

Abstract
A highly enantioselective method for the catalytic addition of terminal 1,3-diynes to aldehydes was developed using our dinuclear zinc ProPhenol (1) system. Furthermore, triphenylphosphine oxide was found to interact synergistically with the catalyst to substantially enhance the chiral recognition. The generality of this catalytic transformation was demonstrated with aryl, α,β-unsaturated and saturated aldehydes, of which the latter were previously limited in alkynyl zinc additions. The chiral diynol products are also versatile building blocks that can be readily elaborated; this was illustrated through highly selective trans-hydrosilylations, which enabled the synthesis of a β-hydroxyketone and enyne. Additionally, the development of this method allowed for the rapid total syntheses of several biologically important diynol-containing natural products.
Introduction
Conjugated diynes are potentially intriguing building blocks due to the unique behavior of acetylenes, especially in transition metal-catalyzed processes.1 The unique juxtaposition of two acetylene units in a 1,3-diyne begets unusual properties, both from a chemical as well as biological point-of-view. Diyne carbinols are of particular interest because of the additional reactivity imparted by the propargyl alcohol, as well as its presence in many fascinating bioactive, fatty acid-derived polyacetylenic natural products (Figure 1).2,3 These optically active propargyl alcohols exhibit a broad array of important biological properties, ranging from antifungal to anticancer activity.2
Figure 1.

Several polyacetylenic natural products with chiral diynol moiety
In spite of the growing recognition of the therapeutic potential of these secondary metabolites, the conventional synthetic routes to these classes of molecules are limited and lengthy. The most common strategy employed in the total syntheses of these molecules relies upon asymmetric ynone reductions to form the chiral alcohol, followed by copper-catalyzed Cadiot-Chodkiewicz cross-coupling to introduce the second acetylene unit (eq 1).3
 |
(1) |
 |
(2) |
A complementary strategy for accessing a chiral diynol is the direct addition of a terminal 1,3-diyne into an aldehyde (eq 2). This rapid approach would set the stereocenter of the secondary alcohol and introduce the diyne moiety in a single chemical operation. While there are many effective catalyst systems for the asymmetric nucleophilic addition of alkynes into aldehydes,4 no known protocols exist for which the analogous transformation can be performed with a diyne. In spite of the structural similarities between an alkyne and 1,3-diyne, due to hyperconjugative effects5 the two acetylenic species possess inherently differential reactivity. Notably, the uniqueness of the 1,3-diyne was observed in practice by Carreira and coworkers: use of their Zn(OTf)2/N-methylephedrine6a system allows for the catalytic asymmetric synthesis of chiral propargyl alcohols,6b however, for the coupling of diynes, a super-stoichiometric quantity (4 equiv each) of both zinc and chiral inducing agent were required.7
Stimulated by our interest in exploring the synthetic potential of diyne carbinols as building blocks, their presence as key structural elements in biological metabolites and the difficulties associated with their asymmetric syntheses, we set out to determine if our ProPhenol (1) catalyst, which has been successfully employed in the alkynylation of aldehydes,8 could also effect a catalytic asymmetric addition of terminal 1,3-diynes to carbonyl groups. Moreover, we wanted to identify a modular diyne nucleophile which could serve as a 1,3-butadiyne equivalent. This would provide access to a lynchpin approach to rapidly generate both stereo- and structural complexity about both ends of the diyne. Herein, we report the details of our efforts to develop a catalytic asymmetric diynylation reaction and the synthetic utility of this transformation.
Results and Discussion
Proposed catalytic cycle
It is believed that a dinuclear zinc ProPhenol (1)-catalyzed terminal 1,3-diyne addition would proceed in a fashion similar to the known alkynylation (Scheme 1).8a The chiral ligand 1 could react with 2 equivalents of a diynylmethylzinc species to generate the dinuclear zinc acetylide A. We hypothesize that in intermediate A the Zn acetylide is also coordinated to the nitrogen of the prolinol moiety and oxygen of the phenol. These hypervalent interactions serve to give the acetylide more anionic character, that is the nucleophile is an activated diynyl zincate species.9 Furthermore, the active catalyst contains a Lewis acidic Zn alkoxide which can bind an aldehyde (B). Preorganization of the diyne and aldehyde allows for a directed addition reaction to form the propargyl zinc alkoxide C. Transmetalation between C and another molecule of diynylmethylzinc liberates the desired product and turns over the catalytic cycle.
Scheme 1.

