

Abstract
Group VIA calcium-independent phospholipase A2 (GVIA iPLA2) has recently emerged as a novel pharmaceutical target. We have now explored the structure-activity relationship between fluoroketones and GVIA iPLA2 inhibition. The presence of a naphthyl group proved to be of paramount importance. 1,1,1-Trifluoro-6-(naphthalen-2-yl)hexan-2-one (FKGK18) is the most potent inhibitor of GVIA iPLA2 (XI(50) 0.0002) ever reported. Being 195 and >455 times more potent for GVIA iPLA2 than for GIVA cPLA2 and GV sPLA2, respectively, makes it a valuable tool to explore the role of GVIA iPLA2 in cells and in vivo models. 1,1,1,2,2,3,3-Heptafluoro-8-(naphthalene-2-yl) octan-4-one inhibited GVIA iPLA2 with a XI(50) value of 0.001, while inhibiting the other intracellular GIVA cPLA2 and GV sPLA2 at least 90-times less potently. Hexa- and octa-fluoro ketones were also found to be potent inhibitors of GVIA iPLA2; however they are not selective.
Keywords: heptafluoropropyl ketones, inhibitors, polyfluoro ketones, phospholipase A2
Introduction
The phospholipase A2 (PLA2) superfamily consists of many different groups of enzymes that catalyze the hydrolysis of the ester bond at the sn-2 position of various phospholipids.1 The products of the hydrolysis are a free fatty acid and a lysophospholipid, both of which may generate second messengers that play important physiological roles. The PLA2 superfamily currently contains 15 separate, identifiable groups and various subgroups.2,3 The three predominant types of PLA2 found in human tissues are the cytosolic (such as the GIVA cPLA2), the secreted (such as the GIIA and GV sPLA2), and the calcium-independent (such as the GVIA iPLA2) enzymes. GIVA cPLA2 is generally considered a pro-inflammatory enzyme which is the rate-limiting provider of arachidonic acid and lysophospholipids.4 In many cases, the activity of secreted PLA2 has been shown to be dependent on or linked to the activity of GIVA cPLA2.5–7 The calcium-independent Group VIA iPLA2 (GVIA iPLA2), typically referred to in the literature as iPLA2, is actually a group of cytosolic enzymes ranging from 85 to 88 kDa and expressed as several distinct splice variants of the same gene.8 GVIA iPLA2 has long been proposed as a homeostatic enzyme involved in basal metabolism within the cell.9–15 However, a number of studies suggest that GVIA iPLA2 also plays important roles in numerous cell types, although they may differ from cell to cell. Recent review articles discuss the role of GVIA iPLA2 in signaling and pathological conditions (for example, cancer and ischemia).16–20
The GVIA iPLA2 enzyme contains a consensus lipase motif, Gly-Thr-Ser*-Thr-Gly, with the catalytic serine confirmed by site-directed mutagenesis.8,21 Both of the intracellular enzymes GIVA cPLA2 and GVIA iPLA2 share the same catalytic mechanism utilizing a serine residue as the nucleophile. The various inhibitor classes of both enzymes are summarized in a recent review article.22 Arachidonyl trifluoromethyl ketone has been shown to function as a tight binding, reversible inhibitor of both GIVA and GVIA PLA2,23,24 while methyl arachidonyl fluorophosphonate functions as an irreversible inhibitor of both enzymes.25 Bromoenol lactone (BEL) (1, Figure 1) has previously been considered to be a selective and irreversible GVIA iPLA2 inhibitor and has been widely applied to study potential biological roles for GVIA iPLA2.11,26 Turk et al. have recently studied the inactivation mechanism of GVIA iPLA2 by 1 (BEL)27 and they concluded that it is likely that this inhibitor affects multiple enzymes and should be used with appropriate caution when studying potential roles of GVIA iPLA2. Our laboratories have previously reported on the development of 2-oxoamide inhibitors targeting GIVA cPLA2.28–32 We have demonstrated that 2-oxoamides containing a free carboxyl group are selective inhibitors of GIVA cPLA2 and most recently we have determined the location of such an inhibitor bound in the active site of GIVA cPLA2 using a combination of deuterium exchange mass spectrometry and molecular dynamics.33 2-Oxoamides based on amino acid esters show cross-reactivity for both GIVA cPLA2 and GVIA iPLA2,30,32 while most recently we identified a 2-oxoamide based on a pseudodipeptide which preferentially inhibits GVIA iPLA2.34
Figure 1.

Some known inhibitors of GVIA iPLA2.
The development of selective inhibitors for the three main human PLA2 enzymes is an important goal, and we have synthesized and assayed a variety of polyfluoroketones for their activity on GIVA cPLA2, GVIA iPLA2 and GV sPLA2. We previously found that 1,1,1,2,2-pentafluoro-7-phenylheptan-3-one (FKGK11) 35 (2, Figure 1) is a selective inhibitor of GVIA iPLA2.36 Trifluoromethyl ketone 3 (FKGK2, Figure 1) can be considered to be a pan inhibitor of all three enzymes: GIVA cPLA2, GVIA iPLA2 and GV sPLA2. The tetrafluoro derivative 4 was found to be the most potent GVIA iPLA2 inhibitor, although it is not selective.36 The selective GVIA iPLA2 inhibitor 2 was successfully used to study the role of this enzyme in neurological disorders such as peripheral nerve injury and multiple sclerosis.37,38 We successfully demonstrated that inhibitor 2 causes a beneficial therapeutic effect in experimental autoimmune encephalomyelitis,38 the animal model of multiple sclerosis. This indicates that GVIA iPLA2 is a novel target for the development of new therapies for multiple sclerosis. The recently emerged important pharmaceutical significance of GVIA iPLA2 and the lack of potent and selective GVIA iPLA2 inhibitors, prompted us to extend our studies towards the discovery of such inhibitors. In this work, we report the synthesis of a variety of new fluoroketones and the study of their selectivity on the three main human phospholipases A2s.
Design and Synthesis of Polyfluoroketones
The rationale behind our design of polyfluoroketones was based on the hypothesis that the introduction of more than three fluorine atoms adjacent to a carbonyl group may increase either the carbonyl reactivity or the inhibitor binding affinity to the target enzyme.36 This hypothesis was confirmed and in fact, such a design led to the selective GVIA iPLA2 inhibitor 2 (pentafluoroethyl ketone) and the tetrafluoro derivative 4, which is a potent GVIA iPLA2 inhibitor, although it is not a selective inhibitor. In the present work, our aim was to extend the structure-activity relationship studies on the potency and the selectivity of heptafluoropropyl ketones and analogs of the lead GVIA iPLA2 inhibitors 2, 3 and 4.
For the synthesis of heptafluoropropyl ketones, carboxylic acids 5a,b were converted to chlorides by treatment with oxalyl chloride and then to the target compounds 6a,b using heptafluorobutanoic anhydride and pyridine (Scheme 1). Wadsworth-Horner-Emmons reaction of benzaldehyde (7) with triethyl phosphonocrotonate,39 followed by saponification, led to unsaturated acid 8 (Scheme 2). We previously showed that α,β-unsaturated acids may be converted into pentafluoroethyl ketones by treatment of the corresponding Weinreb amide with (pentafluoroethyl)lithium.40 Following this procedure, the Weinreb amide 9 was converted to the unsaturated pentafluoroethyl ketone 10.
Scheme 1.

Reagents and conditions: (a) (i) (COCl)2, CH2Cl2, (ii) (C3F7CO)2O, pyridine, CH2Cl2.
Scheme 2.

Reagents and conditions: (a) C2H5OOCCH=CHCH2P(=O)(OC2H5)2, LiOH, THF; (b) (i) NaOH, 1,4-dioxane; (c) DMAP, NMM, WSCI•HCl, CH3ONHCH3 •HCl, CH2Cl2; (d) CF3CF2I, CH3Li•LiBr, Et2O.
