

Abstract
The role of epigenetic modifications in the regulation of gammaherpesvirus latency has been a subject of active study for more than 20 years. DNA methylation, associated with transcriptional silencing in mammalian genomes, has been shown to be an important mechanism in the transcriptional control of several key gammaherpesvirus genes. In particular, DNA methylation of the functionally conserved immediate-early replication and transcription activator (RTA) has been shown to regulate Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus Rta expression. Here we demonstrate that the murine gammaherpesvirus (MHV68) homolog, encoded by gene 50, is also subject to direct repression by DNA methylation, both in vitro and in vivo. We observed that the treatment of latently MHV68-infected B-cell lines with a methyltransferase inhibitor induced virus reactivation. In addition, we show that the methylation of the recently characterized distal gene 50 promoter represses activity in a murine macrophage cell line. To evaluate the role of de novo methyltransferases (DNMTs) in the establishment of these methylation marks, we infected mice in which conditional DNMT3a and DNMT3b alleles were selectively deleted in B lymphocytes. DNMT3a/DNMT3b-deficient B cells were phenotypically normal, displaying no obvious compromise in cell surface marker expression or antibody production either in naïve mice or in the context of nonviral and viral immunogens. However, mice lacking functional DNMT3a and DNMT3b in B cells exhibited hallmarks of deregulated MHV68 lytic replication, including increased splenomegaly and the presence of infectious virus in the spleen at day 18 following infection. In addition, total gene 50 transcript levels were elevated in the spleens of these mice at day 18, which correlated with the hypomethylation of the distal gene 50 promoter. However, by day 42 postinfection, aberrant virus replication was resolved, and we observed wild-type frequencies of viral genome-positive splenocytes in mice lacking functional DNMT3a and DNMT3b in B lymphocytes. The latter correlated with increased CpG methylation in the distal gene 50 promoter, which was restored to levels similar to those of littermate controls harboring functional DNMT3a and DNMT3b alleles in B lymphocytes, suggesting the existence of an alternative mechanism for the de novo methylation of the MHV68 genome. Importantly, this DNMT3a/DNMT3b-independent methylation appeared to be targeted specifically to the gene 50 promoter, as we observed that the promoters for MHV68 gene 72 (v-cyclin) and M11 (v-bcl2) remained hypomethylated at day 42 postinfection. Taken together, these data provide the first evidence of the importance of DNA methylation in regulating gammaherpesvirus RTA/gene 50 transcription during virus infection in vivo and provide insight into the hierarchy of host machinery required to establish this modification.
Herpesviruses are large, double-stranded DNA viruses characterized by distinct lytic and latent stages. Upon initial infection, the virus undergoes several rounds of lytic replication, after which it establishes a latent infection in which viral gene transcription is limited and tightly controlled. Intimately associated with the lytic-latent cycle is virus reactivation, the process by which lytic replication is reinitiated from a latent viral genome. The mechanisms leading to virus reactivation are only partially understood but probably involve a combination of extrinsic cellular signals and a complicated intrinsic cellular and viral protein milieu. It is clear, however, that the regulation of lytic replication and, therefore, virus reactivation is key to herpesvirus biology, as the ability to establish a quiescent latent infection is essential to herpesvirus survival in the host.
Among the three families of herpesviruses, (alpha-, beta-, and gammaherpesviruses), the gammaherpesviruses share the propensity to infect lymphocytes. In the case of the human gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), B cells are the primary reservoir for long-term latency (3). Although in vitro infections and transformed cell lines from patients have been valuable tools in the study of gammaherpesvirus biology, the field is limited by the inability to thoroughly study viral pathogenesis in the context of its natural human host. The development of murine herpesvirus 68 (MHV68) as a model system has provided the opportunity to study a naturally occurring gammaherpesvirus in laboratory mice. MHV68 shares significant sequence homology with other gammaherpesviruses, and infection with MHV68 recapitulates many important aspects of human gammaherpesvirus infection, including B-cell tropism, periodic reactivation, and an association with lymphomagenesis in immunocompromised mice (34, 49, 53).
Although KSHV, EBV, and MHV68 encode several unique proteins, one of the most conserved regions in gammaherpesviruses is that encompassing gene 50 in MHV68, also known as Rta (KSHV and EBV). The immediate-early gene 50/Rta protein is a potent transcriptional activator of both viral and cellular genes and has a key role in lytic replication (43). Gene 50/Rta is required for the lytic replication of MHV68, KSHV, and EBV, and its expression is sufficient to induce reactivation in latently infected cell lines (32, 39, 45, 54, 55). Due to its potent ability to drive lytic viral gene transcription, gene 50/Rta expression must be tightly regulated for gammaherpesvirus latency to be established and maintained. There are several mechanisms by which this is accomplished, and evidence exists to support a strong epigenetic component in the regulation of gene 50/Rta transcription (5, 7, 8, 11, 20, 21, 31). DNA methylation in particular was demonstrated to be an important gammaherpesvirus regulatory mechanism for both lytic and latent genes (5, 8). The Rta promoter was shown previously to be methylated in latently KSHV- and EBV-infected B cells, and treatment with methyltransferase inhibitors initiates reactivation in some latently infected cell lines (4, 8).
DNA methylation, originally characterized for bacteria as a means to protect endogenous bacterial genetic material from self-digestion by restriction endonucleases, can occur at several dinucleotide sequences. In mammals, however, the primary modification is the methylation of cytosines coupled with guanines, commonly called CpG dinucleotides. The addition of methyl groups to cytosines is accomplished by mammalian DNA methyltransferases (DNMTs). Five DNMTs of mammals have been characterized, but only three have been shown to have catalytic DNA methyltransferase activity: DNMT1, DNMT3a, and DNMT3b (23). Although DNMT1 has been shown to have the ability to modify unmethylated DNA (known as “de novo” methylation), its primary role is thought to be the maintenance of preexisting methylation marks present on hemimethylated DNA following semiconservative replication. DNMT3a and DNMT3b have therefore been largely classified as the “de novo” methyltransferases and are crucial to the establishment of methylation patterns during embryonic development following the massive wave of demethylation postimplantation (29). DNA methylation is a crucial regulatory mechanism in mammalian cells, and in promoter regions, it is associated with transcriptional repression. It plays a key role in differentiation, and aberrant DNA methylation is strongly associated with tumorigenesis and disease, therefore making it a subject of intense study in the field of cancer genetics.
We have recently described an additional gene 50 exon and promoter in MHV68 and provided evidence that this additional transcriptional unit is conserved in KSHV and EBV (19). The newly indentified MHV68 “distal” promoter is progressively methylated in splenocytes throughout latent infection. Since virion DNA is unmethylated upon entry into the cell (46), we wished to examine the role of DNMT3a and DNMT3b in establishing methylation patterns at the MHV68 distal gene 50 promoter following the infection of B lymphocytes. Using the conditional deletion of Dnmt3a and Dnmt3b alleles specifically in B cells, we demonstrate that the absence of de novo methyltransferases is associated with aberrant lytic replication and hypomethylation of lytic gene promoters during early latent infection. At later times postinfection, lytic replication is resolved and normal latency is established, which is accompanied by the restoration of methylation at the distal gene 50 promoter but not at other lytic gene promoters. These effects were not due to a perturbation of normal immune function as a result of disrupting Dnmt3a and Dnmt3b in B cells, as B- and T-cell surface phenotypes as well as antibody production were unaffected in infected animals harboring the conditional deletion. These observations demonstrate a role for DNMT3a and DNMT3b activity during the establishment of a latent infection and also suggest the existence of alternative de novo methylation mechanisms for the silencing of the distal gene 50 promoter. Together, these data highlight the importance of DNA methylation in regulating gene 50 expression and provide the first direct evidence of the involvement of the de novo methyltransferases DNMT3a and DNMT3b.
MATERIALS AND METHODS
Viruses, tissue culture, and cell treatments.
Wild-type MHV68 (ATCC VR-1465) cultures were propagated in NIH 3T12 fibroblasts, and titers were determined by plaque assay. Mouse embryonic fibroblasts (MEFs) derived from C57BL/6 embryos, RAW264.7 murine macrophages, and NIH 3T12 fibroblasts were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 100 U penicillin per ml, and 100 mg streptomycin per ml. A20-HE1 and A20-HE2 cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 U penicillin per ml, and 100 mg streptomycin per ml. A20-HE1 and A20-HE2 cells were maintained under hygromycin selection as previously described (15). All tissue cultures were performed in a 5% CO2 tissue culture incubator at 37°C.
Methyltransferase inhibitor treatments and Western blots.
Cells were treated with 5-aza-2-deoxycytidine (5azaCdR) (Sigma) (initially dissolved in dimethyl sulfoxide [DMSO] and diluted with sterile distilled, deionized water) for 72 h at the indicated concentration. Cell equivalents were pelleted and resuspended in radioimmunoprecipitation buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA) supplemented with EDTA-free protease inhibitor minitabs (Roche). Cell lysates were frozen at −20°C. Membranes were blocked for 1 h in 5% milk-phosphate-buffered saline (PBS)-1% Tween and then probed overnight with either rabbit MHV68-lytic antisera or chicken-derived Orf59 antibody diluted in 5% milk-PBS-1% Tween. After washing, membranes were again blocked and probed for 1 h with horseradish peroxidase-conjugated donkey anti-rabbit or donkey anti-chicken antibodies. Chemiluminescence was induced with ECL Western blotting reagents (Pierce) and visualized on film.
In vitro methylation, transfections, and reporter assays.
The −250 bp of the distal promoter region were amplified by using primers described previously (19) but instead incorporating PstI and HindIII restriction sites. The insert was cloned into the pCpG reporter vector and propagated in Pir1 chemically competent cells (Stratagene) with zeocin selection. Vectors were methylated or mock methylated in the presence or absence of M.SssI (2.5 U/μg) (NEB) in buffer 2 and 160 μM S-adenosylmethionine (SAM) at 37°C for 4 h. After 2 h, additional SAM was added to 160 μM. DNA was ethanol precipitated and resuspended in endotoxin-free water. RAW264.7 macrophages were plated at 1.5E6 cells per well in supplemented Dulbecco's modified Eagle's medium (see above) and transfected the next day (at 75% confluence) with the indicated vector and 5 ng of a Renilla control plasmid by using LT-1 TransIT reagent (Mirus) according to the manufacturer's instructions. Cells were treated with medium or lipopolysaccharide (LPS) (5 μg/ml) at 24 h and harvested at 48 h. Firefly and Renilla luciferase activities were read by using the Dual luciferase kit (Promega) as previously described (19).
Infections, immunizations, organ harvesting, and preparation.
See below for a description of the generation of CD19-Cre/Dnmt3a/Dnmt3b conditional knockout mice. All MHV68 infections were performed by the intranasal inoculation of mice (between 6 and 12 weeks of age) with 20 μl of a 5E4-PFU/ml viral stock (1,000 PFU) diluted in CMEM. Mice were anesthetized by isoflurane inhalation prior to infection. Mice were sacrificed by isoflurane inhalation and cervical dislocation. Spleens were harvested, and splenocytes were prepared by homogenization and treated with Tris-ammonium chloride for red blood cell elimination as described previously (51). Splenocytes were immediately used for reactivation analyses and genomic DNA isolation or stored in CMEM-10% dimethyl sulfoxide at −80°C until they were prepared for quantitative or limiting-dilution PCR analyses. For lymphocytic choriomeningitis virus (LCMV) infections, mice were infected with 2 × 105 PFU LCMV Armstrong by intraperitoneal infection. Spleens were harvested 8 days postinfection. To analyze nonviral immune responses, mice were immunized with the intraperitoneal injection of either 0.2 ml of 10% defibrinated sheep red blood cells (SRBCs) (Colorado Serum Company) in PBS, 100 μg ovalbumin in complete Freund's adjuvant (OVA-CFA) emulsion (Sigma), or 50 μg nitrophenylacetate conjugated to chicken gammaglobulin (NP-CGG) in alum.