Proposed mechanism for the ProPhenol (1)-catalyzed diynylation
Reaction optimization
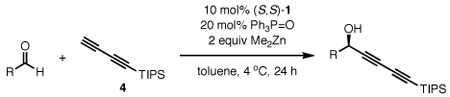
To establish that our dinuclear zinc ProPhenol (1) catalyst system could facilitate the addition of a terminal 1,3-diyne to an aldehyde, we evaluated the reaction of protected penta-2,4-diyn-1-ols with octanal (eq 3). Under the previously developed conditions,8a when tert-butyldimethyl(penta-2,4-diynyloxy)silane7 (2a) was used as the donor, propargyl alcohol 3a was isolated in 48% ee and 68% yield; however, penta-2,4-diynyl ethanoate (2b) furnished the corresponding product 3b with significantly better selectivity (62% ee).
 |
(3) |
Since the non-reactive terminus of the 1,3-diyne is far removed from the active site of the catalyst (see Scheme 1), it was surprising that such a subtle substituent change impacted the selectivity so significantly. Given that 3 equiv of donor was used, we postulated perhaps the Lewis basic acetate of diyne 2b could be interacting with the Zn catalyst. This observation is consistent with previous outcomes where methyl propiolate was shown to be a more effective nucleophile than (trimethylsilyl)acetylene in alkynylations with the ProPhenol (1) catalyst.8a,b Based on these results we sought to identify an exogenous, sub-stoichiometric Lewis base additive that could enhance the enantioselectivity of the diynylation.
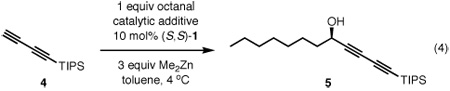
In the evaluation of Lewis base additives for the ProPhenol-catalyzed diynylation, we chose to employ buta-1,3-diynyltriisopropylsilane (4) as the nucleophile. The reason for doing so was two-fold: this particular diyne lacked any Lewis basic sites which could potentially interfere with the action of the additive, and furthermore, the cleavable TIPS group could allow for further functionalization of the diynol product. Diyne 4 was reacted with octanal using 10 mol% ProPhenol ligand 1, 3 equiv of Me2Zn and a catalytic amount of an additive (eq 4). Working under the assumption that our active catalyst was a dinuclear zinc species, we added an equivalent of Lewis base per Zn atom (Table 1).
Table 1.
Evaluation of catalytic Lewis bases in the diynylationa
 | ||||
|---|---|---|---|---|
| entry | additive | mol % | % yieldb | % eec |
| 1 | none | -- | 71 | 50 |
| 2 | 4Å MSd | -- | 59 | 42 |
| 3 | 1,2-DME | 10 | 75 | 52 |
| 4 | EtOAc | 20 | 64 | 55 |
| 5 | 20 | 53 | 57 | |
| 6 | Ph3P=S | 20 | 52 | 58 |
| 7 | Ph3P=O | 20 | 63 | 79 |
Reaction run with 3 equiv of diyne 4.
Isolated yield.
Enantiomeric excess determined by chiral HPLC.
Amount of molecular sieves added based on 100 mg/mmol of octanal.
In the absence of any additive, the catalytic coupling of 4 with octanal yielded propargyl alcohol 5 in only 50% ee (entry 1). The use of 4Å molecular sieves, a common practice in the ProPhenol/dinuclear zinc aldol reactions,10 proved detrimental to the enantioselectivity (entry 2). Since the acetate moiety of diyne 2b had a unique effect on the enantioselectivity, carbonyl-type Lewis bases were primarily examined. Although ethyl acetate and ethylene carbonate gave only modest improvements in the selectivity of the diyne addition (entries 4 and 5, respectively), they proved that a catalytic additive could enhance the reaction. The Trost10a,11 and Shibasaki12 groups previously demonstrated that oxidized phosphines could significantly impact both the reactivity and diastereoselectivity, respectively, of dinuclear zinc-catalyzed aldol reactions. Moreover, Shibasaki and coworkers have repeatedly exploited this phenomenon for 1,2-additions of various nucleophiles to aldehydes with polymetallic catalysts.13 When triphenylphosphine sulfide, which should have a high affinity for zinc, was evaluated diynol 5 was isolated in only 58% ee (entry 6); however, with only 20 mol% triphenylphosphine oxide (TPPO) the enantioselectivity of the diynylation improved to 79% ee (entry 7).
Once TPPO was identified as a beneficial co-catalyst, an array of tertiary phosphine oxides were subsequently examined in the addition of TIPS diyne 4 to octanal (Table 2). Tri(2-furyl)phosphine oxide, which has multiple Lewis basic sites, proved to be less effective than TPPO in the diynylation (68% ee, entry 2). The sterically-demanding tris(2,6-dimethoxyphenyl)phosphine oxide, which the Shibasaki group has utilized quite successfully,13 furnished diynol 5 in only 57% ee (entry 3). A trialkylphosphine oxide was examined, but did not perform as well as TPPO (entry 4). Bidentate phosphine oxides were evaluated, however, they were also found to be suboptimal co-catalysts (entries 5–7), giving enantioselectivities similar to the diynylation with no additive. Thus, triphenylphosphine oxide was found to be the most effective co-catalyst for this transformation.
Table 2.