Various trifluoromethyl, pentafluoroethyl, and heptafluoropropyl ketones 12a–i were synthesized as depicted in Scheme 3. Reaction of furfural (13) with triethyl phosphonocrotonate, followed by hydrogenation and saponification, produced acid 11b. However, treatment of this acid with oxalyl chloride, followed by (C2F5CO)2O and pyridine, led to the formylated derivative 12b. Under these conditions we were unable to prepare the non-formylated derivative.
Scheme 3.

Reagents and conditions: (a) (i) (COCl)2, CH2Cl2, (ii) (CF3CO)2O or (C2F5CO)2O or (C3F7CO)2O, pyridine, CH2Cl2; (b) C2H5OOCCH=CHCH2P(=O)(OC2H5)2, LiOH, THF; (c) H2, 10% Pd/C; (d) NaOH, CH3OH; (e) Br(CH2)3COOEt, K2CO3, acetone.
The synthesis of tetrafluoro derivatives 18a,b was accomplished by procedures developed earlier36 (Scheme 4). On the basis of 1H and 19F NMR data, tetrafluoro derivatives 18a,b appear to be a mixture of ketone-hydrate form.
Scheme 4.

Reagents and conditions: (a) DAST, CH2Cl2 or (CH3OCH2CH2)2NSF3, CH2Cl2; (b) (i) (CH3)3SiCF3, CsF, CH3OCH2CH2OCH3 or (CH3)3SiCF3, TBAF, toluene, (ii) conc. HCl or TBAF, CH3COOH, THF.
Fluoroketones 19–25 (for structures, see Table 1), which were used in the in vitro assays, were prepared as described previously.40
Table 1.
Inhibition of PLA2 by fluoroketones.a
| No | Structure | GVIA iPLA2 | GIVA cPLA2 | GV sPLA2 | ||
|---|---|---|---|---|---|---|
| % Inhibition | XI(50) | % Inhibition | XI(50) | % Inhibition | ||
| 2 | 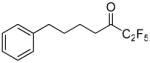 |
99.4 ± 0.1 | 0.0014 ± 0.0001 | N.D. | 28 ± 1 | |
| 6a | 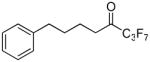 |
99.4 ± 0.0 | 0.0022 ± 0.0001 | 32.6 ± 4.0 | 61.8 ± 6.7 | |
| 6b | 98.4 ± 0.3 | 0.0030 ± 0.0002 | N.D. | N. D. | ||
| 19 | 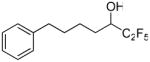 |
94.3 ± 1.5 | 0.0390 ± 0.0024 | 44.8 ± 3.7 | 67.3 ± 7.7 | |
| 20 | 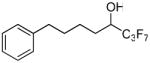 |
97.0 ± 1.3 | 0.0258 ± 0.0016 | 82.0 ± 1.2 | 65.8 ± 1.8 | |
| 10 | 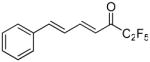 |
96.0 ± 0.8 | 0.0313 ± 0.0025 | 59.0 ± 3.6 | N. D. | |
| 12a | 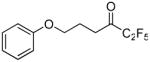 |
99.1 ± 0.5 | 0.0036 ± 0.0001 | N. D. | N. D. | |
| 12b | 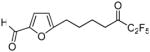 |
84.4 ± 2.1 | 0.0262 ± 0.0006 | N. D. | 41.9 ± 4.4 | |
| 21 | 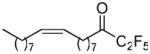 |
95.5 ± 0.4 | 0.0192 ± 0.0007 | 41.4 ± 3.4 | 67.0 ± 6.4 | |
| 22 | 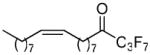 |
80.6 ± 2.5 | 0.0574 ± 0.0030 | 57.0 ± 1.9 | 33.9 ± 18.4 | |
| 23 | 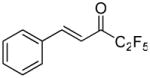 |
96.4 ± 0.6 | 0.0066 ± 0.0005 | N. D. | 76.9 ± 5.3 | |
| 12c | 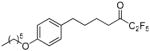 |
98.3 ± 0.2 | 0.0084 ± 0.0006 | 76.1 ± 1.8 | 71.7 ± 3.6 | |
| 12d | 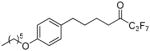 |
95.8 ± 1.3 | 0.0136 ± 0.0006 | 43.7 ± 3.2 | 76.9 ± 2.2 | |
| 12e | 99.4 ± 0.1 | 0.0029 ± 0.0001 | 91.5 ± 0.9 | 0.022 ± 0.001 | 61.4 ± 5.3 | |
| 12f | 99.4 ± 0.1 | 0.0024 ± 0.0001 | 88.8 ± 0.7 | 63.0 ± 6.2 | ||
| 12g | 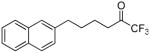 |
99.9 ± 0.1 | 0.0002 ± 0.0000 | 80.8 ± 1.5 | 36.8 ± 7.9 | |
| 12h | 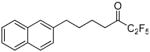 |
99.8 ± 0.0 | 0.0006 ± 0.0000 | 77.1 ± 1.8 | 58.4 ± 5.7 | |
| 12i | 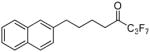 |
99.7 ± 0.2 | 0.0010 ± 0.0001 | 55.8 ± 2.1 | 46.3 ± 10.0 | |
| 18a |  |
97.6 ± 0.2 | 0.0025 ± 0.0001 | 52.1 ± 2.2 | N. D. | |
| 18b | 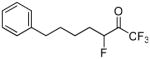 |
98.8 ± 0.2 | 0.0013 ± 0.0000 | 66.0 ± 3.7 | N. D. | |
| 24 | 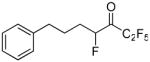 |
100 ± 0.1 | 0.0005 ± 0.0000 | 68.4 ± 1.5 | 39.1 ± 12.6 | |
| 25 | 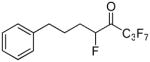 |
100.0 ± 0.1 | 0.0005 ± 0.0000 | 79.9 ± 1.0 | 36.1 ± 8.2 | |
Average percent inhibition and standard error (n=3) reported for each compound at 0.091 mole fraction. XI(50) values determined for inhibitors with greater than 90% inhibition. N.D. signifies compounds with less than 25% inhibition (or no detectable inhibition).
In Vitro Inhibition of GVIA iPLA2, GIVA cPLA2 and GV sPLA2
All synthesized inhibitors were tested for inhibition of human GVIA iPLA2 based on a modification of the previously described mixed micelle-based assay.30 The mixed micelle assay employed herein used 1-palmitoyl-2-arachidonyl-phosphatidylcholine (PAPC) as substrate and the specific conditions employed herein were somewhat different from those employed in the previous mixed micelle assay which employed 1,2-dipalmitoyl-phosphatidylcholine (DPPC) as substrate. This change was made in order to use the same substrate for iPLA2 as for cPLA2, so as to better compare the specificities of both i and cPLA2 toward the same substrate. This also improved the consistency of the standard error in the assay. Using this more refined assay, a XI(50) = 0.0014 was determined for the lead inhibitor 2, lower than that determined previously (0.0073)36. To test the selectivity of the synthesized inhibitors toward GIVA cPLA2, and GV sPLA2, the previously reported mixed micelle-based assays were used.28,29,31 The resulting values of GVIA iPLA2 inhibition are presented in Figure 2 as either percent inhibition or XI(50) values. Initially, the percent of inhibition for each PLA2 enzyme at 0.091 mole fraction of each inhibitor was determined, and XI(50) values were determined for all compounds toward GVIA iPLA2 and for the other two enzymes for all inhibitors that displayed greater than 90% inhibition. However, for two additional iPLA2 inhibitor examples, we also determined their XI(50) toward cPLA2 in order to calculate their relative specificities. The XI(50) is the mole fraction of the inhibitor in the total substrate interface required to inhibit the enzyme by 50%. The inhibition results for all three enzymes are summarized in Table 1.
Figure 2.

GVIA iPLA2 % inhibition at 0.091 mole fraction of inhibitor in mixed micelles (top) and XI(50) values (bottom). Standard error (n = 3) for average % inhibition and XI(50) values for all synthesized compounds is indicated.
