

Abstract
The design and study of two classes of noncompetitive acetylcholinesterase inhibitors (AChEIs) which also function as NSAID prodrugs are reported. The most potent AChEIs disclosed contain an aromatic alkyl-aryl linker between an NSAID and a lipophilic choline mimic and they inhibit acetylcholinesterase (AChE) in the submicromolar range. These agents have the therapeutic potential to dually target inflammation by releasing an NSAID in vivo and activating the cholinergic anti-inflammatory pathway via cholinergic up-regulation.
Pro-inflammatory cytokine up-regulation plays a role in the pathogenesis of a wide range of disorders including osteoarthritis,1 psoriasis,2 multiple sclerosis,3 and other autoimmune disorders.4 Despite decades of research, non-steroidal anti-inflammatory drugs (NSAIDs) are still one of the most commonly used, highly effective treatments for such disorders. However, since chronic NSAID-use often leads to gastrointestinal (GI) side effects, NSAID ester prodrugs have been explored to mask the acidic GI-irritating portion of the NSAID.5 For conditions such as arthritis, topical NSAID prodrugs are particularly useful as their therapeutic action is localized, resulting in minimal systemic side effects.6
Alternately, acetylcholinesterase inhibitors (AChEIs) have implications in the treatment of severe inflammation resulting from sepsis,7 endotoxemia,8 and rheumatoid arthritis,9 as well as in the treatment of neuroinflammation associated with Alzheimer’s disease10–12 and Myasthenia Gravis.13 The administration of CNS-active AChEIs such as galanthamine depletes systemic pro-inflammatory cytokines and ameliorates both central and peripheral inflammation.8 AChEIs seem to suppress inflammation via the cholinergic anti-inflammatory pathway, a mechanism by which the vagus nerve of the CNS regulates the production and release of tumor necrosis factor and other cytokines.14,15
NSAID-AChEI conjugates have recently been explored in order to target neuroinflammation16 and vesicant-induced inflammation.17 These novel agents contain the AChEI, pyridostimine, linked via a hydrocarbon spacer to ibuprofen. When screened against sulfur mustard, the bifunctional agents demonstrated longer and more effective prevention of edema and inflammation than the AChEI alone with a post-treatment anti-inflammatory effect comparable to that of the parent NSAID.17 Furthermore, the same conjugates down-regulated nitric oxide and prostaglandin E2 production and ameliorated autoimmune encephalomyelitis more than either component alone.16 NSAID-AChEIs seem to prove dually effective against inflammatory disorders by activating the cholinergic anti-inflammatory pathway and inhibiting cyclooxygenase. Based on previous findings, two novel classes of noncompetitive AChEIs containing common NSAIDs linked via an ester or an aromatic ester-carbonate backbone to choline bioisosteres have been developed (Figure 1).18 The synthesis and evaluation of these compounds as anti-inflammatory and anticholinesterase agents for topical or oral administration are presented herein.
Figure 1.

Generic Structures of Class 1 (top) and Class 2 (bottom) agents.
Class 1 molecules have recently been designed as both up-regulators of choline and NSAID prodrugs.18 The cholinergic functions (-CO-O-CH2CH2X(CH3)3; X = C, Si, N+) are known bioisosteres of choline which inhibit AChE in both a competitive (as with the silicon derivative) and noncompetitive (as with the carbon derivative) manner.19 It is proposed that AChE binds to the choline mimic and activates the cholinergic anti-inflammatory pathway. Additionally, esterases in the plasma, skin and lipid membranes release the NSAID, thus targeting dual mechanisms of inflammation. Molecules containing NSAIDs directly linked to choline mimics via an ester bond (Figure 1, Class 2) have been included both as a control class as well as an additional class for therapeutic exploration.
Class 1 compounds were obtained by coupling either 3,3-dimethylbutyl carbonochloridate (Ia)20 or 2-(trimethylsilyl)ethyl carbonochloridate (Ib)21 to the phenolic function of p-hydroxybenzyl alcohol. Coupling of II to an NSAID activated with 4-dimethylaminopyridine (DMAP) completed the synthesis (Scheme 1). Overall yields of this procedure ranged from 61 to 76 %, including synthesis of the chloroformate starting materials.