Optimization of the phosphine oxidea
| entry | R3P=O | mol% | % yieldb | % eec |
|---|---|---|---|---|
| 1 | Ph3P=O | 20 | 63 | 79 |
| 2 | (2-furyl)3P=O | 20 | 67 | 68 |
| 3 | TDMPPOd | 20 | 61 | 57 |
| 4 | (n-Bu)3P=O | 20 | 58 | 54 |
| 5 | (R)-BINAPOe | 10 | 74 | 51 |
| 6 | (S)-BINAPOe | 10 | 78 | 47 |
| 7 | 10 | 57 | 48 |
Reaction as shown in eq 4 with 1 equiv of octanal, 3 equiv of diyne 4, 10 mol% 1 and 3 equiv of Me2Zn.
Isolated yield of diynol 5.
Enantiomeric excess of 5 determind by chiral HPLC.
TDMPPO = tris(2,6-dimethoxyphenyl)phosphine oxide.
BINAPO = 2,2'-bis(diphenylphosphoryl)-1,1'-binaphthyl.
Next, optimization of the triphenylphosphine oxide stoichiometry was performed (Table 3). In the coupling of TIPS diyne 4 with octanal, only 20 mol% TPPO (relative to 10 mol% ProPhenol ligand) was required to maximize the enantioselectivity (entry 2). Decreasing the co-catalyst loading led to a marginally less selective diynylation (entry 1), while increasing the amount of TPPO beyond 20 mol% did not further enhance the enantioselectivity (entries 3 and 4).
Table 3.
Optimization of TPPO stoichiometrya
Reaction as shown in eq 4 with 1 equiv of octanal, 3 equiv of diyne 4, 10 mol% 1 and 3 equiv of Me2Zn.
Isolated yield of diynol 5.
Enantiomeric excess of 5 determined by chiral HPLC.
The optimal 2:1 ratio of TPPO:ProPhenol suggests that each zinc atom in the active catalyst is coordinated by a single phosphine oxide. Based on this observation, as well as crystallographic data of the dinuclear zinc catalyst,14 where a molecule of THF is bound to each Zn atom, we hypothesize that the triphenylphosphine oxides are “trans” to one another (Figure 2a). This proposed structure also explains why bidentate phosphine oxides, which cannot achieve the necessary binding geometry, were not beneficial (Table 2, entries 5–7). Furthermore, illustrated in Figure 2b is the putative TPPO/dinuclear zinc catalyst with the diyne and aldehyde substrates bound. It is likely the improved enantioselectivity is a result of the triphenylphosphine oxides reinforcing the chiral pocket created by the diphenylprolinol scaffold.
Figure 2.

a) Top-down, quadrant view of proposed TPPO coordination to the dinuclear zinc catalyst. b) Side view of the dinuclear zinc/TPPO catalyst with diyne and aldehyde substrates bound.
Scope of the diyne addition
Having identified optimal conditions for the asymmetric diyne addition, we then set out to investigate the scope of this transformation (Table 4 and Table 5). In the diynylation of α,β-unsaturated aldehydes, it is noteworthy that utilization of the TPPO co-catalyst allowed for reduction of the amount of diyne and dimethylzinc used from 3 to 2 equivalents without impacting either the reactivity or enantioselectivity. Although excess diyne is still required for good reactivity, the unreacted equivalents could be recovered.
Table 4.
Asymmetric addition of TIPS diyne 4a
 | ||||
|---|---|---|---|---|
| entry | aldehyde | product | % yieldb | % eec |
| 1 | 6a | 84d | 87d | |
| 2 | 6b | 95 (90)d | 98 (98)d | |
| 3 |  |
6c | 86 (89)d | 84 (86)d |
| 4 | 6d | 94 | 87 | |
| 5 | 6e | 97 | 97 | |
| 6 |  |
6f | 91 | 95 |
| 7 | 6g | 89 | 96 | |
| 8 | 6h | 93 | 97 | |
| 9d | 6i | 95 | 63 | |
| 10 | 6j | 58 (69)d | 79 (80)d | |
| 11d | 6k | 72 | 67 | |
| 12d | 6l | 90 | 83 | |
Reaction run with 1 equiv of aldehyde and 2 equiv of diyne 4; unreacted equivalents of 4 could be recovered.
Isolated yields.
Enantiomeric excess determined by chiral HPLC.
Reaction performed with 3 equiv each of 4 and Me2Zn.
Table 5.
Diyne scope in the asymmetric addition to aldehydesa
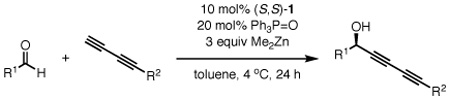 | |||||
|---|---|---|---|---|---|
| entry | aldehyde | R2 | product | % yieldb | % eec |
| 1 | 3a | 76 | 65 | ||
| 2 | 3b | 73 (49)d | 82 (79) d | ||
| 3d | 7a | 93 | 97 | ||
| 4e | 7b | 85 | 88 | ||
| 5e | 7c | 90 | 87 | ||
| 6 | 7d | 98 | 91 | ||
| 7 | 7e | 77 | 73 | ||
| 8 | 7f | >99 | 88 | ||
Reaction performed with 1 equiv of aldehyde and 3 equiv of diyne donor.