The replacement of the pentafluoroethyl group of inhibitor 2 (XI(50) 0.0014) by the heptafluoropropyl group led to inhibitor 6a (XI(50) 0.0022), which resulted in a slightly decreased potency of the GVIA iPLA2 inhibition. Extension of the carbon chain by one carbon atom produced inhibitor 6b, which also resulted in a slightly decreased potency (XI(50) 0.0030). Compounds 19 and 20 which carry a hydroxyl group instead of the carbonyl group of inhibitors 2 and 6a were surprisingly found to be inhibitors of GVIA iPLA2, although not potent. They were also weak inhibitors of the other two PLA2s.
We observed that the insertion of two unsaturated bonds, while keeping the distance between the phenyl and the activated carbonyl group constant, significantly reduced the inhibitory activity of 10 by 22 times. When the carbon atom next to the phenyl group of 2 was replaced by oxygen, compound 12a was 2.5 times less potent than inhibitor 2. At the same time, we found that inhibitor 12a is selective for GVIA iPLA2, since a high mole fraction of the inhibitor (0.091) does not inhibit either GIVA cPLA2 or GV sPLA2 at all. The furan-based inhibitor 12b was not a potent inhibitor.
Pentafluoroethyl derivative 21 based on the oleyl chain inhibited GVIA iPLA2 better than the corresponding palmitoyl derivative.36 Heptafluoropropyl derivative 22 was a weaker inhibitor of GVIA iPLA2 than derivative 21. Both 21 and 22 were able to inhibit weakly the other two enzymes. This data indicates that for the inhibition of GVIA iPLA2 a chain bearing an aromatic ring rather than a long aliphatic saturated or unsaturated chain has to be attached to the activated carbonyl. Reducing the distance between the phenyl and the carbonyl group and inserting a trans double bond led to compound 23, which inhibits GVIA iPLA2 with a XI(50) 0.0066. This derivative does not inhibit at all the other intracellular enzyme GIVA cPLA2 and inhibits weakly GV sPLA2 (77%) at 0.091 mole fraction.
Both the para-hexyloxy substituted pentafluoroethyl and heptafluoropropyl derivatives 12c and 12d inhibited GVIA iPLA2 with XI(50) 0.0084 and 0.0136, respectively. Comparing 12c and 12d with 2 and 6a, it seems that the para-hexyloxy substitution had a negative effect on the GVIA iPLA2 inhibition. Interestingly, when the oxygen atom was moved between the aromatic group and the carbonyl next to the phenyl group, both the trifluoromethyl and the pentafluoroethyl derivatives 12e and 12f strongly inhibited GVIA iPLA2 (XI(50) 0.0029 and 0.0024, respectively). However, all the derivatives bearing a substituent at the para position failed to be selective for any PLA2 enzyme.
The replacement of the phenyl group of inhibitor 2 by a naphthyl group led to excellent results. 1,1,1-Trifluoro-6-(naphthalen-2-yl)hexan-2-one (FKGK18)35 proved to be a very potent inhibitor of GVIA iPLA2 (XI(50) 0.0002), exhibiting 7 times higher inhibition than inhibitor 2. Compound 12g (FKGK18) also inhibited GIVA cPLA2, but at a significant lower level, so we determined its XI(50) which was 0.039 ± 0.001. It is 195 times more potent on GVIA iPLA2 than on GIVA cPLA2. It was also a very weak inhibitor of GV sPLA2 (37% at 0.091 mole fraction) which implies that it is >455 times selective for iPLA2. Both pentafluoroethyl and heptafluoropropyl derivatives 12h and 12i were potent inhibitors of GVIA iPLA2 (XI(50) 0.0006 and 0.0010, respectively). However, both 12h and 12i inhibited weakly GIVA cPLA2 and GV sPLA2. The dose response curves for the inhibition of GVIA iPLA2 by 12g and 12i are presented in Figure 3.
Figure 3.

Dose–response curves for GVIA iPLA2 inhibition by inhibitors 12g and 12i. Inhibition of the activity of human GVIA iPLA2 was tested on mixed-micelles containing 100 μM PAPC and 400 μM Triton X-100. Inhibition curves were generated using Graphpad Prism with a non-linear regression targeted at symmetrical sigmoidal curves based on plots of % inhibition versus log(inhibitor concentration). The reported XI(50) values were calculated from the resultant plots.
Both the tetrafluoro derivatives 18a and 18b were potent inhibitors of GVIA iPLA2 (XI(50) 0.0025 and 0.0013, respectively). 1,1,1,2,2,4-Hexafluoro-7-phenylheptan-3-one 24 (FKGK21)35 and 1,1,1,2,2,3,3,5-octafluoro-7-phenyloctan-4-one 25 (FKGK22)35 were even more potent inhibitors of GVIA iPLA2 (XI(50) 0.0005 both). Comparing 24 and 25 with 2 and 6a, we observe that the insertion of an additional fluorine atom at the α′ position of either the pentafluoroethyl or the heptafluoropropyl ketone results in improved inhibitory potency. However, none of the tetra-, hexa-, and octa-fluoro derivatives proved selective.
The chemical mapping of the GVIA iPLA2 active site through structure-activity studies, carried out in the present and the previous work,36 allow us to understand some of its features, although no crystal structure has been reported. The enzyme-inhibitor complex is likely to be stabilized by an “oxyanion hole” and other features shown in Figure 4. Although long saturated or unsaturated chains may be bound by the enzyme, the presence of an aromatic ring facilitates inhibitor-enzyme binding. The aromatic system, preferably an extended one such as the naphthyl moiety, may be accommodated in an enzyme binding pocket able to create aromatic-aromatic interactions. The aromatic system should have a distance from the carbonyl group corresponding to a four-carbon chain. We propose that the fluorine atoms, apart from their role in increasing the carbonyl reactivity, may contribute to additional interactions of the inhibitor with a “fluorophilic” region of the enzyme. The perfluoroalkyl chain may interact with a “fluorophilic” binding site through a variety of bonds involving fluorine. Recently, it has become clear that fluorine may enhance binding efficacy and selectivity in pharmaceuticals due to a variety of multipolar C-F•••H-N, C-F•••C=O, and C-F•••H-Cα interactions between a fluorinated ligand and protein binding-sites.42,43 The “fluorophilic” binding site of GIVA iPLA2 should be large enough to accommodate even a heptafluoropropyl group.
Figure 4.

Model for the binding mode of fluoroketone inhibitors in the active-site crevice of GVIA iPLA2.
In the present study, we identified five fluoroketones (12g, 12h, 12i, 24 and 25) that are more potent inhibitors of GVIA iPLA2 than the lead inhibitor 2, which has been successfully used in animal models of neurological disorders.37,38 The introduction of one fluorine atom at the α′ position of a pentafluoroethyl or a heptafluoropropyl ketone (compounds 24 and 25) significantly increased the inhibitory potency for GVIA iPLA2. We therefore determined the XI(50) for cPLA2 which was 0.038 ± 0.002. Inhibitor 25 is 76-times and >180-times more potent for GVIA iPLA2 than for GIVA cPLA2 and GV sPLA2, respectively. Inhibitor 12a is also of interest because it inhibits GVIA iPLA2 (XI(50) 0.0036) without affecting at all GIVA cPLA2 and GV sPLA2. The presence of a naphthyl group proved to be of paramount importance. Trifluoromethyl ketone 12g is the most potent inhibitor of GVIA iPLA2 (XI(50) 0.0002) ever reported. Being 195 and >455 times more potent for GVIA iPLA2 than for GIVA cPLA2 and sPLA2, respectively, makes it a valuable tool for ex vivo and in vivo studies.
In conclusion, we developed new, very potent inhibitors of the calcium-independent GVIA iPLA2. Some of them present interesting selectivity over the intracellular GIVA cPLA2 and the secreted GV sPLA2. Applying these inhibitors as tools for studies in animal models, the role of GVIA iPLA2 in various inflammatory diseases may be explored. Since it has become clear that GVIA iPLA2 is a novel target for the development of novel therapies, fluoroketone inhibitors may become leads for the development of novel medicines, in particular for complex neurological disorders such as multiple sclerosis.