Generation of mice in which Dnmt3a and Dnmt3b were specifically knocked out in B lymphocytes.
Mice harboring conditional Dnmt3a (Dnmt3ac/c) (24) or Dnmt3b (Dnmt3bc/c) (12) alleles (C57BL/6 background) were obtained from Taiping Chen at Novartis. Mice were maintained in accordance with university and federal guidelines. In addition, all animal experiments were carried out with the approval of the Emory University Institutional Animal Care and Use Committee (IACUC). Dnmt3ac/c and Dnmt3bc/c mice were interbred, and the F1 generation was bred to obtain Dnmt3a/3bc/c double-conditional knockouts. Double-conditional knockout mice were subsequently bred to CD19+/Cre mice (C57BL/6 background), a kind gift from Klaus Rajewsky (40), to obtain CD19+/Cre Dnmt3a/3bc/c or CD19+/+ Dnmt3a/3bc/c mice. Colonies were maintained by breeding CD19+/Cre/Dnmt3a/3bc/c mice to Dnmt3a/3bc/c mice and screening offspring for the presence of the CD19-driven Cre transgene by using primers 5′-ACGAACCTGGTCGAAATCAGTGCG-3′ and 5′-CGGTCGATGCAACGAGTGATGAG-3′. CD19+/+ Dnmt3a/3bc/c offspring (Cre negative) were used as littermate controls. Mice were screened by using DNA extracted from tail biopsy specimens using the Qiagen blood and tissue kit (Qiagen). Genotyping for the nonrearranged Dnmt3a locus was performed by using primers 5′-TGCAATGACCTCTCCATTGTCAAC-3′ and 5′-GGTAGAACTCAAAGAAGAGGCGGC-3′, and that for Dnmt3b was performed by using primers 5′-AGAGCACTGCACCACTACTGCTGGA-3′ and 5′-CAGGTCAGACCTCTCTGGTGACAAG-3′. The rearranged 1loxP Dnmt3a allele was detected by using primers 5′-CTGTGGCATCTCAGGGTGATGAGC-3′ and 5′-GGTAGAACTCAAAGAAGAGGCGGC-3′, and the rearranged 1loxP allele was detected with primers 5′-GAGCTGCTATATGTGCCTCCCTCAG-3′ and 5′-CAGGTCAGACCTCTCTGGTGACAAG-3′. PCR was performed by using GoFlexiTaq polymerase (Promega) according to the manufacturer's instructions, with the following parameters: 95°C for 5 min; 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s; and 72°C for 7 min.
Flow cytometry analyses.
Single-cell splenocyte samples were prepared as described above. For analysis of cell surface antigen expression, 2 × 106 to 3 × 106 cells per well were washed twice with staining buffer (PBS-0.5% FCS) in tissue culture-treated 96-well plates. Cells were resuspended in 50 μl of staining buffer containing Fc block (anti-CD16/CD32) at 1:50 and blocked for 15 min at 4°C. Cells were washed once with staining buffer and then resuspended in 50 μl of staining buffer containing the appropriate antibody cocktail at a predetermined concentration. Staining was performed for 25 min in the dark at 4°C; cells were then washed twice with staining buffer and resuspended in staining buffer containing 2% paraformaldehyde to fix. Within 3 days of preparation, samples were analyzed by using an LSRII flow cytometer (BD Biosciences), and data were collected by using FACSDiva software (BD Biosciences). Data were analyzed by using FlowJo software (Treestar, Inc.); graphs were generated and statistical analyses were performed by using GraphPad Prism software. Data presented are representative of at least two independent experiments with at least four mice per group.
ELISPOT and enzyme-linked immunosorbent assay (ELISA) analyses.
To analyze antibody-secreting cells (ASCs), splenocytes (NP immunizations) or bone marrow lymphocytes (MHV68 assays) were subjected to enzyme-linked immunospot (ELISPOT) analysis. Splenocytes were obtained as described above; to obtain bone marrow cells, both femurs from individual mice were perfused with medium (CMEM), and cells were collected into tissue culture dishes. Following centrifugation, red blood cell lysis was performed by 10 min of incubation at room temperature in a Tris-ammonium chloride solution (Sigma) as described above for splenocytes. Single-cell suspensions of splenocytes and bone marrow cells were counted, and 1 × 106 cells were used for ELISPOT analysis. ELISPOT analyses s were performed as described previously (42). Briefly, HyBond nitrocellulose plates were prepared by coating membranes with either goat anti-mouse IgM, IgA, and IgG (Southern Biotech) (for total ASCs) or NP-bovine serum albumin (BSA) (Biosearch Technologies) (for NP-specific ASCs). Following blocking, cells in RPMI medium with 10% FCS were plated in serial dilutions (1:3) and incubated for 12 h at 37°C. Plates were washed and incubated with antibodies against the indicated isotype for 1 h at room temperature. Spots representing ASCs were visualized by 3-amino-9-ethylcarbazole (AEC) development following incubation with primary (biotinylated anti-mouse) and secondary (streptavidin-horseradish peroxidase) antibodies. Spots were analyzed by using ImmunoSpot plate reading and ImageAcquistion software (Cell Limited Technologies, Inc.). Each sample was analyzed in duplicate, and each experiment represents data from at least three individual mice. Graphs were generated and statistical analyses were performed by using GraphPad Prism software.
To analyze serum antibody levels, mice were bled by serial tail nicks, and serum was harvested from peripheral blood by centrifugation. Serum was stored at −80°C until analysis. For anti-MHV68 IgG analysis, ELISAs were performed as described previously (18). Data shown are representative of data from at least two independent infections with at least three mice per group. Graphs were generated and statistical analyses were performed by using GraphPad Prism software.
Ex vivo limiting-dilution analyses of latently infected splenocytes.
To determine the frequency of viral genome-positive cells in preparations of splenocytes, serially diluted cells were subjected to nested PCR by using primers to detect gene 50 as previously described (52). To determine the frequency of cells reactivating from latency, single-cell suspensions of splenocytes from mice at day 18 postinfection were plated in 2-fold serial dilutions onto MEFs in 96-well plates as previously described (52). At day 21 postplating, each well was assessed for the presence of a cytopathic effect (CPE). To determine the frequency of cells reactivating, the percentage of wells with CPEs at each dilution was used in a nonlinear regression analysis to calculate the frequency of reactivation per cell by Poisson distribution. Disrupted splenocytes and peritoneal exudate cells (PECs) were also plated in parallel as previously described to determine the contribution of preformed infectious virus to reactivation (51).
RT-PCR and quantitative RT-PCR (qRT-PCR).
Nonquantitative reverse transcription (RT)-PCR to detect spliced gene 50 transcripts following the treatment of cells with the methyltransferase inhibitor 5-aza-2-deoxycytidine was carried out as follows. Total RNA from untreated or treated cells was extracted by using Trizol reagent according to the manufacturer's protocol (Invitrogen). Three micrograms of RNA was treated with DNase and reverse transcribed by using a first-strand cDNA synthesis kit (Invitrogen) and random hexamer primers. Two microliters of cDNA was used in subsequent PCRs with primers for gene 50 transcript and viral DNA polymerase transcripts as previously described (19, 50).
Quantitative RT-PCR to detect the number of copies of gene 50 transcripts in the spleens of MHV68-infected mice at the peak of viral latency was carried out as follows. RNA from total splenocytes (prepared as described above) was isolated by using Trizol reagent (Invitrogen). Three or five micrograms of RNA was treated with DNase I (Invitrogen), according to the manufacturer's instructions, in a total volume of 50 μl. Twenty microliters of DNase-treated RNA was subsequently used for first-strand cDNA synthesis by use of SuperScript II reverse transcriptase (Invitrogen). Five microliters of the cDNA reaction mixture was used in each quantitative amplification reaction mixture. Quantitative PCR was performed by use of iQ Supermix (Bio-Rad) with forward primer 5′-GGAATTTCTGCAGCGATGGCCTCT-3′ and reverse primer 5′-CCTCTTTTGTTTCAGCAGAGACTCCA-3′ at 900 nM. The TaqMan probe was used (at 250 nM) (5′-6-carboxyfluorescein-CTGGCACGGATCGAAGCAGGTCTAC-6-carboxytetramethyl-rhodamine-3′). PCR was performed with the following cycle parameters: 95°C for 3 min; 40 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s; and then 95°C for 1 min. A standard curve was generated by using a spliced E0-E1-E2 PCR product amplified from tetradecanoyl phorbol acetate (TPA)-treated A20-HE2 cDNA under the conditions described below for MHV68 E0-E2 RT-PCR, which was cloned into the pGEMT-Easy vector (Promega). qRT-PCR was performed with a Becton-Dickinson iCycler and analyzed by using Bio-Rad iCycler software.
Bisulfite PCR analyses.
Splenocytes from infected mice at day 18 or day 42 postinfection were prepared as described above, and the CD19+ population was enriched by magnetic-bead sorting using a B-cell isolation kit (Miltenyi Biotech) and the AutoMACS cell separation system (Miltenyi Biotech). CD19+ enrichment was verified by staining with CD19-phycoerythrin (PE) antibody and analysis by flow cytometry; CD19+ cells comprised >85% of total cells following magnetic cell separation. Total genomic DNA was prepared from the enriched CD19+ fraction by phenol-chloroform extraction following overnight proteinase K digestion. Five hundred nanograms of genomic DNA was bisulfite modified using the EZ-DNA methylation kit (Zymo Research) according to the manufacturer's instructions. Bisulfite-modified DNA was amplified by using AmpliTaq Gold DNA polymerase (Applied Biosystems). Each nested and heminested PCR mixture contained 1× AmpliTaq Gold buffer, 3 mM MgCl2, deoxynucleoside triphosphates (0.2 mM each), and forward and reverse primers (0.2 μM each). All reactions were performed with a Becton Dickinson iCycler with the following parameters: 95°C for 10 min (hot start) and 30 cycles of 95°C for 30 s, annealing at the indicated temperature for 30 s, and 72°C for 1 min, followed by a final extension step at 72°C for 7 min. For round 1, 2 μl of bisulfite-modified DNA was used in each 25-μl reaction mixture. One microliter of round 1 product was used as the template for the round 2 reaction. For distal promoter region round 1, forward primer 5′-ATGATGATTTATTAAAGAATTATGTTTTAGGT-3′ and reverse primer 5′-CAACCTCACCAACTTTTACAATAAATA-3′ were used. Round 1 annealing was performed at 49°C. For gene 50 round 2, the forward primer was the same as that used for round 1, and the reverse primer was 5′-CCCTTAATAACCTAATAAAAAACCCAATA-3′. Round 2 annealing was performed at 50°C with 30 cycles. For v-cyclin/v-bcl2 promoter round 1, forward primer 5′-GAGATAATGGTAATATTTATTAAATTATAATAT-3′ and reverse primer 5′-CCCACAACATTCCACCTTCAACAAA-3′ were used for set A at an annealing temperature of 48°C, and forward primer 5′-GGTGTAGTTTGTAGATTGTAGGTTGTT-3′ and reverse primer 5′-CCACAAAATCACCTAAATCTAATCCAAA-3′ were used for set B at an annealing temperature of 50°C. For round 2, forward primer 5′-GGTTTATAGTTAAGTGTATATAGGTTAGTGTGAT-3′ and reverse primer 5′-CAACCTACAATCTACAAACTACACC-3′ were used for set A, and forward primer 5′-GGTGGAATGTTGTGGGGTGT-3′ and reverse primer 5′-CCACAAAATCACCTAAATCTAATCCAAA-3′ were used for set B. The annealing temperature for both v-cyclin/v-bcl2 set A and B round 2 was 55°C. PCR products were visualized by ethidium gel electrophoresis, purified by using the GeneCleanII system as described above, and ligated into the pGEMT-Easy vector (Promega). Plasmid DNA was sequenced by Macrogen USA (Rockville, MD) and analyzed by using SeqMan alignment software (Lasergene).
