

Abstract
The Epstein-Barr virus (EBV) origin of plasmid replication (OriP) is required for episome stability during latent infection. Telomere repeat factor 2 (TRF2) binds directly to OriP and facilitates DNA replication and plasmid maintenance. Recent studies have found that TRF2 interacts with the DNA damage checkpoint protein Chk2. We show here that Chk2 plays an important role in regulating OriP plasmid stability, chromatin modifications, and replication timing. The depletion of Chk2 by small interfering RNA (siRNA) leads to a reduction in DNA replication efficiency and a loss of OriP-dependent plasmid maintenance. This corresponds to a change in OriP replication timing and an increase in constitutive histone H3 acetylation. We show that Chk2 interacts with TRF2 in the early G1/S phase of the cell cycle. We also show that Chk2 can phosphorylate TRF2 in vitro at a consensus acceptor site in the amino-terminal basic domain of TRF2. TRF2 mutants with a serine-to-aspartic acid phosphomimetic substitution mutation were reduced in their ability to recruit the origin recognition complex (ORC) and stimulate OriP replication. We suggest that the Chk2 phosphorylation of TRF2 is important for coordinating ORC binding with chromatin remodeling during the early S phase and that a failure to execute these events leads to replication defects and plasmid instability.
Epstein-Barr virus (EBV) is a human herpesvirus that can persist as a multicopy episome in latently infected B lymphocytes (16, 30). During latent infection, the viral genomes replicate once per cell cycle and utilize the cellular machinery required for chromosomal replication (6, 10, 22, 31, 32, 41, 43). One virally encoded protein, EBNA1, is required for OriP-dependent DNA replication and plasmid maintenance (21, 44; reviewed in references 22 and 38). EBNA1 binds to a family of repeats (FR) and a dyad symmetry element (DS) within OriP (29). The FR confers plasmid maintenance and enhances replication initiation from the DS. The DS consists of two pairs of EBNA1 binding sites flanked by telomere repeat factor (TRF) binding sites (2, 9, 19, 42). The DS has been shown to function similarly to cellular origins that replicate once per cell cycle and assemble a prereplication complex that includes the cellular origin recognition complex (ORC) and minichromosome maintenance (MCM) subunits (6, 10, 31, 32).
ORC recruitment to the DS is thought to play a key role in determining its function as an origin of DNA replication (3, 14). TRF2, along with EBNA1, was shown previously to interact with ORC subunits and to recruit the ORC to the DS (1, 23, 27, 39). The ORC interaction domains in both TRF2 and EBNA1 appear to share an amino acid composition of basic residues, often referred to as arginine-glycine-glycine (RGG) motifs (17). The basic stretches have been implicated in the intermolecular linking activity of EBNA1 and are variably termed linking region 1 (LR1) and LR2 (25). LR1 and LR2 have AT-hook DNA binding activity and confer a metaphase chromosome attachment function to EBNA1, which are thought to be critical for episome maintenance during nuclear membrane breakdown (26, 33). The EBNA1 RGG motifs can also bind RNA (24, 35). Recent studies have shown that both EBNA1 and TRF2 RGG motifs can bind to G-rich RNA capable of forming G-quadruplex structures and that RNA facilitates interactions between these RGG motifs and ORC subunits (27). Although EBNA1 can bind its own RNA in vitro, the identities of the in vivo-bound RNA molecules and their precise function in regulating ORC recruitment or function remain unknown.
Origins of replication are thought to be regulated by cell cycle and DNA damage checkpoint mechanisms (12). A previous study indicated that EBV OriP is subject to a checkpoint regulation that prevents its firing in early S phase (46). This delay in replication firing was dependent on TRF2 and the ability of TRF2 to recruit histone deacetylases (HDACs). Nucleosomes positioned adjacent to TRF2 binding sites at the DS were subject to deacetylation in early S phase (45). The acetylation of these nucleosomes occurred in the mid- to late S phase, when DNA synthesis at OriP could be detected. Based on these observations, we proposed that OriP was subject to checkpoint regulation. Furthermore, treatment with hydroxyurea (HU), which is known to destabilize EBV episomes, disrupted this cell cycle delay in OriP replication (36). It remains unclear how this delay in DNA replication is coordinated with other cell cycle controls and how this might confer genetic stability to EBV episomes.
Origin firing and replication timing in lower eukaryotes can be regulated by checkpoint kinases. Rad53, the budding-yeast orthologue of Chk2, has been implicated in the control of replication timing, since mutations in Rad53 cause late-firing origins to replicate early (34). Similarly, HU treatment leads to an increase in the number of early-firing replication origins in Saccharomyces cerevisiae and humans, although the mechanism regulating this remains controversial (13, 18, 46). Recent studies have shown that Chk2 can bind to TRF2 and that TRF2 can downregulate Chk2 activity (5). Based on these observations, we explored the potential role of Chk2 in the regulation of EBV OriP replication.
MATERIALS AND METHODS
Cell culture.
EBV-positive Raji Burkitt lymphoma cells were maintained in RPMI medium supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate (Cellgro). D98/HR1 cells (EBV-positive [EBV+] adherent cells) and HeLa cells (EBV-negative [EBV−] adherent cells) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate (Cellgro). HCT116 cells was grown in McCoy 5A medium supplemented with 10% fetal bovine serum and antibiotics in a 5% CO2 incubator at 37°C. Hydroxyurea (Sigma) was used at 50 μM. The Chk2 inhibitor debromohymenialdisine (DBH) (7) was purchased from Alexis Biochemicals and used at concentrations recommended by the manufacturer. HeLa, HCT116, and D98/HR1 cells were transfected with Lipofectamine 2000 (Invitrogen, Inc.).
Antibodies.
Rabbit polyclonal anti-EBNA1 and -TRF2 were raised against recombinant full-length EBNA1 and TRF2. Rabbit IgG (Santa Cruz), polyclonal ORC2 (BD Pharmingen), polyclonal acetylated histone H3 (AcH3) (Upstate), polyclonal MCM3 (Abcam), actin (Sigma), EBNA1 (Advanced Biotechnologies, Inc.), and Chk2 (Santa Cruz Biotech) were used according to the manufacturers' suggestions.
