

Abstract
Homeostatic control of neural function can be mediated by the regulation of ion channel expression, neurotransmitter receptor abundance, or modulation of presynaptic release. These processes can be implemented through cell autonomous or intercellular signaling. It remains unknown whether different forms of homeostatic regulation can be coordinated to achieve constant neural function. One way to approach this question is to confront a simple neural system with conflicting perturbations and determine whether the outcome reflects a coordinated, homeostatic response. Here we demonstrate that two A-type potassium channel genes, shal and shaker, are reciprocally, transcriptionally coupled to maintain A-type channel expression. We then demonstrate that this homeostatic control of A-type channel expression prevents target-dependent, homeostatic modulation of synaptic transmission. Thus, we uncover a novel homeostatic mechanism that reciprocally regulates A-type potassium channels and we define a hierarchical relationship between cell-intrinsic control of ion channel expression and target-derived homeostatic control of synaptic transmission.
Introduction
Homeostatic signaling systems are believed to interface with the mechanisms of neural plasticity to achieve stable, yet flexible, neural circuitry (Davis, 2006; Marder and Goaillard, 2006; Turrigiano, 2008). In each example, a perturbation such as activity blockade or neurotransmitter receptor inhibition causes a transient change in neural function. Then, over some period of time, baseline neural function is restored in the continued presence of the perturbation. The means by which homeostatic signaling systems respond to a perturbation and restore neural function are diverse. They include the modulation of ion channel expression, postsynaptic neurotransmitter receptor abundance, and synaptic vesicle release (Davis, 2006; Marder and Goaillard, 2006; Turrigiano, 2008).
Compensatory changes in the abundance of depolarizing or hyperpolarizing ion channels are generally believed to reflect the action of cell-intrinsic, homeostatic mechanisms that control neuronal firing (Marder et al., 1996; Marder and Prinz, 2002). For example, lobster stomatogastric ganglion neurons, when placed in isolated cell culture, will recalibrate the abundance of inward and outward channel densities to reestablish normal neural activity in the absence of synaptic drive (Turrigiano et al., 1995). Since this compensatory reaction restores neural activity, it is considered homeostatic. In addition, this form of compensation occurs in an isolated cell and therefore must reflect cell-intrinsic signaling. More recently, there have been numerous studies demonstrating that the loss or mutation of a single ion channel gene causes a compensatory change in the abundance of other similar ion channels, often restoring neuronal firing properties (Chen et al., 2006; MacLean et al., 2003; Marder et al., 1996; Marder and Goaillard, 2006; Muraro et al., 2008; Nerbonne et al., 2008; Swensen, 2005; Van Wart and Matthews, 2006). Although the initial loss of an ion channel will alter circuit-level neuronal function, the compensatory changes made are generally thought to be the result of cell-autonomous, homeostatic signaling, much like that observed in isolated lobster central neurons (but see; Desai et al., 1999).
There are also several examples where inter-cellular signaling is an essential component underlying the homeostatic control of neuronal function. For example, the homeostatic control of glutamate receptor abundance in response to activity blockade is influenced by intercellular BDNF signaling (Rutherford et al., 1998) and requires glia-derived secretion of TNF-alpha (Kaneko et al., 2008; Stellwagen and Malenka, 2006). At the Drosophila neuromuscular junction (NMJ), inhibition of postsynaptic glutamate receptors induces a compensatory, homeostatic increase in presynaptic neurotransmitter release. This trans-synaptic signaling system includes the Eph receptor, Ephexin and Cdc42 and ultimately converges upon the CaV2.1 calcium channel (Frank et al., 2009). In addition, this form of homeostatic modulation is gated by the presence of low, persistent levels of BMPs in a non-cell autonomous manner (Goold and Davis, 2007).
To date, homeostatic processes that control neural function have been studied, primarily, in isolation. The ability of homeostatic signaling systems to function at the level of an individual cell and at the level of two or more cells within a neural circuit raises a number of interesting questions. For example, what happens when two independent perturbations occur, one inducing cell-intrinsic forms of compensation and another acting to induce inter-cellular or circuit level forms of compensation? Do the compensatory mechanisms function independently or are they coordinated through some master-sensor of neural function? One way to probe this question is to provide sequential, conflicting perturbations to a neural system. If the system achieves an adaptive response that is different from either perturbation alone, restoring normal neural function, this would be consistent with integrated mechanisms of homeostatic compensation. An alternative possibility is that one form of compensation would predominate or occlude the other. This result could define whether homeostatic compensation is favored at the level of the individual cell compared to the surrounding neural circuit, or vice versa.
Beginning with gene identification through a large-scale genetic screen (Dickman and Davis, 2009), we now reveal how two independent and opposing homeostatic signaling systems interact in the Drosophila neuromuscular system. We report the isolation of a subset of potassium channel mutations that block synaptic homeostasis at the NMJ. In defining how these potassium channel mutations block synaptic homeostasis we uncover a second homeostatic signaling system, one that homeostatically and reciprocally couples the expression of A-type potassium channels in Drosophila motoneurons. We then demonstrate that the cell-intrinsic control of ion channel expression prevents the expression of trans-synaptic homeostatic signaling at the NMJ. Taken together, our data argue against coordinated control of independent homeostatic responses. If generalized, these data could influence our view of neurological disease if an initial stress initiates a primary homeostatic response that is restorative, but with consequences for the future capacity of that cell to adapt or respond to additional perturbations (Bernard et al., 2004; Bernard and Johnston, 2003; Bernard et al., 2001; Cossart et al., 2001; El-Hassar et al., 2007; Frohlich et al., 2008; Glykys and Mody, 2006; Mody, 2005).
Results
We recently performed a large-scale, electrophysiology-based forward genetic screen to identify genes that, when mutated, disrupt the homeostatic modulation of presynaptic neurotransmitter release (Dickman and Davis, 2009). This screen was based on the observation that incubation of the Drosophila NMJ with the glutamate receptor antagonist philanthotoxin-433 (PhTx; 4-10μM) for 10min is sufficient to decrease postsynaptic glutamate receptor sensitivity and induce a rapid, compensatory increase in presynaptic neurotransmitter release (Frank et al., 2006; Frank et al., 2009). The increase in presynaptic neurotransmitter release precisely offsets the decrease in postsynaptic receptor function and restores muscle excitation to baseline ‘set-point’ levels, a process referred to as synaptic homeostasis (Frank et al., 2006; Frank et al., 2009; Dickman and Davis, 2009). In this screen, PhTx was applied to the NMJ of individual mutant Drosophila larvae. For each mutant line, we recorded from 3-10 NMJ and calculated the average mEPSP amplitude, EPSP amplitude and quantal content (Dickman and Davis, 2009). This allowed us to quantify the effect of PhTx on postsynaptic receptor sensitivity and to quantify the homeostatic modulation of presynaptic neurotransmitter release for each mutant line.
This screen identified fourteen mutations that appear to block synaptic homeostasis. Remarkably, only three of these mutations fit into a common category, which turned out to be Drosophila potassium channels. In total, mutations that potentially disrupt twenty-three known or predicted potassium channel genes were screened including mutations in shaker, shal, shab, shaw, orc1, KCNC2, eag, slo, and KCNQ (Atkinson et al., 1991, Ganetzky, 1983; Kaplan and Trout, 1969; Koh et al., 2008). The nature of each potassium channel mutation that we screened and data for the average mEPSP and EPSP amplitudes for each of these mutations are presented in Table 1. One potassium channel mutation, shaker14, was found to have an unusually large EPSP amplitude, even in the presence of PhTx, as might be expected for a mutation that broadens the presynaptic action potential (Dickman and Davis, 2009). However, three potassium channel mutants (shal, shab, CG34366) had unusually small EPSP amplitudes in the presence of PhTx (being more than two standard deviations smaller than the distribution mean for all mutations screened; Dickman and Davis, 2009), identifying them as mutations that potentially block synaptic homeostasis.
Table 1. Potassium Channel Mutations Tested in a PhTx-Dependent Electrophysiology Screen.
| Gene (predicted function) | Mutant Allele | Mutation type | Insertion Location | mEPSP + PhTx 2 | EPSP + PhTx 2 | References1 |
|---|---|---|---|---|---|---|
| wild type | N/A | N/A | N/A | 0.51 (0.03) | 32.0 (2.8) N=10 | N/A |
| shaw | UAS-trunc-shaw | Antimorph | N/A | 0.40 (0.15) | 30.4 (5.6) N=4 | Hodge JL, et. al., 2005 |
| ether-a-go-go | eag1 | LOF | N/A | 0.56 (0.14) | 29.8 (4.3) N=5 | Ganetzky and Wu, 1983 |
| slowpoke | slo1 | Functional null | N/A | 0.49 (0.10) | 26.3 (4.3) N=7 | Atkinson et al., 1991 |
| hyperkinetic | hk1 | LOF | N/A | 0.51 (0.11) | 20.2 (4.5) N=6 | Kaplan and Trout, 1969 |
| easily shocked | P(EP1319) | Transposon | NC-exon | 0.50 (0.26) | 35.3 (6.6) N=3 | Flybase |
| CG10864 (K channel) | P(EY12625) | Transposon | Upstream | 0.46 (0.34) | 35.8 (5.7) N=4 | Flybase |
| CG1090 (potassium antiporter) | P(EP3028) | Transposon | Intron | 0.59 (0.21) | 41.4 (6.6) N=4 | Flybase |
| CG3536 (CNG channel) | Pbac(f00046) | Transposon | Exon | 0.38 (0.12) | 35.4 (4.5) N=4 | Flybase |
| CG8713 (potassium channel) | Pbac(e00867) | Transposon | NC-intron | 0.42 (0.27) | 32.9 (3.2) N=3 | Flybase |
| Ih | Pbac(e01599) | Transposon | Intron | 0.43 (0.13) | 34.5 (4.7) N=5 | Flybase |
| KCNQ-channel | P(EY08364) | Transposon | Exon | 0.43 (0.09) | 35.4 (2.8) N=4 | Flybase |
| Mrityu (potassium channel) | P(EY01340) | Transposon | NC-exon | 0.48 (0.17) | 27.9 (4.6) N=5 | Flybase |
| SK (potassium channel) | P(SK-BG01378) | Transposon | Intron | 0.50 (0.10) | 32.4 (3.6) N=4 | Flybase |
| CG30078 (potassium channel) | P(EY01618) | Transposon | Intron | 0.46 (0.11) | 40.2 (3.4) N=5 | Flybase |
| CG12904 (slo-type channel) | Pbac(f03574) | Transposon | Intron | 0.45 (0.07) | 30.3 (5.6) N=4 | Flybase |
| quiver or sleepless | P(EY04063) | LOF | Exon | 0.41 (0.09) | 37.2 (6.1) N=5 | Koh K, et. al., 2008 |
| CG11984 (potassium regulator) | P(d08881) | Transposon | NC-intron | 0.47 (0.14) | 31.9 (4.1) N=4 | Flybase |
| eag-like (K+ channel) | Pbac(f00820) | Transposon | NC-intron | 0.45 (0.08) | 27.9 (2.9) N=6 | Flybase |
| Ork1 | P(d09258) | Transposon | NC-intron | 0.38 (0.20) | 28.1 (6.7) N=3 | Flybase |
| shaker | sh14 | Functional null | N/A | 0.45 (0.18) | 48.4 (7.9) N=5 | Jan and Jan, 1977 |
| CG34366 | Pbac(f04377) | LOF | Intron | 0.37 (0.02) | 14.0 (1.8) N=14 | Flybase |
| shal | Pbac(f00495) | LOF | Intron | 0.6 (0.02) | 12.2 (1.0) N=20 | Flybase |
| shab | Pbac(f05893) | Transposon | NC-intron | 0.49 (0.04) | 17.7 (2.3) N=8 | Flybase |
References are given for previously published genetic lesions. Transposon insertion sites are based upon Flybase annotation as indicated.