RESULTS
Methyltransferase inhibitor treatment reactivates latent MHV68.
It has been demonstrated that reactivation can be induced in some latently EBV- and KSHV-infected B-cell lines following treatment with the methyltransferase inhibitor 5-aza-2-deoxycytidine (5azaCdR), presumably due to the passive demethylation of the viral genome resulting from the inhibition of the maintenance methyltransferase DNMT1 during cell division. To determine the effects of 5azaCdR treatment on latent MHV68 infection, we used murine A20 cell lines latently harboring MHV68 as an in vitro latency model (12). The A20-HE1 and A20-HE2 cell lines were cultured for 72 h either with or without 5azaCdR treatment and analyzed for evidence of lytic antigen expression. Probing with rabbit anti-MHV68 antisera revealed an induction of virus replication-associated antigens in both A20-HE cell lines (Fig. 1A). The specificity of this induction was confirmed by using an antibody specific for the viral DNA polymerase processivity factor encoded by Orf59-untreated cells expressing little or no detectable Orf59 protein, while cells treated with either TPA or 5azaCdR expressed the Orf59 protein (Fig. 1B). Notably, treatment with 5 μM 5azaCdR resulted in levels of the Orf59 protein comparable to that observed following TPA treatment. 5azaCdR-induced reactivation was accompanied by the expression of distal gene 50 promoter-generated transcripts containing the newly characterized gene 50 E0 exon (Fig. 1C). We recently demonstrated that this promoter is methylated in latently infected A20-HE cell lines as well as in a latently MHV68-infected S11 B-lymphoma cell line and in latently infected splenocytes recovered from MHV68-infected mice (19). The treatment of latent MHV68 with methyltransferase inhibitors may therefore induce reactivation in part by facilitating the demethylation of the distal gene 50 promoter. Although this analysis was nonquantitative, it is worth noting that appreciable levels of total (E2-containing) transcripts are detectable in untreated A20-HE2 cells. This was previously observed and most likely reflects low-level spontaneous virus reactivation in this cell line. However, there are very few, if any, detectable transcripts arising from the distal promoter (containing E0) in untreated A20-HE2 cells, which may reflect the presence of CpG methylation in the distal promoter region previously reported (19), which is supported by the increase in levels of E0-containing transcripts following 5azaCdR treatment. In addition, the detection of a band corresponding to an unspliced product in the E1-E2 reaction upon 5azaCdR treatment is intriguing and may indicate the expression of unspliced, antisense transcripts serving to regulate gene 50 transcription during lytic replication.
FIG. 1.

Methyltransferase inhibitor treatment induces MHV68 reactivation. (A) Whole-cell extracts from A20-HE1 or A20-HE2 cells treated for 72 h with 5azaCdR at the indicated concentrations and probed with rabbit-derived MHV68-lytic antisera. (B) Whole-cell extracts from A20-HE2 cells treated for 72 h with 5azaCdR at the indicated concentration and probed with Orf59 antibody. The asterisk in the beta-actin blot indicates residual Orf59 antibody that was not efficiently stripped prior to β-actin antibody hybridization. (C) A20-HE2 cells were treated for 72 h with the indicated concentrations of 5azaCdR. RT-PCR was performed to detect distal gene 50 promoter-initiated transcripts (containing E0) as well as E1 and E2 exon-containing transcripts. RT-PCR was also performed for a lytic transcript (viral DNA polymerase) as a verification of the induction of the MHV68 replication cycle. The asterisks indicate the band corresponding to the characterized spliced gene 50 transcripts (the larger amplification products detected represent either unspliced transcripts or contaminating viral DNA). The products from the RT-PCRs for both untreated and treated cells were electrophoresed in parallel on the same agarose gel alongside other samples unrelated to this experiment; these irrelevant lanes have been removed for presentation, and the amplification products arising from the untreated samples have been juxtaposed with those obtained with RNA prepared from 5azaCdR-treated cells.
Distal gene 50 promoter activity is repressed by methylation.
Methylation of promoters is associated with transcriptional repression and is often studied in promoter regions containing dense CpG distributions. The core region of the proximal gene 50 promoter, defined in murine RAW264.7 macrophages (30), contains no CpGs and, thus, is unlikely to be regulated by DNA methylation. However, the distal gene 50 promoter contains several CpGs that are targeted for methylation in vivo (14). In the murine RAW macrophage cell line, the 250-bp region upstream of gene 50 exon 0 conveys the greatest promoter activity and contains only four CpGs (note that there is an additional CpG within exon 0 that was not included in the reporter construct discussed below) (Fig. 2A). It was unclear whether the low number of CpGs in the distal gene 50 promoter would, when methylated, have a significant impact on promoter activity. To assess directly whether the methylation of these four CpGs could repress distal gene 50 promoter activity, we performed reporter assays by using the pCpG luciferase vector containing the 250-bp distal gene 50 promoter region following in vitro methylation with the methyltransferase M.SssI. The pCpG-Basic vectors are devoid of CpGs (26), thus allowing the targeted methylation of cloned promoter sequences in vitro by using bacterial CpG methyltransferase M.SssI treatment followed by the transfection of target cells. This approach eliminates the arduous and inefficient patch methylation techniques (e.g., methylation cassette assay) necessary with reporter vectors containing CpGs to avoid the repressive effects of a methylated vector backbone (26). Mock methylation (in the absence of M.SssI) did not alter either basal or LPS-induced promoter activity from that of the unmanipulated vector. M.SssI treatment reduced basal distal gene 50 promoter activity and also repressed the strong induction seen following the LPS treatment of transfected cells. This finding suggests that despite the sparse distribution of CpGs within the distal gene 50 promoter region, DNA methylation is still capable of repressing gene 50 promoter activity.
FIG. 2.
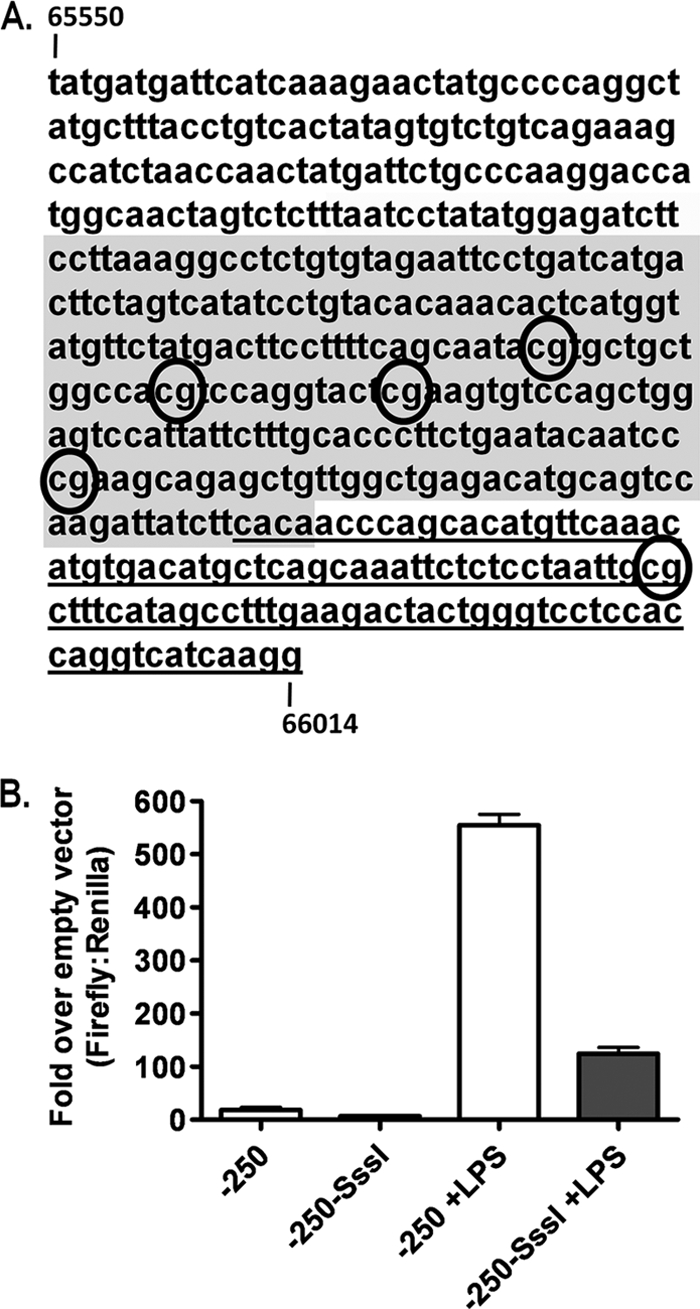
In vitro methylation reduces distal gene 50 promoter activity. (A) Sequence of distal gene 50 promoter from bp 65550 to bp 66014. The CpGs examined by bisulfite analysis are indicated (numbered 1 to 5). Exon 0 is underlined, and the shaded region represents the −250 promoter region examined in B. (B) Reporter assay for distal gene 50 promoter activity in RAW264.7 murine macrophages with or without LPS (5 μg/ml) following M.SssI treatment or mock methylated. Data are representative of data from at least three independent transfections from two independent M.SssI treatments. P values were determined by using an unpaired Student's t test.
Generation of mice lacking functional Dnmt3a and Dnmt3b alleles in B cells.
Our laboratory has studied MHV68 infection of several conditional animals using a recombinant virus expressing Cre-recombinase from a cytomegalovirus (CMV) promoter-driven expression cassette introduced into a phenotypically “neutral” locus of the viral genome located between Orf27 and Orf29b. Since the deletion of Dnmt3a or Dnmt3b results in early-postnatal or embryonic lethality, respectively, we had originally intended to study the effect of the de novo methyltransferase deletion in Dnmt3a2loxP/2loxP or Dnmt3b2loxP/2loxP animals infected with MHV68-Cre, which would result in the deletion of these methyltransferases only in MHV68-infected cells. However, RT-PCR experiments with sorted splenocyte populations revealed that these enzymes are expressed in naïve B cells (data not shown). We therefore reasoned that preexisting DNMT3a and DNMT3b may be present at sufficient levels to methylate the MHV68 genome, rendering the Cre-mediated deletion of the Dnmt3a and Dnmt3b genes ineffective in blocking the methylation of the viral genome. Thus, to eliminate DNMT3a and DNMT3b in naïve B cells prior to infection, we crossed Dnmt3a2loxP/2loxP, Dnmt3b2loxP/2loxP, or Dnmt3a2loxP/2loxP Dnmt3b2loxP/2loxP conditional mice (12, 24) with knock-in mice expressing Cre-recombinase under the control of the CD19 promoter from one of the CD19 alleles (CD19+/Cre). Genotyping confirmed the presence of rearranged Dnmt3a and Dnmt3b alleles in CD19+/Cre Dnmt3a2loxP/2loxP Dnmt3b2loxP/2loxP mice (which we will refer to as CD19+/Cre Dnmt3a/3bc/c mice), while only the intact, unmodified 2loxP alleles were detected in CD19+/+ Dnmt3a2loxP/2loxP Dnmt3a3b2loxP/2loxP littermate controls (which we will refer to as CD19+/+ Dnmt3a/3bc/c mice) (Fig. 3).
FIG. 3.