Plasmids and siRNA.
The oriP plasmid wild type (wt) was described previously and consists of oriP sequences, EBNA1, enhanced green fluorescent protein (GFP) (eGFP), and hygromycin genes as a pREP10 (Invitrogen) derivative (9). FLAG-TRF2 S20A and S20E mutants were generated by site-directed mutagenesis of the pCMV-3xFLAG-TRF2 wild type. Glutathione S-transferase (GST)-TRF2 and its truncation mutants were generated as described previously (1). Further details with regard to plasmids are available upon request. siChk2 was purchased as SmartPools from Dharmacon, Inc., and siCon was nontargeting small interfering RNA (siRNA) from Dharmacon (catalog number D-001810-01). siRNAs were introduced into cells by use of DharmaFECT 1 reagent according to the manufacturer's recommendations. Briefly, ∼3 × 106 cells were plated in antibiotic-free medium onto 10-cm plates 12 to 16 h prior to transfection. Cells were transfected twice within 24 h with a 100 nM final concentration of siRNA, and the transfected cells were cultured and continuously transfected every 3 days. Plasmid DNA and siRNA cotransfections were performed by use of Lipofectamine 2000 reagent (Invitrogen) using 2 to 5 μg of plasmid DNA and a 100 nM final concentration of siRNA for 1.5 × 106 cells, which were seeded onto 6-cm plates 12 to 16 h prior to transfection.
Genome maintenance assay.
Cells (approximately 1 × 106 cells per sample) were collected and resuspended in 100 μl SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris [pH 8.0]). After a brief sonication, immunoprecipitation (IP) dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris [pH 8.0], 167 mM NaCl) was added to 1 ml and then incubated with proteinase K for 2 to 3 h at 50°C. Three hundred microliters was removed and subjected to phenol-choloroform extraction and ethanol precipitation. Precipitated DNA was then assayed by real-time PCR using primers for the DS region of EBV and normalized by the cellular DNA signal at the actin gene locus.
Replication timing assays.
Replication timing assays were described previously (46). Briefly, cells were incubated with 50 μM bromodeoxyuridine (BrdU) for 30 min, stained with propidium iodide (PI), and subjected to fluorescence-activated cell sorting (FACS), where they were separated into cell cycle (G1, S1, S2, S3, S4, and G2/M) fractions. DNA was extracted by SDS-protease K digestion followed by phenol-chloroform extraction and isopropanol precipitation. DNA was sonicated to an average 700-bp fragment length, heat denatured, and then subjected to immunoprecipitation with anti-BrdU (stock, 25 μg/ml; BD Pharmingen) combined with rabbit anti-mouse IgG antibody (Sigma). Immunoprecipitated DNA was extracted and quantified by real-time PCR using the standard-curve method and normalized to the input of each fraction. The data were then normalized to the total signal (from G1 and S1 to S4 to G2/M and set as 100%). At least three independent IPs were performed for each data point. The error bars represent standard deviations from three real-time PCRs from the three IP experiments.
Primers used for real-timer PCR were as follows: forward primer ATGTAAATAAAACCGTGACAGCTCAT and reverse primer TTACCCAACGGGAAGCATATG for the DS, forward primer GCCATGGTTGTGCCATTACA and reverse primer GGCCAGGTTCTCTTTTTATTTCTG for actin, and forward primer GTGCACAGCGCCAGGTTA and reverse primer GTGCACAGCGCCAGGTTA for lamin B2.
Kinase assay.
Purified human Chk2 was purchased as a recombinant protein expressed and purified from baculovirus-infected Sf9 cells (Invitrogen). GST fusion proteins were expressed in Escherichia coli cells and purified by glutathione-Sepharose and glutathione (10 mM) elution. Eluted proteins were dialyzed into D100 buffer at a concentration of ∼0.4 mg/ml. Kinase reaction mixtures included 50 nM Chk2, 10 nM GST fusion protein, 5 mM MgCl2, 100 mM KCl, 10 mM Tris-Cl (pH 7.5), 1 mM dithiothreitol (DTT), 10 μM ATP, and 0.1 nM [γ-32P]ATP. Kinase reaction mixtures were incubated for 30 min at 30°C. Reactions were then analyzed by SDS-PAGE and autoradiography.
Additional methods.
Transient DNA replication assays were described previously (8, 9). Chromatin IP (ChIP) assays were performed as described previously (45). Centrifugal elutriation was performed as described previously (11, 45). Cell cycle progression and length of S phase were measured by BrdU pulse-labeling combined with propidium iodide staining as described previously (4). The RNA electrophoretic mobility shift assay (EMSA) and ORC2 recruitment assay were described previously (27). Quantifications of data from EMSA and kinase assays were performed by PhosphorImager and ImageQuant analyses. For EMSA, the percent bound was calculated as the fraction of probe bound relative to the free probe for each lane. For GST pulldown assays, (see Fig. 6B), 0.5 μg of purified GST and GST-TRF2 derivatives was prebound to 30 ml of glutathione-Sepharose and then incubated with 0.5 ml of HeLa nuclear extract (∼5 mg/ml). HDAC activity was measured as described previously (46) by using a kit from BioVision according to the manufacturer's instructions.
FIG. 6.

TRF2 phosphomimetic mutant S20E abrogates RNA binding, ORC recruitment, and OriP DNA replication. (A) GST, GST-TRF2(1-90), GST-TRF2(1-90)(S20A), and GST-TRF2(1-90)(S20E) were assayed by EMSA for RNA binding to a G-rich probe, (UUAGGG)8 (left), or a C-rich probe, (CCCUAA)8 (right). (B) GST, GST-TRF2(1-90), GST-TRF2(1-90)(S20A), and GST-TRF2(1-90)(S20E) were incubated with HeLa nuclear extracts (0.5 ml at 5 mg/ml), washed extensively, and then assayed by SDS-PAGE for the recruitment of ORC2 (top) or control PCNA (middle). Loading controls for GST (bottom) or Coomassie staining is indicated below. Std, standard. (C) Transient DNA replication assays were performed with HCT116 cells with an OriP plasmid coexpressing the EBNA1 protein. Cells were cotransfected with a cytomegalovirus (CMV)-Flag vector or CMV-FLAG-TRF2(FL) or the S20A, S20E, or ΔB mutant and then assayed at 72 h posttransfection for OriP replication. Recovered plasmid DNA was digested with DpnI plus BamHI (top) or BamHI alone (bottom) and assayed by Southern blotting. (D) Quantification of at least three independent transfection assays represented in C. (E) Western blot analysis of FLAG-TRF2 proteins (anti-FLAG) transfected for the replication assays shown in C. EBNA1 and actin served as transfection and loading controls, respectively.
