

Abstract
Epidemiological studies have shown that advancing age is associated with an increased prevalence of cardiovascular disease (CVD). Vascular smooth muscle cells (VSMC) comprise the major arterial cell population, and changes in VSMC behavior, function, and redox status with age contribute to alterations in vascular remodeling and cell signaling. Over two decades of work on aged animal models provide support for age-related changes in VSMC and/or arterial tissues. Enhanced production of reactive oxygen species (ROS) and insufficient removal by scavenging systems are hallmarks of vascular aging. VSMC proliferation and migration are core processes in vascular remodeling and influenced by growth factors and signaling networks. The intrinsic link between gene regulation and aging often relates directly to transcription factors and their regulatory actions. Modulation of growth factor signaling leads to up- or downregulation of transcription factors that control expression of genes associated with VSMC proliferation, inflammation, and ROS production. Four major signaling pathways related to the transcription factors, AP-1, NF-κB, FoxO, and Nrf2, will be reviewed. Knowledge of age-related changes in signaling pathways in VSMC that lead to alterations in cell behavior and function consistent with disease progression may help in efforts to attenuate age-related CVD, such as atherosclerosis. Antioxid. Redox Signal. 12, 641–655.
Introduction
For more than half a century, scientists have suspected that reactive oxygen species (ROS) play a major role in the aging process and the pathogenesis of age-related diseases. Investigators have examined the role of the redox state in vascular smooth muscle cells (VSMC) in the pathogenesis of vascular disease (16, 68). Numerous studies underscore the importance of dysregulated oxidant and antioxidant balance in advancing age (77) and in the development and progression of atherosclerosis in both animal models and in humans (119). Vascular smooth muscle cells present in atherosclerotic lesions often proliferate and show increased expression of genes for growth factors and other molecules involved in extracellular matrix remodeling (83, 101). Proliferation of VSMC is part of the initiation and the progression of atherosclerosis (97) and may occur in response to injury or as a result of aberrant apoptosis (15). This review will focus on several redox-sensitive signaling pathways known to change with age and to influence VSMC growth and behavior.
The study of age-related changes in VSMC redox status, behavior, and function has primarily been conducted in animal models of aging using the monkey, rabbit, rat, or mouse. These models permit extrapolation to the human, especially in the case of age-associated changes in arterial remodeling. The Fischer 344/Brown Norway F1 hybrid (F344xBN) rat, a well-characterized rat strain approved for aging research by the National Institutes on Aging (NIH-NIA), has been widely used to collect data from arterial tissues where VSMC represent the majority of the cell population. Furthermore, it has been well-established that early passages of explanted VSMC from these models retain their in vivo age-related characteristics (41, 105).
Redox Status with Advancing Age
A role for ROS in many aspects of age-related vascular remodeling has been described. VSMC contain numerous sources of ROS with the major forms being superoxide anion (O2•−), which targets heme groups, Fe–S clusters, cysteine residues, or other electron transfer units, and hydrogen peroxide (H2O2). The short half-life of O2•− limits the likelihood that it serves a paracrine role in the vasculature; however, its more stable metabolite, H2O2, is a viable candidate for this role due to its ability to diffuse freely across the vascular wall. To determine the influence of aging on multiple markers of oxidative stress in the aorta, young and aged experimental animals were studied. Significant increases in O2•− were observed in aortas of old F344xBN rats (93), Wistar–Kyoto rats (WKY) and stroke-prone spontaneously hypertensive rats (SHRSP) (40). These findings were highly correlated with increases in medial thickness, total protein nitration, and NADPH oxidase, suggesting that the aging rat aorta may be suitable for unraveling the molecular events that lead to age-associated oxidative stress. Recently, an interesting longest-living rodent, the naked mole rat (NMR), was used in a study of vascular aging. The maximum lifespan potential of the NMR is over 28 years compared to ∼3 years for the F344xBN rat. O2•− and H2O2 production significantly increased with age in 24-month F344 rat arteries whereas they did not change substantially in vessels from NMR animals of the same age (17), implying that successful clearance of O2•− and H2O2 in vessels may contribute to the delay in vascular aging in the NMR.
A correlation between aging and the accumulation of oxidatively damaged proteins, lipids, and nucleic acids has been reported (106). This raises the possibility that the accumulation of oxidized proteins during aging reflects a loss of apoptotic capacity (i.e., oxidatively modified proteins persist mainly in cells that have escaped apoptosis). Oxidative modification of proteins causes the introduction of a carbonyl group into the protein, leading to loss of catalytic or structural function of the affected proteins. Therefore, increased levels of oxidatively modified proteins during aging will have deleterious effects on cellular and organ function (57). Figure 1 shows the increased protein carbonyl content in VSMC isolated from old compared to young F344 rats.
FIG. 1.

Protein carbonyls in VSMC from young and old rats. In the upper panel, representative blots show protein carbonyl staining obtained with the OxyBlot™ kit in VSMC explanted from a young (6 mon) and an old (24 mon) rats. lane 1: VSMC grown in control medium with 5 mM glucose; lane 2: 3 h exposure to TNF-α (5 ng/ml); lanes 3 and 4: 4 days of exposure to 12.5 or 25 mM of glucose to induce oxidative stress; lane 5: 3 h exposure to 30 μM of H2O2. The lower panel shows the total protein on the same membranes stained with India ink.
In young healthy cells, the accumulation of oxidatively damaged proteins is prevented by the rapid elimination of such proteins through proteolytic systems, including the lysosomal cathepsins, calcium-activated calpains, and the 20S and 26S proteasomes. However, aging causes a dramatic decrease in the ability of cell lysates to degrade damaged protein because of an impairment in proteasome structure and function (103). There is still a need to confirm the existence of age-related changes in proteasome function in VSMC.
In addition to reports of the contribution of oxidized proteins to the aging process, it is known that the absolute effects of cholesterol on annual mortality rates from ischemic heart disease are much greater in the older population. For example, the absolute difference in the annual risk of death from ischemic heart disease for a 1 mmol/L difference in total cholesterol was ∼10 times greater at 80–89 years than at 40–49 years of age (89). The central concept is that oxidative modification of low density lipoproteins (LDL) promotes a proinflammatory response, recruitment of macrophages, and the development of atherosclerotic lesions. The presence of oxidized products of LDL (ox-LDL) in lesions significantly increases with advancing age (21).
ROS generated by the mitochondria are important in the regulation of signaling pathways that contribute to CVD. VSMC isolated from older animals appear to be more vulnerable to glucose-related mitochondrial injury independent of osmolarity (30). Because repair of mitochondrial proteins often requires new protein synthesis, damage to mtDNA is likely to lower this threshold. Thus, the accumulation of mtDNA damage over a lifetime may increase the susceptibility to the development of pathology (39). Mitochondria isolated from aortas of F344 rats exhibit higher rates of oxy-radical production (O2•− and H2O2) in old animals than do those from young animals. Electron microscopy and confocal microscopy also revealed that a decline of mitochondrial biogenesis occurs in aortas with aging. The expression of the mitochondrial biogenesis factors (including mitochondrial transcription factor A and peroxisome proliferator-activated receptor-γ-coactivator-1) in aged vessels was decreased. The vascular expression of complex I, III, IV, and cytochrome c oxidase enzyme activity were also significantly lower with age (110). These changes all coalesce to influence mitochondrial function that may contribute to age-related differences in oxidative stress.
Imbalance Between Pro-Oxidants and Antioxidants in Vascular Aging
Cells have developed a number of antioxidant defense systems to maintain antioxidant capacity in response to oxidative stress. Under normal physiological conditions, ROS quenching by antioxidant enzymes is sufficient to maintain the restitution of antioxidant/pro-oxidant equilibrium following an oxidative challenge. Conversely, when the production of ROS exceeds endogenous antioxidant capacity, oxidative or nitrosative stress results in abnormal physiological responses, with subsequent severe damage to proteins, lipids, and DNA (68, 109). In vascular aging, upregulation of pro-oxidants and downregulation of antioxidants results in an imbalance leading to an increase in ROS. The age-dependent changes in pro-oxidants and antioxidants reported in VSMC or aortic tissues are summarized in Table 1 and described in more detail below.
Table 1.