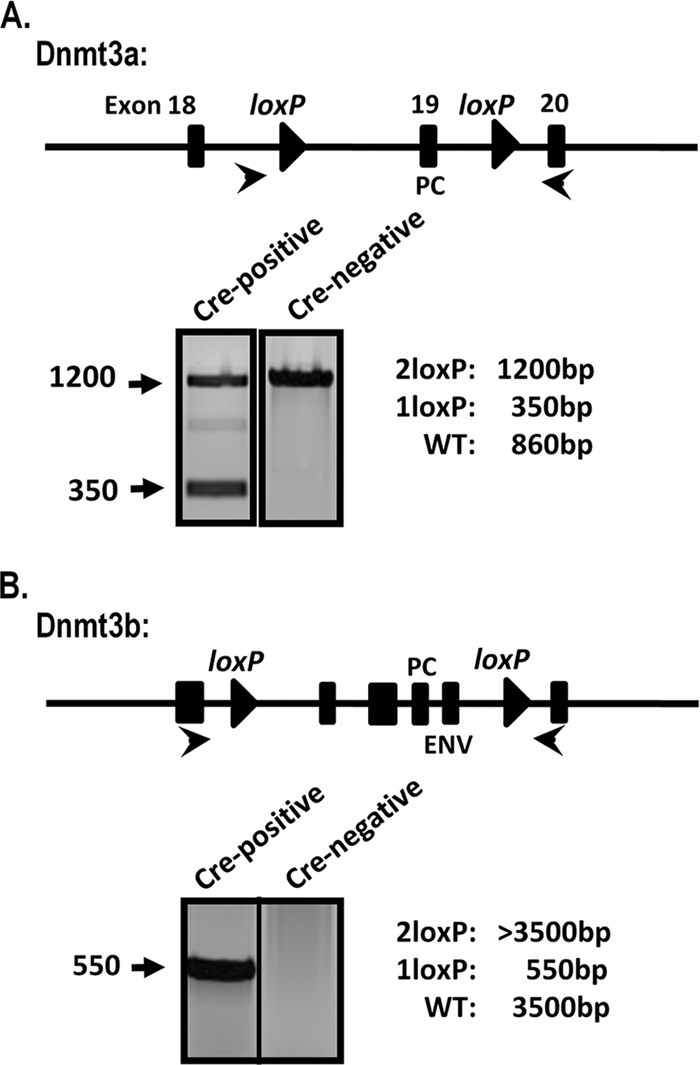
Rearrangement of Dnmt3a and Dnmt3b alleles in CD19+/Cre Dnmt3a/3bc/c splenocytes. Total genomic DNA was prepared from splenocytes from CD19+/Cre Dnmt3a/3bc/c (Cre-recombinase-positive) mice or littermate controls (Cre-recombinase negative). The schematic indicates the location of Dnmt3a or Dnmt3b exons (rectangles) and loxP sites (triangles). Cre-mediated rearrangement deletes exons encoding key DNMT catalytic domains: PC (Pro-Cys) and ENV (Glu-Asn-Val). PCR was performed with the primers indicated (arrows), and representative reactions are shown. WT, wild type.
Loss of DNMT3a and DNMT3b expression in B cells does not alter global B- or T-cell responses during primary immunization or viral infection.
Upon the initiation of studies with CD19+/Cre Dnmt3a/3bc/c mice, we were concerned that phenotypes observed upon MHV68 infection might relate to perturbations in B-cell function upon the loss of DNMT3a and/or DNMT3b expression. DNA methylation was shown to be important for several aspects of B-cell development, particularly variable diversity-joining segments (VDJ) rearrangements and allelic exclusion during light-chain selection (41). However, less is known about the role of DNA methylation in mature or activated B cells. One study implicated a role for DNA methylation in regulating activation-induced cytidine deaminase (AID) expression in germinal-center B cells (16). In addition, an independent study using a Cre-recombinase-expressing retrovirus to mediate the deletion of Dnmt3a and Dnmt3b in hematopoietic stem cells (HSCs) revealed a requirement for either DNMT3a or DMNT3b (but not DNMT3a or DNMT3b alone) in maintaining the self-renewal capacity (47). This study, however, examined the effect of the deletion of Dnmt3a and Dnmt3b at an early stage of differentiation, while in CD19+/Cre Dnmt3a/3bc/c mice, the Dnmt3 alleles are deleted at the pro-B-cell stage, prior to the migration of B cells from the bone marrow to the periphery. Several studies demonstrated the importance of B cells in the control of MHV68 infection (9, 13, 17, 51): it is formally possible that the deletion of Dnmt3a or Dnmt3b would affect some aspect of mature B-cell development or function and thereby compromise our ability to assess the specific role of DNA methylation in regulating MHV68 transcription in infected cells directly. We therefore examined B-cell phenotypes in naïve CD19+/Cre Dnmt3a/3bc/c mice to ensure that normal B-cell development had occurred and in the context of nonviral immunogens or viral infection to verify that B-cell effector functions were intact. Naïve or mock-immunized (PBS) CD19+/Cre Dnmt3a/3bc/c mice displayed no obvious differences in total B- and T-cell numbers relative to those of littermate controls (CD19+/+ Dnmt3a/3bc/c) (Fig. 4A and B). Primary immunization with either OVA-CFA or SRBCs revealed no gross alterations in either B-cell activation (CD69hi), germinal-center formation (GL7+ CD95+), or class switching (IgD−) compared to littermate controls (Fig. 4A). Importantly, the Dnmt3a and Dnmt3b alleles are intact in T cells from CD19+/Cre Dnmt3a/3bc/c mice. However, to determine if unidentified alterations in B-cell function (e.g., aberrant cytokine production) may have altered global T-cell responses, we also assessed the total T-cell number and activation status (Fig. 4B). Total CD4+ and CD8+ T-cell populations were equivalent in CD19+/Cre Dnmt3a/3bc/c mice versus littermate controls, as were the percentages of cells bearing an activated effector phenotype (CD44hi CD62Llo). As these immunogens were nonreplicating and nonpathogenic, we also examined global B- and T-cell responses following infection of mice with either MHV68 or the arenavirus lymphocytic choriomeningitis (LCMV) to determine if any immune defects could be detected in the context of viral infection (Fig. 4A and B). Notably, as shown in Fig. 4A and B, MHV68 and LCMV infections also failed to reveal any gross alterations in the expansion or activation B- and T-cell populations.
FIG. 4.
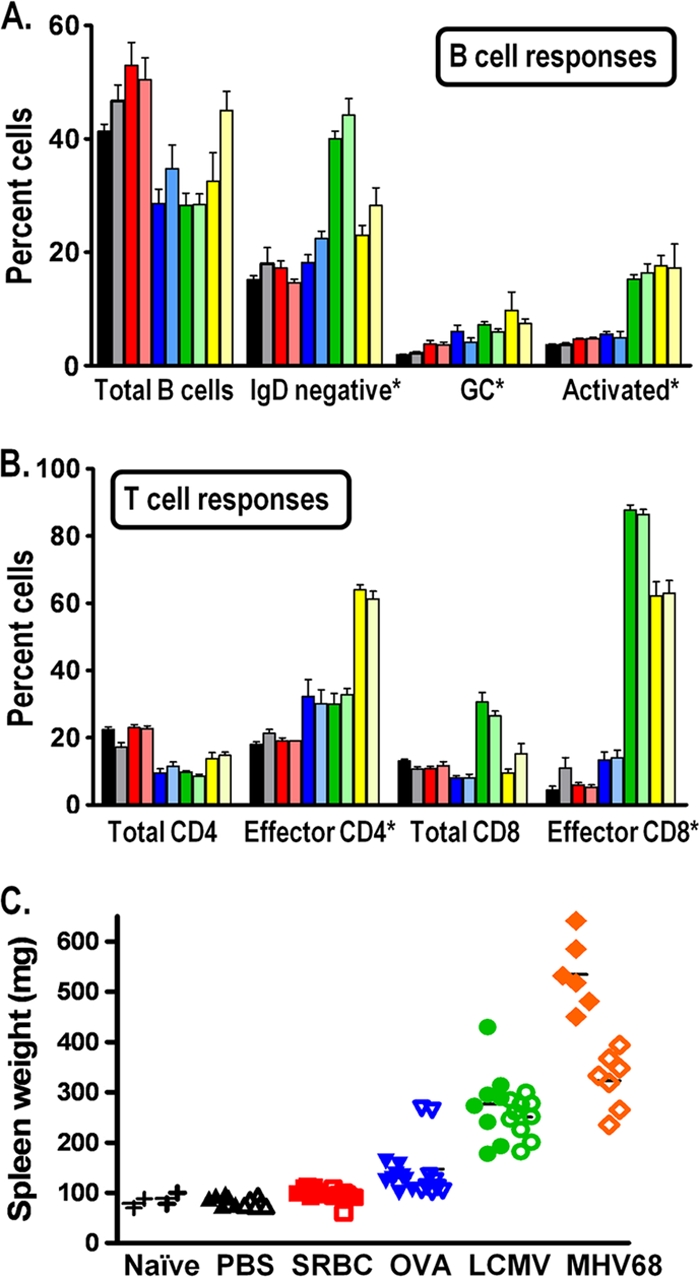
Global immune responses are not altered in CD19+/Cre Dnmt3a/3bc/c mice. (A and B) B-cell (A) and T-cell (B) responses of CD19+/Cre Dnmt3a/3bc/c mice were examined. Splenocytes recovered from naïve mice (black/gray bars), mice immunized with SRBCs (day 18) (red bars) or OVA-CFA (day 14) (blue bars), or mice infected with LCMV Armstrong (day 8) (green bars) or MHV68 (day 18) (yellow bars) were examined by flow cytometry for the indicated populations. The darker bars correspond to cells recovered from mice lacking the expression of Dnmt3a and Dnmt3b in B cells (CD19+/Cre Dnmt3a/3bc/c mice), while the lighter bars indicate cells recovered from littermate control Dnmt3a/Dnmt3b-sufficient mice (CD19+/+ Dnmt3a/3bc/c mice). All populations were gated on live cells. The percentage of total B cells, total CD4+ T cells, and total CD8+ T cells are given relative to the numbers of total splenocytes. The asterisks in A and B indicate the gating of a specific population of splenocytes as follows: the percentages of specific B-cell populations (GC [germinal-center] B cells [GL7+/CD95+] and activated B cells [CD69high]) are given relative to numbers of total CD19+ cells (A), while the percentages of effector CD4+ T cells (CD44high/CD62Llow) and effector CD8+ T cells (CD44high/CD62Llow) are given relative to the numbers of either total CD4+ T cells or total CD8+ T cells, respectively (B). (C) Spleen weights from naïve mice or mice immunized or infected as in described above (A and B).
Although no obvious alterations in the global immune responses were detected in MHV68-infected mice, upon the harvesting of spleens from MHV68-infected CD19+/Cre Dnmt3a/3bc/c mice, we noted a dramatic increase in splenomegaly relative to that of the CD19+/+ Dnmt3a/3bc/c littermate controls (Fig. 4C). Importantly, this increased splenomegaly was not seen in the context of naïve CD19+/Cre Dnmt3a/3bc/c mice or mice immunized with nonviral agents (Fig. 4C). Splenomegaly is a common feature of EBV-induced infectious mononucleosis as well as early MHV68 infection and is associated with inflammatory viral infection and a demand for increased antigen clearance and antibody production. As discussed above, enlarged spleens from MHV68-infected CD19+/Cre Dnmt3a/3bc/c mice shared a cellular composition similar to that from infected littermate control CD19+/+ Dnmt3a/3bc/c mice (Fig. 4A and B). Increased splenomegaly therefore does not appear to be a result of the expansion of any specific B- or T-cell subset. As discussed below, this finding supports the likelihood that it arises as a consequence of prolonged MHV68 replication in CD19+/Cre Dnmt3a/3bc/c mice. To determine if this phenotype was MHV68 specific rather than a general response to viral infections, we also assessed spleen weights from mice infected with LCMV Armstrong at the peak of splenomegaly (day 8 postinfection) (Fig. 4C). Notably, spleen weights were comparable between LCMV-infected CD19+/Cre Dnmt3a/3bc/c mice and littermate controls, suggesting that the enhanced splenomegaly can be attributed to an aspect of MHV68 infection uniquely affected by the absence of DNMT3a and DNMT3b in B cells.