RESULTS
Chk2 depletion inhibits OriP DNA replication and episome maintenance.
To test the hypothesis that Chk2 regulates OriP function, we first assayed transient DNA replication of OriP plasmids in cells where Chk2 was depleted by siRNA (Fig. 1A to C). Transient DNA replication was assayed in HCT116 cells transfected with OriP plasmids that coexpress the EBNA1 protein. Replicated DNA can be detected by Southern blot analysis of DpnI-resistant plasmid DNA recovered 72 h posttransfection. We found that siChk2 depletion caused a ∼4-fold reduction in DNA replication relative to that of control siRNA-transfected cells (Fig. 1A and B). Chk2 depletion was confirmed by Western blotting with antibody to Chk2 (Fig. 1C). Chk2 depletion did not alter EBNA1 or TRF2 expression (Fig. 1C), which could potentially account for an indirect effect of Chk2 depletion on OriP replication. These findings suggest that Chk2 contributes to OriP replication activity.
FIG. 1.

Chk2 depletion inhibits OriP DNA replication and plasmid maintenance. (A) HCT116 cells were cotransfected with siControl or siChk2 siRNA and with OriP plasmid-coexpressing EBNA1 and assayed for transient DNA replication. Plasmid DNA, shown in duplicates, was digested with BamHI (bottom) or DpnI plus BamHI (bottom) and visualized by Southern blotting. (B) Replication efficiency was quantified for at least three independent transfections as the ratio of resistant DpnI to total plasmid DNA recovered (BamHI-linearized DNA), and replication for siCon was normalized to 100%. (C) Western blot control to assay siChk2 depletion and EBNA1, TRF2, and actin expression levels in cells shown in A. (D) HeLa cells were selected for GFP-expressing OriP plasmids and then treated with (+) or without (−) HU (50 μM) for 4 or 15 days, as indicated. OriP plasmids were then extracted from the same number of cells and assayed by Southern blotting. (E) Same as above (D) except that cells were transfected with control or Chk2-targeting siRNA every 3 days for 4 or 15 days. No HU was included in these experiments. (F) The siRNA-transfected cells shown in E were analyzed by Western blotting for Chk2 (top) or actin (bottom) at 4 and 15 days after transfection of the OriP plasmid, as indicated. (G) Raji cells were nucleofected with siRNA for Chk2 or control siRNA every third day for 6 days. The EBV genome copy number was measured by real-time PCR analysis of the EBV DS region relative to cellular actin DNA. qPCR, quantitative PCR.
Chk2 depletion was also assayed for its effect on OriP plasmid maintenance (Fig. 1D and E). OriP plasmids were transfected into HeLa cells and then assayed at 4 or 15 days posttransfection by Southern blot analysis for plasmid maintenance (Fig. 1D and E). As a control, we treated cells with 50 μM HU, which was shown previously to promote the loss of OriP plasmids (Fig. 1D). HU caused an ∼80% reduction in OriP plasmid maintenance by day 15. No apparent change in the level of total OriP plasmids was observed by day 4, indicating that similar levels of OriP plasmids were used for the plasmid maintenance assay in the presence or absence of HU. A similar loss of OriP plasmid maintenance was observed for cells transfected with Chk2 siRNA but not for cells transfected with control siRNA by day 15 (Fig. 1E). The depletion of Chk2 was monitored by Western blotting at day 4 (Fig. 1F, left) and day 15 (Fig. 1F, right). Chk2 depletion was also assayed for its effect on EBV genome copy numbers with the Raji Burkitt lymphoma cell line (Fig. 1G). We found that siChk2 produced a ∼50% loss of EBV genome copy numbers at 6 days post-siRNA transfection relative to the siControl. These results suggest that Chk2 depletion leads to the loss of OriP plasmids and a reduction in EBV genome copy numbers in latently infected cells.
Chk2 depletion advances the replication timing of OriP.
Previous studies have found that OriP replication is delayed in S phase and that this delay correlates with episome stability. To determine if Chk2 was important for this cell cycle regulation, we assayed the replication timing of EBV genomes in D98/HR1 cells after siRNA depletion of Chk2 (Fig. 2A). D98/HR1 cells were used for these experiments because they carry EBV episomes and can be transfected more efficiently than lymphoblastoid cell lines. As previously observed, in control siRNA-transfected cells, EBV DNA replicated in mid- to late S phase (S3 and S4) (Fig. 2B, left), while lamin B of cellular origin replicated in early S phase (S1) (Fig. 2C, left). In contrast, in cells transfected with siChk2 RNA, EBV replication was enriched in earlier-S-phase fractions (S1 and S2) (Fig. 2B, right), while there was no change in lamin B replication timing (Fig. 2C, right). These findings indicate that the siRNA depletion of Chk2 disrupts the normal delay in replication timing of EBV DNA in latently infected D98/HR1 cells.
FIG. 2.