Scheme 1.

Synthesis of Class 1 agents. a) Et3N, THF, −10 to 25°C; b) NSAID, EDC HCl, DMAP, CH2Cl2, 0 to 25°C.
In the simple-ester series (Figure 1, Class 2), standard coupling conditions [1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC HCl)-DMAP] were employed for the carbon and silicon derivatives in a one step, moderate- to high-yielding procedure (55–97 %). Synthesis of the nitrogen-based NSAID-choline esters of Class 2 (11, 14, and 17) has been previously described by J. Young et al.22 and will not be discussed further. Using Young’s method, products were isolated in moderate yields over three steps (30–40 %). All Class 2 agents were purified using non-chromatographic methods such as crystallization and extraction.
Following synthesis and characterization, all compounds were evaluated for anticholinesterase activity using Ellman’s method23 with tacrine,24 one of the most potent commercial AChEIs, as a reference (Table 1). AChE contains a 20 Å deep gorge stretching from the enzyme surface to the active-site serine residue.25 This structural moiety is often termed the aromatic or hydrophobic gorge since it is lined with Trp, Tyr and Phe residues.25 A peripheral binding site termed the peripheral anionic site (PAS) is located at the top of the gorge, near the surface of the enzyme. Keeping in mind the nature of AChE, it was predicted that the highly lipophilic and aromatic Class 1 and 2 agents would interact with the Trp, Tyr and Phe residues at and near the PAS and inhibit enzyme activity.
Table 1.
Anticholinesterase activities and Clog P values of Class 1 and 2 agents.
| Class 1 (Ester-Carbonate Series) | Class 2 (Ester Series) | ||||||
|---|---|---|---|---|---|---|---|
| Compound | IC50 (μM)a | % AChE Recoveryb | Clog Pc | Compound | IC50 (μM)a | % AChE Recoveryb | Clog Pc |
| 1 | 1.93 ± 0.64 | 101 ± 8 | 7.37 | 9 | 24.6 ± 14.5 | 107 ± 17 | 6.47 |
| 2 | 1.19 ± 0.20 | 88 ± 11 | 7.53 | 10 | 25.7 ± 4.9 | 92 ± 11 | 6.64 |
| 3 | 1.74 ± 1.01 | 99 ± 4 | 6.50 | 11 | >2000 | 99 ± 5 | 0.42 |
| 4 | 0.83 ± 0.15 | 98 ± 3 | 6.67 | 12 | 19.7 ± 1.7 | 117 ± 13 | 5.61 |
| 5 | 2.29 ± 0.94 | 78 ± 2 | 7.87 | 13 | 13.9 ± 0.3 | 100 ± 1 | 5.77 |
| 6 | 0.72 ± 0.13 | 86 ± 1 | 8.03 | 14 | >1000 | 93 ± 13 | −0.45 |
| 7 | 0.51 ± 0.02 | 87 ± 11 | 8.41 | 15 | 9.75 ± 0.89 | 80 ± 2 | 6.97 |
| 8 | 1.36 ± 0.13 | 104 ± 23 | 8.58 | 16 | 3.32 ± 0.36 | 73 ± 4 | 7.14 |
| 17 | 230 ± 32 | 101 ± 6 | 0.92 | ||||
| 18 | 2.69 ± 0.15 | 99 ± 4 | 7.52 | ||||
| Tacrine HCl | 0.055 ± 0.005 | 95 | 19 | 2.66 ± 0.25 | 90 ± 10 | 7.68 | |
Source of AChE is electric eel (Electrophorus electricus). Each condition was run in duplicate.
EeAChE was incubated for 30 min with 55 μM inhibitor at 25°C. Enzyme activity was assayed following gel filtration. Values reported are averages of at least two experiments.
Values calculated using ChemBioDraw Ultra 11.0.
