

Abstract
We synthesized a series of peptide amphiphiles (PAs) with systematically modified amino acid sequences to control the mechanical properties of the nanofiber gels they form by self-assembly. By manipulating the number and position of valines and alanines in the peptide sequence we found that valines increase the stiffness of the gel while additional alanines decrease the mechanical properties. Vitreous ice cryo-transmission electron microscopy shows that all PA molecules investigated here form nanofibers 8–10 nm in diameter and several micrometers in length. We found through Fourier transform IR experiments a strong correlation between gel stiffness and hydrogen bond alignment along the long axis of the fiber. Molecules that form supramolecular structures with the highest mechanical stiffness were found by circular dichroism to self-assemble into β-sheets with the least amount of twisting and disorder, a result which is consistent with IR experiments. Molecular control of mechanical stiffness in three-dimensional artificial peptide amphiphile matrices offers a chemical strategy to control biological phenomena such as stem cell differentiation and cell morphology.
Introduction
Great strides have been made in the molecular design of biomedical materials1 through the development of tissue scaffolds composed of natural biopolymers, synthetic polymers, and peptide based materials.1–5 Self-assembling biomaterials have attracted significant interest as non-invasive, injectable scaffolds, in some cases incorporating bioactive signals,6,7 or serving as delivery vehicles for cell therapy.3 A variety of peptide-based molecules self-assemble into shapes such as ribbons,8–10 cylinders,11,12 tubes4,13 and spheres.14–16 Numerous examples of self-assembly in nature demonstrate how certain molecular features influence properties of these assemblies and are therefore helpful in design of synthetic materials. Recent studies have also shown that matrix stiffness is an important feature in cell biology and can be used to influence cell behavior,17–19 so control over the mechanical properties of a scaffold is an important target in regenerative medicine. Efforts to rationally design self-assembling molecules to control their morphology10,20 and mechanical properties21,22 will contribute to the development of new generations of tissue scaffolds.
Peptide amphiphiles (PAs) are a class of molecules consisting of a hydrophobic non-peptidic tail covalently conjugated to a peptide sequence. A subset of these molecules developed in our laboratory self-assemble into high-aspect-ratio cylindrical nanofibers.4,14,23,24 These nanofibers can reversibly gel in water under appropriate conditions of pH and osmolarity. A typical structure for these PA molecules is shown below,  These molecules consist of three segments: (1) a hydrophobic sequence which is commonly an alkyl tail that drives aggregation through hydrophobic collapse; (2) a β-sheet forming peptide that promotes nanofiber formation; and (3) a peptide segment that contains ionizable side chains and often an amino acid sequence of interest for biological signaling. The ionizable residues may be a part of the bioactive signal and their role is to promote solubility in water and at the same time serve as a switch for self-assembly when their charges are screened by osmolarity or changes in pH. Storrie et al studied the effect of RGDS epitope density on biological adhesion of fibroblasts using nanofibers co-assembled with a non-bioactive PA25. Previous studies have shown that oppositely charged PA molecules with different bioactive sequences can co-assemble into the same nanofiber26,27 and a diacetylene moiety can polymerize the PA nanofibers.28–30 These results indicated ordering of the alkyl tail within the nanofiber structure, and other spectroscopic investigations demonstrated aqueous solvation within the nanofibers near their hydrophobic cores.28,31 Fourier transform IR (FTIR) and circular dichroism spectroscopic studies revealed a degree of twisting or disorder in the β-sheets,28,32,33 and Paramonov et al showed that hydrogen bonding close to the hydrophobic core was necessary to form peptide amphiphile nanofibers and had the biggest impact on gel properties.33 Understanding and controlling mechanical properties of cell scaffolds is important because there is increasing evidence demonstrating that cells respond to the mechanical properties of their environment.34 Modifying substrate stiffness influences cell differentiation,17–19 cell morphology,35 neurite branching,36 proliferation37 and growth.38 However, many of these studies use a two-dimensional culture environment to examine cell behavior, which may not adequately model the in vivo cellular microenvironment and have limited applications in regenerative medicine. Peptide amphiphiles offer a versatile, three-dimensional platform for regeneration which can display bioactive epitopes,39 bind proteins26 and self-assemble into hierarchical structures with extracellular matrix components.40