Chk2 depletion or Chk2 inhibition by DBH alters OriP replication timing and function. (A) D98/HR1 cells were transfected with siControl or siChk2 RNA and assayed by Western blotting to verify that Chk2 was depleted at 72 h posttransfection. (B and C) siControl- or siChk2-transfected D98/HR1 cells were pulse-labeled with BrdU, stained with PI, and then fractionated into different cell cycle stages by using FACS. BrdU-labeled DNA specific for the DS (B) or cellular lamin B2 (C) was quantified by anti-BrdU immunoprecipitation and PCR. Cell cycle fractions are indicated. (D and E) A replication timing assay was performed with D98/HR1 cells treated with DBH or control DMSO for 48 h prior to BrdU pulse-labeling, FACS, and IP-PCR analysis. BrdU incorporation was assayed at the EBV DS (D) or at the cellular lamin B2 origin (E). (F) EBV genome copy numbers were assayed in D98/HR1 cells treated with DBH or the DMSO control after 6 days of treatment. The copy number was measured by real-time PCR analysis of the EBV DS relative to cellular actin DNA.
The Chk2 kinase inhibitor DBH advances replication timing of EBV.
To determine whether the kinase activity of Chk2 might be involved in these activities, we tested the effects of DBH on the replication timing and episome maintenance of EBV (Fig. 2D to F). DBH is a pharmacological inhibitor of Chk2 and, to a lesser extent, Chk1 but does not inhibit ATM or ATR kinases (7). D98/HR1 cells were treated with DBH or control dimethyl sulfoxide (DMSO) and then assayed for replication timing (Fig. 2D and E). We found that DS replication occurred in the S3 and S4 fractions in DMSO-treated cells (Fig. 2D, left) but was advanced to the S2 in DBH-treated cells (Fig. 2D, right). DBH treatment did not have any detectable effect on control lamin B replication timing (Fig. 2E). These findings suggest that Chk2 kinase activity is important for the regulation of the replication timing of OriP. To explore whether Chk2 kinase activity was also important for EBV genome maintenance, we assayed the effects of DBH treatment on EBV copy numbers in Raji Burkitt lymphoma cells. We found that DBH treatment for 6 days led to a partial (∼42%) loss of EBV genomes relative to DMSO control-treated cells (Fig. 2F). This suggests that Chk2 kinase activity is important for EBV genome maintenance and copy number control.
Chk2 depletion alters histone deacetylation at DS.
Previous studies have shown that nucleosomes positioned at the DS undergo cell cycle-dependent histone deacetylation, which is correlated with origin activation (45). We therefore examined whether histone H3 acetylation was altered in cells depleted of Chk2 (Fig. 3). D98/HR1 cells were transfected with siChk2 or siControl and then assayed by ChIP assays for the binding of EBNA1, TRF2, acetyl histone H3, or ORC2 at the EBV DS (Fig. 3A) or the control OriLyt region (Fig. 3B). We found that siChk2 had no significant effect on EBNA1 binding to the DS. However, Chk2 depletion caused a ∼4.5-fold increase in histone H3 acetylation at the DS, with no detectable change at OriLyt. We also observed a 2-fold increase in TRF2 binding at the DS, consistent with the recent finding that Chk2 phosphorylation of TRF2 can inhibit TRF2 DNA binding (5). These findings suggest that Chk2 can alter both TRF2 binding and histone H3 acetylation at the DS. Since HDACs 1 and 2 can interact with TRF2, we assayed whether Chk2 depletion altered the binding of these proteins to TRF2. HDAC2 binding to TRF2 was readily detected in TRF2 immunoprecipitations, but the binding was not altered by Chk2 depletion (Fig. 3C). However, we did observe that Chk2 depletion lead to a decrease in total HDAC activity relative to that of control siRNA-transfected cells (Fig. 3D). These results suggest that Chk2 may regulate histone acetylation at OriP through its effect on TRF2 DNA binding as well as through a global effect on HDAC activity.
FIG. 3.

Chk2 depletion abrogates histone deacetylation at the DS. (A and B) D98/HR1 cells were transfected with siControl or siChk2 RNA and then assayed by ChIP for binding of EBNA1, AcH3, TRF2, ORC2, or control IgG at EBV regions for the DS (A) or OriLyt (B). ChIP DNA was quantified by real-time PCR as a relative percentage of the input. (C) D98/HR1 cells were transfected with siChk2 (+) or siControl (−) and then assayed for HDAC2 abundance by Western blotting (input) or after immunoprecipitation with IgG control or anti-TRF2 antibody, as indicated. Control Western blots for TRF2 are indicated at the bottom. The asterisk indicates a cross-reacting IgG band from the immunoprecipitating antibody. (D) HDAC activity assay from total cell extracts derived from siControl- or siChk2-transfected D98/HR1 cells.
Chk2 associates with TRF2 in a cell cycle-dependent manner.
Recent studies have shown that TRF2 can interact directly with Chk2 (5). Since Chk2 appears to regulate the cell cycle control of EBV replication timing, we tested whether the interaction of Chk2 with TRF2 was cell cycle dependent (Fig. 4A and B). We first demonstrated that TRF2 interacts with Chk2 using immunoprecipitation assays of extracts derived from asynchronously growing Raji cells (Fig. 4A). To evaluate the cell cycle dependence of this interaction, Raji cells were fractionated into six cell cycle stages (G1, S1, S2, S3, S4, and G2/M) by using centrifugal elutriation. Total levels of input Chk2 did not change significantly at different cell cycle stages (Fig. 4B, left). Interestingly, we found that the Chk2 interaction with TRF2 was most enriched in the S1 phase of the cell cycle (Fig. 4B, middle). No Chk2 was detected in immunoprecipitates with control IgG (Fig. 4B, right), indicating that the IP with the TRF2 antibody (Fig. 4B, middle) is specific. These findings indicate that the Chk2 association with TRF2 is cell cycle dependent and enriched mostly in the early S phase of the cell cycle.
FIG. 4.