Listed in Table 1 are IC50 values for each compound against AChE. As a control, the parent NSAIDs were also screened for activity and were deemed inactive compared to most of the test compounds (IC50 > 1000 μM, data not shown). A direct comparison of Class 1 and 2 activities reveals that Class 1 compounds are more potent than Class 2 by at least a factor of 4, in all cases. The most potent Class 1 inhibitor (7, Figure 2) has an IC50 comparable to that of the AChEI, galanthamine, a natural alkaloid which has been approved for the treatment of Alzheimer’s disease.24 Since Class 2 compounds were found less active than Class 1, it is likely that the lipophilic aromatic linker is needed to maximize potential π-π stacking interactions26 with the Trp, Tyr and Phe residues of the aromatic gorge.25 The ester-carbonates (Class 1) are also at least 10 times more lipophilic than the corresponding simple esters, which may also increase hydrophobic interactions with AChE (refer to Table 1 for Clog P values). The more hydrophilic nitrogen derivatives of Class 2 (11, 14, and 17) were at least 20 times less potent than their carbon and silicon derivatives, further enhancing the argument that hydrophobic interactions at and near the active-site gorge may be responsible for the observed activities.
Figure 2.
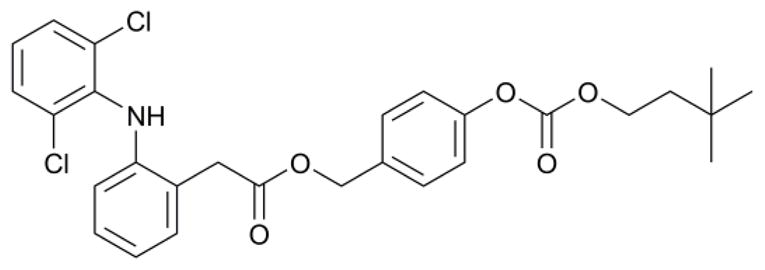
Structure of 7, the most potent AChEI discussed herein (IC50 = 0.51 μM).
Overall, the most active AChEIs in both classes (7, 18, and 19) are all derivatives of the NSAID, diclofenac. This trend is likely due to the high lipophilicity of diclofenac which strengthens interactions with the hydrophobic residues of AChE. Furthermore, the chlorine substituents on one of the aromatic rings of diclofenac may enhance π-π stacking interactions with aromatic residues lining the gorge (likely Trp84, Phe330, or Trp279 in Torpedo californica AChE), as suggested by previous SAR and computational studies performed by Holzgrabe et al.26 and Sussman et al.27
The degree of reversibility of AChE-inhibitor interactions was investigated for all inhibitors. Following an incubation period of AChE with the test agents, gel filtration resulted in near-complete restoration of enzyme activity (80–100 %, see Table 1). It was important to demonstrate the reversible nature of our compounds because many of the known irreversible AChEIs are nerve agents, toxins, or pesticides and are not considered for therapeutic use.28
The mechanism of AChE inhibition was further investigated by generating Lineweaver-Burk plots.29 Briefly, the reciprocal of the reaction velocity (ν−1) was plotted against the reciprocal of the substrate concentration ([acetylthiocholine, ATCh]−1) after assaying enzyme activity for varying inhibitor and ATCh concentrations. All compounds exhibited reversible, noncompetitive inhibition, with quadrant III interceptions (for detailed assay method and example Lineweaver-Burk plots, see Supporting Information). These analyses support the reversibility studies previously discussed. More in depth investigations which address whether compounds 1–19 are interacting with the PAS of AChE or an alternate site are ongoing.
To evaluate Class 1 and 2 agents as oral or topical prodrugs, hydrolytic release of the NSAID was followed by HPLC under various conditions. Gradual release of the parent NSAID was observed for both Class 1 and 2 agents in human plasma (80 % plasma in PBS), with half-lives not exceeding 8 h (Table 2). Negligible hydrolysis was observed in the PBS control during the time of the half-life determination. The ester-carbonate prodrugs released the parent drug more slowly (t1/2 of 2–8 h) compared with the ester prodrugs (t1/2 of 5–100 min). Compound 14 was completely hydrolyzed after 10 min, suggesting that the choline esters (11, 14, and 17) would not make suitable prodrugs.