Experimental Section
Synthesis of Fluoroketone Inhibitors
Melting points were determined on a Buchi 530 apparatus and are uncorrected. Nuclear magnetic resonance spectra were obtained on a Varian Mercury spectrometer (1H NMR recorded at 200 MHz, 13C NMR recorded at 50 MHz, 19F NMR recorded at 188 MHz) and are referenced in ppm relative to TMS for 1H NMR and 13C NMR and relative to TFA as an internal standard for 19F NMR. Thin layer chromatography (TLC) plates (silica gel 60 F254) and silica gel 60 (230–400 mesh) for flash column chromatography were purchased from Merck. Visualization of spots was effected with UV light and/or phosphomolybdic acid, in EtOH stain. Tetrahydrofuran, toluene, and Et2O were dried by standard procedures and stored over molecular sieves or Na. All other solvents and chemicals were reagent grade and used without further purification. All tested compounds possessed ≥ 95% purity as determined by combustion analysis. Intermediate 11a was prepared by known methods,44 and its spectroscopic data were in accordance with those in the literature.
General Procedure for the Synthesis of Heptafluoropropyl Ketones
Oxalyl chloride (0.38 g, 3 mmol) and N,N-dimethylformamide (40 μL) were added to a solution of carboxylic acid (1 mmol) in dry dichloromethane (40 mL). After 3 h stirring at room temperature, the solvent and excess reagent were evaporated under reduced pressure and the residue was dissolved in dry dichloromethane (10 mL). Pyridine (0.64 mL, 8 mmol) and heptafluorobutanoic anhydride (1.5 mL, 6 mmol) were added dropwise to this solution at 0 °C consecutively. After stirring at 0 °C for 30 min and at room temperature for 1.5 h, the reaction mixture was cooled again at 0 °C and water (2 mL) was added dropwise. After stirring for 30 min at 0 °C and another 30 min at room temperature, the reaction mixture was diluted with dichloromethane (10 mL). The organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure, and the residual oil was purified by flash column chromatography [EtOAc-petroleum ether (bp 40–60 °C) 5/95].
1,1,1,2,2,3,3-Heptafluoro-8-phenyloctan-4-one (6a).40
Yield 59%; yellowish oil. 1H NMR (CDCl3): δ 7.32-7.15 (5H, m, Ph), 2.77 (2H, t, J = 6.2 Hz, CH2), 2.65 (2H, t, J = 6.6 Hz, CH2), 1.71-1.59 (4H, m, 2 × CH2). 13C NMR: δ 194.0 (t, JC-CF2 = 26 Hz, CO), 141.6 (Ph), 130.0-103.5 (m, 2 × CF2, CF3), 128.4 (Ph), 128.3 (Ph), 125.9 (Ph), 37.7 (CH2), 35.4 (CH2), 30.3 (CH2), 21.9 (CH2). 19F NMR: δ −9.4 (CF3), −49.9 (CF2), −55.4 (CF2). MS (ESI) m/z (%): 329 [(M-H)−, 100].
1,1,1,2,2,3,3-Heptafluoro-9-phenylnonan-4-one (6b)
Yield 76%; yellowish oil. 1H NMR (CDCl3): δ 7.38-7.15 (5H, m, Ph), 2.74 (2H, t, J = 6.2 Hz, CH2), 2.63 (2H, t, J = 6.6 Hz, CH2), 1.78-1.60 (4H, m, 2 × CH2), 1.42-1.35 (2H, m, CH2). 13C NMR: δ 194.4 (t, JC-CF2 = 26 Hz, CO), 142.4 (Ph), 130.2-103.5 (m, 2 × CF2, CF3), 128.6 (Ph), 128.5 (Ph), 126.4 (Ph), 38.1 (CH2), 35.9 (CH2), 31.3 (CH2), 29.9 (CH2), 22.5 (CH2). 19F NMR: δ −9.4 (CF3), −49.9 (CF2), −55.4 (CF2). MS (ESI) m/z (%): 343 [(M-H)−, 100]. Anal. (C15H15F7O) C, H.
1,1,1,2,2,3,3-Heptafluoro-8-(4-hexyloxyphenyl)octan-4-one (12d)
Yield 62%; yellowish oil. 1H NMR (CDCl3): δ 7.05 (2H, d, J = 8.2 Hz, Ph), 6.87 (2H, d, J = 8.2 Hz, Ph), 3.91 (2H, t, J = 6.6 Hz, OCH2), 2.74 (2H, t, J = 7.7 Hz, CH2), 2.56 (2H, t, J = 7.7 Hz, CH2), 1.78-1.22 (12H, m, 6 × CH2), 0.88 (3H, t, J = 6.2 Hz, CH3). 13C NMR: δ 194.2 (t, JC-C-F = 26.0 Hz, CO), 157.6 (Ph), 132.0 (Ph), 130.2-103.5 (m, 2 × CF2, CF3), 129.1 (Ph), 114.5 (Ph), 68.0 (CH2O), 38.0 (CH2), 34.8 (CH2), 31.8 (CH2), 30.8 (CH2), 29.5 (CH2), 25.9 (CH2), 22.8 (CH2), 22.1 (CH2), 14.3 (CH3). 19F NMR: δ −9.4 (CF3), −49.9 (CF2), −55.4 (CF2). Anal. (C20H25F7O2) C, H.
1,1,1,2,2,3,3-Heptafluoro-8-(naphthalen-2-yl)octan-4-one (12i)
Yield 45%; yellowish oil. 1H NMR (CDCl3): δ 7.90-7.20 (7H, m, Ph), 2.85-2.70 (4H, m, 2 × CH2), 1.85-1.70 (4H, m, 2 × CH2). 13C NMR: δ 194.2 (t, JC-CF2 = 26 Hz, CO), 139.3 (Ph), 133.8 (Ph), 132.3 (Ph), 128.4 (Ph), 127.9 (Ph), 127.7 (Ph), 127.4 (Ph), 126.7 (Ph), 126.2 (Ph), 125.9 (Ph), 125.0-102.0 (m, CF3, 2 × CF2), 37.7 (CH2), 35.6 (CH2), 30.2 (CH2), 22.0 (CH2). 19F NMR: δ −8.8 (CF3), −50.0 (CF2), −55.5 (CF2). MS (ESI) m/z (%): 379 [(M-H)−, 100]. Anal. (C18H15F7O) C, H.
(2E,4E)-N-Methoxy-N-methyl-5-phenylpenta-2,4-dienamide (9).45
To a stirred solution of carboxylic acid (175 mg, 1 mmol) in CH2Cl2 (7 mL), DMAP (122 mg, 1 mmol), N,O-dimethyl hydroxyamine hydrochloride (98 mg, 1 mmol), NMM (0.11 mL, 1 mmol) and WSCI•HCl (192 mg, 1 mmol) were added consecutively at room temperature. The reaction mixture was left stirring for 18 h. It was then washed with an aqueous solution of HCl 1N (3 × 10 mL), brine (1 × 10 mL), an aqueous solution of NaHCO3 5% (3 × 10 mL) and brine (1 x 10 mL). The organic layer was dried (Na2SO4) and concentrated under reduced pressure. The amide purified by flash chromatography eluting with the appropriate mixture of EtOAc-petroleum ether (40–60 °C) 1/9 to afford the desired product. Yield 67%; white solid. 1H NMR (CDCl3): δ 7.55-7.20 (6H, m, Ph, CH), 6.90-6.80 (2H, m, 2 × CH), 6.57 (1H, d, J = 15 Hz, CH), 3.70 (3H, s, CH3O), 3.25 (3H, s, CH3). 13C NMR: δ 167.0 (CO), 143.2 (CH), 139.6 (CH), 136.2 (Ph), 128.7 (Ph), 126.9 (Ph), 126.8 (CH), 119.0 (CH), 61.7 (CH3O), 32.3 (CH3). MS (ESI) m/z (%): 218 (M+, 100).