Isolated yields.
Enantiomeric excess determined by chiral HPLC.
Reaction was performed with 2 equiv of diyne and Me2Zn.
Catalyst loading was increased to 20 mol% 1 and 40 mol% TPPO.
Under the standard catalytic conditions (10 mol% ProPhenol ligand 1 and 20 mol% TPPO), aromatic aldehydes were suitable electrophiles; for instance TIPS diyne 4 reacted with 2-furfural in 98% ee and 95% yield (Table 4, entry 2). Acrolein, a typically difficult substrate, was a competent acceptor, furnishing propargyl alcohol 6c in up to 86% ee and 89% yield (entry 3). Substitution at either the α- or β-positions of the aldehyde was also well-tolerated. In spite of the increased steric congestion at the α-position, the diynylation of methacrolein provided alcohol 6d in 87% ee and 94% yield (entry 4). Sensitive functional groups such as a vinylcyclopropane (6e: 97% ee, entry 5) and allylic dimethylacetal (6f: 95% ee, entry 6) were unperturbed under the reaction conditions. The coupling of diyne 4 with a simple trans-unsaturated aldehyde such as trans-2-decenal led to diynol 6h being formed in 97% ee (entry 8); however, discrimination of the enantiotopic π-faces of a cis-α,β-unsaturated aldehyde by the dinuclear zinc catalyst was less effective (entry 9).
Saturated aldehydes were also suitable electrophiles in the enantioselective diynylation (Table 4, entries 10–12). The coupling of 4 with 4-(4-methoxybenzyloxy)butanal using 10 mol% (S,S)-1, 20 mol% TPPO and 3 equiv of Me2Zn furnished the chiral alcohol in up to 80% ee (entry 10). Even bulky α-substituted aldehydes proceeded with interesting levels of enantiomeric excess: cyclohexane carboxaldehyde furnished the corresponding diynol in 67% ee, while 2,2-dimethyl-4-pentenal reacted with 4 in 83% ee (entries 11 and 12, respectively).
Having demonstrated the effectiveness of TIPS diyne 4 as a nucleophile in the asymmetric addition, we then examined the substitution tolerated on the donor (Table 5). As previously shown (eq 3), the presence of primary propargyl ethers was tolerated. While the enantiomeric excesses were vastly improved with the addition of 20 mol% triphenylphosphine oxide, the selectivity was still highly dependent on the protecting group used. The tert-butyldimethylsilyl protected diyne 2a now coupled with octanal in 65% ee and 76% yield (3a, entry 1), whereas the analogous reaction with penta-2,4-diynyl ethanoate 2b proceeded in 82% ee and 73% yield (3b, entry 2). More importantly, the mild nature of the reaction conditions allow for the presence of an activated ester with no side-effects. Use of an α,β-unsaturated aldehyde was also successful: the reaction of trans-2-nonenal with acetate-protected diyne 2b furnished diynol 7a in 97% ee and 93% yield (entry 3). More elaborate linear saturated aldehydes were also suitable acceptors for the diyne addition: donor 2b was used to quickly access chiral propargyl alcohols 7b and 7c, precursors to strongylodiols A and B,15 with good reactivity and enantioselectivity (entries 4 and 5, respectively).
Unfunctionalized 1,3-diynes were also viable donors with our dinuclear zinc catalyst system. Buta-1,3-diynylcylclohexane reacted with an α,β-unsaturated aldehyde to furnish diynol 7d in 91% ee (entry 6) and octanal to afford 7e in 73% ee (entry 7). The electronic nature of the diyne was varied by extending the conjugation of the π-system; addition of a cyclohexenediyne into trans-4-methyl-2-pentenal afforded propargyl alcohol 7f quantitatively in 88% ee (entry 8).
Selective functionalization of the diynols
Given the versatile nature of the alkyne, we envisioned our optically active diyne carbinols could be useful synthetic building blocks. Previous studies in the Trost group demonstrated the use of [Cp*Ru(MeCN)3][PF6] (Cp* = pentamethylcyclopentadienyl) as a catalyst for the regioselective trans-hydrosilylation of propargyl alcohols.15 Furthermore, the resulting vinylsilane is a useful functional handle, in particular it can be rendered to a ketone.16 Given the highly selective nature of this ruthenium catalyst, we were interested in using it to exploit the directing ability of the propargyl alcohol to distinguish between the two alkynyl units and chemoselectively functionalize the proximal alkyne (Scheme 2).
Scheme 2.