Antibody responses are intact in mice lacking expression of DNMT3a and DNMT3b in B cells.
As mentioned above, the majority of studies linking B cells and DNA methylation have involved the generation of the B-cell receptor and antibody production, and experiments have demonstrated that the IgG subtype dominates the antibody response to MHV68 infection (44). To address the possibility that IgG production was compromised in CD19+/Cre Dnmt3a/3bc/c mice, we used MHV68 infection or the well-established NP-CGG system to examine primary and secondary B-cell responses, respectively. At both 30 days following primary immunization with NP-CGG and 4 days following secondary immunization with NP, ELISAs revealed no gross differences in the levels of serum NP-specific IgG (data not shown). To ensure that normal serum levels were not reflective of disparate antibody production levels by individual cells, the total numbers of antibody-secreting cells were determined by ELISPOT analysis (Fig. 5A). Following secondary immunization, the numbers of both NP-specific and total IgG-secreting cells were comparable between CD19+/Cre Dnmt3a/3bc/c mice and littermate controls (Fig. 5A). This indicates that total, as well as antigen-specific, IgG production was uncompromised in CD19+/Cre Dnmt3a/3bc/c mice and that these mice can mount both primary and secondary antibody responses to a specific immunogen.
FIG. 5.
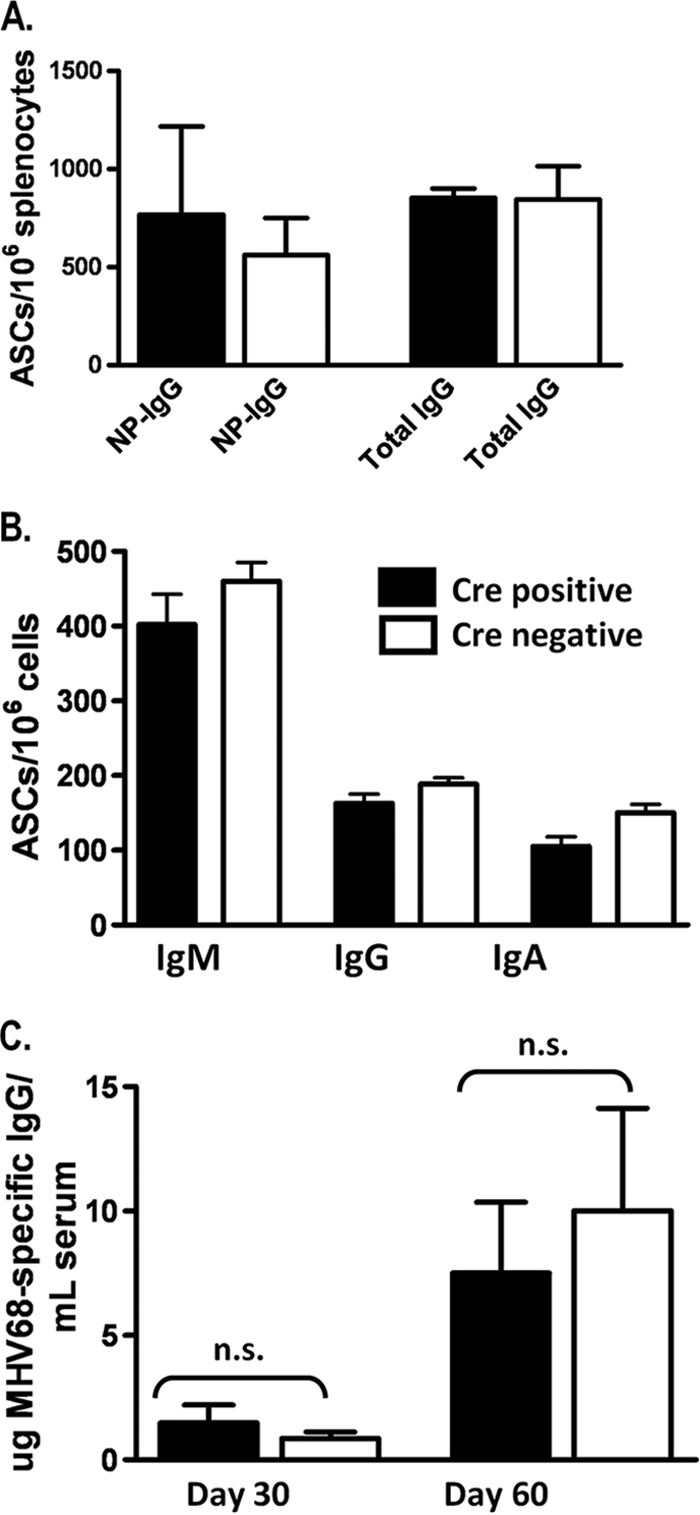
The antibody response of CD19+/Cre Dnmt3a/3bc/c mice is not grossly altered. (A) Numbers of total and NP-specific IgG-secreting B cells (from spleen) were determined by ELISPOT analysis 4 days following secondary immunization. (B) Numbers of IgM-, IgG-, or IgA-secreting cells were determined by ELISPOT analysis at day 18 following MHV68 infection. (C) MHV68-specific serum IgG levels were determined by ELISA at the indicated times postinfection. Data were analyzed by an unpaired Student's t test. n.s., not significant.
To formally address the possibility that B-cell function might be compromised in CD19+/Cre Dnmt3a/3bc/c mice following MHV68 infection, we performed ELISPOT and ELISA analyses of MVH68-infected mice (Fig. 5B and C). At day 18 postinfection, total numbers of splenic IgM-, IgG-, and IgA-secreting cells were similar between CD19+/Cre Dnmt3a/3bc/c and CD19+/+ Dnmt3a/3bc/c mice, indicating that non-MHV68-specific antibody production is intact in CD19+/Cre Dnmt3a/3bc/c mice in the context of MHV68 infection (Fig. 5B). The frequency of MHV68-specific ASCs was below the limit of detection at this time, so to determine MHV68-specific IgG responses in CD19+/Cre Dnmt3a/3bc/c and littermate control mice, we performed serum ELISAs at later times postinfection (Fig. 5C). Notably, no statistically significant differences in MHV68-specific IgG levels were observed at day 30 or 60 postinfection (Fig. 5C), indicating that levels of virus-specific antibody production are largely unaffected by the loss of DNMT3a and DNMT3b expression.
To further investigate MHV68 infection in the absence of DNMT3a and DNMT3b expression in B cells, we assessed virus infection in the spleens of infected mice at the peak of latency (day 18) to determine whether there was any obvious perturbation of the infected B-cell subsets. We have previously shown that the majority of infected B cells in the spleen at the peak of latency exhibit a germinal-center phenotype (10, 53). Our initial analysis of the germinal-center response in MHV68-infected CD19+/Cre Dnmt3a/3bc/c mice and littermate controls failed to detect any difference between DNMT3a/3b-sufficient and DNMT3a/3b-deficient mice (Fig. 4A). Furthermore, using a recently described recombinant MHV68 that expresses yellow fluorescent protein (YFP), allowing the identification of B-cell subsets infected at early times after the establishment of latency (10), we determined that a similar frequency of virus-infected B cells exhibits a germinal-center phenotype in CD19+/Cre Dnmt3a/3bc/c mice and littermate control mice (Fig. 6C).
FIG. 6.

Evidence of ongoing lytic replication in CD19+/Cre Dnmt3a/3bc/c mice during early latency. (A) Limiting-dilution PCR analysis to determine frequencies of genome-positive cells. (B) Limiting-dilution analysis of intact and disrupted cells plated onto MEF monolayers to determine frequencies of cells reactivating from latency and to assess the contribution of preformed infectious virus to CPE. Genome and reactivation frequencies are elevated in Cre-positive mice, and the CPE observed upon the plating of disrupted cells indicates ongoing lytic replication. (C) Evidence that the MHV68-positive reservoir is not altered in CD19+/Cre Dnmt3a/3bc/c mice. Mice were infected with MHV68-YFP virus, and splenocytes were analyzed at day 18 for YFP expression and germinal-center markers (CD95 and GL7). The majority of YFP-positive (MHV68-infected) cells bear hallmarks of germinal-center B cells in both CD19+/Cre Dnmt3a/3bc/c and CD19+/+ Dnmt3a/3bc/c mice.
Although the experiments described above are not exhaustive, we were unable to find any evidence that the deletion of Dnmt3a and Dnmt3b following CD19 promoter-driven Cre-recombinase expression significantly alters B-cell development or B-cell responses, nor does it appear to significantly change the general scope of the lymphocyte immune response. We therefore conclude from these control experiments that the phenotypes observed for CD19+/Cre Dnmt3a/3bc/c mice upon MHV68 infection are likely attributable solely to the consequences of the loss DNMT3a and DNMT3b function in the context of virus-infected B cells.
Deletion of the de novo methyltransferases DNMT3a and DNMT3b in B cells results in prolonged MHV68 replication in the spleen.
The single deletion of either Dnmt3a or Dnmt3b in B cells had no impact on any parameter of MHV68 latent infection examined, including the establishment of latency and reactivation from splenocytes harvested at day 18 following MHV68 infection (data not shown). However, analysis of the frequency of virus-infected splenocytes harvested from MHV68-infected double-conditional knockout mice (CD19+/Cre Dnmt3a/3bc/c mice) revealed a 5-fold increase in viral genome-positive cells in splenocytes from CD19+/Cre Dnmt3a/3bc/c mice relative to those in splenocytes from their littermate (CD19+/+ Dnmt3a/3bc/c) controls (1/200 in littermate control mice versus 1/40 in the double-knockout mice) (Fig. 6A). Furthermore, the frequency of cells reactivating the virus was also augmented about 7-fold in the double-knockout CD19+/Cre Dnmt3a/3bc/c mice (Fig. 6B). In this assay, mechanically disrupted cells were plated in parallel with intact cells to control for the presence of preformed infectious virus, which is largely absent in the spleen by day 16 postinfection in wild-type mice. As expected, in the CD19+/+ Dnmt3a/3bc/c littermate control splenocytes, there was little or no preformed infectious virus detected, whereas disrupted splenocytes from CD19+/Cre Dnmt3a/3bc/c mice contained significant levels of preformed infectious virus (Fig. 7B). This abnormally high level of preformed infectious virus most likely contributes to both the increased frequency of reactivation and viral genome-positive splenocytes in the CD19+/Cre Dnmt3a/3bc/c splenocytes as well as increased splenomegaly and suggests that the cessation of MHV68 replication is altered in the absence of DNMT3a and DNMT3b.
FIG. 7.