(4E,6E)-1,1,1,2,2-Pentafluoro-7-phenylhepta-4,6-dien-3-one (10)
To a stirring solution of the Weinreb amide 9 (78 mg, 0.36 mmol) in Et2O (5 mL) at −78 °C was added pentafluoroiodoethane (0.7 mL, 1.80 mmol) followed by dropwise addition of a MeLi · LiBr solution 1.6 M in ether (1.2 mL, 1.80 mmol). The reaction mixture was stirred at −78 °C for 3 h. Once the reaction was finished, the reaction mixture was poured into H2O and acidified with a 10% solution of KHSO4. The layers were separated and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic layers were washed with a 5% solution of NaHCO3 (40 mL) and dried over MgSO4. The organic solvent was evaporated in vacuo and the residue was purified by column chromatography, eluting with EtOAc-petroleum ether (40–60 °C) 2/98. Yield 90%; yellow oil. 1H NMR (CDCl3): δ 7.74 (1H, dd, J1 = 15.0 Hz, J2 = 10.6 Hz, CH), 7.56-7.44 (2H, m, Ph), 7.42-7.32 (3H, m, Ph), 7.25-6.88 (2H, m, CH), 6.65 (1H, d, J = 15.4 Hz, CH). 13C NMR: δ 182.1 (t, JC-C-F = 25.4 Hz, CO), 149.8 (CH), 146.7 (CH), 135.3 (Ph), 130.4 (Ph), 129.0 (Ph), 127.9 (Ph), 125.9 (CH), 119.9 (CH), 130.0-107.0 (m, CF2, CF3). 19F NMR: δ −4.3 (CF3), −46.0 (CF2). MS (ESI) m/z (%): 276 (M−, 100). Anal. (C13H9F5O) C, H.
Synthesis of Pentafluoroethyl Ketones
The synthesis of pentafluoroethyl ketones was carried out following the procedure described above for heptafluoropropyl ketones, except that pentafluoropropionic anhydride was used instead of heptafluorobutanoic anhydride. The products were purified by flash column chromatography [EtOAc-petroleum ether (bp 40–60 °C) 1/9].
1,1,1,2,2-Pentafluoro-6-phenoxyhexan-3-one (12a)
Yield 60%; yellowish oil. 1H NMR (CDCl3): δ 7.40-7.20 (2H, m, Ph), 7.00-6.83 (3H, m, Ph), 4.02 (2H, t, J = 7 Hz, OCH2), 3.02 (2H, t, J = 6.6 Hz, CH2CO), 2.30-2.10 (2H, m, CH2). 13C NMR: δ 194.0 (t, JC-C-F = 26.4 Hz, CO), 158.5 (Ph), 133.4 (Ph), 129.5 (Ph), 121.0 (Ph), 125.0-110.0 (m, CF3), 114.4 (CH), 106.8 (tq, JC-F2 = 265 Hz, JC-CF3 = 38 Hz, CF2), 68.0 (CH2O), 34.1 (CH2), 22.3 (CH2). 19F NMR: δ − 4.1 (CF3), −45.6 (CF2). MS (ESI) m/z (%): 281 [(M-H)−, 100]. Anal. (C12H11F5O2) C, H.
5-(6,6,7,7,7-Pentafluoro-5-oxoheptyl)furan-2-carboxaldeyde (12b)
Yield 34%; yellowish oil. 1H NMR (CDCl3): δ 9.49 (1H, s, CHO), 7.16 (1H, d, J = 3.8 Hz, arom), 6.26 (1H, d, J = 3.6 Hz, arom), 2.78-2.74 (4H, m, 2 × CH2), 1.76-169 (4H, m, 2 × CH2). 13C NMR: δ 193.9 (t, JC-C-F = 26.4 Hz, CO), 177.0 (CHO), 162.7 (arom), 151.9 (arom), 123.7 (arom), 117.6 (qt, JC-F3 = 286 Hz, JC-CF2 = 34 Hz, CF3), 109.2 (arom), 106.8 (tq, JC-F2 = 265 Hz, JC-CF3 = 38 Hz, CF2), 36.9 (CH2), 28.1 (CH2), 26.4 (CH2), 21.7 (CH2). 19F NMR: δ − 4.0 (CF3), −45.5 (CF2). MS (ESI) m/z (%): 299 [(M+H)+, 100]. Anal. (C12H11F5O3) C, H.
1,1,1,2,2-Pentafluoro-7-(4-hexyloxyphenyl)heptan-3-one (12c)
Yield 61%; yellowish oil. 1H NMR (CDCl3): δ 7.06 (2H, d, J = 8.4 Hz, Ph), 6.82 (2H, d, J = 8.4 Hz, Ph), 3.93 (2H, t, J = 6.6 Hz, OCH2), 2.75 (2H, t, J = 6.6 Hz, CH2), 2.57 (2H, t, J = 6.2 Hz, CH2), 1.77-1.62 (6H, m, 3 × CH2), 1.44-1.27 (6H, m, 3 × CH2), 0.90 (3H, t, J = 6.8 Hz, CH3). 13C NMR: δ 194.2 (t, JC-C-F = 26.4 Hz, CO), 157.5 (Ph), 133.4 (Ph), 129.2 (Ph), 117.6 (qt, JC-F3 = 286 Hz, JC-CF2 = 34 Hz, CF3), 114.4 (Ph), 106.8 (tq, JC-F2 = 265 Hz, JC-CF3 = 38 Hz, CF2), 68.0 (CH2O), 37.2 (CH2), 34.5 (CH2), 31.8 (CH2), 31.6 (CH2), 29.3 (CH2), 27.5 (CH2), 25.7 (CH2), 22.6 (CH2), 14.0 (CH3). 19F NMR: δ − 4.1 (CF3), −45.6 (CF2). MS (ESI) m/z (%): 379 [(M-H)−, 100]. Anal. (C19H25F5O2) C, H.
1,1,1,2,2-Pentafluoro-6-(4-octylphenoxy)hexan-3-one (12f)
Yield 70%; yellowish oil. 1H NMR (CDCl3): δ 7.10 (2H, d, J = 8 Hz, Ph), 6.81 (2H, d, J = 8 Hz, Ph), 3.99 (2H, t, J = 6.6 Hz, CH2), 3.00 (2H, t, J = 6.6 Hz, CH2), 2.57 (2H, t, J = 6.2 Hz, CH2), 2.41-2.14 (2H, m, CH2), 1.64-1.58 (2H, m, CH2), 1.38-1.21 (10H, m, 5 × CH2), 0.91 (3H, t, J = 6.8 Hz, CH3). 13C NMR: δ 194.0 (t, JC-CF2 = 26 Hz, CO), 156.6 (Ph), 135.5 (Ph), 129.1 (Ph), 117.8 (qt, JC-F3 = 287 Hz, JC-CF2 = 34 Hz, CF3), 114.4 (Ph), 106.8 (tq, JC-F2 = 267 Hz, JC-CF3 = 38 Hz, CF2), 65.8 (CH2O), 35.4 (CH2), 34.5 (CH2), 31.5 (CH2), 30.6 (CH2), 29.3 (CH2), 25.7 (CH2), 22.6 (CH2), 21.9 (CH2), 14.2 (CH3). 19F NMR: δ −4.2 (CF3), −45.6 (CF2). MS (ESI) m/z (%): 393 [(M-H)−, 100]. Anal. (C20H27F5O2) C, H.
1,1,1,2,2-Pentafluoro-7-(naphthalen-2-yl)heptan-3-one (12h)
Yield 38%; yellowish oil. 1H NMR (CDCl3): δ 7.88-7.28 (7H, m, Ph), 2.83-2.78 (4H, m, 2 × CH2), 1.80-1.74 (4H, m, 2 × CH2). 13C NMR: δ 194.4 (t, JC-CF2 = 26 Hz, CO), 139.4 (Ph), 133.9 (Ph), 132.4 (Ph), 128.4 (Ph), 127.9 (Ph), 127.6 (Ph), 127.4 (Ph), 126.7 (Ph), 126.2 (Ph), 125.9 (Ph), 118.1 (qt, JC-F3 = 287 Hz, JC-CF2 = 35 Hz, CF3), 107.2 (tq, JC-F2 = 265 Hz, JC-CF3 = 38 Hz, CF2), 37.4 (CH2), 35.9 (CH2), 30.5 (CH2), 22.2 (CH2). 19F NMR: δ −4.1 (CF3), −45.5 (CF2). MS (ESI) m/z (%): 329 [(M-H)−, 100]. Anal. (C17H15F5O) C, H.