Expression of Pro- or Antioxidants Changes with Aging (in VSMC or Aorta Tissue)
| Name | Pro-/anti-oxidants | Functions | Changes with aging | References |
|---|---|---|---|---|
| Nox, Nox1,2,4 | Pro- | O2• production | ↑ | 71, 86 |
| XO | Pro- | O2• production | ↑ | 82 |
| NOS, iNOS | Pro- & Anti- | Nitric oxide synthesis | ↑ | 13, 18, 25 |
| GPx | Anti- | H2O2 → H2O | ↑ | 22 |
| GCL, GCLC | Anti- | Glutathione synthesis | ↑ | 62 |
| HO, HO1 | Anti- | Heme → Fe2+, CO | ↑ | 81 |
| MnSOD | Anti- | O2• → H2O2 | ↓ | 60, 70, 120 |
| Cu2+ Zn2+ dismutase | Anti- | O2• → H2O2 | ↓ | 25 |
| Catalase | Anti- | H2O2 → H2O | ↓ | 22 |
| NO | Pro- & Anti- | ↓ | 35, 110 | |
| GSH | Anti- | H2O2 → H2O | ↓ | 60, 103 |
The most widely studied enzymatic sources of O2•− in the vascular wall are NADPH oxidase (Nox) and xanthine oxidase (XO). A primary source of pro-oxidants in the aortic wall is several isoforms of Nox that produce free radicals through the reaction:
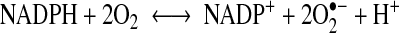 |
(1) |
Most vascular cells express multiple Nox proteins, including gp91phox (also known as Nox2), as well as Nox1, Nox4, and Nox5 (68). In a study of aortic segments from young and old Wistar rats, the activity and expression of Nox subunits gp91phox and p22phox were found to increase with age (86). Furthermore, NADPH oxidase 4 (Nox4) expression was augmented in VSMC isolated from aortas of old rats (71). The XO system is another source of pro-oxidants in the aorta. XO activity in aging rats was significantly higher and paralleled the approximately twofold increase in free radical generation (82). Uncoupled nitric oxide synthase (NOS) (67, 73) and enzymes of the mitochondrial electron transport chain (113) are other potential sources of free radicals in aortas. Nitric oxide (NO) is unique in that it exhibits opposing functions in the vasculature (91). Being a free radical, NO has both pro- and antioxidant properties (88). NO can be protective against oxidative injury, depending on the specific conditions. The pro-oxidant reactions of NO occur with superoxide, whereas the antioxidant effects of NO are related to direct reactions with alkoyl and peroxyl radical intermediates generated during lipid peroxidation. Vascular aging is also associated with progressively reduced NO-mediated vascular relaxation. This is best demonstrated by decreased NO-bioavailability in various aging rat models (35, 80, 111). For example, a clear impairment of the NO-dependent vasodilation was observed in old wild-type B6 (control) and B6xOla (ALDH-2−/−) mice compared to middle-aged animals of the same type. O2•−, as the primary radical generated, inactivated free endothelium-derived NO to form the highly reactive ONOO− that decreases NO bioavailability (121). VSMC are important targets for endothelium-derived NO normally produced by vascular endothelial cells, which constitutively express an endothelial NO synthase, eNOS. VSMC can also produce NO through expression of an inducible NO synthase (iNOS) under inflammatory conditions (123). The cytokine-regulated iNOS converts O2 and L-arginine to NO and L-citrulline. Since NO negatively modulates nitric oxide synthesis (36, 37), decreased NO bioavailability in VSMC from old animals is usually accompanied by an upregulation of signaling for iNOS expression (12, 25) through the NF-kB pathway (123).
Among the cellular pathways involved in the protection against oxidative and nitrosative stress, antioxidant enzymes [such as the superoxide dismutase (SOD) family of CuZnSOD (SOD1), MnSOD (SOD2), and ECSOD (SOD3)], catalase, and glutathione-related enzymes, all actively operate to counteract deleterious consequences of free radical damage. In addition, the vitagene system has been invoked as the group of genes that control maintenance and repair processes in the body in a network that promotes processes associated with longevity (10). The vitagenes encode for cytoprotective heat shock protein 70 (Hsp70), heme oxygenase-1 (HO-1), thioredoxin reductase (TrxR), peroxiredoxin (Prx), and Sirtuin, all of which are actively involved in the process of detoxification.
A reduction in SOD is a sign of vascular aging. Studies of the rat aorta of male Wistar rats revealed significantly higher levels in O2•− and significantly lower levels in SOD activity at 18 and 24 months of age (70). We also reported downregulation of MnSOD expression and activity in explanted VSMC from old F344 rats (62). In addition to the family of SOD, catalase was reduced with aging in aortic tissue from old F344 rats (22).
GSH is a cysteine-containing tripeptide with reducing and nucleophilic properties that plays an important role in cellular protection against oxidative damage. GSH-related enzymes include glutathione peroxidase (Gpx) and glutathione-S-transferase (GST), as well as the enzymes for GSH synthesis. Most of these enzymes belong to the group of Phase II detoxification enzymes, shown in Fig. 2. Reduction of GSH content has been reported in the aorta and VSMC from aged animals (104). However, this was accompanied by an increase in GSH-related phase II enzyme activity, such as Gpx (22, 104) in the aorta and γ-glutamate cysteine ligase catalytic subunit (GCLC) in VSMC (60).
FIG. 2.

GSH-related enzymes. GSH synthesis: First, γ-glutamylcysteine is synthesized from L-glutamate and cysteine via the enzyme GCL, containing GCLC and GCLM subunits. Second, glycine is added to the C-terminal of glu-cys via the enzyme GS. GSH exists in two forms: reduced (GSH) and oxidized (GSSG). In the reduced state, the thiol group of cysteine is able to donate an electron to other unstable molecules, such as ROS to form H2O2, or it may readily react with another GSH to form glutathione disulfide (GSSG). Gpx catalyzes the GSH-dependent reduction of H2O2 to harmless H2O. GSH can be regenerated from GSSG by the NADPH-dependent enzyme glutathione reductase (Gr). GSH also reacts with various electrophiles, physiological metabolites, and xenobiotics. These reactions are initiated by GST where GSH serves as a cofactor in conjugation reactions to form a glutathione S-conjugate (GSX).
Age-Related Changes in VSMC Function During Vascular Remodeling
Besides the evidence for age-related changes in redox status and the possible contribution of oxidant–antioxidant imbalance in VSMC described above, another specific pathophysiological mechanism that underlies atherosclerosis is remodeling of the vasculature. In healthy humans (carefully screened to exclude cardiovascular disease), the large elastic arteries become dilated with advancing age and the intima thickens. The thickness of the arterial wall, as indexed by the thickness of the intimal and medial layers, increases in a linear fashion nearly threefold between the ages of 20 and 90 years, even in the absence of atherosclerotic plaques (79). In seeking evidence from a nonhuman primate, Wang and his colleagues studied aged (20.0 ± 1.9 years) male monkeys without atherosclerosis compared to young (6.4 ± 0.7 years) monkeys and found that intimal thickness increased by threefold with numerous VSMC and increased extracellular matrix in the old (116). Similar results were described recently in human aortic samples obtained from those who died of noncardiovascular events (117). The described age-related pathology in the thoracic aorta of the F344 rat is similar to that found in humans. VSMC comprise the medial layer of blood vessels and represent a dynamic component of the vasculature. Aortic medial VSMC from older rats are larger in size and fewer in number than those in the aorta from young adult rats. Some of these cells appear to have undergone an age-associated phenotypic modulation toward a dedifferentiated and synthetic state. VSMC migration from the medial to the intimal compartment is a plausible mechanism for the increased number of VSMC within the diffusely thickened intima of central arteries as animals age (74) (Fig. 3). The distribution of actively growing explanted VSMC from young and old animals through the cell cycle was examined at 24 hs after subcultivation. The VSMC from older animals showed a higher percentage of their population in the S phase compared to young VSMC (∼20% vs. ∼9%, respectively) and a significantly decreased percentage in the G0/GI phase (∼76% vs. ∼83% respectively) (41).
FIG. 3.

Effects of progressive aging in rat aorta. Representative micrographs showing cross sections of 3-, 6-, 12-, 18-, and 24-month-old Fischer 344/Brown Norway F1 hybrid (F344xBN) rat thoracic aorta. Sections from plastic-embedded tissues were stained with Lee's methylene blue. The internal elastic lamina was identified as the innermost continuous elastic lamina, which separated the intima from the media. The outer boundary of media is defined by dense staining of smooth muscle relative to adventitial connective tissue. Note the increasing thickness of the tunica intima and media and irregularity of the intimal surface with age. Arrows indicate typical intimal protrusions, which are first observed at 6 months of age. Reproduced with permission from AJP-Heart Circ Physiol 293: H2634–H2643, 2007. www.ajpheart.org
Exaggerated responsiveness to injury is also commonly found in old age. From a study that assessed the development of the neointima in a model of mechanical injury to vessels in aging (18 months) and young (2 months) mice, older age was associated with exaggerated neointimal formation (112). An increase in stiffness of the central arterial wall was also observed and accompanied by an increase of central arterial wall thickness after arterial injury. The stiffness of the arterial wall was also modulated by interactions between VSMC and the extracellular matrix (79). Although a decline in many functions seen during aging has been correlated with a decrease in growth capacity of many cells, VSMC have been shown to proliferate excessively with aging (85), synthesizing excess extracellular matrix and inflammatory cytokines. These events occurred simultaneously with the well-documented age-dependent endothelial dysfunction in vascular beds. These key changes in VSMC function during age-related vascular remodeling coalesce to increase susceptibility to the development of atherosclerosis. The next important consideration is the identification of potential mechanisms responsible for these changes during age-related vascular remodeling.
Age-Related Upregulation of Growth Factor and Hormone Signaling in VSMC
In general, growth factors and hormones are the most potent activators that stimulate VSMC growth, migration, and extracellular matrix synthesis. These include platelet-derived growth factor receptor (PDGFR), angiotensin II (Ang II), transforming growth factor-β (TGF-β), insulin-like growth factor receptor (IGF-1R), epidermal growth factor receptor (EGFR), and fibroblast growth factor (FGF). ROS also act as second messengers and mediate many pathophysiological processes that coordinate activation of multiple growth factors/hormones and their respective receptors through direct activation of redox-sensitive signaling networks (16, 68) or indirectly modulate the activation of growth factors through promotion of global DNA hypomethylation or changes in sirtuin-dependent histone modification (26, 27, 34, 95). However, direct evidence that these latter functions are operable in VSMC is still needed.
Ang II signaling has been widely linked to an age-associated increase in the migratory capacity of VSMC and to the proinflammatory features of arterial aging. Ang II increases within the aged arterial wall and activates matrix metalloproteinase type II (MMP2) (50, 116). Ang II appears to initiate growth-promoting signal transduction through ROS-sensitive tyrosine kinases. Previous findings suggest a transactivation of tyrosine kinases cross-talk by Ang II, including the EGFR (28), IGF-1R (108), and PDGFR (45).