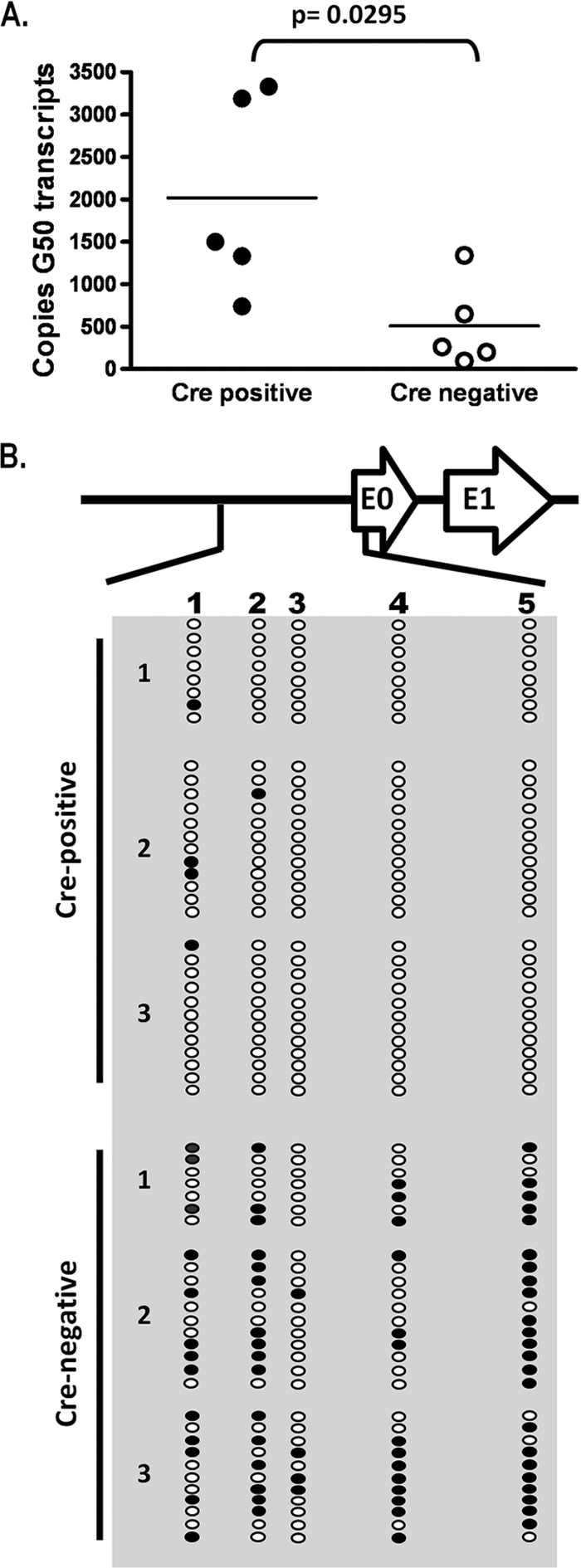
Elevated gene 50 transcription is accompanied by hypomethylation of the gene 50 promoter at day 18. (A) Quantitative RT-PCR for total gene 50 (G50) (exon 2) transcripts on cDNA generated from total splenocyte RNA. Transcript levels are elevated in Cre-positive mice relative to levels of Cre-negative littermate controls. P values were determined by using an unpaired Student's t test. (B) Bisulfite PCR analysis of the distal gene 50 promoter region at day 18. Total genomic DNA was extracted from magnetically purified CD19+ splenocytes from mice at day 18 postinfection and subjected to bisulfite PCR analysis. The region analyzed corresponds to bp 65550 to 66014 upstream of gene 50 exon 0 (Fig. 2). Each column represents an individual CpG dinucleotide, and each row represents an individual PCR clone. Filled circles represent methylated cytosines, while open circles represent unmethylated cytosines. Three individual mice were analyzed for Cre-positive and Cre-negative groups.
The distal gene 50 promoter is hypomethylated in CD19+/Cre Dnmt3a/3bc/c splenocytes during early latency.
Because gene 50 encodes the primary protein required for the induction of virus replication, we used qRT-PCR to examine splenocytes for evidence of increased gene 50 transcription in CD19+/Cre Dnmt3a/3bc/c mice compared to littermate controls at day 18 postinfection. Notably, total gene 50 transcript levels were elevated in CD19+/Cre Dnmt3a/3bc/c mice relative to those of CD19+/+ Dnmt3a/3bc/c littermate controls (Fig. 7A), suggesting that in the absence of DNMT3a and DNMT3b, the distal gene 50 promoter is more transcriptionally active at day 18 postinfection than in DNMT3a- and DNMT3b-sufficient B cells.
We hypothesized that the presence of infectious virus and increased gene 50 transcript levels in the spleens of CD19+/Cre Dnmt3a/3bc/c mice at day 18 postinfection were a consequence of the compromised methylation of gene 50 in Dnmt3a/Dnmt3b-null B cells. To assess this, we used bisulfite PCR to determine the methylation status of the four CpGs within the core distal gene 50 promoter as well as the single CpG located near the 5′ end of exon 0 (Fig. 2A) in CD19+ splenocytes from CD19+/Cre Dnmt3a/3bc/c mice and CD19+/+ Dnmt3a/3bc/c littermate control mice (Fig. 7B). Nearly half of all CpGs in the distal gene 50 promoter were methylated in B cells recovered from littermate control mice, while less than 4% of these sites were methylated in B cells recovered from the CD19-Cre-expressing mice (Fig. 7B). The presence of one or two methylated CpGs in several clones, versus two or more sites in clones from CD19+/+ Dnmt3a/3bc/c mice, suggests that these amplicons are derived from hypomethylated viral genomes within infected cells rather than preformed virions within the cell, which would be devoid of any CpG methylation.
Restoration of wild-type latency and distal gene 50 promoter methylation at later times postinfection.
Previous studies with transgenic mice and recombinant MHV68 overexpressing the gene 50 protein demonstrated that the overexpression of the gene 50 protein, or a failure to properly control lytic replication, results in a failure to establish either long-term MHV68 latency or persistent, high-level viral replication and associated pathologies (9, 13, 33). We therefore analyzed MHV68 genome loads and reactivation at day 42 postinfection to determine if increased lytic replication at day 18 postinfection in CD19+/Cre Dnmt3a/3bc/c mice had compromised aspects of latent infection as well as to determine whether ongoing virus replication in the spleen was still detectable. Notably, there was little or no evidence of MHV68 reactivation from splenocytes of either CD19+/Cre Dnmt3a/3bc/c or CD19+/+ Dnmt3a/3bc/c mice at day 42 postinfection, indicating that lytic replication had been largely silenced from that seen for CD19+/Cre Dnmt3a/3bc/c mice at day 18 postinfection (data not shown). Furthermore, by day 42 postinfection, the frequency of viral genome-positive cells in splenocytes recovered from CD19+/Cre Dnmt3a/3bc/c mice was equivalent to that from CD19+/+ Dnmt3a/3bc/c littermate controls (Fig. 8A). This suggested that at some point between days 18 and 42 postinfection, the transcription of gene 50 had been controlled in such a way that allowed the efficient establishment and maintenance of MHV68 latency, even in the absence of the de novo methyltransferases DNMT3a and DNMT3b.
FIG. 8.
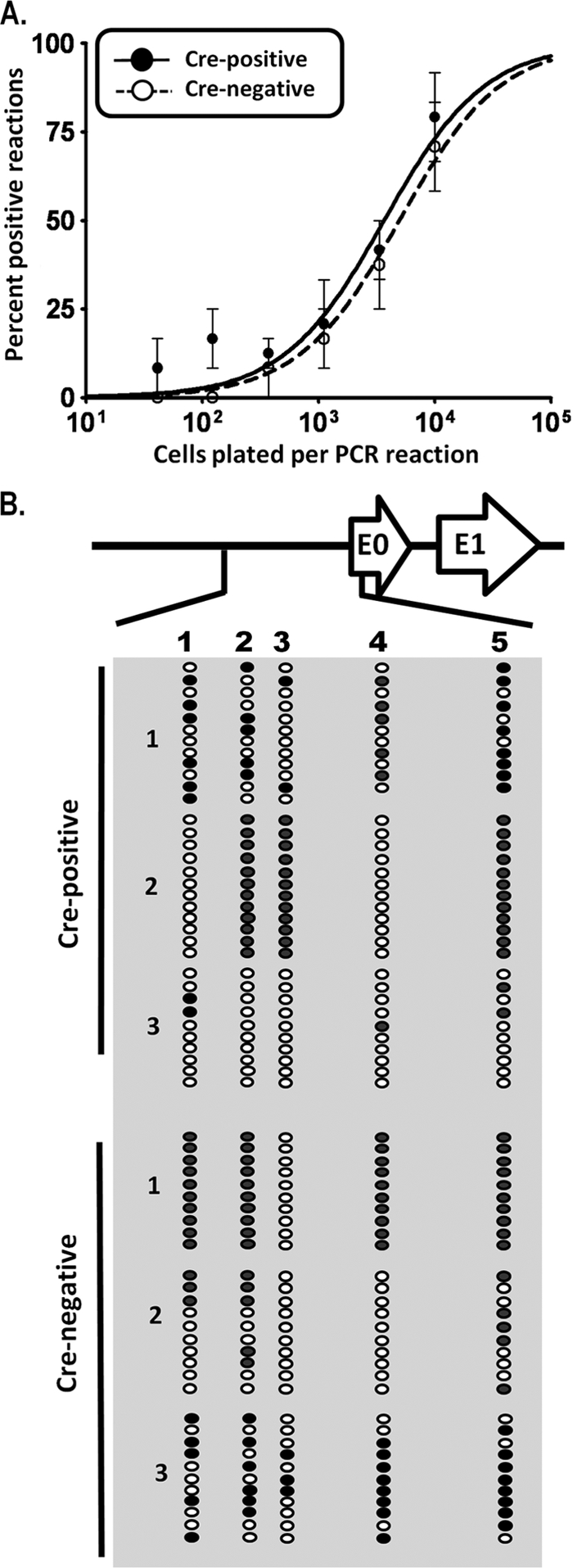
Normal genome frequency at day 42 postinfection is accompanied by restoration of gene 50 promoter methylation. (A) Limiting-dilution analysis to determine frequencies of genome-positive cells in Cre-positive and Cre-negative mice. Reactivation was negligible for both groups at this time. (B) Bisulfite PCR analysis of the distal gene 50 promoter region at day 42 postinfection. Total genomic DNA was extracted from purified CD19+ splenocytes isolated from mice at day 42 postinfection and subjected to bisulfite PCR analysis as described in the legend to Fig. 7.
To determine if the cessation of aberrant lytic replication in the spleens of MHV68-infected CD19+/Cre Dnmt3a/3bc/c mice correlated with the appearance of the CpG methylation of the distal gene 50 promoter, we again performed bisulfite PCR on CD19+ splenocytes recovered from mice at day 42 postinfection. Surprisingly, the extent of methylation of the distal gene 50 promoter was nearly equivalent between CD19+/Cre Dnmt3a/3bc/c mice and littermate controls, with the exception of a single infected CD19+/Cre Dnmt3a/3bc/c mouse (mouse 3) (Fig. 8B). This finding is interesting in that it supports the existence of a DNMT3a/DNMT3b-independent mechanism of de novo DNA methylation (perhaps mediated by the maintenance methyltransferase Dnmt1). Notably, consistent with a DNMT3a/DNMT3b-independent mechanism being involved in methylating the distal gene 50 promoter, the pattern of CpG methylation in CD19+/Cre Dnmt3a/3bc/c mice is distinct from that observed for littermate controls (overall, more methylation observed at CpG 3 and less observed at CpG 4) (Fig. 8B). The observation that the distal gene 50 promoter becomes methylated in DNMT3a- and DNMT3b-deficient B cells further strengthens the hypothesis that this epigenetic mechanism plays an important role in regulating MHV68 gene 50 promoter activity such that selective pressure exists to methylate and thus silence gene 50 expression to allow the establishment of a latent infection.
Dnmt3a- and Dnmt3b-independent methylation is targeted specifically to the distal gene 50 promoter.
