with MglA and/or SspA. We showed earlier that adding MglA and PmrA together did not result in a supershift, which would have been an indication of an interaction between the two proteins (Fig. 2). This in vitro assay does not contain many of the components that would be present in vivo (e.g., SspA and RNA polymerase), so we attempted to coprecipitate PmrA with MglA and SspA from whole-cell lysates. Soluble fractions were prepared from lysates of F. novicida ΔpmrA (JSG2845), F. novicida ΔmglA (JSG2250), and F. novicida His-SspA (JSG2970) strains. His-PmrA was added to the soluble fraction of the F. novicida ΔpmrA (Fig. 7, lane 1). Once precipitated with His-bind resin, PmrA and associated proteins were detected by Western blotting. PmrA was detected by an anti-PmrA antiserum (Fig. 7A, lane 1) and by an anti-His antibody (Fig. 7C, lane 1). MglA was detected in the PmrA precipitated sample by anti-MglA anti-sera (Fig. 7B, lane 1), demonstrating that MglA coprecipitates with PmrA. Similarly, His-MglA was added to the soluble fraction from the F. novicida ΔmglA strain. Precipitation with His-bind resin allowed detection of MglA by the use of an anti-MglA antiserum (Fig. 7B, lane 2) and the anti-His antibody (Fig. 7C, lane 2). PmrA precipitated with MglA and was detected with anti-PmrA antiserum (Fig. 7A, lane 2). This confirms that PmrA and MglA coprecipitate. Finally, SspA was precipitated from the soluble fraction of the F. novicida His-SspA strain and detected with the anti-His antibody (Fig. 7C, lane 3). PmrA was present in the SspA precipitated sample (Fig. 7A, lane 3), as was MglA (Fig. 7B, lane 3). Using the identical protocol without adding His-tagged proteins did not result in immunoprecipitation of PmrA, MglA, or SspA (data not shown). Additionally, adding purified His-tagged Salmonella PhoP to an F. novicida ΔpmrA whole-cell soluble fraction and precipitating using His-bind resin did not coprecipitate MglA or SspA (data not shown). The interaction of PmrA, MglA, and SspA does not appear to be dependent on DNA because the proteins coprecipitate in the presence of DNase (data not shown). These data indicate that PmrA, MglA, and SspA are parts of the same protein complex.
FIG. 7.
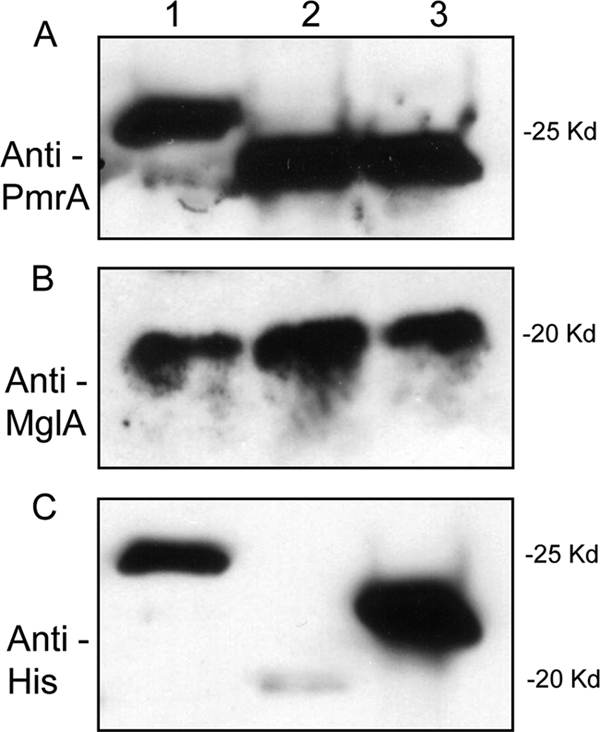
Coprecipitation. PmrA, MglA, and SspA coprecipitate. Western blot analysis of soluble lysates to which His-PmrA or His-MglA was added and precipitated with His-bind resin. (A) Anti-PmrA sera. (B) Anti-MglA sera. (C) Anti-His monoclonal antibody. Lane 1, His-PmrA added to an F. novicida ΔpmrA soluble fraction and precipitated with His-bind resin; lane 2, His-MglA added to an F. novicida ΔmglA soluble fraction and precipitated with His-bind resin; lane 3, His-SspA from F. novicida with chromosomally encoded His-tagged sspA precipitated with His-bind resin.