Data are in mV (±SEM). Wild type mEPSP and EPSP amplitudes without PhTx are 1.05 (0.03) and 32.4 (2.35) mV n=16.
Red text highlights those mutations with EPSP amplitudes, recorded in the presence of PhTx, that are more than two standard deviations smaller than the distribution mean for all mutations tested in the presence of PhTx. Blue text is two standard deviations greater than the distribution mean. Sample size for mEPSP and EPSP amplitudes are the same in all cases.
Abbreviations: LOF = loss of function; NC = non-coding; CNG = cyclic nucleotide; slo = slowpoke; trunc = truncated.
To investigate why three independent potassium channel mutations emerged from our genetic screen, we first determined whether the observed defects in synaptic homeostasis could be a secondary consequence of altered baseline transmission or impaired synapse morphology. First, we find that NMJ morphology is normal in all three mutants and, therefore, cannot account for impaired synaptic homeostasis (Supplemental Fig. 1). Second, we find that baseline synaptic transmission in the absence of PhTx is not severely perturbed in these three mutants (see below for additional quantitative information), indicating that a large disruption of baseline transmission cannot account for impaired synaptic homeostasis. It appears, therefore, that three independent potassium channel mutants disrupt either the induction or expression of synaptic homeostasis at the Drosophila NMJ.
The demonstration that potassium channel mutations block synaptic homeostasis was a surprise since an increase in neuromuscular excitability is the predicted phenotype of these channel mutations. To define why these potassium channel mutants might disrupt synaptic homeostasis we first focused our attention on a single gene, shal. We chose to focus on shal because it is known to be expressed in Drosophila neurons but not muscle, and it is known to mediate a rapidly activating and inactivating A-current in Drosophila motoneurons (Baker and Salkoff, 1990; Baro et al., 1996; Birnbaum et al., 2004; Jan et al., 1977; Solc and Aldrich, 1988; Tsunoda and Salkoff, 1995). In addition, shal is highly conserved throughout evolution and mutations in shal have been linked to altered neural plasticity and neurological disease including chronic pain, epilepsy and heart arrhythmia (Birnbaum et al., 2004; Castro et al., 2001). Thus, defining how shal mutations disrupt synaptic homeostasis may have widespread implications.
Shal localizes to the motoneuron axon initial segment and is absent from the NMJ
We identified two transposon insertions in the shal gene as well as a deficiency chromosome that uncovers the shal locus (Fig. 1A). To identify whether these mutations are protein null and to explore where the Shal channel resides within Drosophila motoneurons, we took advantage of a previously developed Shal antibody (Baro et al., 2000). In wild-type animals Shal protein is highly expressed in central neuropil (possibly dendritic arborizations) and within the initial portion of the motor nerve as it exits the ventral nerve cord (VNC) (Fig. 1B). The presence of immuno-staining in the initial portion of the motor nerve strongly suggests that Shal protein is present in Drosophila motoneurons, consistent with prior physiological analyses (Tsunoda and Salkoff, 1995). Protein localization within the motoneurons tapers dramatically over the first 120μm of axon (measured from the origin of the motor nerve at a site adjacent to the neuropil within the central nervous system). Ultimately, after ∼120μm Shal expression decreases to background levels (Fig. 1B and D). No detectable staining is observed at the neuromuscular junction (data not shown) indicating that Shal is restricted to the dendrites and axon initial segment of motoneurons. Consistent with this conclusion, we find no effect of the Shal-specific toxin, phrixotoxin, on neuromuscular synaptic transmission in wild-type animals (Supplemental Fig. 2) (Gasque et al., 2005). Finally, the specificity of the antibody staining data is confirmed by staining the shal495 mutant animal with anti-Shal. Anti-Shal staining is absent in this homozygous mutant and when this mutation is placed in trans to a deficiency that uncovers the shal locus (Fig. 1C-D). These results also indicate that the shal495 mutation is a protein null. Consistent with this conclusion, we assayed shal expression by quantitative real time polymerase chain reaction (qPCR) comparing wild type and shal495 (see methods). We find that shal expression is decreased by 97.7% (± 0.86, compared to wild type) in the shal495 homozygous mutant background (see also below for additional quantification). The localization of shal to the axon initial segment, at or near the site of action potential initiation in motoneurons, is consistent with shal being important for the control of motoneuron excitability.
Figure 1. Analysis of Shal localization and current in wild type and shal mutants.
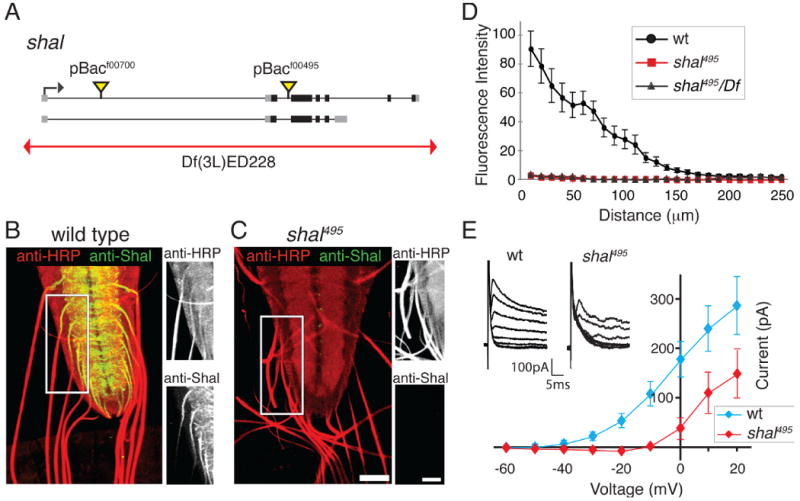
(A) Diagram of the Drosophila shal gene locus (black bars are coding sequence, grey boxes mRNA), indicating the sites of transposon insertion and a deficiency chromosome (red line). (B-C) Representative images of Shal protein (green) within the ventral nerve cord and peripheral axons in wt (B) and shal495 (C). HRP (red) labels the neuronal membrane (scale bar = 40 microns). Side panels show the axon initial segment at higher magnification for each color channel (scale bar = 64 microns). Shal is highly expressed in the neuropil and in peripheral axons as they exit the ventral nerve cord. Anti-Shal staining is absent in shal495 mutants. (D) Quantification of Shal staining intensity in the axon initial segment as a function of distance from the ventral nerve cord in wt (black), shal495 (red), and shal495/Df (grey). Shal is reduced to background in the first ∼120μm of axon. (E) Average IA recorded at the motoneuron soma as a function of voltage step for wt (blue) and shal495 (red). Inset shows representative subtracted IA traces from wt and shal495. Mutations in shal result in a reduced IA compared to wt. See also Supplemental Figure 2 for analysis of the effects of PaTx on EPSP amplitude recorded at the NMJ. Data are presented as the mean ± SEM in this and all subsequent figures.
Absence of an A-current in the motoneuron soma of shal mutants
We next examined the presence of the A-type current in Drosophila motoneuron soma comparing wild type and shal mutant animals to determine if loss of Shal protein eliminates the somatic A-type current in our shal mutant animals. Compared to wild type, there is a dramatic reduction in the A-current in shal mutant animals (Fig. 1E). At a holding potential of -10mV we observe a 100pA A-type current in wild type and the complete absence of an A-type current in the shal mutant. However, at higher holding potentials, we observe the emergence of a rapidly inactivating current in the shal mutant animals that reaches approximately 50% wild type levels at the highest holding potentials tested. One possibility is that this represents residual Shal protein not detected by the antibody. We consider this unlikely because shal expression is effectively eliminated in the shal mutant (assayed by qPCR, see above). An alternative explanation is that the residual A-current in shal mutants is due to the activity of a different channel that is not localized to the soma but which could be activated at an electrotonically distant site (dendrite or axon) when high voltage steps are applied to the soma. Finally, this current could reflect the activity of KCNC2-type channels that are characterized by high voltage activation (Rudy and McBain, 2001) and are encoded in the Drosophila genome (see below). Regardless, our data indicate that there has not been a dramatic, compensatory replacement of an A-current at or near the motoneuron soma.
shal mutants have a mild deficit in baseline transmission
Having identified a protein null mutation in shal we performed a detailed characterization of baseline synaptic transmission in this mutant (Fig. 2 and Supplemental Table 1). There is no significant change in mEPSP amplitude comparing wild type (w1118) with shal495/+ or shal495 homozygous mutants or shal495/Df (Fig. 2A and C). There is a small, statistically significant, deficit in EPSP amplitude observed in the shal495 mutant and shal495/Df when compared to wild type. We observe corresponding changes in quantal content (Fig. 2 D, E) (see methods). This mild effect on synaptic electrophysiology is also observed across a range of external calcium concentrations (Fig. 2B). The mild decrease in baseline synaptic transmission is unexpected for several reasons. First, loss of an A-type potassium channel would be expected to broaden the action potential and potentiate release. Second, we cut the nerve and stimulate below the level where Shal protein is no longer present in the axon. Thus, it is unclear why loss of Shal would have any effect on synaptic transmission at the NMJ. Below we identify compensatory changes at the nerve terminal that could reasonably explain this result.
Figure 2. shal mutants exhibit mild deficits in baseline transmission.