Synthesis of Trifluoromethyl Ketones
The synthesis of trifluoromethyl ketones was carried out following the procedure described above for heptafluoropropyl ketones, except that trifluoroacetic anhydride was used instead of heptafluorobutanoic anhydride. The products were purified by flash column chromatography [EtOAc-petroleum ether (bp 40–60 °C) 3/7].
1,1,1-Trifluoro-5-(4-octylphenoxy)pentan-2-one (12e)
Yield 32%; yellowish oil. 1H NMR (CDCl3): δ 7.10 (2H, d, J = 8 Hz, Ph), 6.80 (2H, d, J = 8 Hz, Ph), 3.99 (2H, t, J = 6.6 Hz, OCH2), 2.95 (2H, t, J = 6.6 Hz, CH2), 2.54 (2H, t, J = 6.2 Hz, CH2), 2.20-2.10 (2H, m, CH2), 1.61-1.51 (2H, m, CH2), 1.28-1.21 (10H, m, 5 × CH2), 0.88 (3H, t, J = 6.8 Hz, CH3). 13C NMR: δ 193.9 (t, JC-CF2 = 26 Hz, CO), 156.5 (Ph), 135.5 (Ph), 129.3 (Ph), 115.8 (q, JC-F = 292 Hz, CF3), 114.2 (Ph), 65.8 (CH2O), 35.0 (CH2), 33.1 (CH2), 31.9 (CH2), 31.7 (CH2), 31.6 (CH2), 31.5 (CH2), 29.5 (CH2), 22.6 (CH2), 22.4 (CH2), 14.0 (CH3). 19F NMR: δ −1.5 (s, CF3). MS (ESI) m/z (%): 343 [(M-H)−, 100]. Anal. (C19H27F3O2) C, H.
1,1,1-Trifluoro-6-(naphthalen-2-yl)hexan-2-one (12g)
Yield 39%; yellowish oil. 1H NMR (CDCl3): δ 7.81-7.29 (7H, m, Ph), 2.81-2.73 (4H, m, 2 × CH2), 1.79-1.73 (4H, m, 2 × CH2). 13C NMR: δ 194.4 (t, JC-C-F = 26 Hz, CO), 139.4 (Ph), 133.9 (Ph), 132.3 (Ph), 128.4 (Ph), 127.9 (Ph), 127.7 (Ph) 127.4 (Ph), 126.7 (Ph), 126.2 (Ph), 125.9 (Ph), 115.8 (q, JC-F = 292 Hz, CF3), 36.2 (CH2), 35.6 (CH2), 30.3 (CH2), 22.0 (CH2). 19F NMR: δ −1.5 (s, CF3). MS (ESI) m/z (%): 279 [(M-H)−, 100]. Anal. (C16H15F3O) C, H.
5-(Furan-2-yl)pentanoic acid (11b).46
A suspension of aldehyde 13 (0.096 g, 1 mmol), triethyl 4-phosphonocrotonate (0.37 g, 1.5 mmol), lithium hydroxide (0.036 g, 1.5 mmol), and molecular sieves (beads, 4–8 mesh, 1.5 g/mmol aldehyde) in dry tetrahydrofuran (10 mL) was refluxed under argon for 24 h. The reaction mixture was then cooled to room temperature, filtered through a thin pad of celite and the solvent evaporated under reduced pressure. The residual oil was purified by chromatography on silica gel eluting with ether-petroleum ether (bp 40–60 °C) 1/9. A mixture of the unsaturated ester (135 mg, 0.7 mmol) in dry 1,4-dioxane (7 mL) and 10% palladium on activated carbon (0.07 g) was hydrogenated for 12 h under atmospheric conditions. After filtration through a pad of celite, the solvent was removed in vacuo to give the saturated compound. The solution of the saturated ester in methanol (1.4 mL) was treated with sodium hydroxide 1N (1 mL, 1 mmol). The mixture was stirred at room temperature for 12 h, acidified with 1N HCl and extracted with EtOAc (3 × 10 mL). The solvent was removed in vacum to afford the saturated acid. Yield 66%; white solid; mp 40–41 °C. 1H NMR (CDCl3): δ 10.00 (1H, br, COOH), 7.29-7.27 (1H, m, arom), 6.27-6.25 (1H, m, arom), 6.00-5.97 (1H, m, arom), 2.64 (2H, t, J = 6.4 Hz, CH2), 2.36 (2H, t, J = 6.0 Hz, CH2), 1.73-1.63 (4H, m, 2 × CH2). 13C NMR: δ 178.4 (CO), 155.5 (arom), 140.9 (arom), 110.0 (arom), 104.9 (arom), 33.8 (CH2), 27.6 (CH2), 27.4 (CH2), 24.1 (CH2).
(2E,4E)-5-Phenylpenta-2,4-dienoic acid (8).47
Benzaldehyde was treated with triethyl 4-phosphonocrotonate and the resulting ester was saponified as described above. Yield 76%; white solid; mp 165–166 °C. 1H NMR (CDCl3): δ 7.58-7.22 (6H, m, Ph, CH), 7.05-6.90 (2H, m, CH), 6.00 (1H, d, J = 15 Hz, CH). 13C NMR: δ 169.3 (CO), 145.5 (CH), 140.6 (CH), 136.4 (Ph), 128.9 (Ph), 128.4 (Ph), 127.1 (Ph), 126.2 (CH), 121.1 (CH).
4-(4-Octyl-phenoxy)butyric acid ethyl ester (15)
A mixture of p-octyl-phenol (206 mg, 1 mmol), K2CO3 (415 mg, 3 mmol) and ethyl 4-bromobutyrate (215 mg, 1.1 mmol) in acetone (7.6 mL) was refluxed overnight. The reaction mixture was then cooled to room temperature and the solvent evaporated under reduced pressure. The residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 1/9. Yield 71%; colorless oil. 1H NMR (CDCl3): δ 7.07 (2H, d, J = 8.8 Hz, Ph), 6.81 (2H, d, J = 8.8 Hz, Ph), 4.17 (2H, q, J = 7 Hz, OCH2CH3), 3.92 (2H, t, J = 6.6 Hz, OCH2), 2.60-2.45 (4H, m, 2×CH2), 2.18-2.05 (2H, m, CH2CH2COO), 1.65-1.42 (2H, m, CH2), 1.38-1.21 (13H, br, 5 × CH2, CH3), 0.90 (3H, t, J = 6.8 Hz, CH3). 13C NMR: δ 173.4 (CO), 157.1 (Ph), 135.3 (Ph), 129.4 (Ph), 114.4 (Ph), 66.9 (CH2O), 60.5 (OCH2CH3), 35.3 (CH2), 32.1 (CH2), 32.0 (CH2), 31.5 (CH2), 31.0 (CH2), 29.7 (CH2), 29.5 (CH2), 24.9 (CH2), 22.9 (CH2), 14.5 (CH3), 14.3 (CH3). Anal. (C20H32O3) C, H.
α-Fluorination of α-Hydroxy Methyl Esters
Compound 16a or 16b (1 mmol) was added to a solution of DAST (0.14 mL, 1 mmol) in dry dichloromethane (0.2 mL) at −78 °C. After stirring for 2 h at −78 °C and another 3 h at room temperature, the reaction mixture was quenched with saturated aqueous NaHCO3 (2.5 mL). The organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure, and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7.