Selective synthetic manipulations of the chiral diynol
Subjection of a diynol to a hydrosilylation/oxidation sequence would furnish the corresponding aldol adduct, a β-hydroxy ynone. The direct aldol reaction with methyl ynones was previously accomplished using the ProPhenol dinuclear zinc catalyst,17 however, the addition was limited to α,α-disubstituted aldehydes. Moreover, conventional aldol reaction methods involving electron-deficient aldehydes give competing elimination to form the unsaturated ketone. In the context of the asymmetric addition, our diyne nucleophile would serve as a methyl ynone surrogate for a challenging class of aldol reactions (eq 5). Treatment of furyl diynol 6b with ethoxydimethylsilane and 5 mol% [Cp*Ru] afforded the cyclic vinylsiloxane 8 quantitatively as observed by 1H NMR spectroscopy. It is of note that this hydrosilylation proceeded with exquisite chemo- and regioselectivity; the other possible isomers were not observed. Subsequent Tamao-Fleming oxidation of intermediate 8 with KHF2 and mCPBA furnished the desired β-hydroxyketone 9 in 70% overall yield from diyne 6b.
Given the trans-selectivity of the ruthenium-catalyzed hydrosilylation, we also envisioned employing this as a method for the partial reduction of alkynes to trans-olefins (Scheme 2, eq 6).18 Treatment of diynol 7d with benzyldimethylsilane (BDMS-H) and 2 mol% [Cp*Ru(MeCN)3][PF6] furnished the resulting vinylsilane 10 exclusively. Replacing the bulky triisopropylsilyl group on the diyne terminus (see eq 5) with a smaller cyclohexyl substituent did not impact the chemoselectivity, suggesting the hydrosilylation is indeed directed by the propargyl alcohol. Facile protodesilylation was achieved by treatment of 10 with tetrabutylammonium fluoride in THF to afford enyne 11 in 75% yield over 2 steps.
Natural product synthesis
The versatility of the asymmetric diynylation was also established through the total syntheses of a variety of optically active polyacetylenic natural products. Strongylodiols A (12) and B (13) were isolated from an Okinawan marine sponge of the genus Strongylophora and exhibited micromolar activity against several human cancer cell lines.19 As shown earlier, penta-2,4-diynyl ethanoate (2b) was an effective nucleophile for the coupling with saturated aldehydes and offered quick access to two strongylodiol precursors (Table 5, entries 4 and 5). While these strongylodiols have been previously synthesized,7,20 the overall efficiency of the ProPhenol-catalyzed transformation allows for a more atom-economical21 approach. Following the key diynylation step, strongylodiol A (12) was isolated in quantitative yield from 7b through a base-mediated cleavage of the primary propargyl acetate (Scheme 3, eq 7); strongylodiol B (13) was also synthesized in a similar manner (eq 8).
Scheme 3.

Total syntheses of strongylodiols A and B
As previously mentioned, we chose to use TIPS diyne 4 because it is a surrogate for 1,3-butadiyne. We sought to demonstrate the versatility of this nucleophile in the total synthesis of panaxytriol (14), an active component of red ginseng which is commonly used in traditional Asian medicine.22 Aside from its well-known analeptic and erythropoietic properties, panaxytriol is also cytotoxic against a variety of cancer cell lines.23 Furthermore, recent studies have shown that the anti-cancer properties of 14 are a result of a cytoprotective mechanism.24,25 Due to its important biological properties, many total syntheses of panaxytriol have been accomplished,26 where the C3 stereocenter has been derived from sugar degradation,26a,b,e enzymatic resolution26d or asymmetric ynone reduction,26f,g but not through direct asymmetric diyne addition (Scheme 4).
Scheme 4.

Total synthesis of (3R,9R,10R)-panaxytriol (14)a
aConditions: (a) Bu4N+F−, THF, 0 °C, >99%; (b) 1) BuLi, THF, −78 °C, then BF3•Et2O; 2) 16, THF; 3) Bu4N+F−, 7%.
Treatment of diynol 6c, formed in 86% ee from the addition of TIPS diyne 4 into acrolein, with TBAF revealed terminal diyne 15. Next, double deprotonation of 15 with BuLi generated the dianion which was complexed with BF3•Et2O to form the diynyl borate. The reaction of this acetylide with siloxy epoxide 16 led to regioselective ring-opening of the epoxide. An in situ quench of the ring-opened intermediate with TBAF furnished (3R,9R,10R)-panaxytriol in 70% yield. This lynchpin diynylation strategy not only allowed for rapid enantioselective construction of the diyne carbinol, but also facile coupling of two chiral fragments without the need to discretely pre-activate either piece. Through our sequence, (3R,9R,10R)-panaxytriol was prepared in 70% yield over three steps from acrolein.
In the synthesis of panaxytriol, a chiral diynol was coupled with a fragment containing its own chirality; however, we were also interested in constructing a molecule using sequential asymmetric diynylations. 8-Hydroxyheptadeca-1-en-4,6-diyn-3-yl ethanoate (17), an unnamed natural product, was an ideal target with which to demonstrate this principle (Figure 3). Isolated from the Tanzanian medicinal plant Cussonia zimmermannii, polyacetylene 17 exhibited potent antiprotozoal activity and also cytotoxicity.27 Although the structure was elucidated by NMR spectroscopy, no stereochemical assignment was reported. As this molecule had not been previously synthesized, a total synthesis would serve to confirm the structural assignment and establish the absolute and relative stereochemistry. Given the structural similarity of 17 with (3S,8S)-falcarindiol,28 we hypothesized the target compound also possesses the (3S,8S)-configuration.