DISCUSSION
F. tularensis is an intracellular pathogen whose virulence is closely tied to the in vivo expression of the genes borne within a pathogenicity island. Though the regulation of this island is poorly understood, it is clear that the FPI is important for escape from the phagosome and replication within macrophages. PmrA regulates the FPI and is required for intramacrophage survival and virulence (20). MglA and SspA are also regulators of the FPI and required for virulence and FPI gene transcription by physically interacting with the RNA polymerase (6). The data presented here show that PmrA is a DNA binding protein whose interaction with DNA is enhanced by Asp51 phosphorylation mediated primarily by the histidine kinase KdpD. This aspartate residue is required for phosphorylation of PmrA, normal replication within macrophages, and full virulence. Furthermore, PmrA coprecipitates with MglA and SspA from whole-cell lysates.
Primer extension was used to determine the start of transcription for two PmrA regulated genes: pmrA and pdpD. The pdpD gene is regulated by PmrA, MglA, and SspA; however, pmrA is positively autoregulated by PmrA but not affected by MglA or SspA. The start of transcription for pdpD was identified as the thymine 32 bases upstream of its ATG. This identifies the promoter as being located in the 101-bp intergenic region between pdpD and FTN1326. Due to the lack of significant intergenic spaces, it is likely that pdpD is the lead gene of the putative operon including iglABCD. The identical 101 bases are upstream of pdpD in the fully virulent F. tularensis Schu4 strain, indicating that the start of transcription and the promoter are the same for both species. The start of transcription of pmrA is located 40 bases upstream of its ATG. There is a 441-nucleotide intergenic region upstream of pmrA to the end of FTN1466, the first 324 bases of which are identical in F. tularensis Schu4, suggesting that the pmrA promoter is conserved. The pmrA operon contains five genes, consisting of pmrA, lepB, rnc, truB, and rnr (24). To our knowledge this is the first detailed characterization of a regulated promoter in Francisella.
His-PmrA binds to the promoter fragments of both pmrA and pdpD, and treating His-PmrA with acetyl phosphate increased binding to both fragments. The pmrA promoter region required approximately 45 nanomoles of purified protein to observe a shift in labeled DNA, while the pdpD promoter region required a greater amount (∼80 nanomoles) of protein to visualize binding. This indicates that PmrA has a greater affinity for its own promoter than it does for the promoter of pdpD, which may explain why MglA and SspA are not required for pmrA activation (3, 6, 20). Perhaps this increased affinity obviates the need for interaction with MglA/SspA, which may be required to recruit RNA polymerase to lower affinity promoters. EMSA experiments performed with PmrA homologs have used similar amounts of protein to shift labeled promoter fragments (28). From scanning the fragments and the likely binding locations on these fragments, we were unable to identify a consensus binding site. The identification and characterization of additional promoters will undoubtedly aid the defining of such a sequence. Further complicating the issue was our finding that smaller, overlapping fragments of the shifted promoters did not bind His-PmrA, suggesting the involvement of multiple, nontandem binding sites (see Fig. S3 in the supplemental material). We also tested the ability of His-MglA to bind to these same promoter fragments, but no binding was observed with as much as 320 nanomoles of purified protein. A supershift was not observed when both purified His-MglA and His-PmrA were used together in these assays. This suggested either that PmrA and MglA do not physically interact or that their physical interaction is dependent upon other molecules (e.g., SspA, RNA polymerase, or FevR). Based on other data gained here and discussed below, we favor the latter hypothesis.