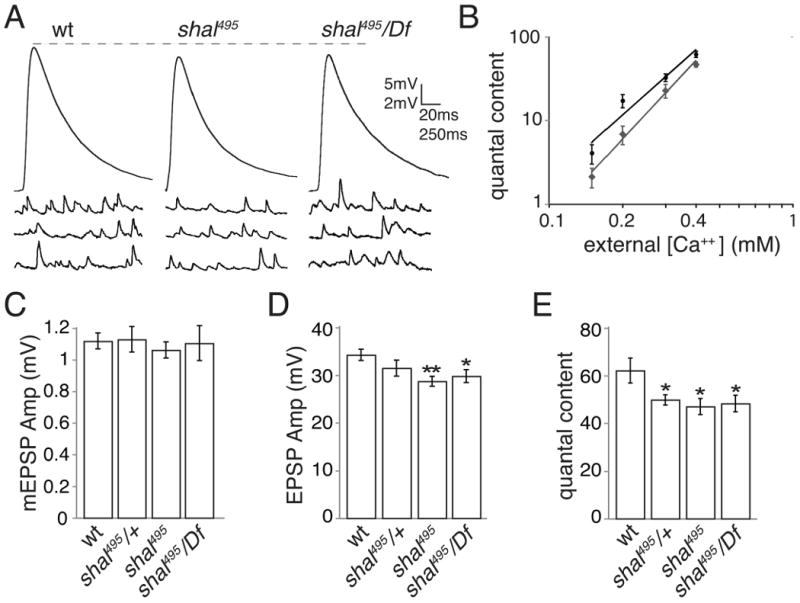
(A) Representative EPSP and mEPSP traces for wt, shal495, and shal495/Df. (B) Extracellular calcium concentration is plotted against quantal content on a logarithmic scale. Quantal content values were corrected for nonlinear summation as done previously (Frank et al., 2006, 2009). (C-E) Average mEPSP (C), EPSP (D), and quantal content (corrected for non-linear summation) (E) are shown for wt, shal495/+, shal495, and shal495/Df. EPSP amplitudes in shal495 and shal495/Df are significantly reduced compared to wt (p < 0.05 for shal495/Df compared to wild type and p< 0.01 for shal495 compared to wild type, Student's t-test). Corresponding reductions in quantal content were observed. * p<0.05; ** p<0.01. All values are listed in Supplemental Table 1.
The rapid induction and sustained expression of synaptic homeostasis are blocked in shal mutants
We next analyzed synaptic homeostasis in greater detail in the shal mutants. First, we repeated the application of PhTx to wild type and shal mutant animals. In shal mutant animals, mEPSP amplitudes are similarly suppressed by application of PhTx, but EPSP amplitudes fail to recover to wild-type levels (Fig. 3A and Supplemental Table 1). Calculation of quantal content demonstrates that the normal, homeostatic enhancement of presynaptic release is completely blocked in the shal495 mutant. Statistically identical defects in synaptic homeostasis were observed in four different shal mutant combinations (Fig. 3B-C). Together, these data demonstrate that the rapid induction of synaptic homeostasis is blocked by mutations that eliminate Shal.
Figure 3. shal mutants block the acute induction of synaptic homeostasis.
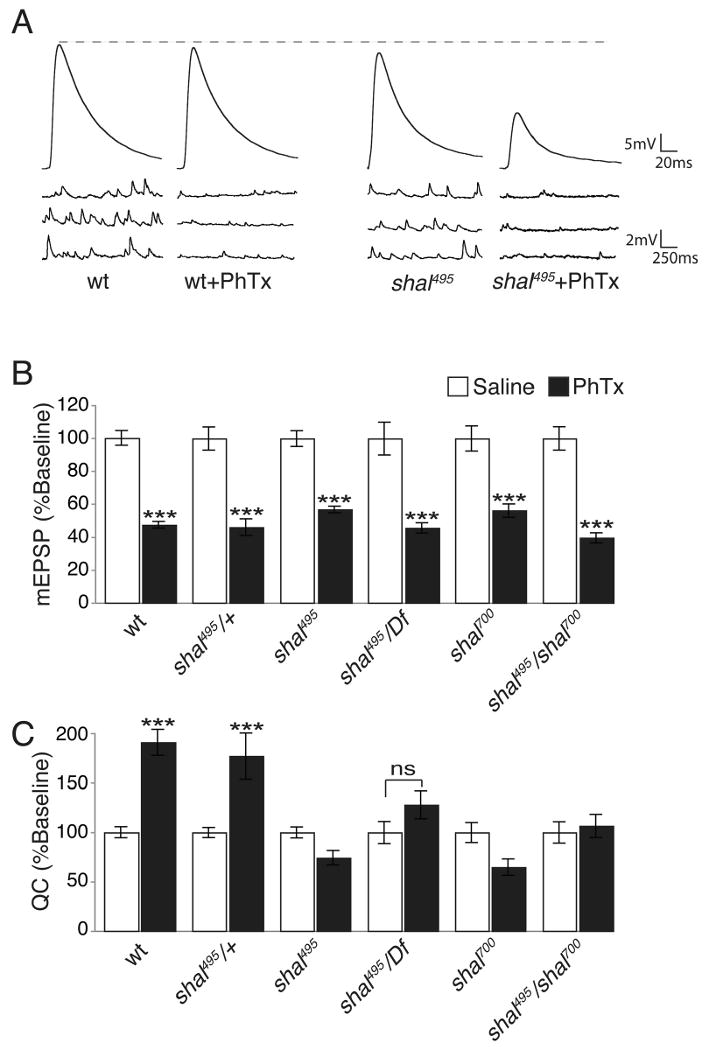
(A) Representative EPSP and mEPSP traces for wt and shal495 in saline and following a 10 min PhTx incubation. Wild type EPSPs return to baseline following PhTx incubation, shal495 EPSPs do not. (B) Average mEPSP values, normalized to their own baseline for the indicated genotypes, in the absence of PhTx (white bars) and following PhTx incubation (black bars). (C) Average quantal content normalized to baseline as in (B). All statistical comparisons are made within single genotypes. Mutations in shal prevent a homeostatic increase in quantal content following PhTx incubation. *** indicates p < 0.001 (Student's t-test). Absolute values are listed in Supplemental Table 1.
In order to determine whether shal mutations also block the persistent expression of synaptic homeostasis we placed the shal495 mutation in the GluRIIASP16 (GluRIIA) mutant background. It has been previously demonstrated that mEPSP amplitudes are decreased in the GluRIIA mutant throughout larval development and that there is a robust homeostatic increase in presynaptic release (Frank et al., 2006; Petersen et al., 1997). Here we demonstrate that the expression of synaptic homeostasis in the GluRIIA mutant is blocked in the shal mutant (Fig. 4 and Supplemental Table 1). This result was confirmed by demonstrating a block of synaptic homeostasis when the shal495/Df allelic combination is placed in the GluRIIA mutant background. Thus, shal is required for both the rapid induction and persistent expression of synaptic homeostasis. The rapid induction of synaptic homeostasis is a local phenomenon that can occur at the isolated NMJ (Frank et al., 2006) raising the question why loss of Shal, which is not present at the NMJ, blocks this process.
Figure 4. shal mutants block sustained expression of synaptic homeostasis.
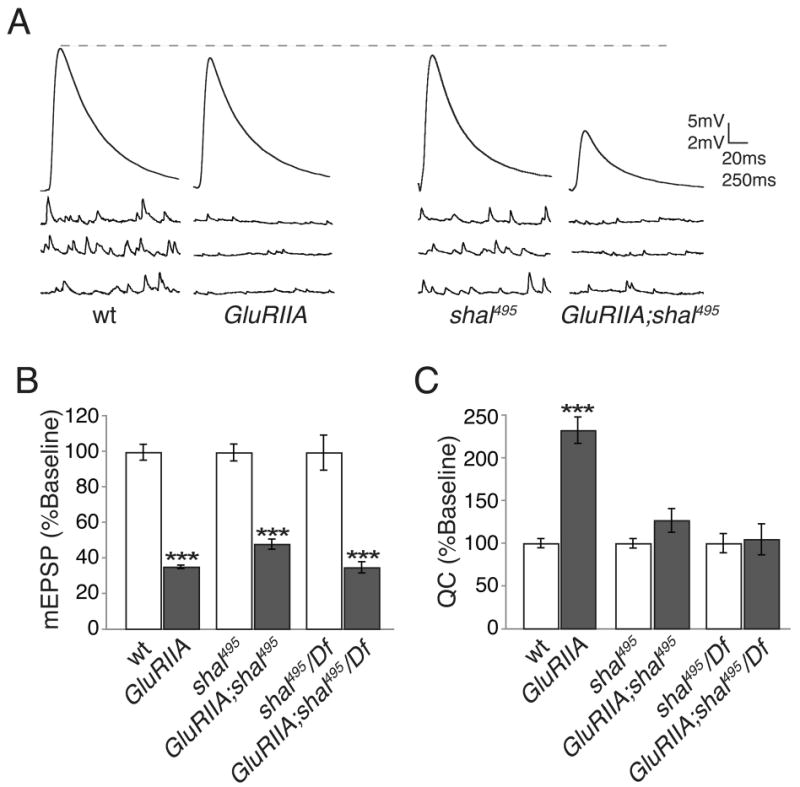
(A) Representative traces for the indicated genotypes. The GluRIIA mutants have reduced mEPSP amplitudes while EPSP amplitudes remain equivalent to wild type due to a presynaptic increase in quantal content. Mutations in shal block the homeostatic increase in quantal content when placed in the GluRIIA mutant background, resulting in a smaller EPSP. (B-C) Quantification of average mEPSP amplitude (B) and quantal content (C) for wild type and shal mutations alone (white bars) and when placed in the GluRIIA mutant background (grey bars). Values are normalized to the genotypic baseline in the absence of GluRIIA. All statistical comparisons are made within a given genetic background, with or without the presence of the GluRIIA mutation. *** indicates p < 0.001 (Student's t-test). Absolute values are listed in Supplemental Table 1.
Homeostatic Coupling of IA Channel Expression in Drosophila
In systems as diverse as the lobster stomatogastric ganglion and the mouse hippocampus, loss or over-expression of an individual ion channel has been observed to drive compensatory changes in the expression of other ion channels (Chen et al., 2006; MacLean et al., 2003; Nerbonne et al., 2008; Swensen, 2005). This has been referred to as a form of cell-intrinsic homeostatic compensation that stabilizes neural activity (Marder et al., 1996; Marder and Prinz, 2002). Drosophila motoneurons express two channels encoding A-type currents, Shal and Shaker (Wei et al., 1990). In vertebrates, loss of KV4.2 (Shal) initiates a compensatory increase in the KV1 (Shaker) current, though ion channel expression was not determined in this study (Chen et al., 2006). Here we test whether there is homeostatic coupling between IA currents in Drosophila motoneurons.