Chk2 binds TRF2 in a cell cycle-dependent manner and phosphorylates TRF2 in vitro. (A) Asynchronous Raji cell cultures were subjected to immunoprecipitation with antibodies to TRF2 or control IgG and then assayed by Western blotting for Chk2. Input lysates (shown in the left lane) represent 10% of starting material for IP. IB, immunoblot. (B) Raji cells were cell cycle fractionated using centrifugal elutriation and then subjected to IP with antibodies to TRF2 or control IgG, as indicated. Input lanes represent 10% of starting material. The amount of the Chk2 protein was assayed by Western blotting with anti-Chk2 antibody. The asterisks indicate the nonspecific cross-reaction with the IgG heavy and light chains from IP. (C) Coomassie blue staining of SDS-PAGE gels was used to assess the purity of the baculovirus-derived Chk2 kinase (left) or the bacterially derived GST, GST-TRF2(1-90), and GST-TRF2(FL) proteins used for kinase assays. (D) The purified Chk2 protein was assayed at 0 nM, 50 nM (+), or 200 nM (++) for its ability to phosphorylate GST, GST-TRF2(1-90), or GST-TRF2(FL), as indicated. 32P-labeled TRF2 fusion proteins are indicated by arrows, and the arrowhead indicates a GST-TRF2 breakdown product. Chk2 autophosphorylation is indicated by asterisks.
Chk2 can phosphorylate the TRF2 amino-terminal domain.
Our data suggest that Chk2 kinase activity is important for OriP regulation. Since Chk2 can interact directly with TRF2, we assayed whether TRF2 is a substrate for Chk2 phosphorylation. Others have shown previously that Chk2 phosphorylation can inhibit TRF2 DNA binding (5). Examination of the TRF2 protein sequence revealed a Chk2 consensus site in the amino-terminal domain of TRF2. We therefore expressed and purified full-length TRF2 [TRF2(FL)] and the TRF2 amino-terminal domain at amino acids (aa) 1 to 90 [TRF2(1-90)] as GST fusion proteins from E. coli (Fig. 4C). GST, GST-TRF2(1-90), and GST-TRF2(FL) were incubated with purified Chk2, which was expressed and purified from baculovirus-infected Sf9 cells, and assayed by in vitro kinase reactions (Fig. 4D). We found that GST-TRF2(1-90) and GST-TRF2(FL) were substrates for Chk2, while the GST protein alone was not subject to Chk2-dependent phosphorylation.
TRF2 S20 is a Chk2 phosphoacceptor site.
We observed that a Chk2 consensus site (LXXRXXS/T) exists in the amino-terminal domain of TRF2 (S20) as well as within the RGG motifs of EBNA1 and the histone H3 amino-terminal tail (Fig. 5A). To determine if S20 is a potential Chk2 phosphoacceptor site in the TRF2 amino-terminal domain, we generated point mutations of S20 to alanine (S20A) or glutamic acid (S20E). These point mutations were purified as GST fusion proteins containing TRF2(1-90) as either the wt or the S20A or S20E mutant (Fig. 5B), and they were assayed as kinase substrates for the purified Chk2 protein in vitro (Fig. 5C). We found that wt GST-TRF2(1-90), but not the S20A or S20E mutant, could be phosphorylated by Chk2 in the in vitro kinase assay with purified Chk2. These results indicate that TRF2 amino acid residue S20 is subject to Chk2 phosphorylation in vitro.
FIG. 5.

Serine 20 in TRF2 can be phosphorylated by Chk2 in vitro. (A) Candidate Chk2 phosphorylation sites in TRF2 (S20), EBNA1 (S350), and histone H3 (T11) are aligned with the known consensus sites for Chk2 phosphorylation. (B) GST, GST-TRF2(1-90), GST-TRF2(1-90)(S20A), and GST-TRF2(1-90)(S20E) were purified from E. coli cells and visualized by Coomassie blue staining after SDS-PAGE. (C) The purified Chk2 protein at 0 nM, 50 nM (+), or 250 nM (++) was assayed for its ability to phosphorylate GST, GST-TRF2(1-90), GST-TRF2(1-90)(S20A), and GST-TRF2(1-90)(S20E), as indicated. The arrows indicate TRF2(1-90), and the arrowhead indicates a GST-TRF2 breakdown product. Chk2 autophosphorylation is indicated in the upper panel.
TRF2 S20 is important for RNA binding, ORC recruitment, and OriP DNA replication.
To investigate the potential role of S20 phosphorylation in the biochemical function of TRF2, we compared the abilities of wt GST-TRF2(1-90) and the S20A and S20E mutants to bind G-rich RNA (Fig. 6A) or recruit the ORC from HeLa nuclear extracts (Fig. 6B). Previous studies have implicated these biochemical functions in replication and maintenance activities of EBNA1 at OriP (27). We found that wt GST-TRF2(1-90) and the S20A mutant were capable of binding G-rich RNA by EMSA, while the GST-TRF2(1-90) S20E mutant was compromised for this activity (Fig. 6A, left). GST alone had no ability to bind G-rich RNA, and none of the TRF2 fusion proteins were capable of binding C-rich RNA (Fig. 6A, right). We also found that wt GST-TRF2(1-90) and the S20A mutant were capable of recruiting ORC2 from HeLa nuclear extracts, while the GST-TRF2(1-90) S20E mutant was reduced ∼3-fold for this interaction (Fig. 6B, top). GST alone did not recruit ORC2, and none of the GST fusion proteins recruited PCNA, indicating that the interaction with the ORC was specific for wt GST-TRF2 and the S20A mutant. Since S20E can partially mimic the charge of the phosphorylated serine, these data suggest that S20 phosphorylation will disrupt the TRF2 interaction with G-rich RNA and its ability to recruit the ORC.
The effect of S20 substitution mutations on TRF2 function in OriP DNA replication was assayed by a transient OriP DNA replication assay (Fig. 6C to E). Previous studies have shown that the ectopic expression of TRF2 can stimulate OriP DNA replication but that a deletion in the TRF2 basic domain (TRF2ΔB) had no stimulatory effect (1, 8). As expected, the ectopic expression of full-length TRF2 stimulated OriP replication 1.8-fold relative to vector-transfected cells. The ectopic expression of the TRF2 S20A mutant also stimulated OriP replication, while the TRF2 S20E mutant and TRF2ΔB had no effect on OriP replication (Fig. 6C and D). The expression levels of FLAG-tagged TRF2 proteins were assayed by Western blotting with anti-FLAG antibody (Fig. 6E). TRF2 mutants were expressed at levels comparable to those of full-length TRF2, and no change in the EBNA1 protein was detected. These findings indicate that the TRF2 S20E mutant, which disrupts RNA binding and ORC recruitment in vitro, fails to stimulate OriP replication in cells.