PDGF and its receptor (PDGFR) are potent mitogenic and migratory factors that regulate tyrosine phosphorylation of a variety of signaling proteins via activation by intracellular ROS, namely H2O2. In a study to examine the effect of aging on neointimal formation in a mouse model of mechanical vascular injury, aging mice were found to have exaggerated neointimal development after injury. Medial VSMC from aging aortas expressed more PDGFR-α compared to VSMC from young counterparts (112). It was also shown that there was a progressive increase with age in aortic mRNA levels of PDGFR-β in aged Wistar rats (99). PDGFR signaling transactivates the release of FGF2 and subsequently activates the FGFR1 transduction pathways in human VSMC in an autocrine and paracrine fashion. Both contribute to the second wave of extracellular signal-regulated kinase (ERK) activation (75).
Both active TGF-β1 and its receptor increase within the aged aortic wall, particularly within the thickened intima of F344xBN rats. TGF-β1 receptor-mediated SMAD signaling is also increased in VSMC from old rat aortas; phosphorylated SMAD2/3 and 4 increased, whereas the level of the inhibitory SMAD7 protein decreased with age (118). These growth factors (Ang II, PDGF, and TGF-β) also differentially contribute to the regulation of Nox activity through Nox1, Nox2, and Nox4 (16, 56, 68) and thereby influence the production of ROS in VSMC.
IGF-1 is another potent mitogen for VSMC. Several in vitro studies of both animal and human VSMC show that IGF-1 induces cell cycle changes leading to proliferation and migration. The action of IGF-1 is mediated through specific membrane receptors, IGF-1 receptor (IGF-1R), which is highly expressed in VSMC of intact arteries and in cultured VSMC. A 2.8-fold increase of IGF-1R mRNA based on a microarray analysis done during the development of progressive aortic vasculopathy in aged aortas has been described in the F344 rat model of aging (74). Recently, constitutive activation of IGF-1R (β-chain) expression and its tyrosine kinase activity was also reported in VSMC explanted from the old F344 rats (59). IGF-1/IGF-1R stimulates VSMC proliferation and migration by activating both mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways (90).
In addition to the IGF-1/IGF-1R axis, IGF-1 can also act synergistically with PDGFR (72) and basic fibroblast growth factor (6) to stimulate VSMC proliferation. IGF-1R also has a transactivation effect to mediate Ang II-signaling (5). In contrast, estrogen has been shown to downregulate IGF-1/ IGF-1R expression in VSMC (100), and this may account for the higher age-related IGF-1R level in women after menopause because postmenopausal women have lower levels of estrogen.
Redox-Sensitive Signaling Pathways in VSMC: Influence of Age
Aging naturally involves all life processes and multiple subtle changes work in concert to elicit the characteristics of aging. Thus, a broad range of biological information can be expected to contribute to our understanding of the aging process. The signaling networks that modulate all the key changes in VSMC during the aging process, including those we have reviewed above, may give insight into the mechanisms responsible for vascular aging. We will focus on four related transcriptional regulatory pathways that most often contribute to changes in VSMC redox status and function.
Activator protein-1 (AP-1): role in VSMC proliferation and migration
c-fos and c-jun are early response genes that are induced by a variety of external stresses and encode protein subunits (Jun homodimers or Jun/Fos heterodimers) that comprise the AP-1 transcription factor (3). The AP-1 transcription factor is important in regulating genes involved in cell cycle progression, inflammation, and apoptosis. For example, activation of early response genes is involved in the transition from the GI into the S phase of the cell cycle (53). Upregulation of c-fos and c-jun, as reflected by increases in mRNA, protein, or AP-1 DNA binding activity, are markers of the immediate early gene response that plays a key role in the control of terminal cell differentiation, proliferation and apoptosis in a number of cell types (52).
Age-related differences in the activation of AP-1 have been shown to contribute to the age-dependent increase in VSMC proliferation and migration. As depicted in Fig. 4, binding of growth factors to their receptors induces receptor dimerization and autophosphorylation, which, in turn, leads to the activation of transcription factors, such as AP-1, via activation of ERK1/2. Activation of growth factor receptors also leads to tyrosine phosphorylation of src-homology2/collagen-α (Shc) proteins, followed by activation of oncogenic Ras, which then phosphorylates Raf (c-Raf1, B-Raf, or A-Raf ) and ERK kinase (MEK) to activate ERK1/2 (51). ERK can also be activated through association with the p85 regulatory subunit in the PI3K pathway (84). Regardless of the initiating pathway, activated ERK1/2 is translocated into the nucleus to either phosphorylate, and thereby potentiate the transcriptional activity of ternary complex factors (TCF), which can bind to fos promoters and initiate Fos transcription, or directly phosphorylate the Fos proteins (102). Regulation of the Jun N-terminal kinase (JNK) pathway is extremely complex and influenced by many MAPK kinase kinases (MKKK). The diversity of MKKK allows for a wide range of stimuli leading to differential responses in the activation of Jun proteins. Ultimately, the combination of Fos and Jun proteins in the AP-1 transcription factor complex regulates a wide range of cellular processes, including cell proliferation, death, survival, and differentiation. It is well known that AP-1 promotes cell proliferation through upregulation of its target genes for GI cyclins, such as cyclin D1 (102), cyclin E (120), and their corresponding CDKs and through downregulation of a CDK inhibiter, p21cip-1 (p21) (46). Proliferating cell nuclear antigen (PCNA), a cell cycle-regulated protein possessing DNA-polymerase-δ ability (107), is also a target for AP-1 regulation (65). In addition to the cyclins, the MMPs, key regulators of cell migration, are direct targets of the ERK/Fos pathway, and include membrane-type1 MMP (MT1-MMP) (47) MMP-2, and MMP-9 (7, 76). MMPs influence VSMC behavior through cleavage of both matrix and nonmatrix substrates (83). AP-1 is also related to transcriptional upregulation of Nox, p22 phox subunit in human VSMC (69).
FIG. 4.

Aging-related upregulation of AP-1 signaling in VSMC. Aging is associated with an increase in growth factors that bind to receptors and lead to phosphorylation of the Shc/RAS pathway or the p85 regulatory subunit of the PI3K. This in turn leads to phosphorylation of ERK1/2. Activated ERK1/2 is translocated into the nucleus to either phosphorylate, and thereby potentiate the transcriptional activity of TCF that can bind to fos promoters and initiate Fos transcription, or directly phosphorylate the Fos proteins. Regulation of the JNK pathway is influenced by many MKKKs. After phosphorylation by MKK4/7, JNK also enters the nucleus. The combination of Fos and Jun proteins forms the AP-1 transcription factor, which regulates a wide range of genes for cellular processes involved in VSMC proliferation, migration, ROS production, and extracellular matrix degradation.
Gennaro et al. (32) have observed in VSMC isolated from the aorta of young and old rabbits that the proliferative index after serum stimulation was significantly increased in old compared to young and associated with a significant and specific age-dependent increase in ERK1/2 activation. These results were further confirmed in vivo using a model of balloon injury in rabbit iliac arteries (32). The activation of ERK1/2 but not JNK and p38 was also observed in explanted VSMC from old F344 rats compared to young rats (61). An age-dependent increase in c-fos activity and cyclin A expression was found in VSMC from young and old New Zealand rabbits (96). An age-dependent increase in cyclin D in aortas from F344 rats (74), as well as an increase aortic MMP-2 and MT1-MMP activities in aging primates (116), have also been reported. In summary, these findings confirm an age-related activation of the ERK/AP-1 pathway leading to upregulation of a number of target genes associated with the promotion of VSMC proliferation and migration.
Nuclear factor-kappa B (NF-κB): Role in VSMC inflammatory responses
NF-κB is a transcription factor encoded by members of the Rel gene family. The transcription factor resides in the cytoplasm, where it is sequestered as a complex with IκB, a family of inhibitory proteins that mask the NF-κB nuclear localization signal (NLS) and prevents its activity. Currently five members of the NF-κB family of transcription factors: p50, p52, p65 (RelA), c-Rel, and RelB, and six IκBs have been identified (43), emphasizing the complex mechanisms for NF-κB regulation. NF-κB regulates the expression of genes encoding multiple functions that include inflammatory and immune modulatory proteins, as well as genes that regulate cellular differentiation, survival, and proliferation (43).
In most cells, activation of the NF-κB pathway is mediated by an IκB kinase (IKK) complex, which phosphorylates IκB and targets it for proteasomal degradation (42). The NF-κB complex then enters the nucleus to modulate target gene expression. Signaling to IKK proceeds through the TNF Receptor Associated Factor family (TRAF) and TRAF-interacting protein (RIP) complexes, generally in conjunction with TGF-β activated kinase 1 (TAK1), leading to canonical NF-κB signaling, or through TRAFs and NF-κB-inducing kinase (NIK), leading to the noncanonical NF-κB pathway (43).
Age-related differences in the activation of the NF-κB pathway in VSMC have been described by a number of investigators. Greater activation of NF-κB (93) and NIK/IKK/IκB pathways (124) were found in the aortas of old F344 rats compared to young rats. The increase in NF-κB activity in response to proinflammatory stimuli was significantly higher in VSMC explanted from old rats compared to their young counterparts (31, 123).