First, we tested shaker RNA expression comparing wild type and shal mutant animals. Currently, antibodies are not available to Shaker in Drosophila. Therefore, we tested mRNA expression using qPCR. We find an increase in shaker expression in the shal mutant (252% ± 30.7 compared to wild type) assaying mRNA derived from dissected central nervous systems (Fig. 5). As a control, we document a statistically significant decrease in shaker expression when we drive expression of a UAS-shaker-RNAi (shakerRNAi) in the nervous system confirming that shaker is expressed presynaptically and that we can accurately measure both an increase and decrease in shaker expression via qPCR (Fig. 5). Thus, in the shal mutants, there is an up-regulation of neuronal shaker expression that could alter channel abundance in the motor axon and presynaptic nerve terminal where Shaker normally resides (Gho and Ganetzky, 1992; Martinez-Padron and Ferrus, 1997; Sheng et al., 1993).
Figure 5. shaker and shal expression are transcriptionally linked.
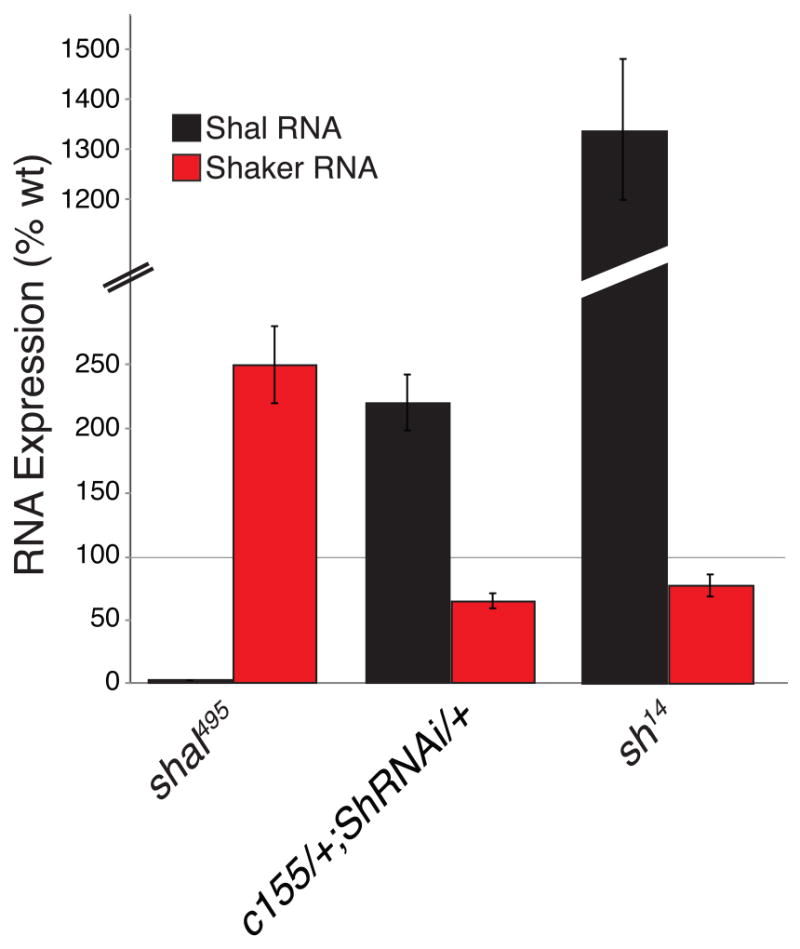
shal RNA expression (black bars) and shaker RNA expression (red bars) were measured in the following genotypes: shal495, neuronal expression of shakerRNAi (c155-gal4/+; shakerRNAi/+), as well as shaker14 (sh14) mutants. shakerRNAi is shortened to shRNAi for display. All bars are represented as percent of wild type animals. Mutations in shal lead to an increase in shaker RNA expression. Neuronal RNAi knockdown of shaker results in an increase in shal RNA expression. shal expression is dramatically increased in sh14 mutants.
Next we tested whether IA channel expression is reciprocally coupled, something that is unknown in any system. We show that neuronal expression of shakerRNAi knocks down shaker expression and that this causes an increase in shal mRNA (223% ± 22.4 increase compared to wild type) (Fig. 5). We recognize that the knockdown of shaker expression is incomplete and sought to repeat this experiment in a shaker mutation. Unfortunately, molecular null mutations in shaker are no longer commonly available. Therefore, we repeated our experiment and assayed shal expression in a shaker14 mutation, which is a functional null (Lichtinghagen et al., 1990). Consistent with the results of shaker knockdown, we find that shal expression is dramatically increased in the shaker14 mutant (Fig. 5). This result confirms a reciprocal regulation of IA channel expression in Drosophila motoneurons.
Finally, as a control, we asked whether the increase in Shaker transcription occurs in the GluRIIA; shal double mutant, just as it does in the shal mutant. We find a robust, statistically significant increase in shaker (189 ± 12.5% increase compared to wild type; p<0.05) in the GluRIIA; shal double mutant that is not statistically different from that observed in shal alone. In the double mutant animals, EPSP amplitudes are significantly smaller than that observed in the shal mutant alone. Thus, the magnitude of the postsynaptic EPSP amplitude does not strongly influence the compensatory change in shaker transcription, consistent with the hypothesis that the compensatory regulation ion channel expression is cell intrinsic.
A compensatory increase in presynaptic Shaker blocks synaptic homeostasis in the shal mutant background
Shaker is expressed at the presynaptic nerve terminal of Drosophila motoneurons (Ganetzky and Wu, 1982; Jan et al., 1977; Wu et al., 1983). One possibility, therefore, is that loss of Shal initiates an increase in presynaptic Shaker and this is the cause of impaired synaptic homeostasis. Such an effect could also explain reduced baseline transmission in the shal mutant. If this is the case, then a shaker mutation might restore synaptic homeostasis when placed in the background of the shal mutant. To test this hypothesis, we generated double mutant animals harboring mutations in both shal and shaker.
As shown previously, shaker mutant NMJs have normal mEPSP amplitudes (p>0.3 compared to wild type) and a dramatic increase in the average EPSP amplitude (Fig. 6A and Supplemental Table 1). At the extracellular calcium concentration used (0.3mM Ca2+), the increase in EPSP amplitude can be primarily accounted for by an increase in presynaptic release due, most likely, to broadening of the presynaptic action potential. When PhTx is applied for 10min under these conditions, wild-type animals show a decrease in mEPSP amplitude and a robust homeostatic increase in presynaptic release (Fig. 6). The shaker mutant animals also show a decrease in mEPSP amplitude and a robust homeostatic increase in presynaptic release. Only shal mutant animals fail to show homeostatic compensation (Fig. 6).
Figure 6. Synaptic homeostasis is restored by loss of Shaker.
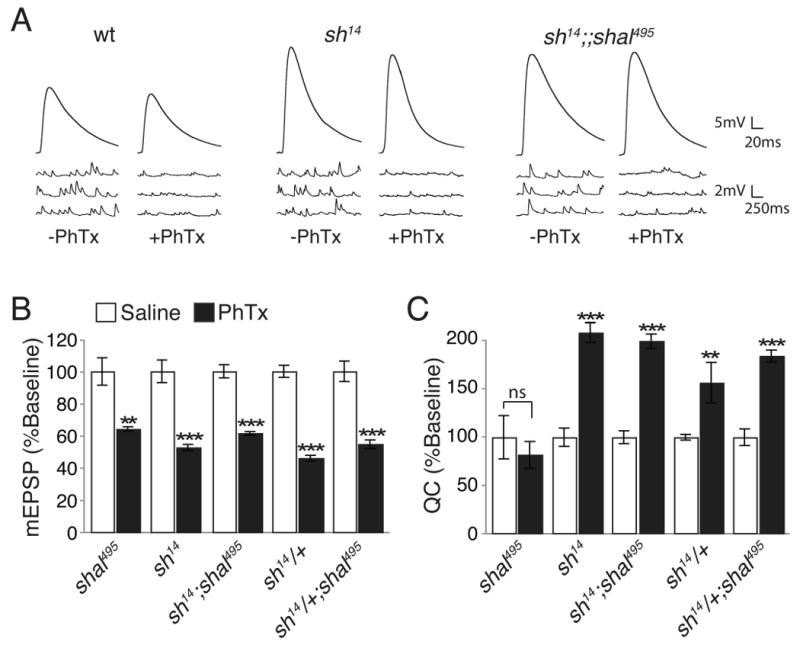
(A) Representative mEPSP and EPSP traces, with and without PhTx, recorded in 0.3mM calcium for the indicated genotypes. (B-C) Average mEPSP amplitudes (B) and quantal content (C) with (black) and without (white) PhTx incubation for each genotype. Values are normalized to each genotypic baseline. Comparisons are made within a single genotype. ** indicates p < 0.01; *** indicates p<0.001 (Student's t-test). The compensatory increase in quantal content is restored with the removal of one or both copies of shaker. Absolute values are listed in Supplemental Table 1.
We next assayed baseline transmission and homeostatic compensation in two mutant combinations, the shaker-shal double mutant and shal animals that harbor a heterozygous mutation in shaker (sh/+). We observe robust homeostatic compensation in the shaker-shal double mutant in contrast to shal mutants alone (Fig. 6). A quantitatively identical result is observed when only a single copy of shaker is removed, indicating that the restoration of synaptic homeostasis is sensitive to the dosage of shaker (Fig. 6). These data support the hypothesis that the compensatory increase in shaker transcription observed in shal mutants could be responsible for blocking synaptic homeostasis. It should be noted, however, that there is an increase in baseline transmission caused by the shaker mutation, both in the heterozygous and homozygous condition (Fig 6A and Supplemental Table 1).
To further investigate whether increased presynaptic Shaker is responsible for the block of synaptic homeostasis in shal mutants, we used RNAi to knock down shaker specifically in presynaptic neurons. We expressed shakerRNAi in presynaptic neurons in shal mutant animals, achieving significant shaker knockdown by qPCR (69.19 ± 6.11%; see methods) and hypothesized that synaptic homeostasis would again be restored. This is precisely what we observed (Fig. 7). This experiment is important for two additional reasons. First, this demonstrates that neuronal Shaker knockdown rescues synaptic homeostasis in shal. Second, there is no statistically significant change in baseline synaptic transmission caused by neuronal Shaker knockdown (p > 0.1; Supplemental Table 1). Since presynaptic shaker knockdown restores synaptic homeostasis in shal without a change in baseline transmission, we can conclude that the restoration of synaptic homeostasis is caused by preventing an increase in shaker expression, and is not secondary to a large increase in baseline transmission. These data strongly support the hypothesis that increased levels of presynaptic Shaker are responsible for the block of synaptic homeostasis observed in shal mutant animals.
Figure 7. Presynaptic knockdown of Shaker is sufficient to restore homeostatic compensation.