DISCUSSION
Chk2 is a key regulator of S-phase progression and DNA replication function. We therefore tested its role in regulating functional properties of EBV OriP. We found that the siRNA depletion of Chk2 leads to a loss of OriP-dependent DNA replication and plasmid maintenance (Fig. 1). We also show that Chk2 depletion and the pharmacological inhibition of Chk2 kinase activity advance the replication timing of EBV genomes (Fig. 2) and increase the histone H3 acetylation at the DS (Fig. 3). The pharmacological inhibition of Chk2 kinase similarly advanced the replication timing of EBV genomes and reduced the EBV copy number in latently infected cells (Fig. 2D to F). The potential mechanism through which Chk2 regulates these activities was also investigated. We found that Chk2 binds TRF2 in a cell cycle-dependent manner, with an increase in binding in the early S phase (Fig. 4A and B). The purified recombinant Chk2 protein phosphorylated purified full-length TRF2 and amino-terminal-domain proteins in vitro (Fig. 4C and D). The substitution mutation of TRF2 S20 to alanine or glutamic acid abrogated phosphorylation by Chk2 (Fig. 5). Finally, we found that the TRF2 S20E mutant was compromised in interactions with G-rich RNA (Fig. 6A) for the recruitment of the ORC from nuclear extracts (Fig. 6B) and the stimulation of OriP DNA replication activity (Fig. 6C). Based on these experimental results, we propose that Chk2 regulates OriP plasmid maintenance through the direct phosphorylation of the TRF2 amino-terminal domain, which in turn regulates the TRF2 interaction with ORC subunits (Fig. 7).
FIG. 7.
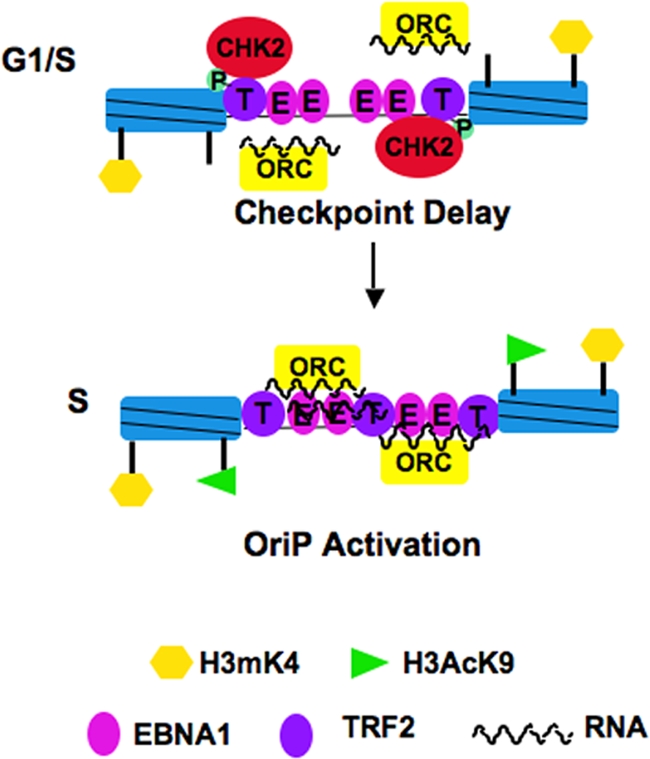
Speculative model for Chk2 regulation of OriP. Chk2 is shown to associate with phosphorylated TRF2 in the G1/S phase. Phosphorylated TRF2 is compromised in RNA binding and ORC recruitment, which may prevent replication initiation and provide a Chk2-dependent checkpoint delay in replication initiation. In the mid-S phase, Chk2 dissociates from TRF2 and returns OriP to a competent replication origin.
EBV genome stability during latent infection depends on numerous factors, including efficient DNA replication, chromatin assembly, epigenetic modifications, metaphase chromosome attachment, and sister-chromatid segregation. How all of these events are coordinated with host cell DNA replication and cellular division is not completely understood. A recent study has shown that cell cycle timing of replication determines the histone modification pattern, with early-S-phase replication correlating with acetylated histones and late-S-phase replication conferring deacetylated histones (20). We have recently found that OriP replication has a programmed cell cycle delay that prevents it from replicating among the earliest origins, like the cellular origin at the lamin B locus (46). This delay in replication firing may be important for establishing the appropriate chromatin modifications necessary for gene expression control during latency. In lower eukaryotes, replication timing is controlled by the S-phase checkpoint protein Rad53 and by the histone deacetylase Rpd3 (34, 40). Therefore, we explored whether OriP replication timing was regulated by human orthologues of Rad53, namely, Chk2, and whether this function involved the human orthologues of Rpd3, namely, HDAC1 and HDAC2. Since OriP also contains telomere repeat factors, we investigated the potential interactions between Chk2 and TRF2 in the regulation of OriP replication function, timing, and chromatin structure. Based on our findings, we suggest that Chk2-TRF2-HDAC interactions constitute an S-phase checkpoint axis that is required for OriP replication function and stable maintenance.
TRF2 is a constitutive component of the DS of OriP (8, 9). One function of TRF2 at telomeres is to protect telomere repeat DNA from DNA damage recognition and repair (28). TRF2 interacts with several DNA damage checkpoint proteins, including ATM and the more recently discovered Chk2 (5, 15). In both cases, TRF2 has been implicated in the downmodulation of the DNA damage response at telomeres. TRF2 may function at OriP similarly to its proposed function at telomeres, namely, to inhibit a DNA damage response to DNA structures formed during replication initiation at the DS. Consistent with this model, the DNA damage recognition proteins MRE11 and NBS1 physically associate with the DS and function in DS replication and OriP plasmid maintenance (11). TRF2 may prevent ATM and Chk2 activation of a DNA damage response at the DS during the early stages of the S phase. This may be necessary to prevent cell cycle arrest as well as to prevent unwarranted recombination or resection of viral DNA by cellular DNA repair machinery.