By amino acid alignment with known response regulators from other Gram-negative bacteria, the PmrA phosphorylation site was predicted to be D51. A His-PmrA(D51A) mutant protein was constructed to test this prediction. We demonstrated that His-PmrA but not His-PmrA(D51A) is phosphorylated by the enteric histidine kinase CheA and F. novicida wild-type membrane fractions. The His-PmrA(D51A) mutant still bound DNA, demonstrating that the substitution did not result in a nonfunctional protein. However, from our analysis, it is not known if PmrA and PmrA(D51A) bind to the same promoter DNA sequence or if the binding of His-PmrA(D51A) is specific. Regardless, His-PmrA(D51A) binding to promoter regions does not productively stimulate gene transcription (Fig. 5). The role of the putative histidine kinases in the Francisella genome in phosphorylating PmrA was also investigated. These data indicated that KdpD is the histidine kinase primarily responsible for phosphorylating PmrA; however, there appeared to be some cross talk or target promiscuity, as His-PmrA was phosphorylated to a small degree in the absence of KdpD. In enterohemorrhagic E. coli, the sensor kinase QseC has been shown to phosphorylate KdpE, demonstrating communication between the Kdp and the Qse TCS (12). A kdpD transposon mutant, like a strain carrying a mutation in its downstream target PmrA, is deficient for replication within macrophages (Fig. 6A). In addition, also like PmrA, microarray analysis comparing wild-type bacteria to the kdpD mutant indicates that KdpD regulates the PmrA operon and the FPI (data not shown) and KdpD is a virulence determinant uncovered using an in vivo negative selection screen (29). Membrane fractions missing the putative sensor kinase FTN1453 resulted in increased PmrA phosphorylation compared to the wild type, suggesting that it regulated itself or acted as a phosphatase. Interestingly, F. tularensis Schu4 has no FTN1453 homolog.
Growth of a pmrA null strain complemented with pmrA(D51A) within macrophages was diminished compared to a strain complemented with native pmrA; however, some replication of the pmrA(D51A) complemented strain was observed compared to a pmrA null mutant. Similarly, the PmrA(D51A) complemented strain was more virulent in the mouse model than were strains devoid of PmrA. This is likely a result of PmrA(D51A) overexpression such that increased copy number coupled with the residual PmrA(D51A) DNA binding resulted in modest expression of the PmrA regulon, resulting in some replication within macrophages and retention of virulence.
Coimmunoprecipitation experiments reported here showed that PmrA, MglA, and SspA are a part of the same protein complex. In addition to the F. novicida data presented, PmrA also coprecipitates with MglA and SspA from an F. tularensis Schu4 whole-cell soluble fraction (data not shown). This was predicted because the amino acid sequences of MglA and SspA are nearly identical between F. novicida and F. tularensis Schu4, and the PmrA protein sequence is identical between these subspecies. The data presented, however, do not indicate whether PmrA binds to MglA, SspA, or both proteins. We predict that RNA polymerase is a part of this complex; however, other binding partners/complex components may also exist. We did not examine the interaction of FevR (PigR) with PmrA, as there is conflicting data in the literature concerning the likelihood of its function as a DNA binding protein or its ability to interact with MglA/SspA. The most recent data do suggest a physical interaction of FevR (PigR) with MglA/SspA that is facilitated by the alarmone ppGpp and thus present potential new interacting partners. The sequence of protein binding for initiating transcription is also unclear. PmrA does bind promoters in the absence of MglA and SspA, but the EMSA data suggest that MglA will not interact with unbound or DNA-bound PmrA in vitro without the presence of other factors. MglA may need to interact with SspA before it can bind PmrA, or Francisella RNA polymerase may need to interact with MglA and/or SspA before binding to PmrA.