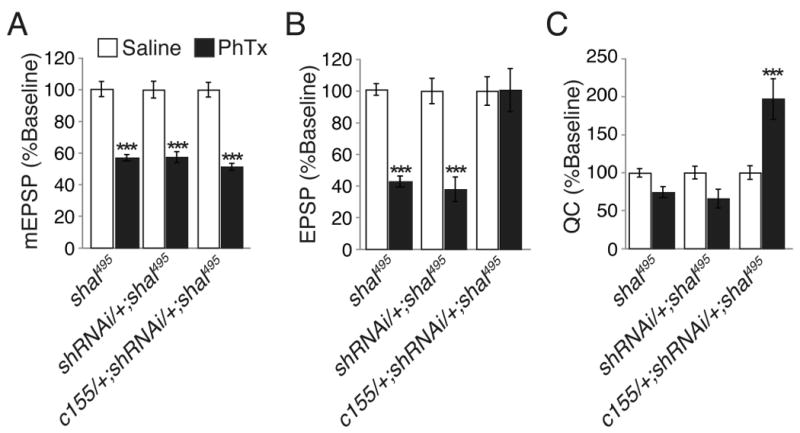
(A-C) Average mEPSP amplitude (A), EPSP amplitude (B), and quantal content (C) in the presence (black) or without (white) PhTx incubation for each of the indicated genotypes including neuronal expression of shakerRNAi in the shal mutant background (c155-gal4/+; UAS-shakerRNAi/+; shal495). Note that shakerRNAi is shortened to shRNAi for display. Values are normalized to each genotypic baseline. When shakerRNAi was driven with the neuronal driver c155-gal4 in the shal mutant background, a homeostatic increase in quantal content was restored. Statistical comparisons were made within single genotypes, comparing the presence or absence of PhTx. *** indicate p < 0.001 (Student's t-test). Absolute values are listed in Supplemental Table 1.
Transgenic overexpression of shaker blocks synaptic homeostasis
To this point we have provided molecular and genetic evidence that increased levels of presynaptic Shaker in the shal mutant cause a block of synaptic homeostasis. To further test this model we asked whether transgenic overexpression of shaker is sufficient to block synaptic homeostasis in an otherwise wild type background. We used the motoneuron-specific driver Ok6-gal4 to express a previously published, modified Shaker potassium channel termed Electrical KnockOut-222 (EKO) (White et al., 2001). Presynaptic expression of EKO does not alter the PhTx-dependent decrease in mEPSP amplitude, but completely blocks the homeostatic increase in presynaptic release normally observed in wild type (Fig. 8). Thus, increased Shaker is sufficient to block the acute expression of synaptic homeostasis. It should be noted that expression of EKO decreases evoked release by ∼55% compared to wild type (Supplemental Table 1). However, we have previously identified other mutations in synaptic genes that disrupt synaptic transmission to an equal or greater extent compared to EKO expression but do not block synaptic homeostasis (Goold and Davis, 2007). Furthermore, a 10-fold decrease in extracellular calcium concentration, reducing transmission below that observed in EKO, also does not block synaptic homeostasis (Frank et al., 2006). Thus, a decrease in baseline transmission is not correlated with impaired synaptic homeostasis. Together, our data further support the model that a compensatory increase in synaptic Shaker is responsible for the defect in synaptic homeostasis observed in the shal mutant background.
Figure 8. Overexpression of Shaker presynaptically blocks homeostatic compensation.
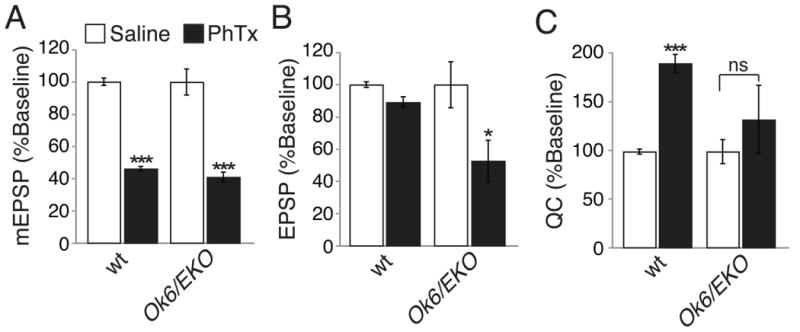
(A-C) Average mEPSP amplitude (A), EPSP amplitude (B), and quantal content (C) in the presence (black) and without (white) PhTx incubation for wild type (wt) and when the modified Shaker transgene (EKO) is overexpressed in motoneurons (Ok6-gal4/+; EKO/+). Values are normalized to each genotypic baseline. No homeostatic increase in quantal content is observed in animals overexpressing the modified Shaker channel, EKO in motoneurons. All statistical comparisons are made within single genotypes, in the presence or absence of PhTx. *** indicates p < 0.001, * indicates p < 0.05 (Student's t-test). Absolute values are listed in Supplemental Table 1.
Increased Shaker blocks the expression versus the induction of synaptic homeostasis
Shaker function is accessible to pharmacological inhibition, allowing us to test whether elevated Shaker blocks the induction versus the expression of synaptic homeostasis. To do so, we asked whether acute pharmacological inhibition of Shaker can restore synaptic homeostasis in the shal mutant. We tested a range of 4-AP concentrations in wild type and selected a concentration (25μM) that produces only a modest change in EPSP amplitude in wild type (27%). When 25μM 4-AP is applied following application of PhTx to the shal mutant NMJ, a robust homeostatic increase in presynaptic release is observed (Fig. 9). This homeostatic increase in presynaptic release is significantly greater than the increase in baseline transmission observed when 4-AP is applied to shal mutants alone (Supplemental Table 1). In addition, we show that application of PhTx to wild type causes a homeostatic increase in quantal content and there is no further increase in quantal content when 4-AP is co-applied with PhTx (Figure 9 D-F). When taken together with the genetic experiments described above, we conclude that elevated levels of synaptic Shaker impair synaptic homeostasis in the shal mutant background. Furthermore, since acute application of 4-AP restores homeostatic compensation when applied after PhTx it demonstrates that increased Shaker can mask the expression of previously induced synaptic homeostasis (Fig. 9). This implies that the homeostatic control ion channel abundance in individual cells can supercede or prevent expression of additional forms of homeostatic compensation.
Figure 9. Acute pharmacological inhibition of Shaker restores synaptic homeostasis.
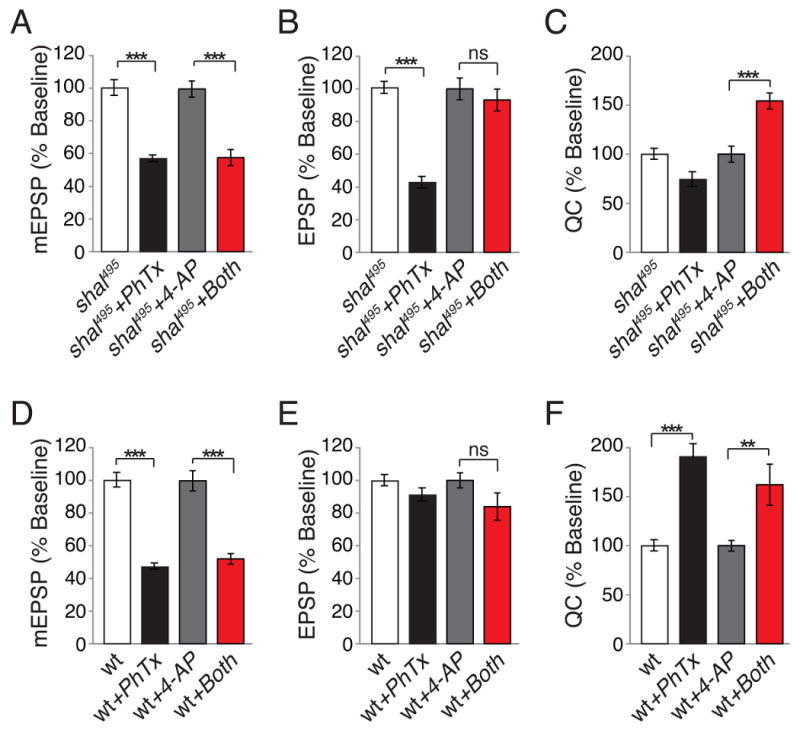
(A-C) Average mEPSP amplitude (A), EPSP amplitude (B), and quantal content (C) for shal495 in the presence of PhTx (black), without PhTx incubation (white), in the presence of 4-AP alone (gray) and in the presence of both 4-AP and PhTx (red). Addition of 4-AP following PhTx incubation reveals a robust, homeostatic increase in quantal content in shal495 (C). (D-F) Same as above, but for recordings made in wild-type controls. Wild-type animals show robust homeostatic compensation following PhTx incubation. There is no further increase in quantal content with co-application of PhTx and 4-AP. Values are normalized to baseline (shal + PhTx is normalized to shal, while shal + both 4-AP and PhTx is normalized to shal + 4-AP alone). *** indicate p < 0.001, ** indicates p < 0.01 (One-Way ANOVA with Bonferroni post test). Absolute values are listed in Supplemental Table 1.
Impaired synaptic homeostasis in an additional, novel potassium channel mutant
Finally, we sought to address two additional questions. First, is altered Shaker expression a common form of compensation that would adjust for loss of any neuronal potassium current? Second, we sought to control for the possibility that shaker knockdown might non-specifically restore homeostatic compensation to any given mutant background. We are able to address both of these issues by analysis of an addition potassium channel mutation isolated in our genetic screen. In our genetic screen, we identified a transposon insertion within the coding region of CG34366 (CG343664377) that decreases gene expression, assayed by qPCR, by 38.01% (± 6.85) compared to wild type. The CG34366 gene encodes the Drosophila homolog of the human KCNC2 (KV3.2) potassium channel (Fig. 10A). This channel is expressed in the embryonic Drosophila central nervous system but no genetic or functional analyses have yet been performed (Hodge et al., 2005). The KV3 potassium channels are widely expressed in the mammalian nervous system (Rudy and McBain, 2001) and can be localized to both the cell soma and synaptic terminals (Goldberg et al., 2005; Itri et al., 2005; Rudy and McBain, 2001). These channels have positively shifted voltage dependencies and very fast deactivation kinetics. The KV3 channels facilitate action potential repolarization, sometimes being referred to as the fast delayed rectifier, and are necessary for the fast repetitive firing observed in numerous neuronal types including purkinje cells and neurons with the globus pallidus and superchiasmatic nucleus (Goldberg et al., 2005; Hernandez-Pineda et al., 1999; Itri et al., 2005; Rudy and McBain, 2001). The KV3.1/KV3.2 double knockout mice show broadened action potentials, increased synaptic transmission and associated decrease in paired-pulse ratios (Goldberg et al., 2005). This is consistent with the required function of these channels in action potential repolarization and subsequent synaptic transmission.