One of the major conceptual advances in our understanding of the pathogenesis of age-associated CVD has been the insight that age-related oxidative stress may promote vascular inflammation. The role of inflammation and white blood cells in vascular remodeling has become increasingly apparent (55). Inflammation is considered to be a critical initial step in the development of atherosclerosis during aging (20). Csiszar et al. (20) have reported that plasma levels of the proinflammatory cytokine, tumor necrosis factor-alpha (TNF-α), significantly increased with aging. Gene expression profiling from coronary arteries isolated from young adult and aged rats also showed that expression of TNF-α, interleukin (IL)-1β, IL-6, IL-6Rα, and IL-17 genes were all significantly increased in aged compared to young vessels. Immunofluorescent double labeling demonstrated that in aged vessels, IL-1β and IL-6 are predominantly localized in the endothelium, whereas TNF-α and IL-17 are localized in VSMC (19). Furthermore, anti-TNF-α treatment in old rats exerted an anti-aging, vasculoprotective effect (18). As depicted in Fig. 5, TNF-α signaling via members of the TNF receptor superfamily, such as TNFR1, bind to TRAF, and induce TRAF and NIK degradation, thereby activating IKK. IKK, in turn, induces processing of the NF-κB family member p100 into p52 and, thus, activation of p52-containing NF-κB complexes (heterodimer of RelB and p52). Activation of NF-κB could also be initiated through Toll-like receptors (TLRs) bound to ligands, recruiting Toll/IL-1R (TIR)-containing adaptor molecules, MyD88 and/or TIR domain-containing adaptor inducing interferon (TRIF), and subsequently activating the IKK complex (44). TLRs are not only expressed in immune cells but also expressed in the vascular system. Therefore, TLRs could be a key link between vascular remodeling and the immune system (29). These receptors of innate immunity recognize multiple ligands including bacterial lipopolysaccharide (LPS). In the vascular system, TLRs are thought to be activated by oxidized products of LDL (ox-LDL) (29). In addition, Ang II has been reported to stimulate NF-κB nuclear translocation in VSMC via Ang II type-I receptor (AT1) and Ang II type-II receptor (AT2) (38). Ang IV, a product of angiotensin degradation, was reported to activate the NF-κB pathway via AT4 receptors and this activation increased expression of NF-κB-relevant proinflammatory factors (14, 38, 98).
FIG. 5.

Aging-related upregulation of NF-κB signaling in VSMC. Inactive NF-κB resides primarily in the cytoplasm as a complex with inhibitory IκB proteins. When the pathway is activated, the IkB protein is degraded and the NF-κB complex enters the nucleus. NF-κB can be activated by proinflammatory cytokines, such as TNF-α that increases with age. TNF signaling via members of the TNF receptor superfamily, such as TNFR1, leads to binding and induction of TRAF and subsequently, to IKK activation, which phosphorylates IkB and in turn, induces processing of the NF-κB family member p100 into p52, and the formation of the RelB and p52 heterodimers. On the other hand, growth factors may enhance IL-1β–induced persistent activation of NF-κB through enhancing ERK phosphorylation. Furthermore, NF-κB could be activated through TLRs bound to a ligand, such as ox-LDL, which is increased in the vascular wall with aging. This recruits adaptor molecules, MyD88 and/or TRIF, and subsequently activates the IKK complex and induces processing of the p65 and p50 heterodimer. Finally, NF-κB can be activated through Ang II ligation with AT1 and AT2 receptors. Ultimately, translocation of NF-κB into the nucleus leads to modulation of gene expression, including those involved in the synthesis of inflammatory chemokines, adhesion molecules, survival signals, and multiple enzymes.
There are a considerable number of genes regulated by NF-κB and whose products will influence VSMC. These include cyclooxygenase-1 (Cox), which converts lipid from LDL particles into inflammatory lipids; chemokines, which attract monocytes; MMP-9 adhesion molecules that may facilitate VSMC migration; and tissue factor plasminogen activator inhibitor 1 (PAI-1). NF-κB is also a primary regulator of the expression of the iNOS (33). All three isoforms of NOS are expressed in the vasculature. The predominant isoform of NOS detectable in VSMC is iNOS that replaces eNOS as the main source of NO production after endothelial dysfunction (54). As noted earlier, a role for ERK1/2 in the regulation of NF-κB has been elucidated in VSMC. Jiang et al. reported that IL-1β induced expression of NF-κB-dependent genes such as iNOS in the presence of growth factors and was dependent on ERK1/2 enhancement of NF-κB activation (48). These studies were extended to show that ERK activation could differentially affect NF-κB dependent genes (iNOS, Cox-2, vascular cell adhesion molecule, and MnSOD) by modulating the IL-1β-induced persistent NF-κB activation (49). Aortic VSMC from old rats have much higher basal levels of iNOS mRNA or show a much greater increase in iNOS mRNA when exposed to the pro-inflammatory cytokine IL-1β, as compared to VSMC from the aortas of young animals (13). Migration and proliferation of VSMC are also mediated by a family of low-molecular-weight cytokines that are under NF-κB control and implicated in many aspects of vascular biology. For example, rat aortic monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 increase with age and alter vascular smooth muscle cell function in F344 rats (105). The same age-related change of MCP-1 was reported to occur within the human arterial wall (117). Inflammation-related adhesion molecules are considered to be another NF-κB target related to vascular inflammation and have been shown to increase with age in humans. The gene products of these molecules were demonstrated to be increased with age in rodent coronary arteries and the aorta, specifically, intercellular adhesion molecule (ICAM-1) (123) and vascular cell adhesion molecule (VCAM-1) (63, 74, 124). Finally, increased expression of PAI-1 contributes to VSMC survival and resistance to apoptosis (14).
Forkhead box O (FoxO): Role in antioxidant defense
FoxO transcription factors are emerging as a convergence point for signaling in response to stimulation by growth factors, nutrients, and oxidative stress. FoxO factors primarily act as important downstream targets of the insulin/IGF-1 signaling pathway that control FoxO protein levels, subcellular localization, DNA-binding, and transcriptional activity. FoxO factors consist of four members, FoxO 1 (FKHR), FoxO 3 (FKHRL1), FoxO 4 (AFX), and FoxO 6. Together, they coordinate a wide range of cellular functions related to the control of the cell cycle, apoptosis, DNA repair, defense against oxidative stress, and aging per se.
FoxO regulation is achieved through post-translational modification of the FoxO proteins, including phosphorylation, acetylation, ubiquitination (11) (Fig. 6). Akt (protein kinase B) has been shown to directly phosphorylate FoxO factors. The FoxO factors are also phosphorylated by serum glucocorticoid-inducible kinase-1 (SGK1) and IKK. Akt and SGK1 both phosphorylate Thr-32; SGK1 preferentially phosphorylates Ser-315, whereas Akt phosphorylates Ser-253 (8). These characteristics emerge as the mechanism whereby differential activation of FoxO factors in response to a variety of stimuli occurs. Phosphorylation at these three conserved sites by Akt and SGK causes the sequestration of FoxO factors in the cytoplasm, thereby preventing FoxO factors from transactivating their target genes. FoxO proteins are also phosphorylated by other protein kinases, such as JNK, which phosphorylates FoxO under conditions of oxidative stress and is mediated by the small GTPase Ral (24). This phosphorylation causes the translocation of FoxO from the cytoplasm to the nucleus, thus opposing Akt's action (115). FoxO proteins also bind to co-activator or co-repressor complexes and become acetylated or deacetylated [i.e., acetylation by CBP/p300 and deacetylation by Sirtuin 1(Sirt1) (9)]. Sirt1, a mammalian homolog of Sir2, has been shown to promote resistance to environmental stress and suppress apoptosis through a FoxO-dependent mechanism in mammalian cells (66). The deacetylase Sirt1 increases FoxO DNA binding ability that promotes the expression of p27 and MnSOD and changes FoxO-dependent responses from apoptosis-promoting to cell cycle arrest and stress-resistance in response to oxidative stress. Finally, FoxO proteins can be poly-ubiquitinated and targeted for protein degradation (4). The unique phosphorylation, acetylation, and ubiquitination status of FoxO under specific environmental conditions may provide a triple layer control of specificity in the regulation of subsets of FoxO target genes.
FIG. 6.

Aging-related downregulation of FoxO signaling in VSMC. FoxO factors primarily reside in the nucleus where they transactivate target genes related to cell cycle arrest, apoptosis, and the synthesis of antioxidant enzymes. Akt has been demonstrated to directly phosphorylate FoxO factors. In addition, FoxO factors can be phosphorylated by SGK1 and IKK at different residues. Phosphorylation of FoxO causes the sequestration of FoxO factors in the cytoplasm and in turn, degradation by ubiquitination, thereby preventing FoxO factors from transactivating their target genes. FoxO proteins are also phosphorylated by other protein kinases, such as JNK, which phosphorylate FoxO under conditions of oxidative stress and mediated by the small GTPase, Ral. This phosphorylation causes the translocation of FoxO from the cytoplasm to the nucleus, thus opposing Akt's action. Age-related activation of growth factor receptors, such as IGF-1R, and Akt signaling causes the export of FoxO from the nucleus, thereby downregulating FoxO functions. There is no apparent age effect on JNK signaling. Secondarily, FoxO may be deacetylated by Sirt1, which switches FoxO-dependent responses from apoptosis-promoting to cell cycle arrest and stress-resistance in response to oxidative stress.
FoxO factors regulate multiple aspects of cellular function in vascular tissues. Recent studies elucidated the role of FoxO in the maintenance of vascular homoeostasis and suppression of aberrant vascular outgrowth. An inhibitory role of FoxO proteins was found in injury-induced neointimal hyperplasia in vivo through the expression of p27 (2, 87). As age-related vascular dysfunction and damage are closely correlated with increased oxidative stress and increased growth factor levels, FoxO transcription factors were among the usual suspects in the search for key regulators of vascular cell function under these conditions (23). Akt content and phosphorylation were found to be significantly increased in aortic tissue (92, 94) and in explanted VSMC (62) from aged compared to young F344 rats. Because FoxO upregulates several antioxidant and cytoprotective enzymes, such as MnSOD, catalase, and GADD45, an increase in Akt activity, but not JNK activity, with advancing age is likely responsible for the downregulation of FoxO, as well as antioxidant capacity in VSMC (59). Interestingly, the age-dependent increase in IGF-1R signaling in VSMC seems to preferentially inhibit MnSOD. Suppression of IGF-1R with a specific inhibitor, AG1024, upregulated MnSOD expression, but inhibition of PI3K by LY294002, which blocks most growth factor signaling, failed to change MnSOD levels (59). This provides additional support that FoxO factors respond differently to different stimuli.