Methyl 5-Phenyl-2-fluoropentanoate (17a)
Yield 60%; yellowish oil. 1H NMR (CDCl3): δ 7.35-7.02 (5H, m, Ph), 4.98 (1H, dt, JH-F = 48.2 Hz, JH-H = 6.2 Hz, CHF), 3.78 (3H, s, CH3O), 2.67 (2H, t, J = 6.6 Hz, PhCH2), 2.04-1.78 (4H, m, 2 × CH2). 13C NMR: δ 170.2 (d, JC-CF = 23.5 Hz, CO), 141.3 (Ph), 128.4 (Ph), 128.3 (Ph), 125.9 (Ph), 88.8 (d, JC-F = 183.3 Hz, CHF), 52.1 (CH3), 35.1 (CH2), 31.8 (d, JC-CF = 20.8 Hz, CH2CHF), 25.9 (CH2). 19F NMR: δ −114.1 (CF). MS (ESI) m/z (%): 212 [(M+H)+,100]. Anal. (C12H15FO2) C, H.
Methyl 6-Phenyl-2-fluorohexanoate (17b)
Yield 52%; colourless oil. 1H NMR (CDCl3): δ 7.35-7.16 (5H, m, Ph), 4.86 (1H, dt, JH-F = 48.2 Hz, JH-H = 6.2 Hz, CHF), 3.78 (3H, s, CH3O), 2.64 (2H, t, J = 6.6 Hz, PhCH2), 2.10-1.46 (6H, m, 3 × CH2). 13C NMR: δ 170.2 (d, JC-CF = 23.5 Hz, CO), 141.9 (Ph), 128.2 (Ph), 128.1 (Ph), 125.6 (Ph), 88.8 (d, JC-F = 183.3 Hz, CHF), 52.0 (CH3), 35.4 (CH2), 32.0 (d, JC-CF = 20.8 Hz, CH2CHF), 30.7 (CH2), 23.8 (d, JC-C-CF = 3.0 Hz CH2). 19F NMR: δ −114.1 (CF). Anal. (C13H17FO2) C, H.
Synthesis of 1,1,1,3-Tetrafluoro Ketones
Method A
A solution of compound 17a or 17b (1 mmol) and trifluoromethyltrimethylsilane (283 μL, 1.92 mmol) in ethylene glycol dimethyl ether (0.92 mL) at 0 °C was treated with cesium fluoride (4 mg). After stirring for 30 min at 0 °C and another 18 h at 25 °C the reaction mixture was treated with concentrated HCl (1 mL). After stirring for another 18 h at 25 °C, the reaction mixture was diluted with EtOAc (10 mL). The organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure, and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7.
Method B
A solution of compound 17a or 17b (1 mmol) and trifluoromethyl-trimethylsilane (1 mL, 6.9 mmol) in toluene (9 ml) at μ78 °C was treated with TBAF 1.0 M (45 μL) in THF. After stirring for 2 h at 25 °C the intermediate silyl ether was formed and then it was treated by TBAF 1.0 M (1.2 mmol) in THF and glacial (3 drops) acetic acid. The reaction mixture stirred for 30 min at 25 °C and diluted with EtOAc (10 mL). The organic phase was washed first with saturated solution of K2CO3 and then with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure, and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7.
1,1,1,3-Tetrafluoro-6-phenylhexan-2-one (in equilibrium with 1,1,1,3-tetrafluoro-6-phenyl-2,2-gem-hexanodiol) (18a)
Yield 47% (Method A), 93% (Method B); yellowish oil. 1H NMR (CDCl3): δ 7.34-7.15 (5H, m, Ph), 5.23 (1/4H, dm, JH-F = 48.2 Hz, CH), 4.65 (3/4H, dm, JH-F = 48.2 Hz, CH), 3.74 (3/4H, s, OH), 3.49 (3/4H, s, OH), 2.68 (2H, t, J = 6.2 Hz, CH2), 1.90-1.10 (4H, m, 2 × CH2). 13C NMR: 141.6 (Ph), 128.4 (Ph), 126.1 (Ph), 125.9 (Ph), 122.6 (q, JC-F3 = 286 Hz, CF3), 92.4 (d, JC-F = 175 Hz, CF), 92.2 [m, C(OH)2], 35.4 (CH2), 31.8 (d, JC-F = 20 Hz, CH2), 27.6 (d, JC-F = 20 Hz, CH2). 19F NMR: δ 1.6 (CF3), −5.3 (CF3), −120.9 (CHF). MS (ESI) m/z (%): 247 [(M-H)−, 100].
1,1,1,3-Tetrafluoro-7-phenylheptan-2-one (in equilibrium with 1,1,1,3-tetrafluoro-7-phenyl-2,2-gem-heptanodiol) (18b)
Yield 45% (Method A), 94% (Method B); yellowish oil. 1H NMR (CDCl3): δ 7.32-7.15 (5H, m, Ph), 5.20 (1/6H, dm, JH-F = 48.2 Hz, CH), 4.63 (5/6H, dm, JH-F = 48.2 Hz, CH), 2.64 (2H, t, J = 7.4 Hz, CH2), 1.84-1.80 (2H, m, CH2), 1.74-1.42 (4H, m, 2 × CH2). 13C NMR: 142.1 (Ph), 128.3 (Ph), 125.8 (Ph), 125.3 (Ph), 122.6 (q, JC-F3 = 286 Hz, CF3), 92.3 (d, JC-F = 175 Hz, CF), 92.2 [m, C(OH)2], 35.6 (CH2), 31.0 (CH2), 27.8 (d, JC-F = 20 Hz, CH2), 24.6 (d, JC-C-F = 2.6 Hz, CH2). 19F NMR: δ 1.6 (CF3), −5.3 (CF3), −120.8 (CHF). MS (ESI) m/z (%): 261 [(M-H)−, 100].
In Vitro PLA2 Assays
Phospholipase A2 activity was determined using the previously described modified Dole assay28 with buffer and substrate conditions optimized for each enzyme as described previously.29,31,34 The specific assay conditions employed for the studies reported in this manuscript for each enzyme follow: (i) GIVA cPLA2 substrate mixed-micelles were composed of 400 μM Triton X-100, 97 μM PAPC, 1.8 μM 14C-labeled PAPC, and 3 μM PIP2 in buffer containing 100 mM HEPES pH 7.5, 90 μM CaCl2, 2 mM DTT and 0.1 mg/ml BSA; (ii) GVI iPLA2 substrate mixed-micelles were composed of 400 μM Triton X-100, 98.3 μM PAPC, and 1.7 μM 14C-labeled PAPC in buffer containing 100 mM HEPES pH 7.5, 2 mM ATP and 4 mM DTT; and (iii) GV sPLA2 substrate mixed-micelles were composed of 400 μM Triton X-100, 99 μM DPPC, and 1.5 μM 14C-labeled DPPC in buffer containing 50 mM Tris pH 8.0 and 5 mM CaCl2.
In Vitro PLA2 Inhibition Studies
Initial screening of compounds at 0.091 mole fraction inhibitor in mixed-micelles was carried out. Compounds displaying 25% or less inhibition of the assays were considered to have no inhibitory affect (designated N.D.). We report average percent inhibition (and standard error, n = 3) for compounds displaying less than 90% enzyme inhibition. If the percent inhibition was greater than 90%, we determined its XI(50) by plotting percent inhibition vs. inhibitor mole fraction (typically 7 concentrations between 0.00091 and 0.091 mole fraction). Inhibition curves were modeled in Graphpad Prism 5.0 using non-linear regression targeted at symmetrical sigmoidal curves based on plots of % inhibition versus log (inhibitor concentration), to calculate the reported XI(50) and associated error values.
Acknowledgments
This work was supported by the European Social Fund and National Resources (G.K.) and by NIH GM 20,501 (E.A.D.).