Figure 3.

Structural similarity of 17 and falcarindiol
Since (S,S)-ProPhenol (1) affords the (R)-diynol, use of the (R,R)-enantiomer was required to prepare ent-6c with the correct stereochemistry (Scheme 5). The absolute configuration of the diyne addition was verified by the O-methylmandelate ester method.29,30 Treatment of ent-6c with acetic anhydride, followed by cleavage of the TIPS group with TBAF, furnished 18 in 70% yield. The resulting terminal diyne was then utilized in a subsequent ProPhenol-catalyzed diynylation; this double-barreled approach allows for the rapid construction of two challenging stereocenters. Under the reaction conditions with catalytic (R,R)-1 and TPPO, diyne 18 coupled with decyl aldehyde to furnish polyacetylene 17 in 67% yield.
Scheme 5.

Total synthesis of (3S,8S)-17a
aConditions: (a) 1) Ac2O, Et3N, 10 mol% DMAP, CH2Cl2; 2) Bu4N+F−, AcOH, THF, 0 °C, 70% over 2 steps; (b) decyl aldehyde, 20 mol% (R,R)-1, 40 mol% TPPO, 3 equiv Me2Zn, toluene, 4 °C, 67%.
The measured optical rotation of the synthetic natural product was consistent with the literature report, confirming our hypothesis that naturally occurring 17 is of the (3S,8S)-configuration. Furthermore, the stereochemistry of the propargyl alcohol at C8 was verified using the O-methylmandelate ester method;30 this derivitization also revealed the diastereoselectivity (>8.2:1 d.r.) of the second addition. Also of note is that the chiral acetoxy group in diyne 18 did not appear to interfere with the stereoselectivity of the second diynylation. Since the formation of the two stereogenic centers was catalyst-controlled the other diastereomers of 17 can be rapidly accessed simply by interchanging the ProPhenol ligand.
Conclusion
In conclusion, a catalytic and enantioselective process for terminal 1,3-diyne additions to aldehydes has been developed. Notably, in combination with the ProPhenol (1) catalyst, triphenylphosphine oxide served to enhance the chiral recognition of the dinuclear zinc acetylide species, a unique effect that was not previously observed in our aldol additions. The ProPhenol-catalyzed diynylation exhibited good generality with a variety of silyl- and alkyl-substituted diyne nucleophiles. Furthermore, we evaluated a range of aldehydes and demonstrated that aryl, α,β-unsaturated and the potentially enolizable saturated aldehydes are all well-tolerated by the dinuclear zinc catalyst system.
The chiral diynols formed were also used as synthetic building blocks; by taking advantage of the directing effect of the propargyl alcohol, the proximal alkyne moiety could be manipulated in a highly chemo- and regioselective fashion. Through a hydrosilylation/Tamao-Fleming oxidation sequence, we showed that the diyne carbinol is a β-hydroxy ynone surrogate. Additionally, the 1,3-diyne could be partially reduced to the olefin in a trans-selective manner to furnish highly functionalized allylic alcohols.
The synthetic utility of this method was further demonstrated in the total syntheses of four biologically active polyacetylenic natural products. The cytotoxic strongylodiols A and B (12 and 13, respectively) were synthesized in a straightforward manner using a catalytic asymmetric addition of diyne 2b into the corresponding saturated aldehydes with good levels of enantioselectivity. Employment of a lynchpin strategy, where silyldiyne 4 served as a 1,3-butadiyne surrogate, enabled the rapid preparation of (3R,9R,10R)-panaxytriol (14) in only 6 steps in the longest linear sequence. Although the diynylation with saturated aldehydes did not always yield the highest enantioselectivities, they were still effective substrates as exemplified in the expedient total synthesis of the antifungal (3S,8S)-8-hydroxyheptadeca-1-en-4,6-diyn-3-yl ethanoate (17), where we verified the structure and assigned the stereochemistry of the natural product. Most importantly, use of this lynchpin approach allowed for the direct and facile generation of molecular complexity around the butadiyne moiety.
Experimental Section
Typical procedure for asymmetric diyne addition into aldehydes
A flame-dried microwave vial equipped with a stir bar was charged with triphenylphosphine oxide (8.7 mg, 0.031 mmol, 0.2 equiv), ProPhenol ligand 1 (10 mg, 0.016 mmol, 0.1 equiv) and toluene (1.0 mL) under argon. The TIPS diyne 4 (77 µL, 0.31 mmol, 2 equiv) was added via syringe, followed by dimethylzinc (261 µL, 0.31 mmol, 2 equiv, 1.2 M in toluene). The alkynyl zinc solution was stirred at room temperature for 30 minutes, then the reaction solution was cooled to 0 °C and the appropriate aldehyde was added (0.16 mmol, 1 equiv). The reaction was left to proceed under argon at 4 °C. After 24 h, the reaction solution was quenched with aqueous, saturated NH4Cl (2 mL) and stirred vigorously for 10 min. The toluene phase was separated and the aqueous phase was extracted with Et2O (4 × 2 mL). The combined organic fractions were dried over sodium sulfate, filtered and concentrated on a rotary evaporator. The crude product was then purified by flash column chromatography on silica gel.