Figure 10. Impaired synaptic homeostasis in a Drosophila KV3.2-like potassium channel mutation is not rescued by neuronal expression of shakerRNAi.
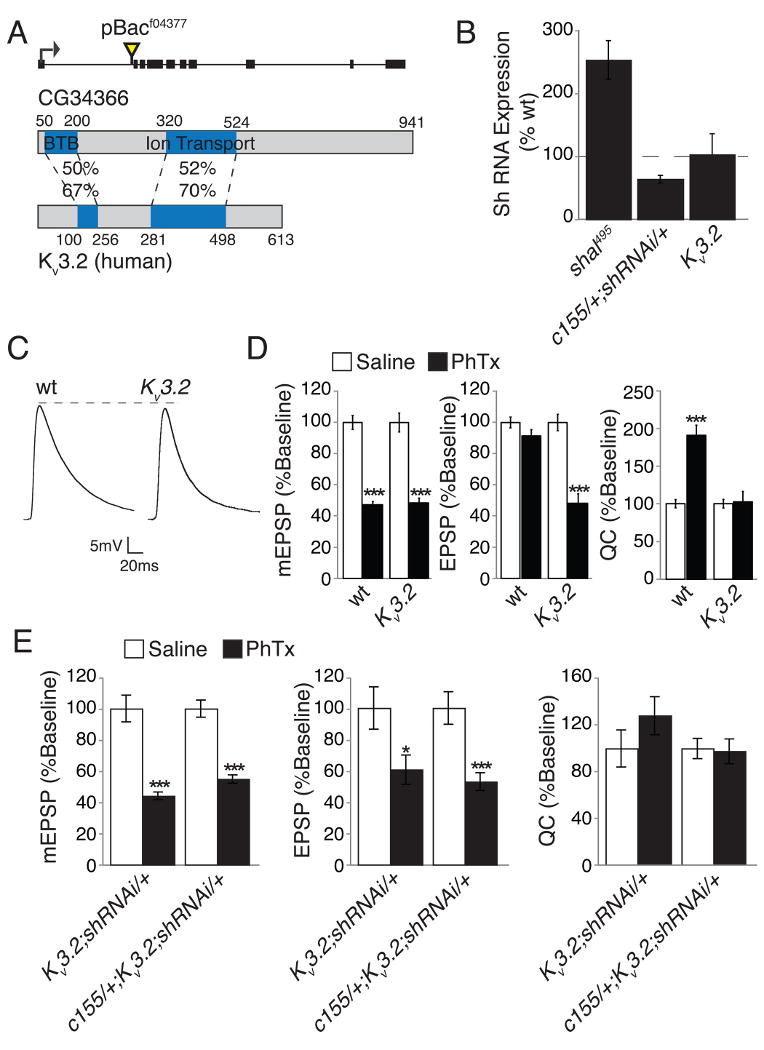
(A) Diagram of the Drosophila CG34366 gene locus indicating the site of transposon insertion. Black boxes indicate coding sequence. CG34366 shows homology to human KV3.2. Blue regions indicate known domains. Sequence identity (top) and similarity (bottom) are given. (B) Shaker RNA expression as a percent of wild type is given for shal495 and animals with neuronal Shaker knockdown (c155-gal4/+; shakerRNAi/+) and in the CG343664377 mutant as indicated (note CG343664377 is indicated as KV3.2 for purposes of display). Shaker expression is unchanged in the CG343664377 mutant. (C) Representative EPSP traces for wt and CG343664377. The CG343664377 EPSP amplitude is slightly reduced compared to wild type (p < 0.01 Student's t-test – see Supplemental Table 1 for average values). (D) Average mEPSP, EPSP, and quantal content without (white bars) and with (black bars) 10 min PhTx incubation for wild type and CG343664377. The CG343664377 mutation shows no homeostatic increase in quantal content. All values are also listed in Supplemental Table 1. (E) Average mEPSP amplitude, EPSP amplitude, and quantal content in the presence (black) and without (white) PhTx incubation for the indicated genotypes including CG343664377 mutant with presynaptic Shaker knockdown (c155-gal4/+; CG343664377; UAS-shakerRNAi/+) and control (CG343664377; UAS-shakerRNAi/+). Values are normalized to the appropriate genotypic baseline. All statistical comparisons are made within single genotypes. Presynaptic knockdown of shaker does not rescue synaptic homeostasis in CG34366 mutants. *** indicate p < 0.001, * indicates p < 0.05 (Student's t-test). Absolute values are listed in Supplemental Table 1.
We first analyzed baseline synaptic transmission in the KV3.2 mutant (Fig. 10C-D). The KV3.2 mutants show modest changes in baseline synaptic transmission (Fig. 10C; Supplemental Table 1). The average amplitude of spontaneous miniature release events is slightly decreased compared to wild type (p<0.05) and there is an associated, statistically significant decrease in EPSP amplitude (p < 0.01). However, there is no deficit in quantal content compared to wild type, underscoring the mild nature of these effects (Fig. 10C-D and Supplemental Table 1). There is also no change in muscle resting membrane potential (-69±0.4mV in wild type versus - 70.1±1.3mV in the KV3.2 mutant) and only a slight change in muscle input resistance (10.5±0.7MΩ in wild type versus 8.5±0.7MΩ in the KV3.2 mutant, p=0.05) that is within normal genotypic variation (see Supplemental Table 1). We next confirmed that there is a block of synaptic homeostasis following application of PhTx to the KV3.2 mutant animals. Application of PhTx causes a significant decrease in mEPSP amplitude, similar to that observed in wild type. However, there is no compensatory increase in presynaptic transmitter release, confirming a block of synaptic homeostasis identical to that observed in shal mutants (Fig. 10C-D and Supplemental Table 1).
Next, we performed a series of experiments to determine if loss of KV3.2 causes an increase in synaptic shaker, as observed in the shal mutant. First, we tested for a change in shaker expression via qPCR. However, there is no significant change in shaker transcript (Fig. 10B). We also performed a genetic test of altered Shaker abundance in KV3.2 mutants. We knocked down shaker expression presynaptically in the KV3.2 mutant and asked whether this would restore synaptic homeostasis, as it did in the shal mutant background. However, shaker knockdown (via RNAi expression) did not restore synaptic homeostasis in KV3.2 animals (Fig. 10E and Supplemental Table 1). From these data we are able to draw two conclusions. First, these data demonstrate that the rescue of synaptic homeostasis in shal mutants by neuronal expression of shaker RNAi is specific and not a consequence of increased transmission. Second, altered shaker expression is not a generalized compensatory response. This result emphasizes that shaker and shal expression seem to be specifically coupled in a homeostatic manner. It remains to be determined whether loss of KV3.2 directly prevents synaptic homeostasis, or whether there is a unique compensatory response in KV3.2 mutant animals that also blocks expression of synaptic homeostasis. An answer to this question will be the subject of future studies.
Discussion
An electrophysiology-based forward genetic screen identified three potassium channel mutations, including mutations in shal and Drosophila KV3.2, that block the expression of synaptic homeostasis following inhibition of postsynaptic glutamate receptor function. We have focused on how mutations in a single potassium channel, shal, lead to a blockade of synaptic homeostasis. We demonstrate that loss of shal induces a compensatory increase in shaker expression, and vice versa, suggesting homeostatic maintenance of A-type channel abundance in Drosophila motoneurons. The compensatory increase in shaker expression is remarkable, however, because it does not replace the A-type current recorded at the motoneuron soma (Fig. 1). Rather, increased Shaker functions to restrict neurotransmitter release from the motoneuron terminal, decreasing baseline release and blocking any further homeostatic enhancement of presynaptic release. There are several implications. First, our data demonstrate that the unique subcellular localization of each ion channel will determine how any compensatory change in ion channel abundance affects neural activity and synaptic transmission. Second, it appears that cell-autonomous control of intrinsic excitability can occlude the expression of subsequent intercellular homeostatic signaling. This suggests a hierarchical control of cell-intrinsic excitability compared to circuit level homeostatic regulation. This also calls into question the concept of a master, homeostatic sensor of neuronal activity. Finally, we define a form of compensation that may largely preserve neuronal output properties without restoring cellular excitation at the level of the cell soma.
Blocking the expression of synaptic homeostasis by increased Shaker
Here we demonstrate that a compensatory increase in Shaker expression is necessary and sufficient to block the subsequent expression of synaptic homeostasis following postsynaptic GluR inhibition. In a shal mutant we observe a ∼250% increase in shaker expression (Fig. 5). If we prevent this increase in Shaker expression in any of three different ways, 1) genetically by introducing shaker mutations (Fig. 6), 2) transgenically through neuron-specific dsRNA knockdown of shaker (Fig. 7), or 3) pharmacologically (Fig. 9), then we restore synaptic homeostasis in the shal mutant. Furthermore, acute block of Shaker by 4-AP following PhTx provides evidence that increased Shaker levels block the expression of synaptic homeostasis, not the induction of this form of homeostatic plasticity (Fig. 9). Finally, we demonstrate that exogenous overexpression of a Shaker transgene (EKO) is sufficient to block synaptic homeostasis in an otherwise wild type background (Fig. 8). Thus, the compensatory increase in Shaker expression in the shal mutant blocks subsequent expression of synaptic homeostasis.
We also provide numerous experiments that argue against the possibility that loss of Shaker rescues synaptic homeostasis through a non-specific potentiation of synaptic transmission. First, neuronal expression of shaker RNAi in the shal mutant background reduces shaker transcript (∼70% reduction) and restores synaptic homeostasis without potentiating baseline transmission. Second, pharmacological inhibition of Shaker was performed using 4-AP concentrations that have a minimal effect on baseline synaptic transmission (∼27% change), yet synaptic homeostasis is restored. Finally, synaptic homeostasis is also blocked in the KV3.2 mutant, but there is no change in shaker expression nor does presynaptic knockdown of shaker in the KV3.2 mutant rescue synaptic homeostasis (Fig. 10). We conclude that the increased shaker expression is specific to the shal mutant and that reducing shaker expression or function in the shal mutant is sufficient to reveal the expression of synaptic homeostasis in the shal mutant.