The response regulator PmrA is the first characterized DNA binding protein for Francisella. Phosphorylation of PmrA enhances promoter binding, affecting gene regulation. PmrA is phosphorylated at Asp51 primarily by the putative histidine kinase KdpD, forming the first TCS described for Francisella species. The KdpD/PmrA TCS plays an important role in regulating the FPI, affecting replication in macrophages and virulence. Phosphorylation of PmrA is complicated in F. novicida by the presence of a secondary KdpD target, KdpE, and an additional kinase, FTN1453, both of which negatively affect the phosphorylation state of PmrA. Likely due to the absence of FTN1453 and KdpE in the fully virulent F. tularensis Schu4 strain, phosphorylation of PmrA was increased, which may result in comparatively higher expression of the PmrA regulon. Further investigation into the protein-protein interactions of factors at FPI promoters, as well as PmrA-regulated expression of FPI genes in F. tularensis, will provide further insight into the molecular pathogenesis of tularemia.
Supplementary Material
Acknowledgments
We thank Daniel Wozniak for providing purified CheA and Denise Monack of Stanford University for generously providing the F. novicida His-sspA strain. Mike Zianni and the Plant Microbe Genetics Facility at The Ohio State University were instrumental in performance of the primer extension experiments.
This work was supported by funding from The Region V “Great Lakes” Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (NIH award 1-U54-AI-057153) and by NIH/NIAID award 1-T32-AI-065411, an NRSA training grant administered by the Center for Microbial Interface Biology at The Ohio State University.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 15 March 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Baron, G. S., and F. E. Nano. 1998. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol. Microbiol. 29:247-259. [DOI] [PubMed] [Google Scholar]
- 2.Brotcke, A., and D. M. Monack. 2008. Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect. Immun. 76:3473-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brotcke, A., D. S. Weiss, C. C. Kim, P. Chain, S. Malfatti, E. Garcia, and D. M. Monack. 2006. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 74:6642-6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchan, B. W., R. L. McCaffrey, S. R. Lindemann, L. A. Allen, and B. D. Jones. 2009. Identification of migR, a regulatory element of the Francisella tularensis live vaccine strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect. Immun. 77:2517-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charity, J. C., L. T. Blalock, M. M. Costante-Hamm, D. L. Kasper, and S. L. Dove. 2009. Small molecule control of virulence gene expression in Francisella tularensis. PLoS Pathog. 10:e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charity, J. C., M. M. Costante-Hamm, E. L. Balon, D. H. Boyd, E. J. Rubin, and S. L. Dove. 2007. Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 3:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clemens, D. L., B. Y. Lee, B., and M. A. Horwitz. 2005. Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect. Immun. 73:5892-5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis, J., P. C. Oyston, M. Green, and R. W. Titball. 2002. Tularemia. Clin. Microbiol. Rev. 15:631-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fortier, A. H., S. J. Green, T. Polsinelli, T. R. Jones, R. M. Crawford, D. A. Leiby, K. L. Elkins, M. S. Meltzer, and C. A. Nacy. 1994. Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol. Ser. 60:349-361. [PubMed] [Google Scholar]
- 10.Gallagher, L. A., E. Ramage, M. A. Jacobs, R. Kaul, M. Brittnacher, and C. Manoil. 2007. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. U. S. A. 104:1009-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavrilin, M. A., I. J. Bouakl, N. L. Knatz, M. D. Duncan, M. W. Hall, J. S. Gunn, and M. D. Wewers. 2006. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1β processing and release. Proc. Natl. Acad. Sci. U. S. A. 103:141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Gunn, J. S., E. L. Hohmann, and S. I. Miller. 1996. Transcriptional regulation of Salmonella virulence: a PhoQ periplasmic domain mutation results in increased net phosphotransfer to PhoP. J. Bacteriol. 178:6369-6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes, D. T., M. B. Clarke, K. Yamamoto, D. A. Rasko, and V. Sperandio. 2009. The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog. 5:e1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keim, P., A. Johansson, and D. M. Wagner. 2007. Molecular epidemiology, evolution, and ecology of Francisella. Ann. N. Y. Acad. Sci. 1105:30-66. [DOI] [PubMed] [Google Scholar]