Table 2.
Hydrolysis of Class 1 and 2 Compounds in Human Plasma.a
| Class 1 (Ester-Carbonate Series) | Class 2 (Ester Series) | ||
|---|---|---|---|
| Compound | t1/2 (min)b | Compound | t1/2 (min)b |
| 1 | 204 | 12 | 38 |
| 3 | 135 | 13 | 63 |
| 4 | 253 | 14 | < 5 |
| 5 | 468 | 18 | 111 |
| 7 | 357 | ||
Hydrolysis measured in 80 % human plasma in PBS at 37 °C.
Half-lives determined by plotting the semi-log of prodrug disappearance.
Hydrolysis in acetate buffer was also determined at the pH on the surface of the skin (pH 5). No significant hydrolysis was observed for up to seven days under acidic conditions, suggesting that the ester and ester-carbonate linkages should be resistant to cleavage on the surface of the skin. Preliminary hydrolysis data suggest that Class 1 and 2 agents will act as NSAID prodrugs in vivo. Class 1 compounds demonstrate a more gradual NSAID-release when compared to Class 2. Additionally, they are more lipophilic than their Class 2 counterparts, making them more suitable as topical prodrugs.
Novel AChEIs have been developed with the potential to dually target inflammation by activating the cholinergic anti-inflammatory pathway and releasing an NSAID, in vivo. Lineweaver-Burk plots indicate that all test compounds are reversible, noncompetitive inhibitors of AChE. The most potent inhibitors (Class 1) contain a lipophilic aromatic spacer and are 4 times more active than those with solely an ester linkage between the anticholinergic and anti-inflammatory functionalities (Class 2). Of the compounds screened, diclofenac derivative 7 was most active likely due to its high lipophilicity, aromaticity and the chlorine-substituted ring.26,27 Hydrolysis data suggest that these agents should be effective as either oral or topical NSAID prodrugs.
Supplementary Material
Acknowledgments
This work was funded in part by the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (award #U54AR055073). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government. The authors would like to thank Professors Sam Niedbala and Jebrell Glover for use of their instrumentation.
Footnotes
Synthetic procedures, physical characterization of compounds, in vitro assay methods and Lineweaver-Burk plots can be found in the online version of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Özgüney I. Expert Opin Pharmacother. 2008;9:1805–1816. doi: 10.1517/14656566.9.10.1805. [DOI] [PubMed] [Google Scholar]
- 2.Deeva I, Mariani S, De Luca C, Pacifico V, Leoni L, Raskovic D, Kharaeva Z, Korkina L, Pastore S. Wide-spectrum profile of inflammatory mediators in the plasma and scales of patients with psoriatic disease. Cytokine. 2009 doi: 10.1016/j.cyto.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 3.Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death & Diff. 2009 doi: 10.1038/cdd.2009.179. [DOI] [PubMed] [Google Scholar]
- 4.Kunz M, Ibrahim S. Mediators Inflamm. 2009;2009:1–20. doi: 10.1155/2009/979258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannon J. Pharmacology for Chemists. 2. Oxford University Press; New York: Concepts of Pharmacology; pp. 13–17. [Google Scholar]
- 6.Trommer H, Neubert R. Skin Pharmacol Physiol. 2006;19:106–121. doi: 10.1159/000091978. [DOI] [PubMed] [Google Scholar]