As discussed above, the area encompassing MHV68 lytic genes, particularly gene 50, is dramatically CpG suppressed, suggesting that these regions or the viral genome has been subjected to intense DNA methylation over the course of MHV68 evolution, resulting in the loss of CpGs. In an effort to further investigate the importance of methylation in the gene 50 promoter region, we examined the promoter regions of two other lytic genes, Orf72 and M11, encoding the viral D-type cyclin and bcl2, respectively. We have confirmed promoter activity in the intergenic region between these two viral genes in the context of v-cyclin (1), and unpublished observations suggested that the expression of v-bcl2 (encoded on the opposite strand of the genome) shares this promoter located between the 5′ ends of the two genes (Fig. 9). At day 18 postinfection, this region is extensively methylated in CD19+/+ Dnmt3a/3bc/c littermate control mice and, much like the distal gene 50 promoter, is hypomethylated in CD19+/Cre Dnmt3a/3bc/c mice (Fig. 9A). However, while the extent of methylation in the distal gene 50 promoter increased with ongoing latent infection, the intergenic region between Orf72 and M11 in CD19+Cre+ Dnmt3a/3bc/c mice remained hypomethylated at day 42 (Fig. 9B). These results suggest that, unlike the distal gene 50 promoter, methylation of this region is not required for the efficient establishment of long-term latency. Furthermore, these results argue that there is likely selective pressure to methylate the distal gene 50 promoter for the maintenance of latency. Finally, the observation that the Orf72/M11 intergenic region remains hypomethylated at day 42 postinfection argues strongly against the appearance of methylation at the distal gene 50 promoter arising from the MHV68 infection of a subset of B cells in which the conditional Dnmt3a and Dnmt3b genes were not inactivated by Cre-recombinase-mediated deletion.
FIG. 9.

Bisulfite PCR analysis of the v-cyclin and v-bcl2 promoter region at days 18 and 42 postinfection. Total genomic DNA was extracted from purified CD19+ splenocytes from mice at day 18 (A) or day 42 (B) postinfection and subjected to bisulfite PCR analysis. The region analyzed corresponds to bp 102548 to 103915, between the 5′ ends of each gene. Each column represents an individual CpG dinucleotide, and each row represents an individual PCR clone. Filled circles represent methylated cytosines, while open circles represent unmethylated cytosines. Dashes represent CpGs whose methylation status was indeterminate upon sequence analysis. Three individual mice were analyzed for Cre-positive and Cre-negative groups.
DISCUSSION
The analysis of MHV68 infection of mice with a deficiency in DNMT3a and DNMT3b expression in B cells provides direct evidence that DNA methylation plays an important role in the regulation of MHV68 gene 50 promoter activity. The demethylation of the viral genome is associated with reactivation from latently infected cells, and concomitantly, methylation is associated with distal gene 50 promoter silencing and reduced transcriptional activity both in vitro and in vivo. Because DNA in infectious herpesvirus virions has been shown to be unmethylated, we hypothesized that the methylation of the MHV68 genome would require the action of the de novo methyltransferases DNMT3a and DNMT3b. The global deletion of either Dnmt3a or Dnmt3b results in embryonic lethality or death soon after birth. We therefore sought to examine the effect of the deletion the de novo methyltransferases in B cells, the primary targets of MHV68 latency, by the CD19 promoter-driven Cre-recombinase-mediated deletion of Dnmt3a and Dnmt3b. We found evidence of aberrant lytic replication in splenocytes from mice with a conditional deletion of these enzymes, as evidenced by splenomegaly, the presence of ongoing persistent virus replication, and increased gene 50 transcription during the early stages of latent infection (day 18 postinfection). The distal gene 50 promoter, which has been shown to be methylated throughout latent infection, was hypomethylated in B cells lacking DNMT3a and DNMT3b, supporting in vitro data demonstrating that DNA methylation can repress distal gene 50 promoter activity. Importantly, aberrant lytic replication was resolved in these animals by day 42 postinfection, accompanied by the restoration of distal gene 50 promoter methylation to wild-type levels. The latter observation indicates that there is a DNMT3a- and DNMT3b-independent mechanism for methylating the distal gene 50 promoter. In contrast, the promoters for two other lytic viral genes (v-cyclin and v-bcl2) remained hypomethylated at both days 18 and 42 postinfection. Furthermore, the failure to initially control MHV68 lytic replication was most likely not due to compromised immune function as a result of the deletion of Dnmt3a and Dnmt3b in B cells, as immune responses appeared normal in CD19+/Cre Dnmt3a/3bc/c mice compared littermate controls. Taken together, these data are in favor of a model in which DNA methylation is an important mechanism regulating gene 50 expression and that alternative mechanisms exist to specifically target de novo methylation to the distal gene 50 promoter in the absence of DNMT3a and DNMT3b.
Relative importance of methylation in regulating gene 50 transcription.
The splenomegaly, increased gene 50 transcription, presence of infectious virus, and hypomethylation of the distal gene 50 promoter at day 18 all support an important role for the DNMT3a- and DNMT3b-mediated methylation of the viral genome. We were surprised, however, to find that distal gene 50 promoter methylation was restored at later times postinfection and is associated with the resolution of aberrant, persistent virus replication. Two possible explanations for this finding are that (i) the MHV68 genome is targeted for methylation by a DNMT3a- and DNMT3b-independent mechanism (perhaps mediated by DNMT1) and (ii) the observed methylation of the MHV68 genome at late times postinfection arises in cells in which the Cre-mediated deletion of the Dnmt3a and Dnmt3b alleles has been inefficient (i.e., not all alleles have been deleted in an appropriate time frame). We favor the former explanation, since the intergenic region between the v-cyclin and v-bcl2 genes remained hypomethylated at day 42 postinfection, indicating the absence of de novo methyltransferase activity in MHV68-infected B cells.
What evidence exists for selective pressure to methylate the gene 50 promoter? As discussed above, the MHV68 genome is highly CpG suppressed overall. However, the gene 50 locus is even more so, indicating that this region has been subjected to methylation as the virus has evolved, leading to an eventual loss of CpG dinucleotides through the mutagenesis of CpGs to TpGs (19). Also, we demonstrate that the distal gene 50, v-cyclin, and v-bcl2 promoter regions are hypomethylated at day 18 in DNMT3a/DNMT3b-deficient mice. However, while gene 50 promoter methylation is restored to near-wild-type levels by day 42 postinfection, the intergenic region between the v-cyclin- and v-bcl2-coding exons does not become methylated in these mice, suggesting that the methylation and transcriptional repression of this locus are not the primary means of negative regulation. Also, the intergenic region between v-cyclin and v-bcl2 is more CpG dense, perhaps suggesting that the methylation of this segment of the genome has been less aggressive as MHV68 has evolved relative to the gene 50 region. This targeted repression is logical, as the expression of gene 50 alone is necessary and sufficient for MHV68 reactivation, while v-cyclin and v-bcl2 have less influential roles in terms of driving virus reactivation/replication in B cells.
The role of DNA methylation in EBV latency has been studied at great length. Numerous studies have shown that methylation is a key component in determining specific latent-gene promoter usage, which corresponds to distinct latency programs in infected cells. We cannot formally exclude the possibility that the disruption of Dnmt3a and Dnmt3b affects the methylation of other MHV68 latency promoters, thereby contributing to altered kinetics in the resolution of lytic replication. For example, our laboratory has demonstrated that MHV68 LANA is transcribed from multiple promoters as part of a multicistronic transcript analogous to EBV latency transcripts containing EBNA-1 (2). Notably, KSHV LANA was shown to negatively regulate gene 50 (27, 28); although this relationship has not been corroborated for MHV68 LANA and gene 50, it is possible that altered LANA transcription in the absence of DNMT3a and DNMT3b results in an altered form of latency permitting prolonged lytic gene expression. Indeed, bisulfite PCR analyses show that the terminal-repeat region containing the MHV68 LANA p1 promoter is densely methylated in latent MHV68-infected cell lines (K. S. Gray and S. H. Speck, unpublished observations). Our reporter assays demonstrating that methylation directly represses the distal gene 50 promoter, in conjunction with the high degree of CpG suppression, support a direct role for DNA methylation in specifically regulating gene 50 transcription. However, given the wealth of data demonstrating the importance of DNA methylation in regulating EBV latency programs, it is most likely that this modification is key to regulating both lytic and latent gammaherpesvirus gene expression.
Gene 50/Rta/Rp promoter methylation and reactivation: similarities and differences.
Gene 50 promoter methylation may serve another purpose distinct from transcriptional repression in the context of MHV68 latency. Recent intriguing studies demonstrated that the Zta protein of EBV not only is capable of activating a methylated viral promoter but in some cases actually appears to preferentially do so (5, 22, 25). These studies are particularly enlightening given a study addressing the role of DNMT3b in EBV promoter methylation. Tao et al. (48) previously demonstrated the hypomethylation of both latent and lytic viral promoters in lymphoblastoid cell lines (LCLs) generated from cells of patients with immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome, a rare disorder arising from deleterious mutations in the Dnmt3b gene. Those authors were surprised to find that, despite the relative hypermethylation of the Rp promoter, LCLs generated from normal cells produced higher levels of Rp-driven transcripts than those generated from ICF patients. Recent studies demonstrating that methylation actually enhances the Zta-mediated transactivation of Rp help to explain those observations. Our observations are similar to those reported by Tao et al. in that the lack of functional DNMT3a and DNMT3b results in the hypomethylation of the MHV68 distal gene 50 promoter. However, they are strikingly dissimilar in that this hypomethylation is associated with increased gene 50 transcription. In addition, there was no observable phenotype for the single-conditional-deletion mice, which may simply reflect a redundancy of Dnmt3 function that was observed previously by other murine methyltransferase studies (36). Unlike KSHV and EBV, a Zta/bZIP protein homolog has not been identified for MHV68. However, the highly CpG-suppressed MHV68 genome suggests that the virus has been extensively methylated over time, therefore requiring the evolution of a methylation-resistant reactivation mechanism similar to that of its human counterparts. We have demonstrated that the proximal gene 50 promoter, which lacks any CpGs in the core promoter sequences, drives gene 50 transcription first during MHV68 reactivation (19), but the precise cellular signals initiating this process are currently unknown. Is the gene 50 product encoded by the proximal-promoter-driven transcript capable of transactivating the methylated distal gene 50 promoter? Or does it induce sufficient viral replication to overwhelm the host methylation machinery and thus passively dilute methylation such that the distal promoter is once again rendered active? Further characterization of the distal gene 50 promoter will partially answer these questions and is currently being pursued.
Another interesting question addresses the effect of reactivation on methylation status throughout the duration of latent infection. We demonstrated that methylation accumulates in the MHV68 distal gene 50 promoter region over time. Studies with EBV and KSHV have also described methylated latent genomes in both normal and tumor cells (8, 14, 35, 37, 38). One model proposes that gammaherpesviruses infect naïve B lymphocytes and, thus, are exposed to the unique environment of the germinal center following B-cell activation. Germinal-center B cells express high levels of AID; DNA in these cells is therefore highly susceptible to mutagenesis, especially at methylated cytosines. In the context of an actual infection, it could be that periodic reactivation serves as a “reset” mechanism for viral methylation patterns to avoid potentially deleterious mutations that may result from the repeated activation of latently infected memory B cells. If key regulatory CpGs in the MHV68 distal gene 50 promoter were methylated in this setting, either during the establishment of latency or during a reactivation episode, it could lead to a deleterious loss of what appears to be an important gene 50 regulatory mechanism.