While TRF2 may prevent the Chk2 activation of a DNA damage response, our data also suggest that Chk2 modifies TRF2 functions at OriP. We and others found that Chk2 can phosphorylate TRF2 (5). Buscemi et al. previously found that the Chk2 phosphorylation of TRF2 inhibited TRF2 DNA binding to telomere repeat DNA, and this finding was consistent with our observation that TRF2 binding was enhanced in siChk2-depleted cells (Fig. 3A) (5). We found that Chk2 could phosphorylate TRF2 at amino acid S20 in the amino-terminal basic domain (Fig. 5). A phosphomimetic substitution mutation of S20 to glutamic acid inhibited TRF2 binding to G-rich RNA, TRF2 recruitment of the ORC, and the stimulation of OriP replication activity (Fig. 6). These findings suggest that Chk2 may restrict TRF2 function at the DS by inhibiting its ability to recruit the ORC. The transient interaction of Chk2 with TRF2 in the early S phase would restrict this repression to the early stages of the S phase, when Chk2 binds TRF2 (Fig. 4B). This interpretation is consistent with our observation that Chk2 depletion leads to an advance in the replication timing of OriP. It is also consistent with the role of Rad53 (the yeast orthologue of Chk2) in the regulation of replication timing in budding yeast (34).
How does the Chk2 phosphorylation of TRF2 inhibit early-S-phase firing of OriP and enhance episome stability? First, it is also likely that Chk2 phosphorylates other components of the DS, including consensus sites found in EBNA1 RGG motif 2 and in the basic amino-terminal tail of histone H3 (Fig. 5A). One possible mechanism is that the Chk2 phosphorylation of TRF2 (and possibly EBNA1) inhibits the recruitment of the ORC. This inhibition may be limited to the early stages of the S phase, when Chk2 binds most efficiently to TRF2. Other mechanisms are also possible, including Chk2 phosphorylation of histone H3 and an inhibition of nucleosome remodeling during early S phase. Alternatively, TRF2 may be subject to other cell cycle controls, including its ability to be phosphorylated by the Aurora C kinase (37). How this transient inhibition of replication initiation enhances episome stability is not known. We speculate that early-S-phase initiation increases the risk of replication errors or genomic catastrophe if conditions are not optimal. Chk2 may function as a sensor to prevent OriP replication initiation in cells where nucleotide pools are low or other forms of DNA damage may have occurred. This may also explain how HU treatment destabilizes EBV episomal maintenance. Low doses of HU advance the replication timing of OriP and increase histone H3 acetylation at the DS similar to that observed for cells where Chk2 has been depleted (36). We speculate that low doses of HU inactivate Chk2 checkpoint function at OriP. Future studies will be required to address this speculation. Despite these limitations, we conclude that Chk2 associates with OriP through an interaction with TRF2, modulates histone acetylation and ORC recruitment, and enhances OriP-dependent episome maintenance.
Acknowledgments
We thank Andreas Wiedmer for technical support and the Wistar Cancer Center Core Facilities for their assistance.
This work was funded by NCI CA93606 to P.M.L., a UPENN virology training grant to J.N., and a Lymphoma Research Foundation fellowship to J.Z.
Footnotes
Published ahead of print on 3 March 2010.
REFERENCES
- 1.Atanasiu, C., Z. Deng, A. Wiedmer, J. Norseen, and P. M. Lieberman. 2006. ORC binding to TRF2 stimulates OriP replication. EMBO Rep. 7:716-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bashaw, J. M., and J. L. Yates. 2001. Replication from oriP of Epstein-Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J. Virol. 75:10603-10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell, S. P. 2002. The origin recognition complex: from simple origins to complex functions. Genes Dev. 16:659-672. [DOI] [PubMed] [Google Scholar]
- 4.Bhaskara, S., B. J. Chyla, J. M. Amann, S. K. Knutson, D. Cortez, Z. W. Sun, and S. W. Hiebert. 2008. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 30:61-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buscemi, G., L. Zannini, E. Fontanella, D. Lecis, S. Lisanti, and D. Delia. 2009. The shelterin protein TRF2 inhibits Chk2 activity at telomeres in the absence of DNA damage. Curr. Biol. 19:874-879. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri, B., H. Xu, I. Todorov, A. Dutta, and J. L. Yates. 2001. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 98:10085-10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curman, D., B. Cinel, D. E. Williams, N. Rundle, W. D. Block, A. A. Goodarzi, J. R. Hutchins, P. R. Clarke, B. B. Zhou, S. P. Lees-Miller, R. J. Andersen, and M. Roberge. 2001. Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J. Biol. Chem. 276:17914-17919. [DOI] [PubMed] [Google Scholar]
- 8.Deng, Z., C. Atanasiu, J. S. Burg, D. Broccoli, and P. M. Lieberman. 2003. Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein-Barr virus OriP. J. Virol. 77:11992-12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng, Z., L. Lezina, C. J. Chen, S. Shtivelband, W. So, and P. M. Lieberman. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol. Cell 9:493-503. [DOI] [PubMed] [Google Scholar]
- 10.Dhar, S. K., K. Yoshida, Y. Machida, P. Khaira, B. Chaudhuri, J. A. Wohlschlegel, M. Leffak, J. Yates, and A. Dutta. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287-296. [DOI] [PubMed] [Google Scholar]
- 11.Dheekollu, J., Z. Deng, A. Wiedmer, M. D. Weitzman, and P. M. Lieberman. 2007. A role for MRE11, NBS1, and recombination junctions in replication and stable maintenance of EBV episomes. PLoS One 2:e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Early, A., L. S. Drury, and J. F. Diffley. 2004. Mechanisms involved in regulating DNA replication origins during the cell cycle and in response to DNA damage. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359:31-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge, X. Q., D. A. Jackson, and J. J. Blow. 2007. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 21:3331-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerbi, S. A., Z. Strezoska, and J. M. Waggener. 2002. Initiation of DNA replication in multicellular eukaryotes. J. Struct. Biol. 140:17-30. [DOI] [PubMed] [Google Scholar]