Supplementary Material
Acknowledgment
We thank the NSF for their generous support of our programs. We also thank Sigma-Aldrich for donating ProPhenol ligand. V.S.C. acknowledges Dr. J. P. Lumb for insightful discussions.
Footnotes
Supporting Information Available: Detailed experimental procedures and characterization data for all new compounds. This material is available free of charge on the Internet at http://pubs.acs.org.
REFERENCES
- 1.Stang PJ, Diederich F, editors. Modern Acetylene Chemistry. Weinheim, Germany: VCH; 1995. [Google Scholar]
- 2.Minto RE, Blacklock BJ. Prog. Lipid Res. 2008;47:233–306. doi: 10.1016/j.plipres.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shun ALKS, Tykwinski RR. Angew. Chem. Int. Ed. 2006;45:1034–1057. doi: 10.1002/anie.200502071. [DOI] [PubMed] [Google Scholar]
- 4.For a recent review, see: Trost BM, Weiss AH. Adv. Synth. Catal. 2009;351:963–983. doi: 10.1002/adsc.200800776.
- 5.Jarowski PD, Wodrich MD, Wannere CS, Schleyer PvR, Houk KN. J. Am. Chem. Soc. 2004;126:15036–15037. doi: 10.1021/ja046432h. [DOI] [PubMed] [Google Scholar]
- 6.(a) Frantz DE, Fässler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806. [Google Scholar]; (b) Anand NK, Carreira EM. J. Am. Chem. Soc. 2001;123:9687–9688. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]
- 7.Reber S, Knöpfel TF, Carreira EM. Tetrahedron. 2003;59:6813–6817. [Google Scholar]
- 8.(a) Trost BM, Weiss AH, von Wangelin AJ. J. Am. Chem. Soc. 2006;128:8–9. doi: 10.1021/ja054871q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Weiss AH. Org. Lett. 2006;8:4461–4464. doi: 10.1021/ol0615836. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Weiss AH. Angew. Chem. Int. Ed. 2007;46:7664–7666. doi: 10.1002/anie.200702637. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, O’Boyle BM. J. Am. Chem. Soc. 2008;130:16190–16192. doi: 10.1021/ja807127s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Trost BM, O’Boyle BM, Hund D. J. Am. Chem. Soc. 2009;131:15061–15074. doi: 10.1021/ja906056v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitamura M, Suga S, Kawai K, Noyori R. J. Am. Chem. Soc. 1986;108:6071–6072. doi: 10.1021/ja00279a083. [DOI] [PubMed] [Google Scholar]
- 10.For the seminal publication, see: (a) Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003–12004. For a recent example and references therein: (b) Trost BM, Hitce J. J. Am. Chem. Soc. 2009;131:4572–4573. doi: 10.1021/ja809723u.
- 11.(a) Trost BM, Silcoff ER, Ito H. Org. Lett. 2001;3:2497–2500. doi: 10.1021/ol0161211. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Terrell LR. J. Am. Chem. Soc. 2003;125:338–339. doi: 10.1021/ja028782e. [DOI] [PubMed] [Google Scholar]
- 12.Yoshikawa N, Kumagai N, Matsunaga S, Moll G, Ohshima T, Suzuki T, Shibasaki M. J. Am. Chem. Soc. 2001;123:2466–2467. doi: 10.1021/ja015580u. [DOI] [PubMed] [Google Scholar]
- 13.For leading references: Yamagiwa N, Abiko Y, Sugita M, Tian J, Matsunaga S, Shibasaki M. Tetrahedron: Asymmetry. 2006;17:566–573.