A recent study by Yang et al. examined DNA methylation and histone acetylation in the MHV68 gene 50 promoter region in the MHV68 latent cell line S11 and in cells from infected animals (56). In agreement with our previous observations, they found that the promoter is progressively methylated during the establishment of latency but concluded that acetylation, not demethylation, is primarily responsible for the reexpression of the gene 50 protein during reactivation from latency. Growing evidence exists to support a role for DNA methylation in perpetuating the placement of repressive chromatin modifications (6). Therefore, it may well be that the methylation of the distal gene 50 promoter is a key step in the initiation of heterochromatin formation in this region, thereby setting the stage for the repressed transcriptional state required for B-cell latency. If this is true, it is important to understand the basic mechanism by which initial DNA methylation is established at the gene 50 promoter. Our study aims to directly examine the mechanism underlying the development of a repressed gene 50 promoter prior to the establishment of latent gammaherpesvirus infection and provides clues as to the molecule players involved in this process.
In summary, these studies have demonstrated that DNA methylation appears to be an important mechanism regulating MHV68 gene 50 promoter activity. There is a role for DNMT3a and/or DNMT3b in establishing the methylation of gene 50 during early latency, but an alternative mechanism exists to allow for the restoration of gene 50 distal promoter methylation and the resolution of persistent virus replication. The latter likely plays a critical role in facilitating the establishment of long-term latency. Further studies are required to elucidate the contribution of other epigenetic mechanisms to the regulation of this key viral replicating gene and may provide valuable information regarding the regulation of gammaherpesvirus latency.
Acknowledgments
This research was supported by NIH R01 grants CA052004 and CA058524. S.H.S. was also supported by NIH R01 grants CA087650, AI058057, and AI073830.
We thank Klaus Rajewsky for generously providing the CD19+/Cre mice and Taiping Chen for providing the Dnmt3a and Dnmt3b conditional mice. We give much additional thanks to Maja Klug and Micheal Rehli for their kind gift of the pCpG vectors. We thank Chelsey Goins for help with the NP-CGG immunizations and ELISPOT analyses, Ben Youngblood for help with the LCMV infections, and members of the Speck and Mocarski laboratories for helpful comments.
Footnotes
Published ahead of print on 3 March 2010.
REFERENCES
- 1.Allen, R. D., III, M. N. DeZalia, and S. H. Speck. 2007. Identification of an Rta responsive promoter involved in driving gammaHV68 v-cyclin expression during virus replication. Virology 365:250-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen, R. D., III, S. Dickerson, and S. H. Speck. 2006. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 80:2055-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barozzi, P., L. Potenza, G. Riva, D. Vallerini, C. Quadrelli, R. Bosco, F. Forghieri, G. Torelli, and M. Luppi. 2007. B cells and herpesviruses: a model of lymphoproliferation. Autoimmun. Rev. 7:132-136. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Sasson, S. A., and G. Klein. 1981. Activation of the Epstein-Barr virus genome by 5-aza-cytidine in latently infected human lymphoid lines. Int. J. Cancer 28:131-135. [DOI] [PubMed] [Google Scholar]
- 5.Bhende, P. M., W. T. Seaman, H. J. Delecluse, and S. C. Kenney. 2005. BZLF1 activation of the methylated form of the BRLF1 immediate-early promoter is regulated by BZLF1 residue 186. J. Virol. 79:7338-7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cedar, H., and Y. Bergman. 2009. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10:295-304. [DOI] [PubMed] [Google Scholar]
- 7.Chang, L. K., and S. T. Liu. 2000. Activation of the BRLF1 promoter and lytic cycle of Epstein-Barr virus by histone acetylation. Nucleic Acids Res. 28:3918-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, J., K. Ueda, S. Sakakibara, T. Okuno, C. Parravicini, M. Corbellino, and K. Yamanishi. 2001. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. U. S. A. 98:4119-4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christensen, J. P., R. D. Cardin, K. C. Branum, and P. C. Doherty. 1999. CD4(+) T cell-mediated control of a gamma-herpesvirus in B cell-deficient mice is mediated by IFN-gamma. Proc. Natl. Acad. Sci. U. S. A. 96:5135-5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins, C. M., J. M. Boss, and S. H. Speck. 2009. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J. Virol. 83:6484-6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Countryman, J. K., L. Gradoville, and G. Miller. 2008. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol. 82:4706-4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodge, J. E., M. Okano, F. Dick, N. Tsujimoto, T. Chen, S. Wang, Y. Ueda, N. Dyson, and E. Li. 2005. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 280:17986-17991. [DOI] [PubMed] [Google Scholar]
- 13.Dutia, B. M., C. J. Clarke, D. J. Allen, and A. A. Nash. 1997. Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J. Virol. 71:4278-4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott, J., E. B. Goodhew, L. T. Krug, N. Shakhnovsky, L. Yoo, and S. H. Speck. 2004. Variable methylation of the Epstein-Barr virus Wp EBNA gene promoter in B-lymphoblastoid cell lines. J. Virol. 78:14062-14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forrest, J. C., and S. H. Speck. 2008. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 82:7688-7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujimura, S., T. Matsui, K. Kuwahara, K. Maeda, and N. Sakaguchi. 2008. Germinal center B-cell-associated DNA hypomethylation at transcriptional regions of the AID gene. Mol. Immunol. 45:1712-1719. [DOI] [PubMed] [Google Scholar]
- 17.Gangappa, S., S. B. Kapadia, S. H. Speck, and H. W. Virgin IV. 2002. Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J. Virol. 76:11460-11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gargano, L. M., J. M. Moser, and S. H. Speck. 2008. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 82:3853-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray, K. S., R. D. Allen III, M. L. Farrell, J. C. Forrest, and S. H. Speck. 2009. Alternatively initiated gene 50/RTA transcripts expressed during murine and human gammaherpesvirus reactivation from latency. J. Virol. 83:314-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gwack, Y., H. Byun, S. Hwang, C. Lim, and J. Choe. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75:1909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gwack, Y., S. Hwang, C. Lim, Y. S. Won, C. H. Lee, and J. Choe. 2002. Kaposi's sarcoma-associated herpesvirus open reading frame 50 stimulates the transcriptional activity of STAT3. J. Biol. Chem. 277:6438-6442. [DOI] [PubMed] [Google Scholar]
- 22.Heather, J., K. Flower, S. Isaac, and A. J. Sinclair. 2009. The Epstein-Barr virus lytic cycle activator Zta interacts with methylated ZRE in the promoter of host target gene egr1. J. Gen. Virol. 90:1450-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeltsch, A. 2006. Molecular enzymology of mammalian DNA methyltransferases. Curr. Top. Microbiol. Immunol. 301:203-225. [DOI] [PubMed] [Google Scholar]
- 24.Kaneda, M., M. Okano, K. Hata, T. Sado, N. Tsujimoto, E. Li, and H. Sasaki. 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429:900-903. [DOI] [PubMed] [Google Scholar]
- 25.Karlsson, Q. H., C. Schelcher, E. Verrall, C. Petosa, and A. J. Sinclair. 2008. Methylated DNA recognition during the reversal of epigenetic silencing is regulated by cysteine and serine residues in the Epstein-Barr virus lytic switch protein. PLoS Pathog. 4:e1000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klug, M., and M. Rehli. 2006. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1:127-130. [DOI] [PubMed] [Google Scholar]
- 27.Lan, K., D. A. Kuppers, and E. S. Robertson. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468-3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lan, K., D. A. Kuppers, S. C. Verma, and E. S. Robertson. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585-6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, E. 2002. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 3:662-673. [DOI] [PubMed] [Google Scholar]
- 30.Liu, S., I. V. Pavlova, H. W. Virgin IV, and S. H. Speck. 2000. Characterization of gammaherpesvirus 68 gene 50 transcription. J. Virol. 74:2029-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luka, J., B. Kallin, and G. Klein. 1979. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94:228-231. [DOI] [PubMed] [Google Scholar]
- 32.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 33.May, J. S., H. M. Coleman, B. Smillie, S. Efstathiou, and P. G. Stevenson. 2004. Forced lytic replication impairs host colonization by a latency-deficient mutant of murine gammaherpesvirus-68. J. Gen. Virol. 85:137-146. [DOI] [PubMed] [Google Scholar]
- 34.Nash, A. A., B. M. Dutia, J. P. Stewart, and A. J. Davison. 2001. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:569-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niller, H. H., H. Wolf, and J. Minarovits. 2009. Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin. Cancer Biol. 19:158-164. [DOI] [PubMed] [Google Scholar]
- 36.Okano, M., D. W. Bell, D. A. Haber, and E. Li. 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247-257. [DOI] [PubMed] [Google Scholar]
- 37.Paulson, E. J., J. D. Fingeroth, J. L. Yates, and S. H. Speck. 2002. Methylation of the EBV genome and establishment of restricted latency in low-passage EBV-infected 293 epithelial cells. Virology 299:109-121. [DOI] [PubMed] [Google Scholar]
- 38.Paulson, E. J., and S. H. Speck. 1999. Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J. Virol. 73:9959-9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlova, I. V., H. W. Virgin IV, and S. H. Speck. 2003. Disruption of gammaherpesvirus 68 gene 50 demonstrates that Rta is essential for virus replication. J. Virol. 77:5731-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rickert, R. C., J. Roes, and K. Rajewsky. 1997. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25:1317-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlissel, M. S. 2004. Regulation of activation and recombination of the murine Igkappa locus. Immunol. Rev. 200:215-223. [DOI] [PubMed] [Google Scholar]
- 42.Slifka, M. K., and R. Ahmed. 1996. Limiting dilution analysis of virus-specific memory B cells by an ELISPOT assay. J. Immunol. Methods 199:37-46. [DOI] [PubMed] [Google Scholar]
- 43.Staudt, M. R., and D. P. Dittmer. 2007. The Rta/Orf50 transactivator proteins of the gamma-herpesviridae. Curr. Top. Microbiol. Immunol. 312:71-100. [DOI] [PubMed] [Google Scholar]
- 44.Stevenson, P. G., and P. C. Doherty. 1998. Kinetic analysis of the specific host response to a murine gammaherpesvirus. J. Virol. 72:943-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szyf, M., L. Eliasson, V. Mann, G. Klein, and A. Razin. 1985. Cellular and viral DNA hypomethylation associated with induction of Epstein-Barr virus lytic cycle. Proc. Natl. Acad. Sci. U. S. A. 82:8090-8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tadokoro, Y., H. Ema, M. Okano, E. Li, and H. Nakauchi. 2007. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 204:715-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tao, Q., H. Huang, T. M. Geiman, C. Y. Lim, L. Fu, G. H. Qiu, and K. D. Robertson. 2002. Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum. Mol. Genet. 11:2091-2102. [DOI] [PubMed] [Google Scholar]
- 49.Tarakanova, V. L., F. Suarez, S. A. Tibbetts, M. A. Jacoby, K. E. Weck, J. L. Hess, S. H. Speck, and H. W. Virgin IV. 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J. Virol. 79:14668-14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Virgin, H. W., IV, R. M. Presti, X. Y. Li, C. Liu, and S. H. Speck. 1999. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J. Virol. 73:2321-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. W. Virgin IV. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weck, K. E., S. S. Kim, H. W. Virgin IV, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willer, D. O., and S. H. Speck. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77:8310-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu, T. T., L. Tong, T. Rickabaugh, S. Speck, and R. Sun. 2001. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J. Virol. 75:9262-9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu, T. T., E. J. Usherwood, J. P. Stewart, A. A. Nash, and R. Sun. 2000. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 74:3659-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang, Z., H. Tang, H. Huang, and H. Deng. 2009. RTA promoter demethylation and histone acetylation regulation of murine gammaherpesvirus 68 reactivation. PLoS One 4:e4556. [DOI] [PMC free article] [PubMed] [Google Scholar]