Why does increased expression of Shaker, at or near the synaptic terminal block the expression of synaptic homeostasis? We presume that increased expression of Shaker in the shal mutant causes a decrease in action potential width. Unfortunately, it is not possible to record the presynaptic action potential from the synaptic terminal because the terminal is embedded within the muscle and is otherwise surrounded by the muscle basal lamina. There are several possible ways that a narrower action potential could block expression of synaptic homeostasis. One possibility is that synaptic homeostasis requires an increase in action potential duration and this is prevented by increased Shaker expression. If so, it is unlikely that Shaker is the direct target of this homeostatic signaling system because homeostatic compensation is observed in the shaker mutant background (Fig. 6). Alternatively, a narrower action potential could prevent recruitment of newly inserted presynaptic calcium channels. Genetic data indicate that synaptic homeostasis involves a change in calcium influx at a fixed number of active zones and this could be achieved by an increase in the number of presynaptic calcium channels (Frank et al., 2006; Frank et al., 2009).
Homeostatic control of ion channel expression: Restoring neural activity versus constraining neuronal output
The transcriptional coupling of shaker and shal would seem to be a homeostatic mechanism since both channels encode A-type potassium currents. However, these channels localize to different subcellular compartments. Thus, increased Shaker expression should not homeostatically restore wild-type motoneuron excitability since the somatic A-current remains absent. Rather, increased Shaker seems to inhibit presynaptic neurotransmitter release and may thereby guard against inappropriately enhanced glutamatergic transmission. This effect differs from current homeostatic hypotheses because baseline neural activity is not re-established, but neural output is constrained within reasonable limits.
The importance of channel localization during homeostatic compensation is also highlighted by recent studies in vertebrate central neurons. It was recently demonstrated that KV4.2 knockout animals lack dendritically recorded A-type currents in hippocampal neurons (Chen et al., 2006). The absence of a dendritic A-type current potentiates back propagating action potentials and enhances LTP (Chen et al., 2006). Thus, at the level of the neuronal dendrite, this is an example of failed homeostatic compensation. However, this study also documents a compensatory increase in somatically recorded KV1-type currents (Chen et al., 2006). It seems plausible that the observed compensatory increase in somatic KV1-type currents could counteract increased dendritic excitability and, thereby, homeostatically restrain neural output. This possibility is supported by data from additional studies examining KV4.2 knockouts in other neuronal cell types (Nerbonne et al., 2008). In these studies, neuronal firing properties measured at the soma are largely normal in the KV4.2 knockout despite the absence of the dendritic A-type current.
Homeostatic coupling of specific ion channel pairs
Here we demonstrate that shal and shaker, which encode A-type potassium channels, are reciprocally, homeostatically coupled. What drives the compensatory change in ion channel expression following loss of a given ion channel? One possibility, suggested by prior research in other systems (see below) is that the neuron senses a persistent change in cellular activity and initiates a homeostatic response that modulates the expression of other ion channels. Our data are consistent with an activity-dependent model. Knockdown of shaker expression (65% of the wild type level) leads to a 223% increase in shal expression. Remarkably, we observe a 1300% increase in shal expression in the shaker14 mutant, which is a point mutation resulting in a non-functional channel (Lichtinghagen et al., 1990). In the shaker14 experiment, the mutant shaker transcript continues to be expressed at 80% wild type levels (Fig. 5). Thus, the degree to which shal expression is increased correlates with the severity of altered channel function rather than the loss of shaker message. This suggests that altered channel function or altered neural activity could be the trigger for the compensatory response. These data also raise an interesting question. If the expression of one ion channel, such as shal, is specifically coupled to the expression of another channel, such as shaker, how could this be achieved by a general monitor of neural activity?
Several studies have now documented that prolonged inhibition of an ion channel, or genetic ablation of an ion channel, can lead to increased expression of a different ion channel with overlapping function, again suggesting coupling between specific pairs of ion channels. For example, loss of NaV1.6 causes increased expression of NaV1.1 in purkinje cells (Burgess et al., 1995) and increased expression of NaV1.2 in retinal ganglion cells (Van Wart and Matthews, 2006). Similarly, loss of A-type potassium currents in KV4.2 (the vertebrate shal homolog) knockout animals causes a compensatory increase in both IK and ISS that preserves action potential shape and neuronal firing properties (Nerbonne et al., 2008). In these examples, the compensatory changes in sodium or potassium channel expression seem to homeostatically maintain appropriate neuronal firing properties. These studies support the hypothesis that ion channels are free variables that can be adjusted by a homeostatic monitor of neural activity and that specific pairs of ion channels may be homeostatically coupled (Marder and Bucher, 2007; Marder and Goaillard, 2006; Schulz et al., 2006).
An alternate form of regulation has been suggested by work in lobster stomatogastric neurons where there is evidence for an activity-independent mechanism that couples shal and Ih expression (MacLean et al., 2003). In this system, overexpression of shal leads to increased Ih current (channel expression was not tested). However, overexpression of a pore-blocked shal also leads to increased Ih current. Thus, altered neural function does not appear to be the trigger for a compensatory change in Ih current. Rather, the cell could monitor the level shal message or protein and regulate Ih current accordingly. This mechanism would allow for specific coupling of ion channel pairs, but appears to be different from the phenomenon identified in Drosophila motoneurons.
One interesting possibility is that the developmental programs that initially specify the active properties of a given neuron could, later, control ion channel expression in a homeostatic manner. Modeling studies suggest that there are large numbers of physiologically plausible combinations of ion channels that could give rise to a cell with a specific firing property (Prinz et al., 2004). However, if the expression of pairs or combinations of ion channels are somehow coupled, then the parameter space for defining the firing properties of a given cell type would be dramatically simplified (Schulz et al., 2007). It is interesting, therefore, to speculate that the apparent homeostatic compensation for loss of a given ion channel could represent the re-use of an earlier developmental program that initially served to balance the expression of specific pairs or combinations of ion channels during cell fate specification (Borodinsky et al., 2004; Marder and Bucher, 2007; Marder and Goaillard, 2006; Muraro et al., 2008; Schulz et al., 2006; Schulz et al., 2007). It will be important to determine whether there are any general rules by which one might predict how a cell will respond to the altered expression of a specific ion channel or whether all such relationships will be defined in a cell-type specific manner.
Control of intrinsic excitability at the expense of network modulation
The regulation of A-type currents in Drosophila motoneurons occludes trans-synaptic, homeostatic modulation of neurotransmitter release. The consequence is that the postsynaptic muscle target is unable to restore normal synaptic drive from the motoneuron terminal and remains hypo-excitable. Specifically, EPSP amplitudes are significantly smaller in the shal; GluRIIA double mutant animals compared to either shal or GluRIIA alone (Fig. 4). Thus, at the neuromuscular junction, the regulation of motoneuron intrinsic excitability supercedes the homeostatic control of motor unit function.
The homeostatic modulation of synaptic transmission can be induced in seconds to minutes. By contrast, the compensatory control of ion channel expression clearly involves gene transcription and is likely to be induced more slowly. One question is whether, given enough time, the mechanisms of synaptic homeostasis can adjust to the change in ion channel expression observed in the shal mutant background. This does not appear to be the case. The GluRIIA mutation causes a persistent change in postsynaptic receptor function leading to a persistent homeostatic increase in presynaptic release that is present throughout the four days of larval development. Synaptic homeostasis is still blocked in the GluRIIA; shal double mutant and we observe a statistically similar increase shaker transcription.
It is worth emphasizing that the homeostatic modulation of presynaptic release appears to have been executed, unaltered in the shal mutant background because acute application of 4-AP reveals normal homeostatic compensation in the shal mutant. These data argue against the possibility that independent homeostatic signaling systems are somehow coordinated at the level of the motor unit, or perhaps neural circuit. Thus, even though an initial homeostatic action is restorative, any change in the balance of ion conductances that control the action potential could dramatically alter how a cell responds to a future perturbation. It has been speculated in systems ranging from crustacean central neurons to the vertebrate cortex, that normal cell-to-cell differences in ionic conductances recorded from an identified cell type might reflect the activity of homeostatic signaling systems (Marder and Prinz, 2002, Davis, 2006). The question remains, will these different cells respond similarly to future homeostatic pressures?
Experimental Procedures
Electrophysiology
All neuronal recordings were performed on central neurons from wandering third-instar larvae (see Supplemental Methods for additional detail). For muscle recordings, quantal content was calculated for each individual recording by calculating the average EPSP/average mEPSP (Albin and Davis, 2004; Davis et al., 1998; Paradis et al., 2001). EPSPs, mEPSPs, and quantal contents calculated for each recording were then averaged across animals for a given genotype. As a control, all calculated quantal content values were corrected for non-linear summation (Davis and Dickman, 2009). In no case does this correction alter the statistical significance of our comparisons or conclusions. Therefore, the majority of our data are presented as non-corrected values with the exception of data presented in Figure 2, which compares quantal contents across a range of extracellular calcium concentration.
Anatomical Analysis
Third-instar larval preparations were fixed in Bouins, washed and incubated overnight at 4°C in primary antibody: anti-shal (1:500; rabbit; a kind gift from Dr. Ronald Harris-Warrick). Ventral Nerve Cord image stacks were captured using a Zeiss Axioskop 2 microscope and the Zeiss LSM 510 Meta Laser Scanning System. Staining quantification was done using Image J software. Shal fluorescence intensity along the axons was calculated within a 10μm2 box. This box was then moved along the length of the axon in 10μm increments. The box was oriented in the HRP channel to properly visualize the axons and then switched to the shal channel for intensity calculations. Four axons from four animals were used for each genotype. Anatomical visualization of the NMJ to quantify bouton numbers was achieved by staining the NMJ with anti-nc82 (gift from Erich Buchner) and anti-Dlg (Pielage et al., 2008). Bouton numbers were quantified as described previously (Albin and Davis, 2004).
Statistical Analyses
All comparisons were analyzed using both Student's t-test and either a one-way or two-way ANOVA including Bonferroni post-test. In all cases, the conclusions and statistical significance remained the same for both types of analysis. Figure legends indicate which test is depicted in graphical form.
CNS Quantitative RT-PCR
Primer-probes specific for real-time PCR detection of Shal, Shaker, Ribosomal protein L32 (RpL32) were designed and developed by Applied Biosystems. The CNS was removed from 25 third-instar larvae per sample (3-6 samples/genotype). Total RNA was isolated from each sample using the standard Trizol protocol. A DNase digestion removed potential DNA contamination (RQ1 RNase-free DNase Promega) (see supplemental methods for additional detail).
Fly stocks and Genetics
In all experiments, the w1118 strain was used as the wild-type control and animals were raised at 22°C unless otherwise noted. shaker RNAi (shakerRNAi) was obtained from Vienna Stock Center (VDRC stock 23671 and 23673). shakerRNAi; shal495 were crossed to c155-gal4;;shal495 and raised at 30°C in parallel with controls. EKO-222 (a kind gift from Dr. Haig Keshishian) flies were crossed to Ok6-gal4 and raised at 25°C in parallel with controls. All other mutant fly lines were obtained from the Bloomington Drosophila stock center (Bloomington, IN) or the Exelixis Drosophila disruption lines (Harvard University, MA).