Nuclear factor E2–related factor-2 (Nrf2): Role in detoxification of ROS
During vascular aging, ROS generated by Nox, uncoupled eNOS, XO, and other enzymatic sources or occurring as a byproduct of mitochondrial respiration are increased, whereas antioxidant capacity appears limited by reduced antioxidants and related enzymes, including MnSOD, catalase, and GSH. This leads to ROS accumulation and VSMC dysfunction. Fortunately, cells have a feed-back system for detoxification to help in the maintenance of homeostasis. The transcription factor, Nrf2, initiates antioxidant and cytoprotective responses upon oxidative and electrophilic stress. Although this detoxification mechanism may not be expected to restitute the whole antioxidant capacity, it plays a role in delaying the aging process and its study has shed light on cellular protection during aging.
Nrf2 is a member of CNC (cap ‘n’ collar) family of b-Zip transcription factors and an indispensable positive regulator of many antioxidant and phase II detoxifying enzymes. As shown in Fig. 7, under physiological conditions, reduced kelch-like ECH-associated protein 1 (Keap1) binds Nrf2 and retains it in the cytosol, where it has a short half-life and undergoes ubiquitination. Upon exposure to oxidants or electrophilic stress, several reactive cysteine residues in Keap1 are covalently bonded to the electrophiles (64, 122). With the oxidation or adduction of the crucial thiol residues of Keap1, Nrf2 escapes from Keap1-mediated proteasomal degradation, translocates to the nucleus, where it interacts with a RNA polymerase III transcriptional repressor, small Maf protein, or other partners (such as Fos or Jun proteins) and binds to the antioxidant response element/electrophile response element (ARE/EpRE), to cause enhanced transcription of Phase II enzymes (involved in metabolite conjugation) and adaptive response genes (78). ARE/EpRE is a common regulatory element found in the 5′-flanking regions of antioxidant and detoxification enzymes.
FIG. 7.

Aging-related upregulation of Nrf2 signaling in VSMC. Under physiological conditions, reduced kelch-like ECH-associated protein 1 (Keap1) binds Nrf2 and retains it in the cytosol, where it has a short half-life and undergoes ubiquitination. Age-related imbalance between pro-oxidants and antioxidants leads to exposure of Keap1 to accumulated oxidants or electrophilic stress. Keap1 possesses several reactive cysteine residues that are covalently bonded with electrophiles. Upon oxidation or adduction of the crucial thiol residues of Keap1, Nrf2 escapes from Keap1, translocates to the nucleus where it interacts with small Maf or other partners (such as Jun or Fos proteins) and binds to the ARE/EpRE to enhance transcription of Phase II or other adaptive response genes, including enzymes involved in GSH metabolism, NQO1,2, and HO-1.
An increase in nuclear Nrf2 was found in VSMC from old compared to young F344 rats (60). There are a large number of genes regulated by Keap1-Nrf2-ARE, and some of the phase II enzymes were also found to be upregulated in VSMC with aging, including the enzymes involved in GSH metabolism and heme oxygenase1 (HO1) (81), as shown in Table 1. Recently, the Nrf2 factor has been widely investigated as a therapeutic target to reduce VSMC oxidative stress and thereby inhibit VSMC proliferation. For example, transfer of Nrf2 by an adenovirus to a rabbit balloon injury model has resulted in increased antioxidant defenses (58). In the same study, Nrf2 gene transfer effectively reduced oxidative stress determined by an antibody against ox-LDL and inhibited the recruitment of macrophages into the vessel wall. Reduction of inflammation and ox-LDL accumulation might thereby influence neointimal formation. The inhibitory effect of nitroalkene derivatives of linoleic acid (nitrolinoleate) on VSMC proliferation was also reported to act through activation of the Keap1/Nrf2 signaling pathway (114).
In summary, the available data in the literature support an independent effect of age on specific redox-sensitive signaling pathways in VSMC. An age-related imbalance between pro-oxidants and antioxidants leads to changes in redox status and thereby to activation of specific redox-sensitive signaling pathways. In turn, changes in signaling pathways promote alterations in VSMC proliferation, migration, and extracellular matrix remodeling. The series of events lead to increased vessel wall thickness, inflammation, and vulnerability to the development of atherosclerosis. To date, the major gaps in this area of research are the demonstration that these mechanisms are operable in the human. Furthermore, investigation of targeted interventions needs to consider the integrated effects on other cells and tissues that interact in the final pathogenesis of disease.
Abbreviations Used
- ALDH-2
aldehyde dehydrogenase
- Ang II
angiotensin II
- AP-1
activator protein-1
- ARE/EpRE
antioxidant response element/electrophile response element
- AT1
Ang II type-I receptor
- CNC
cap ‘n’ collar
- Cox
cyclooxygenase-1
- COX
cytochrome c oxidase enzyme
- CVD
cardiovascular diseases
- EGFR
epidermal growth facor receptor
- eNOS
endothelial NO synthase
- ERK
extracellular signal-regulated kinase
- FGF
fibroblast growth factor
- FoxO
forkhead box O
- F344xBN
Fischer 344/Brown Norway F1 hybrid rat
- GCLC
γ-glutamate cysteine ligase catalytic subunit
- GCLM
γ-glutamate cysteine ligase modulatory subunit
- Gpx
glutathione peroxidase
- Gr
glutathione reductase
- GS
glutathione synthetase
- GSSG
oxidized GSH
- GST
glutathione-S-transferase
- HO1
heme oxygenase1
- H2O2
hydrogen peroxide
- Hsp70
heat shock protein 70
- ICAM-1
intercellular adhesion molecule-1
- IGF-1R
insulin-like growth factor receptor
- IκB
inhibitor of NF-κB
- IKK
IκB kinase
- IL
interleukin
- iNOS
inducible NO synthase
- JNK
Jun N-terminal kinase
- Keap1
Kelch-like ECH-associated protein 1
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemoattractant protein-1
- MEK
MAPK/ERK kinase
- MKKK
MAPK kinase kinase
- MMP
matrix metalloproteinases
- MT1-MMP
membrane-type1 MMP
- NF-κB
nuclear factor-kappa B
- NIK
NF-κB-inducing kinase
- NMR
naked mole rat
- NO
nitric oxide
- Nox
NADPH oxidase
- NQO
NADPH: quinone oxidoreductase
- Nrf2
nuclear factor E2–related factor 2
- O2•−
superoxide anion
- Ox-LDL
oxidized products of LDL
- P21
p21cip-1
- PAI-1
plasminogen activator inhibitor 1
- PCNA
proliferating cell nuclear antigen
- PDGFR
platelet-derived growth factor receptor
- PI3K
phosphatidyl inositol 3-kinase
- Prx
peroxiredoxin
- RIP
TRAF-interacting protein
- ROS
reactive oxygen species
- Shc
Src homologous and collagen-like protein
- SHRSP
stroke-prone spontaneously hypertensive rats
- Sirt 1
sirtuin 1
- SKG1
serum glucocorticoid-inducible kinase-1
- SOD
superoxide dismutase
- TAK1
TGF-beta activated kinase 1
- TCF
ternary complex factors
- TGF-β
transforming growth factor-β
- TIR
Toll/IL-1R
- TLR
Toll-like receptors
- TNF-α
tumor necrosis factor-α
- TRAF
TNF Receptor Associated Factor family
- TRIF
TIR domain-containing adaptor inducing interferon
- TrxR
thioredoxin reductase
- VCAM-1
vascular cell adhesion molecule-1
- VSMC
vascular smooth muscle cells
- WKY
Wistar–Kyoto rats
- XO
xanthine oxidase
References
- 1.Abid M. Yano K. Guo S. Patel VI. Shrikhande G. Spokes KC. Ferran C. Aird WC. Forkhead transcription factors inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia. J Biol Chem. 2005;280:29864–29873. doi: 10.1074/jbc.M502149200. [DOI] [PubMed] [Google Scholar]
- 2.Angel P. Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta Revs Cancer. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 3.Aoki M. Jiang H. Vogt PK. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci USA. 2004;101:13613–13617. doi: 10.1073/pnas.0405454101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aranda R. Doménech E. Rus AD. Real JT. Sastre J. Viña J. Pallardó FV. Age-related increase in xanthine oxidase activity in human plasma and rat tissues. Free Radic Res. 2007;41:1195–1200. doi: 10.1080/10715760701481461. [DOI] [PubMed] [Google Scholar]
- 5.Azar ZM. Mehdi MZ. Srivastava AK. Insulin-like growth factor type-1 receptor transactivation in vasoactive peptide and oxidant-induced signaling pathways in vascular smooth muscle cells. Can J Physiol Pharmacol. 2007;85:105–111. doi: 10.1139/Y06-101. [DOI] [PubMed] [Google Scholar]
- 6.Bayes-Genis A. Conover CA. Schwartz RS. The insulin-like growth factor axis : A review of atherosclerosis and restenosis. Circ Res. 2000;86:125–130. doi: 10.1161/01.res.86.2.125. [DOI] [PubMed] [Google Scholar]
- 7.Bendeck MP. Irvin C. Reidy MA. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ Res. 1996;78:38–43. doi: 10.1161/01.res.78.1.38. [DOI] [PubMed] [Google Scholar]