- 14.Lai, X. H., I. Golovliov, and A. Sjostedt. 2004. Expression of IglC is necessary for intracellular growth and induction of apoptosis in murine macrophages by Francisella tularensis. Microb. Pathog. 37:225-230. [DOI] [PubMed] [Google Scholar]
- 15.Lauriano, C. M., J. R. Barker, S. S. Yoon, F. E. Nano, B. P. Arulanandam, D. J. Hassett, and K. E. Klose. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. U. S. A. 101:4246-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, L. A., M. R. Zianni, and F. R. Tabita. 1999. Inactivation of the monocistronic rca gene in Anabaena variabilis suggests a physiological ribulose bisphosphate carboxylase/oxygenase activase-like function in heterocystous cyanobacteria. Plant Mol. Biol. 40:467-478. [DOI] [PubMed] [Google Scholar]
- 17.Ludu, J. S., O. M. de Bruin, B. N. Duplantis, C. L. Schmerk, A. Y. Chou, K. L. Elkins, and F. E. Nano. 2008. The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 190:4584-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCleary, W. R., and J. B. Stock. 1994. Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 269:31567-31572. [PubMed] [Google Scholar]
- 19.Meibom, K. L., A. L. Forslund, K. Kuoppa, K. Alkhuder, I. Dubail, M. Dupuis, A. Forsberg, and A. Charbit. 2009. Hfq, a novel pleiotropic regulator of virulence associated genes in Francisella tularensis. Infect. Immun. 77:1866-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohapatra, N. P., S. Soni, B. L. Bell, R. Warren, R. K. Ernst, A. Muszynski, R. W. Carlson, and J. S. Gunn. 2007. Identification of an orphan response regulator required for the virulence of Francisella spp. and transcription of pathogenicity island genes. Infect. Immun. 75:3305-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nano, F. E., N. Zhang, S. C. Cowley, K. E. Klose, K. K. Cheung, M. J. Roberts, J. S. Ludu, G. W. Letendre, A. I. Meierovics, G. Stephens, and K. L. Elkins. 2004. A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186:6430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nigrovic, L. E., and S. L. Wingerter. 2008. Tularemia. Infect. Dis. Clin. North Am. 22:489-504. [DOI] [PubMed] [Google Scholar]
- 23.Oyston, P. C., A. Sjostedt, and R. W. Titball. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2:967-978. [DOI] [PubMed] [Google Scholar]
- 24.Sammons-Jackson, W. L., K. McClelland, J. N. Manch-Citron, D. W. Metzger, C. S. Bakshi, E. Garcia, A. Rasley, and B. E. Anderson. 2008. Generation and characterization of an attenuated mutant in a response regulator gene of Francisella tularensis live vaccine strain (LVS). DNA Cell. Biol. 27:387-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santic, M., M. Molmeret, K. E. Klose, and Y. Abu Kwaik. 2006. Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 14:37-44. [DOI] [PubMed] [Google Scholar]
- 26.Sjöstedt, A. 2006. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 8:561-567. [DOI] [PubMed] [Google Scholar]
- 27.Stock, A. M., V. L. Robinson, and P. N. Goudreau. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183-215. [DOI] [PubMed] [Google Scholar]
- 28.Tamayo, R., A. M. Prouty, and J. S. Gunn. 2005. Identification and functional analysis of Salmonella enterica serovar Typhimurium PmrA-regulated genes. FEMS Immunol. Med. Microbiol. 43:249-258. [DOI] [PubMed] [Google Scholar]
- 29.Weiss, D. S., A. Brotcke, T. Henry, J. J. Margolis, K. Chan, and D. M. Monack. 2007. In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U. S. A. 104:6037-6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitchurch, C. B., T. E. Erova, J. A. Emery, J. L. Sargent, J. M. Harris, A. B. Semmler, M. D. Young, J. S. Mattick, and D. J. Wozniak. 2002. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J. Bacteriol. 184:4544-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.