Abbreviations
- ATP
adenosine triphosphate
- BEL
bromoenol lactone
- BSA
bovine serum albumin
- DAST
diethylaminosulfur trifluoride
- DMAP
4-dimethylaminopyridine
- DPPC
1,2-dipalmitoylphosphatidylcholine
- DTT
dithiothreitol
- EtOAc
ethyl acetate
- GIVA cPLA2
Group IVA cytosolic phospholipase A2
- GV sPLA2
Group V secreted phospholipase A2
- GVIA iPLA2
Group VIA calcium-independent phospholipase A2
- HEPES
4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid
- NMM
N-methylmorpholine
- PAPC
1-palmitoyl-2-arachidonylphosphatidylcholine
- PIP2
phosphatidyl inositol (4,5)-bisphosphate
- TBAF
tetra-n-butylammonium fluoride
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- Tris
tris(hydroxymethyl)aminomethane
- TMS
tetramethylsilane
- WSCI•HCl
N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride
References
- 1.Burke JE, Dennis EA. Phospholipase A2 Biochemistry. Cardiovasc Drugs Ther. 2009;23:45–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaloske RH, Dennis EA. The phospholipase A(2) superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50:S237–42. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, Gelb MH. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-α. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 6.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirai Y, Balsinde J, Dennis EA. Localization and functional interrelationships among cytosolic Group IV, secreted Group V, and Ca2+-independent Group VI phospholipase A2s in P388D1 macrophages using GFP/RFP constructs. Biochim Biophys Acta. 2005;1735:119–129. doi: 10.1016/j.bbalip.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem. 1998;273:207–214. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 9.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci USA. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 11.Balsinde J, Dennis EA. Function and inhibition of intracellular calcium-independent phospholipase A2. J Biol Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- 12.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 13.Birbes H, Drevet S, Pageaux JF, Lagarde M, Laugier C. Involvement of calcium-independent phospholipase A2 in uterine stromal cell phospholipid remodelling. Eur J Biochem. 2000;267:7118–7127. doi: 10.1046/j.1432-1327.2000.01814.x. [DOI] [PubMed] [Google Scholar]
- 14.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A(2) Lipids. 2001;36:689–700. doi: 10.1007/s11745-001-0774-9. [DOI] [PubMed] [Google Scholar]
- 15.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, Turk J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2β) indicate a signaling rather than a housekeeping role for iPLA2β. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 16.Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A(2) in activated cells. Cellular Signalling. 2005;17:1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Balsinde J, Perez R, Balboa MA. Calcium-independent phospholipase A2 and apoptosis. Biochim Biophys Acta. 2006;1761:1344–1350. doi: 10.1016/j.bbalip.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 18.Hooks SB, Cummings BS. Role of Ca2+-independent phospholipase A2in cell growth and signaling. Biochem Pharmacol. 2008;76:1059–1067. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkins WP, Barbour SE. Group VI phospholipase A2: Homeostatic phospholipases with significant potential as targets for novel therapeutics. Curr Drug Targets. 2008;9:683–697. doi: 10.2174/138945008785132385. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins CM, Cedars A, Gross RW. Eicosanoid signalling pathways in the heart. Cardiovasc Res. 2009;82:240–249. doi: 10.1093/cvr/cvn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 22.Magrioti V, Kokotos G. Synthetic inhibitors of Group IVA and Group VIA phospholipase A2. Anti-Inflammatory Anti-Allergy Agents. Med Chem. 2006;5:189–203. [Google Scholar]
- 23.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, Tremblay NM, Huang Z, Weech PK, Gelb MH. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 24.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 25.Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca2+-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim Biophys Acta. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- 26.Balsinde J, Dennis EA. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J Biol Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 27.Song H, Ramanadham S, Bao S, Hsu FF, Turk J. A bromoenol lactone suicide substrate inactivates group VIA phospholipase A2 by generating a diffusible bromomethyl keto acid that alkylates cysteine thiols. Biochemistry. 2006;45:1061–1073. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. Novel 2-oxoamide inhibitors of human Group IVA phospholipase A2. J Med Chem. 2002;45:2891–2893. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]
- 29.Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. Inhibition of Group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells and in vivo. J Med Chem. 2004;47:3615–3628. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 30.Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. Differential inhibition of Group IVA and Group VIA phospholipases A(2) by 2-oxoamides. J Med Chem. 2006;49:2821–2828. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker S, Dennis EA, Kokotos G. Structure-activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phopholipase A2. J Med Chem. 2007;50:4222–4235. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 32.Antonopoulou G, Barbayianni E, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. Structure-activity relationships of natural and non-natural amino acid-based amide and 2-oxoamide inhibitors of human phospholipase A2enzymes. Bioorg Med Chem. 2008;16:10257–10269. doi: 10.1016/j.bmc.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burke JE, Babakhani A, Gorfe AA, Kokotos G, Li S, Woods VL, McCammon JA, Dennis EA. Location of inhibitors bound to Group IVA phospholipase A2 determined by molecular dynamics and deuterium exchange mass spectrometry. J Am Chem Soc. 2009;131:8083–8091. doi: 10.1021/ja900098y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbayianni E, Stephens D, Grkovich A, Magrioti V, Hsu YH, Dolatzas P, Kalogiannidis D, Dennis EA, Kokotos G. 2-Oxoamide inhibitors of phospholipase A2 activity and cellular arachidonate release based on dipeptides and pseudodipeptides. Bioorg Med Chem. 2009;17:4833–4843. doi: 10.1016/j.bmc.2009.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.David S, Kalyvas A, Lopez R, Kokotos G, Constantinou-Kokotou V, Baskakis C, Kokotos CG, Stephens D, Dennis EA. WO 2008122119 A1. Perfluoroketone compounds and uses thereof. 2008 Oct 16;
- 36.Baskakis C, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. Synthesis of polyfluoro ketones for selective inhibition of human phospholipase A2 enzymes. J Med Chem. 2008;51:8027–8037. doi: 10.1021/jm800649q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez-Vales R, Navarro X, Shimizu T, Baskakis C, Kokotos G, Constantinou-Kokotou V, Stephens D, Dennis EA, David S. Intracellular phospholipase A2 group IVA and group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain. 2008;131:2620–2631. doi: 10.1093/brain/awn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalyvas A, Baskakis C, Magrioti V, Constantinou-Kokotou V, Stephens D, Lopez-Vales R, Lu JQ, Yong VW, Dennis EA, Kokotos G, David S. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain. 2009;132:1221–1235. doi: 10.1093/brain/awp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takacs JM, Jaber MR, Clement FC, Walters C. A useful procedure for the preparation of (E,E)-2,4-dienoates: Lithium hydroxide-promoted dienylation by 4-phosphonocrotonate. J Org Chem. 1998;63:6757–6760. [Google Scholar]
- 40.Kokotos CG, Baskakis C, Kokotos G. Synthesis of medicinally interesting polyfluoro ketones via perfluoroalkyl lithium reagents. J Org Chem. 2008;73:8623–8626. doi: 10.1021/jo801469x. [DOI] [PubMed] [Google Scholar]
- 41.Huisgen R, Rietz U. Darstellung und cyclisierung der a-[naphthyl-(2)]-fettsauren. Chem Ber. 1957;90:2768–2777. [Google Scholar]
- 42.Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 43.Bohm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, Obst-Sander U, Stahl M. Fluorine in medicinal chemistry. Chem Bio Chem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 44.Lachapelle A, St-Jacques M. Conformational analysis of 3-substituted-2,3,4,5-tetrahydro-1-benzoxepin by 1H and 13C nuclear magnetic resonance. Can J Chem. 1987;65:2575–2594. [Google Scholar]
- 45.Blackburn L, Kanno H, Taylor RJK. In situ alcohol oxidation-Wittig reactions using N-methoxy- N-methyl-2-(triphenylphosphoranylidine)acetamide: application to the synthesis of a novel analogue of 5-oxo-eicosatetraenoic acid. Tetrahedron Lett. 2003;44:115–118. [Google Scholar]
- 46.Crabbe P, Depres JP. Synthesis and properties of 5-bromocyclohepta[b]furan-4-one. J Chem Soc, Perkin Trans. 1980;1:2081–2083. [Google Scholar]
- 47.Dockendorff C, Sahli S, Olsen M, Milhau L, Lautens M. Synthesis of dihydronaphthalenes via aryne Diels-Alder reactions: Scope and diastereoselectivity. J Am Chem Soc. 2005;127:15028–15029. doi: 10.1021/ja055498p. [DOI] [PubMed] [Google Scholar]