- 7.Fodale V, Letterio B. Crit Care Med. 2008;36:622–623. doi: 10.1097/CCM.0B013E31816297CE. [DOI] [PubMed] [Google Scholar]
- 8.Pavlov V, Parrish W, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, Chavan S, Al-Abed Y, Tracey K. Brain Behav Immun. 2009;23:41–45. doi: 10.1016/j.bbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Maanen M, Vervoordeldonk M, Tak P. Nat Rev Rheumatol. 2009;5:229–232. doi: 10.1038/nrrheum.2009.31. [DOI] [PubMed] [Google Scholar]
- 10.Tsuno N. Expert Rev Neurother. 2009;9:591–598. doi: 10.1586/ern.09.23. [DOI] [PubMed] [Google Scholar]
- 11.Guay D. Consult Pharm. 2008;23:598–609. doi: 10.4140/tcp.n.2008.598. [DOI] [PubMed] [Google Scholar]
- 12.Marco L, do Carmo Carreiras M. Recent Pat CNS Drug Discov. 2006;1:105–111. doi: 10.2174/157488906775245246. [DOI] [PubMed] [Google Scholar]
- 13.Brenner T, Nizri E, Irony-Tur-Sinai M, Hamra-Amitay Y, Wirguin I. J Neuroimmunol. 2008;201–202:121–127. doi: 10.1016/j.jneuroim.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Tracey K. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 15.Tracey K. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nizri E, Adani R, Meshulam H, Amitai G, Brenner T. Neurosci Lett. 2005;376:46–50. doi: 10.1016/j.neulet.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 17.Amitai G, Adani R, Fishbein E, Meshulam H, Laish I, Dachir S. J Appl Toxicol. 2006;26:81–87. doi: 10.1002/jat.1111. [DOI] [PubMed] [Google Scholar]
- 18.Laskin JD, Fabio K, Lacey CJ, Young S, Mohanta P, Guillon C, Heindel ND, Huang M-T, Heck DE. Unique Dual-Action Therapeutics. PCT/US2009/005961. U S Patent Pending. 2009 November 3;
- 19.Cohen S, Chishti S, Elkind J, Reese H, Cohen J. J Med Chem. 1985;28:1309–1313. doi: 10.1021/jm00147a033. [DOI] [PubMed] [Google Scholar]
- 20.Hudson H, Koplick A, Poulton D. J Chem Soc, Perkin Trans 2. 1979:57–66. [Google Scholar]
- 21.Berglund A, Akerblom E, Hedin A. 0,209,586. EU Patent. 1986
- 22.Venuti M, Young J, Maloney P, Johnson D, McGreevy K. Pharm Res. 1989;6:867–873. doi: 10.1023/a:1015960522189. [DOI] [PubMed] [Google Scholar]
- 23.Ellman G, Courtney K, Andres V, Feather-Stone R. Biochem Phamacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 24.Guillou C, Mary A, Zafiarisoa Renko D, Gras E, Thal C. Bioorg Med Chem Lett. 2000;10:637–639. doi: 10.1016/s0960-894x(00)00059-7. [DOI] [PubMed] [Google Scholar]
- 25.Shen T, Tai K, Henchman R, McCammon A. Acc Chem Res. 2002;35:332–340. doi: 10.1021/ar010025i. [DOI] [PubMed] [Google Scholar]
- 26.Kapkova P, Stiefl N, Sürig U, Engels B, Baumann K, Holzgrabe U. Arch Pharm Pharm Med Chem. 2003;336:523–540. doi: 10.1002/ardp.200300795. [DOI] [PubMed] [Google Scholar]
- 27.Dvir H, Wond D, Harel M, Barril X, Orozco M, Luque F, Muñoz-Torrero D, Camps P, Rosenberry T, Silman I, Sussman J. Biochemistry. 2002;41:2970–2981. doi: 10.1021/bi011652i. [DOI] [PubMed] [Google Scholar]
- 28.Taylor P. In: The Pharmacological Basis of Therapeutics. Hardman J, Limbird L, Molinoff P, Ruddon R, Gilman A, editors. McGraw-Hill; New York: 1996. pp. 161–176. [Google Scholar]
- 29.(a) Lineweaver H, Burk D. J Am Chem Soc. 1934;56:658–666. [Google Scholar]; (b) Bergmeyer H, Gawehn K. Principles of Enzymatic Analysis. Verlag Chemie; NY: 1978. pp. 36–40. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.