- 8.Brunet A. Park J. Tran H. Hu LS. Hemmings BA. Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunet A. Sweeney LB. Sturgill JF. Chua KF. Greer PL. Lin Y. Tran H. Ross SE. Mostoslavsky R. Cohen HY. Hu LS. Cheng HL. Jedrychowski MP. Gygi SP. Sinclair DA. Alt FW. Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 10.Calabrese V. Cornelius C. Mancuso C. Pennisi G. Calafato S. Bellia F. Bates T. Giuffrida Stella A. Schapira T. Dinkova Kostova A. Rizzarelli E. Cellular stress response: A novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem Res. 2008;33:2444–2471. doi: 10.1007/s11064-008-9775-9. [DOI] [PubMed] [Google Scholar]
- 11.Calnan DR. Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 12.Cernadas MR. de Miguel LS. Garcia-Duran M. Gonzalez-Fernandez F. Millas I. Monton M. Rodrigo J. Rico L. Fernandez P. de Frutos T. Rodriguez-Feo JA. Guerra J. Caramelo C. Casado S. Lopez-Farre A. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ Res. 1998;83:279–286. doi: 10.1161/01.res.83.3.279. [DOI] [PubMed] [Google Scholar]
- 13.Chan GHH. Fiscus RR. Exaggerated production of nitric oxide (NO) and increases in inducible NO-synthase mRNA levels induced by the pro-inflammatory cytokine interleukin-1[beta] in vascular smooth muscle cells of elderly rats. Experimental Gerontology. 2004;39:387–394. doi: 10.1016/j.exger.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y. Budd RC. Kelm RJ., Jr. Sobel BE. Schneider DJ. Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. 2006;26:1777–1783. doi: 10.1161/01.ATV.0000227514.50065.2a. [DOI] [PubMed] [Google Scholar]
- 15.Clarke MCH. Figg N. Maguire JJ. Davenport AP. Goddard M. Littlewood TD. Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 16.Clempus RE. Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csiszar A. Labinskyy N. Orosz Z. Xiangmin Z. Buffenstein R. Ungvari Z. Vascular aging in the longest-living rodent, the naked mole rat. Am J Physiol Heart Circ Physiol. 2007;293:H919–H927. doi: 10.1152/ajpheart.01287.2006. [DOI] [PubMed] [Google Scholar]
- 18.Csiszar A. Labinskyy N. Smith K. Rivera A. Orosz Z. Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-{alpha} treatment in aging. Am J Pathol. 2007;170:388–698. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Csiszar A. Ungvari Z. Koller A. Edwards JG. Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. FASEB J. 2003;17:1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 20.Csiszar A. Wang M. Lakatta EG. Ungvari ZI. Inflammation and endothelial dysfunction during aging: role of NF-{kappa}B. J Appl Physiol. 2008;105:1333–1341. doi: 10.1152/japplphysiol.90470.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Armiento FP. Bianchi A. de Nigris F. Capuzzi DM. D'Armiento MR. Crimi G. Abete P. Palinski W. Condorelli M. Napoli C. Gronholdt ML. Age-related effects on atherogenesis and scavenger enzymes of intracranial and extracranial arteries in men without classic risk factors for atherosclerosis. Editorial Comment. Stroke. 2001;32:2472–2480. doi: 10.1161/hs1101.098520. [DOI] [PubMed] [Google Scholar]
- 22.Demaree SR. Lawler JM. Linehan J. Delp MD. Ageing alters aortic antioxidant enzyme activities in Fischer-344 rats. Acta Physiologica Scandinavica. 1999;166:203–208. doi: 10.1046/j.1365-201x.1999.00552.x. [DOI] [PubMed] [Google Scholar]
- 23.Dzau VJ. Braun-Dullaeus RC. Sedding DG. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 24.Essers MA. Weijzen S. Vries-Smits AM. Saarloos I. de Ruiter ND. Bos JL. Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrini MG. Davila HH. Valente EGA. Gonzalez-Cadavid NF. Rajfer J. Aging-related induction of inducible nitric oxide synthase is vasculo-protective to the arterial media. Cardiovasc Res. 2004;61:796–805. doi: 10.1016/j.cardiores.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Fraga MF. Agrelo R. Esteller M. Cross-talk between aging and cancer: the epigenetic language. Ann NY Acad Sci. 2007;1100:60–74. doi: 10.1196/annals.1395.005. [DOI] [PubMed] [Google Scholar]
- 27.Fraga MF. Esteller M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007;23:413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 28.Frank GD. Eguchi S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: Significance and involvement of EGF receptor transactivation by angiotensin II. Antioxid Redox Signal. 2003;5:771–780. doi: 10.1089/152308603770380070. [DOI] [PubMed] [Google Scholar]
- 29.Frantz S. Ertl G. Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–454. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 30.Fukagawa NK. Li M. Liang P. Russell JC. Sobel BE. Absher PM. Aging and high concentrations of glucose potentiate injury to mitochondrial DNA. Free Radic Biol Med. 1999;27:1437–1443. doi: 10.1016/s0891-5849(99)00189-6. [DOI] [PubMed] [Google Scholar]
- 31.Fukagawa NK. Li M. Timblin CR. Mossman BT. Modulation of cell injury and survival by high glucose and advancing age. Free Radic Biol Med. 2001;31:1560–1569. doi: 10.1016/s0891-5849(01)00736-5. [DOI] [PubMed] [Google Scholar]
- 32.Gennaro G. Menard C. Giasson E. Michaud SE. Palasis M. Meloche S. Rivard A. Role of p44/p42 MAP kinase in the age-dependent increase in vascular smooth muscle cell proliferation and neointimal formation. Arterioscler Thromb Vasc Biol. 2003;23:204–210. doi: 10.1161/01.atv.0000053182.58636.be. [DOI] [PubMed] [Google Scholar]
- 33.Ginnan R. Guikema BJ. Halligan KE. Singer HA. Jourd'heuil D. Regulation of smooth muscle by inducible nitric oxide synthase and NADPH oxidase in vascular proliferative diseases. Free Rad Biol Med. 2008;44:1232–1245. doi: 10.1016/j.freeradbiomed.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldberg AD. Allis CD. Bernstein E. Epigenetics: A landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez Bosc LV. Kurnjek ML. Muller A. Terragno NA. Basso N. Effect of chronic angiotensin II inhibition on the nitric oxide synthase in the normal rat during aging. J Hypertens. 2001;19:1403–1409. doi: 10.1097/00004872-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Griscavage JM. Hobbs AJ. Ignarro LJ. Negative modulation of nitric oxide synthase by nitric oxide and nitroso compounds. Adv Pharmacol. 1995;34:215–234. doi: 10.1016/s1054-3589(08)61088-1. [DOI] [PubMed] [Google Scholar]
- 37.Grumbach IM. Chen W. Mertens SA. Harrison DG. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J Mol Cell Cardiol. 2005;39:595–603. doi: 10.1016/j.yjmcc.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 38.Guo Rw. Yang Lx. Wang H. Liu B. Wang L. Angiotensin II induces matrix metalloproteinase-9 expression via a nuclear factor-kappaB-dependent pathway in vascular smooth muscle cells. Regulatory Peptides. 2008;147:37–44. doi: 10.1016/j.regpep.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Gutierrez J. Ballinger SW. Darley-Usmar VM. Landar A. Free radicals, mitochondria, and oxidized lipids: The emerging role in signal transduction in vascular cells. Circ Res. 2006;99:924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton CA. Brosnan MJ. McIntyre M. Graham D. Dominiczak AF. Superoxide excess in hypertension and aging: A common cause of endothelial dysfunction. Hypertension. 2001;37:529–534. doi: 10.1161/01.hyp.37.2.529. [DOI] [PubMed] [Google Scholar]
- 41.Hariri RJ. Hajjar DP. Coletti D. Alonso DR. Weksler ME. Rabellino E. Aging and arteriosclerosis. Cell cycle kinetics of young and old arterial smooth muscle cells. Am J Pathol. 1988;131:132–136. [PMC free article] [PubMed] [Google Scholar]
- 42.Hayden MS. West AP. Ghosh S. NF-[kappa]B and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 43.Hayden MS. Ghosh S. Shared principles in NF-[kappa]B signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 44.Hayden MS. West AP. Ghosh S. Snapshot: NF-[kappa]B signaling pathways. Cell. 2006;127:1286. doi: 10.1016/j.cell.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Heeneman S. Haendeler J. Saito Y. Ishida M. Berk BC. Angiotensin II induces transactivation of two different populations of the platelet-derived growth factor beta receptor. Key role for the p66 adaptor protein Shc. J Biol Chem. 2000;275:15926–15932. doi: 10.1074/jbc.M909616199. [DOI] [PubMed] [Google Scholar]
- 46.Hennigan RF. Stambrook PJ. Dominant negative c-jun inhibits activation of the cyclin D1 and cyclin E kinase complexes. Mol Biol Cell. 2001;12:2352–2363. doi: 10.1091/mbc.12.8.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itoh Y. MT1-MMP: A key regulator of cell migration in tissue. IUBMB Life. 2006;58:589–596. doi: 10.1080/15216540600962818. [DOI] [PubMed] [Google Scholar]
- 48.Jiang B. Xu S. Brecher P. Cohen RA. Growth factors enhance interleukin-1{beta}-induced persistent activation of nuclear factor-{kappa}B in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2002;22:1811–1816. doi: 10.1161/01.atv.0000037679.60584.3f. [DOI] [PubMed] [Google Scholar]