Supplementary Material
Acknowledgments
We would like to thank Martin Mueller and C. Andrew Frank for reading prior versions of the text and helpful comments, Ed Pym and Catherine Massaro for technical assistance. These studies were supported by NIH Grant number NS39313 to GWD. SBB was supported by an ARCS fellowship. DKD was supported by A. P. Giannini Foundation and Jane Coffin Childs Memorial Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albin SD, Davis GW. Coordinating structural and functional synapse development: postsynaptic p21-activated kinase independently specifies glutamate receptor abundance and postsynaptic morphology. J Neurosci. 2004;24:6871–6879. doi: 10.1523/JNEUROSCI.1538-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- Baker K, Salkoff L. The Drosophila Shaker gene codes for a distinctive K+ current in a subset of neurons. Neuron. 1990;4:129–140. doi: 10.1016/0896-6273(90)90449-p. [DOI] [PubMed] [Google Scholar]
- Baro DJ, Ayali A, French L, Scholz NL, Labenia J, Lanning CC, Graubard K, Harris-Warrick RM. Molecular underpinnings of motor pattern generation: differential targeting of shal and shaker in the pyloric motor system. J Neurosci. 2000;20:6619–6630. doi: 10.1523/JNEUROSCI.20-17-06619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baro DJ, Coniglio LM, Cole CL, Rodriguez HE, Lubell JK, Kim MT, Harris-Warrick RM. Lobster shal: comparison with Drosophila shal and native potassium currents in identified neurons. J Neurosci. 1996;16:1689–1701. doi: 10.1523/JNEUROSCI.16-05-01689.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Bernard C, Johnston D. Distance-dependent modifiable threshold for action potential back-propagation in hippocampal dendrites. J Neurophysiol. 2003;90:1807–1816. doi: 10.1152/jn.00286.2003. [DOI] [PubMed] [Google Scholar]
- Bernard C, Marsden DP, Wheal HV. Changes in neuronal excitability and synaptic function in a chronic model of temporal lobe epilepsy. Neuroscience. 2001;103:17–26. doi: 10.1016/s0306-4522(00)00524-8. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Borodinsky LN, Root CM, Cronin JA, Sann SB, Gu X, Spitzer NC. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature. 2004;429:523–530. doi: 10.1038/nature02518. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Castro PA, Cooper EC, Lowenstein DH, Baraban SC. Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J Neurosci. 2001;21:6626–6634. doi: 10.1523/JNEUROSCI.21-17-06626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JC, Park D, Griffith LC. … characterization of identified motor neurons in the Drosophila third instar larva central …. Journal of neurophysiology. 2004 doi: 10.1152/jn.01115.2003. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Milroy A, Jan YN, Jan LY, Lowenstein DH. Presynaptic localization of Kv1.4-containing A-type potassium channels near excitatory synapses in the hippocampus. J Neurosci. 1998;18:965–974. doi: 10.1523/JNEUROSCI.18-03-00965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 1998;20:305–315. doi: 10.1016/s0896-6273(00)80458-4. [DOI] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The Schizophrenia Susceptibility Gene dysbindin Controls Synaptic Homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hassar L, Milh M, Wendling F, Ferrand N, Esclapez M, Bernard C. Cell domain-dependent changes in the glutamatergic and GABAergic drives during epileptogenesis in the rat CA1 region. J Physiol. 2007;578:193–211. doi: 10.1113/jphysiol.2006.119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich F, Bazhenov M, Sejnowski TJ. Pathological effect of homeostatic synaptic scaling on network dynamics in diseases of the cortex. J Neurosci. 2008;28:1709–1720. doi: 10.1523/JNEUROSCI.4263-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B, Wu CF. Drosophila mutants with opposing effects on nerve excitability: genetic and spatial interactions in repetitive firing. J Neurophysiol. 1982;47:501–514. doi: 10.1152/jn.1982.47.3.501. [DOI] [PubMed] [Google Scholar]
- Gasque G, Labarca P, Reynaud E, Darszon A. Shal and shaker differential contribution to the K+ currents in the Drosophila mushroom body neurons. J Neurosci. 2005;25:2348–2358. doi: 10.1523/JNEUROSCI.4384-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gho M, Ganetzky B. Analysis of repolarization of presynaptic motor terminals in Drosophila larvae using potassium-channel-blocking drugs and mutations. J Exp Biol. 1992;170:93–111. doi: 10.1242/jeb.170.1.93. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol. 2006;95:2796–2807. doi: 10.1152/jn.01122.2005. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Pineda R, Chow A, Amarillo Y, Moreno H, Saganich M, Vega-Saenz de Miera EC, Hernandez-Cruz A, Rudy B. Kv3.1-Kv3.2 channels underlie a high-voltage-activating component of the delayed rectifier K+ current in projecting neurons from the globus pallidus. J Neurophysiol. 1999;82:1512–1528. doi: 10.1152/jn.1999.82.3.1512. [DOI] [PubMed] [Google Scholar]
- Hodge JJ, Choi JC, O'Kane CJ, Griffith LC. Shaw potassium channel genes in Drosophila. J Neurobiol. 2005;63:235–254. doi: 10.1002/neu.20126. [DOI] [PubMed] [Google Scholar]
- Itri JN, Michel S, Vansteensel MJ, Meijer JH, Colwell CS. Fast delayed rectifier potassium current is required for circadian neural activity. Nat Neurosci. 2005;8:650–656. doi: 10.1038/nn1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Voltage-gated and inwardly rectifying potassium channels. J Physiol. 1997;505(Pt 2):267–282. doi: 10.1111/j.1469-7793.1997.267bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY, Dennis MJ. Two mutations of synaptic transmission in Drosophila. Proc R Soc Lond B Biol Sci. 1977;198:87–108. doi: 10.1098/rspb.1977.0087. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan WD, Trout WE., 3rd The behavior of four neurological mutants of Drosophila. Genetics. 1969;61:399–409. doi: 10.1093/genetics/61.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Joiner WJ, Wu MN, Yue Z, Smith CJ, Sehgal A. Identification of SLEEPLESS, a sleep-promoting factor. Science. 2008;321:372–376. doi: 10.1126/science.1155942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtinghagen R, Stocker M, Wittka R, Boheim G, Stuhmer W, Ferrus A, Pongs O. Molecular basis of altered excitability in Shaker mutants of Drosophila melanogaster. EMBO J. 1990;9:4399–4407. doi: 10.1002/j.1460-2075.1990.tb07890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron. 2003;37:109–120. doi: 10.1016/s0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- Marder E, Abbott LF, Turrigiano GG, Liu Z, Golowasch J. Memory from the dynamics of intrinsic membrane currents. Proc Natl Acad Sci U S A. 1996;93:13481–13486. doi: 10.1073/pnas.93.24.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Bucher D. Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu Rev Physiol. 2007;69:291–316. doi: 10.1146/annurev.physiol.69.031905.161516. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Modeling stability in neuron and network function: the role of activity in homeostasis. Bioessays. 2002;24:1145–1154. doi: 10.1002/bies.10185. [DOI] [PubMed] [Google Scholar]
- Martinez-Padron M, Ferrus A. Presynaptic recordings from Drosophila: correlation of macroscopic and single-channel K+ currents. J Neurosci. 1997;17:3412–3424. doi: 10.1523/JNEUROSCI.17-10-03412.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I. Aspects of the homeostaic plasticity of GABAA receptor-mediated inhibition. J Physiol. 2005;562:37–46. doi: 10.1113/jphysiol.2004.077362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro N, Weston A, Gerber A, Luschnig S, Moffat K, Baines R. Pumilio Binds para mRNA and Requires Nanos and Brat to Regulate Sodium Current in Drosophila Motoneurons. Journal of Neuroscience. 2008;28:2099–2109. doi: 10.1523/JNEUROSCI.5092-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol. 2008;586:1565–1579. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron. 2001;30:737–749. doi: 10.1016/s0896-6273(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Pielage J, Cheng L, Fetter RD, Carlton PM, Sedat JW, Davis GW. A presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion. Neuron. 2008;58:195–209. doi: 10.1016/j.neuron.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz AA Bucher D, Marder E. Similar network activity from disparate circuit parameters. Nat Neurosci. 2004;7:1345–52. doi: 10.1038/nn1352. [DOI] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard JM, Marder E. Variable channel expression in identified single and electrically coupled neurons in different animals. Nat Neurosci. 2006;9:356–362. doi: 10.1038/nn1639. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard JM, Marder EE. Quantitative expression profiling of identified neurons reveals cell-specific constraints on highly variable levels of gene expression. Proc Natl Acad Sci U S A. 2007;104:13187–13191. doi: 10.1073/pnas.0705827104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Liao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. 1993;365:72–75. doi: 10.1038/365072a0. [DOI] [PubMed] [Google Scholar]
- Solc CK, Aldrich RW. Voltage-gated potassium channels in larval CNS neurons of Drosophila. J Neurosci. 1988;8:2556–2570. doi: 10.1523/JNEUROSCI.08-07-02556.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Swensen A. Robustness of Burst Firing in Dissociated Purkinje Neurons with Acute or Long-Term Reductions in Sodium Conductance. Journal of Neuroscience. 2005;25:3509–3520. doi: 10.1523/JNEUROSCI.3929-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda S, Salkoff L. Genetic analysis of Drosophila neurons: Shal, Shaw, and Shab encode most embryonic potassium currents. J Neurosci. 1995;15:1741–1754. doi: 10.1523/JNEUROSCI.15-03-01741.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G, LeMasson G, Marder E. Selective regulation of current densities underlies spontaneous changes in the activity of cultured neurons. J Neurosci. 1995;15:3640–3652. doi: 10.1523/JNEUROSCI.15-05-03640.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J Neurosci. 2006;26:7172–7180. doi: 10.1523/JNEUROSCI.1101-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A, Covarrubias M, Butler A, Baker K, Pak M, Salkoff L. K+ current diversity is produced by an extended gene family conserved in Drosophila and mouse. Science. 1990;248:599–603. doi: 10.1126/science.2333511. [DOI] [PubMed] [Google Scholar]
- White BH, Osterwalder TP, Yoon KS, Joiner WJ, Whim MD, Kaczmarek LK, Keshishian H. Targeted attenuation of electrical activity in Drosophila using a genetically modified K(+) channel. Neuron. 2001;31:699–711. doi: 10.1016/s0896-6273(01)00415-9. [DOI] [PubMed] [Google Scholar]
- Wu CF, Ganetzky B, Haugland FN, Liu AX. Potassium currents in Drosophila: different components affected by mutations of two genes. Science. 1983;220:1076–1078. doi: 10.1126/science.6302847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.