- 14.Xiao Y, Wang Z, Ding K. Chem. Eur. J. 2005;11:3668–3678. doi: 10.1002/chem.200401159. [DOI] [PubMed] [Google Scholar]
- 15.(a) Trost BM, Ball ZT, Jöge T. Angew. Chem. Int. Ed. 2003;42:3415–2418. doi: 10.1002/anie.200351587. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Ball ZT. J. Am. Chem. Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trost BM, Ball ZT, Laemmerhold KM. J. Am. Chem. Soc. 2005;127:10028–10038. doi: 10.1021/ja051578h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660–2661. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Frederiksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J. Am. Chem. Soc. 2005;125:3666–3667. doi: 10.1021/ja042435i. [DOI] [PubMed] [Google Scholar]
- 18.Trost BM, Ball ZT, Jöge T. J. Am. Chem. Soc. 2002;124:7922–7923. doi: 10.1021/ja026457l. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe K, Tsuda Y, Yamane Y, Takahashi H, Iguchi K, Naoki H, Fujita T, Van Soest RWM. Tetrahedron Lett. 2000;41:9271. [Google Scholar]
- 20.(a) Yadav JS, Mishra K. Tetrahedron Lett. 2002;43:1739–1741. [Google Scholar]; (b) Kirkham JED, Courtney TDL, Lee D, Baldwin JE. Tetrahedron Lett. 2004;45:5645–5647. [Google Scholar]; (c) Kirkham JED, Courtney TDL, Lee V, Baldwin JE. Tetrahedron. 2005;61:7219–7232. [Google Scholar]
- 21.(a) Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. Angew. Chem. Int. Ed. Engl. 1995;34:259–281. [Google Scholar]; (c) Trost BM. Acc. Chem. Res. 2002;35:695–705. doi: 10.1021/ar010068z. [DOI] [PubMed] [Google Scholar]
- 22.(a) Kitagawa I, Yoshikawa M, Yoshihara M, Hayashi T, Taniyama T. Yakugaku Zasshi. 1983;103:612–622. [PubMed] [Google Scholar]; (b) Kitagawa I, Taniyama T, Shibuya H, Noda T, Yoshikawa M. Yakugaku Zasshi. 1987;107:495–505. doi: 10.1248/yakushi1947.107.7_495. [DOI] [PubMed] [Google Scholar]
- 23.(a) Katano M, Yamamoto H, Matsunaga H, Mori M, Takata K, Nakamura M. Gan to Kagaku Ryoho. 1990;17:1045–1049. [PubMed] [Google Scholar]; (b) Matsunaga H, Saita T, Naguo F, Mori M, Katano M. Cancer Chemother. Pharmacol. 1995;35:291–296. doi: 10.1007/BF00689447. [DOI] [PubMed] [Google Scholar]; (c) Saita T, Katano M, Matsunaga H, Kouno I, Fujito H, Mori M. Bio. Pharm. Bull. 1995;18:933–937. doi: 10.1248/bpb.18.933. [DOI] [PubMed] [Google Scholar]; (d) Kim JY, Lee K-W, Kim S-H, Wee JJ, Kim Y-S, Lee HJ. Planta Med. 2002;68:119–122. doi: 10.1055/s-2002-20240. [DOI] [PubMed] [Google Scholar]
- 24.Halim M, Yee DJ, Sames D. J. Am. Chem. Soc. 2008;130:14123–14128. doi: 10.1021/ja801245y. [DOI] [PubMed] [Google Scholar]
- 25.Ng F, Yun H, Lei X, Danishefsky SJ, Fahey J, Stephenson K, Flexner C, Lee L. Tetrahedron Lett. 2008;49:7178–7179. doi: 10.1016/j.tetlet.2008.09.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Lu W, Zheng G, Cai J. Synlett. 1998:737–738. [Google Scholar]; (b) Lu W, Zheng G, Gao D, Cai J. Tetrahedron. 1999;55:7157–7168. [Google Scholar]; (c) Gurjar MK, Kumar VS, Rao BV. Tetrahedron. 1999;55:12563–12576. [Google Scholar]; (d) Mayer SF, Steinreiber A, Orru RVA, Faber K. J. Org. Chem. 2002;67:9115–9121. doi: 10.1021/jo020073w. [DOI] [PubMed] [Google Scholar]; (e) Yadav JS, Maiti A. Tetrahedron. 2002;58:4955–4961. [Google Scholar]; (f) Yun H, Danishefsky SJ. J. Org. Chem. 2003;68:4519–4522. doi: 10.1021/jo0341665. [DOI] [PubMed] [Google Scholar]; (g) Yun H, Chou T-C, Dong H, Tian Y, Li Y-M, Danishefsky SJ. J. Org. Chem. 2005;70:10375–10380. doi: 10.1021/jo0515475. [DOI] [PubMed] [Google Scholar]; (h) Cho EJ, Lee D. Org. Lett. 2008;10:257–259. doi: 10.1021/ol702651s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Baur R, Simmen U, Senn M, Séquin U, Sigel E. Mol. Pharmacol. 2005;68:787–792. doi: 10.1124/mol.105.011882. [DOI] [PubMed] [Google Scholar]; (b) Senn M, Gunzenhauser S, Brun R, Séquin U. J. Nat. Prod. 2007;70:1565–1569. doi: 10.1021/np0702133. [DOI] [PubMed] [Google Scholar]
- 28.Bernart MW, Cardellina JH, II, Balaschak MS, Alexander MR, Shoemaker RH, Boyd MR. J. Nat. Prod. 1996;59:748–753. doi: 10.1021/np960224o. [DOI] [PubMed] [Google Scholar]
- 29.Trost BM, Belletire JL, Godleski S, McDougal PG, Balkovec JM. J. Org. Chem. 1986;51:2370–2374. [Google Scholar]
- 30.See the Supporting Information for analysis of the O-methylmandelate esters.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.