- 49.Jiang B. Xu S. Hou X. Pimentel DR. Brecher P. Cohen RA. Temporal control of NF-{kappa}B activation by ERK differentially regulates interleukin-1{beta}-induced gene expression. J Biol Chem. 2004;279:1323–1329. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- 50.Jiang L. Wang M. Zhang J. Monticone RE. Telljohann R. Spinetti G. Pintus G. Lakatta EG. Increased aortic calpain-1 activity mediates age-associated angiotensin II signaling of vascular smooth muscle cells. PLoS ONE. 2008;3:e2231. doi: 10.1371/journal.pone.0002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson GL. Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 52.Karin M. Liu Z. Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 53.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 54.Kibbe M. Billiar T. Tzeng E. Inducible nitric oxide synthase and vascular injury. Cardiovasc Res. 1999;43:650–657. doi: 10.1016/s0008-6363(99)00130-3. [DOI] [PubMed] [Google Scholar]
- 55.Korshunov VA. Schwartz SM. Berk BC. Vascular remodeling: Hemodynamic and biochemical mechanisms underlying Glagov's phenomenon. Arterioscler Thromb Vasc Biol. 2007;27:1722–1728. doi: 10.1161/ATVBAHA.106.129254. [DOI] [PubMed] [Google Scholar]
- 56.Lassegue B. Clempus RE. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 57.Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Rad Biol Med. 2002;32:790–796. doi: 10.1016/s0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 58.Levonen AL. Inkala M. Heikura T. Jauhiainen S. Jyrkkanen HK. Kansanen E. Maatta K. Romppanen E. Turunen P. Rutanen J. Yla–Herttuala S. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler Thromb Vasc Biol. 2007;27:741–747. doi: 10.1161/01.ATV.0000258868.80079.4d. [DOI] [PubMed] [Google Scholar]
- 59.Li M. Chiu JF. Gagne J. Fukagawa NK. Age-related differences in insulin-like growth factor-1 receptor signaling regulates Akt/FOXO3a and ERK/Fos pathways in vascular smooth muscle cells. J Cell Physiol. 2008;217:377–387. doi: 10.1002/jcp.21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li M. Liu RM. Timblin CR. Meyer SG. Mossman BT. Fukagawa NK. Age affects ERK1/2 and NRF2 signaling in the regulation of GCLC expression. J Cell Physiol. 2006;206:518–525. doi: 10.1002/jcp.20496. [DOI] [PubMed] [Google Scholar]
- 61.Li M. Mossman BT. Kolpa E. Timblin CR. Shukla A. Taatjes DJ. Fukagawa NK. Age-related differences in MAP kinase activity in VSMC in response to glucose or TNF-alpha. J Cell Physiol. 2003;197:418–425. doi: 10.1002/jcp.10384. [DOI] [PubMed] [Google Scholar]
- 62.Li M. Chiu JF. Mossman BT. Fukagawa NK. Down-regulation of manganese-superoxide dismutase through phosphorylation of FOXO3a by Akt in explanted vascular smooth muscle cells from old rats. J Biol Chem. 2006;281:40429–40439. doi: 10.1074/jbc.M606596200. [DOI] [PubMed] [Google Scholar]
- 63.Li Z. Froehlich J. Galis ZS. Lakatta EG. Increased expression of matrix metalloproteinase-2 in the thickened intima of aged rats. Hypertension. 1999;33:116–123. doi: 10.1161/01.hyp.33.1.116. [DOI] [PubMed] [Google Scholar]
- 64.Liebler DC. Guengerich FP. Elucidating mechanisms of drug-induced toxicity. Nat Rev Drug Discov. 2005;4:410–420. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- 65.Liu YC. Chang HW. Lai YC. Ding ST. Ho JL. Serum responsiveness of the rat PCNA promoter involves the proximal ATF and AP-1 sites. FEBS Lett. 1998;441:200–204. doi: 10.1016/s0014-5793(98)01549-x. [DOI] [PubMed] [Google Scholar]
- 66.Longo VD. Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 67.Loomis ED. Sullivan JC. Osmond DA. Pollock DM. Pollock JS. Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharmacol Exp Ther. 2005;315:1058–1064. doi: 10.1124/jpet.105.091728. [DOI] [PubMed] [Google Scholar]
- 68.Lyle AN. Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology. 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- 69.Manea A. Manea SA. Gafencu AV. Raicu M. Simionescu M. AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: Role of p22phox subunit. Arterioscler Thromb Vasc Biol. 2008;28:878–885. doi: 10.1161/ATVBAHA.108.163592. [DOI] [PubMed] [Google Scholar]
- 70.Marmol F. Sanchez J. Lopez D. Martinez N. Rosello-Catafau J. Mitjavila MT. Puig–Parellada P. Loss of adaptation to oxidative stress as a mechanism for aortic damage in aging rats. J Physiol Biochem. 2007;63:239–247. doi: 10.1007/BF03165787. [DOI] [PubMed] [Google Scholar]
- 71.McCrann DJ. Yang D. Chen H. Carroll S. Ravid K. Upregulation of Nox4 in the aging vasculature and its association with smooth muscle cell polyploidy. Cell Cycle. 2009;8:902–908. doi: 10.4161/cc.8.6.7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meng D. Shi X. Jiang BH. Fang J. Insulin-like growth factor-I (IGF-I) induces epidermal growth factor receptor transactivation and cell proliferation through reactive oxygen species. Free Rad Biol Med. 2007;42:1651–1660. doi: 10.1016/j.freeradbiomed.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 73.Miller JD. Chu Y. Brooks RM. Richenbacher WE. Peta-Silva R. Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller SJ. Watson WC. Kerr KA. Labarrere CA. Chen NX. Deeg MA. Unthank JL. Development of progressive aortic vasculopathy in a rat model of aging. Am J Physiol Heart Circ Physiol. 2007;293:H2634–H2643. doi: 10.1152/ajpheart.00397.2007. [DOI] [PubMed] [Google Scholar]
- 75.Millette E. Rauch BH. Kenagy RD. Daum G. Clowes AW. Platelet-derived growth factor-BB transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc Med. 2006;16:25–28. doi: 10.1016/j.tcm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 76.Moon SK. Cho SH. Kim KW. Jeon JH. Ko JH. Kim BY. Kim CH. Overexpression of membrane sialic acid-specific sialidase Neu3 inhibits matrix metalloproteinase-9 expression in vascular smooth muscle cells. Biochem Biophys Res Commun. 2007;356:542–547. doi: 10.1016/j.bbrc.2007.02.155. [DOI] [PubMed] [Google Scholar]
- 77.Moon SK. Thompson LJ. Madamanchi N. Ballinger S. Papaconstantinou J. Horaist C. Runge MS. Patterson C. Aging, oxidative responses, and proliferative capacity in cultured mouse aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2001;280:H2779–H2788. doi: 10.1152/ajpheart.2001.280.6.H2779. [DOI] [PubMed] [Google Scholar]
- 78.Motohashi H. Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 79.Najjar SS. Scuteri A. Lakatta EG. Arterial aging: Is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454–462. doi: 10.1161/01.HYP.0000177474.06749.98. [DOI] [PubMed] [Google Scholar]
- 80.Napoli C. Aldini G. Wallace JL. de Nigris F. Maffei R. Abete P. Bonaduce D. Condorelli G. Rengo F. Sica V. D'Armiento FP. Mignogna C. de Rosa G. Condorelli M. Lerman LO. Ignarro LJ. Efficacy and age-related effects of nitric oxide-releasing aspirin on experimental restenosis. Proc Natl Am Soc USA. 2002;99:1689–1694. doi: 10.1073/pnas.022639399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ndisang JF. Wu L. Zhao W. Wang R. Induction of heme oxygenase-1 and stimulation of cGMP production by hemin in aortic tissues from hypertensive rats. Blood. 2003;101:3893–3900. doi: 10.1182/blood-2002-08-2608. [DOI] [PubMed] [Google Scholar]
- 82.Newaz MA. Yousefipour Z. Oyekan A. Oxidative stress-associated vascular aging is xanthine oxidase-dependent but not NAD(P)H oxidase-dependent. J Cardiovasc Pharmacol. 2006;48:88–94. doi: 10.1097/01.fjc.0000245402.62864.0a. [DOI] [PubMed] [Google Scholar]
- 83.Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006;69:614–624. doi: 10.1016/j.cardiores.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 84.Nosaka Y. Arai A. Kanda E. Akasaki T. Sumimoto H. Miyasaka N. Miura O. Rac is activated by tumor necrosis factor [alpha] and is involved in activation of Erk. Biochem Biophys Res Commun. 2001;285:675–679. doi: 10.1006/bbrc.2001.5222. [DOI] [PubMed] [Google Scholar]
- 85.Orlandi A. Bochaton-Piallat ML. Gabbiani G. Spagnoli LG. Aging, smooth muscle cells and vascular pathobiology: Implications for atherosclerosis. Atherosclerosis. 2006;188:221–230. doi: 10.1016/j.atherosclerosis.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 86.Oudot A. Martin C. Busseuil D. Vergely C. Demaison L. Rochette L. NADPH oxidases are in part responsible for increased cardiovascular superoxide production during aging. Free Radical Biology and Medicine. 2006;40:2214–2222. doi: 10.1016/j.freeradbiomed.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 87.Park KW. Kim DH. You HJ. Sir JJ. Jeon SI. Youn SW. Yang HM. Skurk C. Park YB. Walsh K. Kim HS. Activated Forkhead transcription factor inhibits neointimal hyperplasia after angioplasty through induction of p27. Arterioscler Thromb Vasc Biol. 2005;25:742–747. doi: 10.1161/01.ATV.0000156288.70849.26. [DOI] [PubMed] [Google Scholar]