- 15.Karlseder, J., K. Hoke, O. K. Mirzoeva, C. Bakkenist, M. B. Kastan, J. H. Petrini, and T. de Lange. 2004. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2:E240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kieff, E. 2007. Epstein-Barr virus and its replication, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 17.Kiledjian, M., and G. Dreyfus. 1992. Primary structure and binding activity of hnRNP U protein: binding RNA through RGG box. EMBO J. 11:2655-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim, S. M., and J. A. Huberman. 2001. Regulation of replication timing in fission yeast. EMBO J. 20:6115-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koons, M. D., S. V. Scoy, and J. Hearing. 2001. The replicator of the Epstein-Barr virus latent cycle origin of DNA replication, oriP, is composed of multiple functional elements. J. Virol. 75:10582-10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lande-Diner, L., J. Zhang, and H. Cedar. 2009. Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol. Cell 34:767-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, M. A., M. E. Diamond, and J. L. Yates. 1999. Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein-Barr virus. J. Virol. 73:2974-2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindner, S. E., and B. Sugden. 2007. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid 58:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindner, S. E., K. Zeller, A. Schepers, and B. Sugden. 2008. The affinity of EBNA1 for its origin of DNA synthesis is a determinant of the origin's replicative efficiency. J. Virol. 82:5693-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu, C. C., C. W. Wu, S. C. Chang, T. Y. Chen, C. R. Hu, M. Y. Yeh, J. Y. Chen, and M. R. Chen. 2004. Epstein-Barr virus nuclear antigen 1 is a DNA-binding protein with strong RNA-binding activity. J. Gen. Virol. 85:2755-2765. [DOI] [PubMed] [Google Scholar]
- 25.Mackey, D., and B. Sugden. 1999. The linking regions of EBNA1 are essential for its support of replication and transcription. Mol. Cell. Biol. 19:3349-3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marechal, V., A. Dehee, R. Chikhi-Brachet, T. Piolot, M. Coppey-Moisan, and J.-C. Nicolas. 1999. Mapping EBNA-1 domains involved in binding to metaphase chromosomes. J. Virol. 73:4385-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norseen, J., A. Thomae, V. Sridharan, A. Aiyar, A. Schepers, and P. M. Lieberman. 2008. RNA-dependent recruitment of the origin recognition complex. EMBO J. 27:3024-3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palm, W., and T. de Lange. 2008. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42:301-334. [DOI] [PubMed] [Google Scholar]
- 29.Rawlins, D. R., G. Milman, S. D. Hayward, and G. S. Hayward. 1985. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell 42:859-868. [DOI] [PubMed] [Google Scholar]
- 30.Rickinson, A. B., and E. Kieff. 2007. Epstein-Barr virus, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 31.Ritzi, M., K. Tillack, J. Gerhardt, E. Ott, S. Humme, E. Kremmer, W. Hammerschmidt, and A. Schepers. 2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J. Cell Sci. 116:3971-3984. [DOI] [PubMed] [Google Scholar]
- 32.Schepers, A., M. Ritzi, K. Bousset, E. Kremmer, J. L. Yates, J. Harwood, J. F. Diffley, and W. Hammerschmidt. 2001. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J. 20:4588-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sears, J., M. Ujihara, S. Wong, C. Ott, J. Middeldorp, and A. Aiyar. 2004. The amino terminus of Epstein-Barr virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol. 78:11487-11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shirahige, K., Y. Hori, K. Shiraishi, M. Yamashita, K. Takahashi, C. Obuse, T. Tsurimoto, and H. Yoshikawa. 1998. Regulation of DNA-replication origins during cell-cycle progression. Nature 395:618-621. [DOI] [PubMed] [Google Scholar]
- 35.Snudden, D. K., J. Hearing, P. R. Smith, F. A. Grasser, and B. E. Griffin. 1994. EBNA-1, the major nuclear antigen of Epstein-Barr virus, resembles ‘RGG’ RNA binding proteins. EMBO J. 13:4840-4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snyder, A. R., J. Zhou, Z. Deng, and P. M. Lieberman. 2009. Therapeutic doses of hydroxyurea cause telomere dysfunction and reduce TRF2 binding to telomeres. Cancer Biol. Ther. 8:1136-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spengler, D. 2007. The protein kinase Aurora C phosphorylates TRF2. Cell Cycle 6:2579-2580. [DOI] [PubMed] [Google Scholar]
- 38.Sugden, B., and E. R. Leight. 2001. Molecular mechanisms of maintenance and disruption of virus latency, p. 3-11. In K. Takada (ed.), Epstein-Barr virus and human cancer, vol. 258. Springer, Heidelberg, Germany. [Google Scholar]
- 39.Tatsumi, Y., K. Ezura, K. Yoshida, T. Yugawa, M. Narisawa-Saito, T. Kiyono, S. Ohta, C. Obuse, and M. Fujita. 2008. Involvement of human ORC and TRF2 in pre-replication complex assembly at telomeres. Genes Cells 13:1045-1059. [DOI] [PubMed] [Google Scholar]
- 40.Vogelauer, M., L. Rubbi, I. Lucas, B. J. Brewer, and M. Grunstein. 2002. Histone acetylation regulates the time of replication origin firing. Mol. Cell 10:1223-1233. [DOI] [PubMed] [Google Scholar]
- 41.Wang, J., S. E. Lindner, E. R. Leight, and B. Sugden. 2006. Essential elements of a licensed, mammalian plasmid origin of DNA synthesis. Mol. Cell. Biol. 26:1124-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yates, J. L., S. M. Camiolo, and J. M. Bashaw. 2000. The minimal replicator of Epstein-Barr virus oriP. J. Virol. 74:4512-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yates, J. L., and N. Guan. 1991. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 65:483-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yates, J. L., N. Warren, and B. Sugden. 1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812-815. [DOI] [PubMed] [Google Scholar]
- 45.Zhou, J., C. M. Chau, Z. Deng, R. Shiekhattar, M. P. Spindler, A. Schepers, and P. M. Lieberman. 2005. Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J. 24:1406-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou, J., A. Snyder, and P. M. Lieberman. 2009. Epstein-Barr virus episome stability is coupled to a delay in replication timing. J. Virol. 83:2154-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]