- 88.Patel RP. Levonen AL. Crawford JH. Darley–Usmar VM. Mechanisms of the pro- and antioxidant actions of nitric oxide in atherosclerosis. Cardiovasc Res. 2000;47:465–474. doi: 10.1016/s0008-6363(00)00086-9. [DOI] [PubMed] [Google Scholar]
- 89.Prospective studies collaboration. Lewington S. Whitlock G. Clarke R. Sherliker P. Emberson J. Halsey J. Dizilbash N. Peto R. Collins R. Blood cholesterol and vascular mortality by age, sex, and blood pressure: A meta-analysis of individual data from 61 prospective studies with 55000 vascular deaths. The Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 90.Radhakrishnan Y. Maile LA. Ling Y. Graves LM. Clemmons DR. Insulin-like growth factor-I stimulates Shc-dependent phosphatidylinositol 3-kinase activation via Grb2-associated p85 in vascular smooth muscle cells. J Biol Chem. 2008;283:16320–16331. doi: 10.1074/jbc.M801687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramachandran A. Levonen AL. Brookes PS. Ceaser E. Shiva S. Barone MC. Darley-Usmar V. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Rad Biol Med. 2002;33:1465–1474. doi: 10.1016/s0891-5849(02)01142-5. [DOI] [PubMed] [Google Scholar]
- 92.Rice KM. Kinnard RS. Wright GL. Blough ER. Aging alters vascular mechanotransduction: Pressure-induced regulation of p70S6k in the rat aorta. Mech Ageing Develop. 2005;126:1213–1222. doi: 10.1016/j.mad.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 93.Rice KM. Preston DL. Walker EM. Blough ER. Aging influences multiple incidices of oxidative stress in the aortic media of the Fischer 344/NNia-á + ù-áBrown Norway/BiNia rat. Free Rad Res. 2006;40:185–197. doi: 10.1080/10715760500464957. [DOI] [PubMed] [Google Scholar]
- 94.Rice KM. Desai DH. Preston DL. Wehner PS. Blough ER. Uniaxial stretch-induced regulation of mitogen-activated protein kinase, Akt and p70 S6 kinase in the ageing Fischer 344 x Brown Norway rat aorta. Exper Physiol. 2007;92:963–970. doi: 10.1113/expphysiol.2007.037275. [DOI] [PubMed] [Google Scholar]
- 95.Richardson B. Impact of aging on DNA methylation. Ageing Res Rev. 2003;2:245–261. doi: 10.1016/s1568-1637(03)00010-2. [DOI] [PubMed] [Google Scholar]
- 96.Rivard A. Principe N. Andres V. Age-dependent increase in c-fos activity and cyclin A expression in vascular smooth muscle cells. A potential link between aging, smooth muscle cell proliferation and atherosclerosis. Cardiovasc Res. 2000;45:1026–1034. doi: 10.1016/s0008-6363(99)00385-5. [DOI] [PubMed] [Google Scholar]
- 97.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 98.Ruiz-Ortega M. Esteban V. Egido Js. The regulation of the inflammatory response through nuclear factor-[kappa]B pathway by angiotensin IV extends the role of the renin angiotensin system in cardiovascular diseases. Trends Cardiovasc Med. 2007;17:19–25. doi: 10.1016/j.tcm.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 99.Sarzani R. Arnaldi G. Takasaki I. Brecher P. Chobanian AV. Effects of hypertension and aging on platelet-derived growth factor and platelet-derived growth factor receptor expression in rat aorta and heart. Hypertension. 1991;18:III93–III99. doi: 10.1161/01.hyp.18.5_suppl.iii93. [DOI] [PubMed] [Google Scholar]
- 100.Scheidegger KJ. Cenni B. Picard D. Delafontaine P. Estradiol decreases IGF-1 and IGF-1 receptor expression in rat aortic smooth muscle cells. Mechanisms for its atheroprotective effects. J Biol Chem. 2000;275:38921–38928. doi: 10.1074/jbc.M004691200. [DOI] [PubMed] [Google Scholar]
- 101.Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;100:S87–S89. [PubMed] [Google Scholar]
- 102.Shaulian E. Karin M. AP-1 as a regulator of cell life and death. Nature Cell Biol. 2002;4:E131. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 103.Shringarpure R. Davies KJA. Protein turnover by the proteasome in aging and disease. Free Rad Biol Med. 2002;32:1084–1089. doi: 10.1016/s0891-5849(02)00824-9. [DOI] [PubMed] [Google Scholar]
- 104.Smith AR. Visioli F. Frei B. Hagen TM. Lipoic acid significantly restores, in rats, the age-related decline in vasomotion. Br J Pharmacol. 2008;153:1615–1622. doi: 10.1038/bjp.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Spinetti G. Wang M. Monticone R. Zhang J. Zhao D. Lakatta EG. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2004;24:1397–1402. doi: 10.1161/01.ATV.0000134529.65173.08. [DOI] [PubMed] [Google Scholar]
- 106.Stadtman ER. Protein oxidation in aging and age-related diseases. Ann NY Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 107.Tan CK. Sullivan K. Li XY. Tan EM. Downey KM. So AG. Autoantibody to the proliferating cell nuclear antigen neutralizes the activity of the auxiliary protein for DNA polymerase delta. Nucleic Acids Res. 1987;15:9299–9308. doi: 10.1093/nar/15.22.9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Touyz RM. Cruzado M. Tabet F. Yao G. Salomon S. Schiffrin EL. Redox-dependent MAP kinase signaling by Ang II in vascular smooth muscle cells: Role of receptor tyrosine kinase transactivation. Can J Physiol Pharmacol. 2003;81:159–167. doi: 10.1139/y02-164. [DOI] [PubMed] [Google Scholar]
- 109.Touyz RM. Reactive oxygen species in vascular biology: Role in arterial hypertension. Expert Rev Cardiovasc Ther. 2003;1:91–106. doi: 10.1586/14779072.1.1.91. [DOI] [PubMed] [Google Scholar]
- 110.Ungvari Z. Labinskyy N. Gupte S. Chander PN. Edwards JG. Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol. 2008;294:H2121–H2128. doi: 10.1152/ajpheart.00012.2008. [DOI] [PubMed] [Google Scholar]
- 111.van der LB. Labugger R. Skepper JN. Bachschmid M. Kilo J. Powell JM. Palacios-Callender M. Erusalimsky JD. Quaschning T. Malinski T. Gygi D. Ullrich V. Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vazquez-Padron RI. Lasko D. Li S. Louis L. Pestana IA. Pang M. Liotta C. Fornoni A. Aitouche A. Pham SM. Aging exacerbates neointimal formation, and increases proliferation and reduces susceptibility to apoptosis of vascular smooth muscle cells in mice. J Vasc Surg. 2004;40:1199–1207. doi: 10.1016/j.jvs.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 113.Viel EC. Benkirane K. Javeshghani D. Touyz RM. Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295:H281–H288. doi: 10.1152/ajpheart.00304.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Villacorta L. Zhang J. Garcia-Barrio MT. Chen Xl. Freeman BA. Chen YE. Cui T. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am J Physiol Heart Circ Physiol. 2007;293:H770–H776. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang MC. Bohmann D. Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 116.Wang M. Takagi G. Asai K. Resuello RG. Natividad FF. Vatner DE. Vatner SF. Lakatta EG. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–1316. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 117.Wang M. Zhang J. Jiang LQ. Spinetti G. Pintus G. Monticone R. Kolodgie FD. Virmani R. Lakatta EG. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50:219–227. doi: 10.1161/HYPERTENSIONAHA.107.089409. [DOI] [PubMed] [Google Scholar]
- 118.Wang M. Zhao D. Spinetti G. Zhang J. Jiang LQ. Pintus G. Monticone R. Lakatta EG. Matrix Mmetalloproteinase 2 activation of transforming growth factor-{beta}1 (TGF-{beta}1) and TGF-{beta}1-type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol. 2006;26:1503–1509. doi: 10.1161/01.ATV.0000225777.58488.f2. [DOI] [PubMed] [Google Scholar]
- 119.Wassmann S. Wassmann K. Nickenig G. Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension. 2004;44:381–386. doi: 10.1161/01.HYP.0000142232.29764.a7. [DOI] [PubMed] [Google Scholar]
- 120.Watanabe G. Howe A. Lee RJ. Albanese C. Shu IW. Karnezis AN. Zon L. Kyriakis J. Rundell K. Pestell RG. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci USA. 1996;93:12861–12866. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wenzel P. Schuhmacher S. Kienhofer J. Muller J. Hortmann M. Oelze M. Schulz E. Treiber N. Kawamoto T. Scharffetter-Kochanek K. Munzel T. Burkle A. Bachschmid MM. Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–289. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamamoto T. Suzuki T. Kobayashi A. Wakabayashi J. Maher J. Motohashi H. Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yan Zq. Sirsjo A. Bochaton-Piallat ML. Gabbiani G. Hansson GK. Augmented expression of inducible NO synthase in vascular smooth muscle cells during aging is associated with enhanced NF-{kappa}B activation. Arterioscler Thromb Vasc Biol. 1999;19:2854–2862. doi: 10.1161/01.atv.19.12.2854. [DOI] [PubMed] [Google Scholar]
- 124.Zou Y. Yoon S. Jung KJ. Kim CH. Son TG. Kim MS. Kim YJ. Lee J. Yu BP. Chung HY. Upregulation of aortic adhesion molecules during aging. J Gerontol A Biol Sci Med Sci. 2006;61:232–244. doi: 10.1093/gerona/61.3.232. [DOI] [PubMed] [Google Scholar]