These molecules consist of three segments: (1) a hydrophobic sequence which is commonly an alkyl tail that drives aggregation through hydrophobic collapse; (2) a β-sheet forming peptide that promotes nanofiber formation; and (3) a peptide segment that contains ionizable side chains and often an amino acid sequence of interest for biological signaling. The ionizable residues may be a part of the bioactive signal and their role is to promote solubility in water and at the same time serve as a switch for self-assembly when their charges are screened by osmolarity or changes in pH. Storrie et al studied the effect of RGDS epitope density on biological adhesion of fibroblasts using nanofibers co-assembled with a non-bioactive PA25. Previous studies have shown that oppositely charged PA molecules with different bioactive sequences can co-assemble into the same nanofiber26,27 and a diacetylene moiety can polymerize the PA nanofibers.28–30 These results indicated ordering of the alkyl tail within the nanofiber structure, and other spectroscopic investigations demonstrated aqueous solvation within the nanofibers near their hydrophobic cores.28,31 Fourier transform IR (FTIR) and circular dichroism spectroscopic studies revealed a degree of twisting or disorder in the β-sheets,28,32,33 and Paramonov et al showed that hydrogen bonding close to the hydrophobic core was necessary to form peptide amphiphile nanofibers and had the biggest impact on gel properties.33 Understanding and controlling mechanical properties of cell scaffolds is important because there is increasing evidence demonstrating that cells respond to the mechanical properties of their environment.34 Modifying substrate stiffness influences cell differentiation,17–19 cell morphology,35 neurite branching,36 proliferation37 and growth.38 However, many of these studies use a two-dimensional culture environment to examine cell behavior, which may not adequately model the in vivo cellular microenvironment and have limited applications in regenerative medicine. Peptide amphiphiles offer a versatile, three-dimensional platform for regeneration which can display bioactive epitopes,39 bind proteins26 and self-assemble into hierarchical structures with extracellular matrix components.40
We report here on a series of PAs with systematically modified β-sheet regions to observe the relationship between amino acid sequence, supramolecular structures and the mechanical properties of the resulting gel. In this work we have used circular dichroism (CD), and fourier transform infrared spectroscopy (FTIR) to investigate the link between the internal structure of PA nanofibers with different amino acid sequences and stiffness of the gels they form. Understanding the molecular factors affecting PA gel stiffness is important in tailoring these matrices for specific biological targets.
Results and Discussion
The six molecules in this study were designed to systematically investigate the effects of altering the number and position of alanines and valines in the β-sheet region. All PAs have three glutamic acids, which is a strong β-sheet blocker and a strong helix former.41 Valine has the highest propensity of any natural amino acid to form β-sheets and favors forming them over any other secondary structure.41,42 Alanine, on the other hand, is a weak β-sheet former42 and has a preference for forming α-helices.41 Changing the number and position of weak and strong β-sheet forming amino acids allowed us to observe the importance of length and sequence in the β-sheet region, while roughly maintaining the overall dimensions of PA molecules the same. All PAs were synthesized using solid-phase peptide synthesis and purified using reverse phase high performance liquid chromatography (HPLC). After purification, the molecules are dissolved in water above pH 7 and are soluble at concentrations of up to 30 mM. PA 1 forms a self-supporting gel at concentrations as low as 5 mM with the addition of two molar equivalents of CaCl2. For the rheological studies, 10 mM PA solutions were gelled with an equal volume of a 20 mM CaCl2 solution, such that the total charge of the Ca2+ ions balanced out the formal net charge of −4 on each PA molecule.
Mechanical Properties
As indicated in Table 1, PAs 1–3 were synthesized to compare β-sheet length while keeping the valine-to-alanine ratio equal. Rheological measurements show that increasing the length of the β-sheet region while keeping the valine-to-alanine ratio constant increases the stiffness of the resulting gel (Figure 1a). To study the effect of valine-to-alanine ratio on gel stiffness, we altered the number of valines and alanines while keeping the length of the β-sheet constant (Figure 1b). Interestingly, the substitution of valine for alanine has a dramatic effect on the stiffness of the gels obtained. For example, replacing two alanine residues (PA 4) with valines (PA 5) increases the storage modulus of the gel by an order of magnitude.
Table 1.
Peptide sequences of β -sheet regions
| PA | β -sheet region |
|---|---|
| PA 1 | VVVAAA |
| PA 2 | VVAA |
| PA 3 | VVVVAAAA |
| PA 4 | VVAAAA |
| PA 5 | VVVVAA |
| PA 6 | AAAVVV |
Figure 1.

Effect of β -sheet modifications on the rheological properties of peptide amphiphiles with the general structure of C16H31O-(Val)x-(Ala)y -(Glu)3. (a) length of (Val)x -(Ala)y, (b) x/y ratio, (c) Val and Ala sequence order and (d) the effects of adding and subtracting valines and alanines. Error bars represent +/− standard deviation, and *p< 0.05.
The position of amino acids in the β-sheet region was also found to affect mechanical properties (Figure 1c). PAs 1 and 6 have β-sheet regions with six amino acids consisting of three valine and three alanine residues, but have valines located at different places in the β-sheet region. Previous literature has shown that hydrogen bonds closer to the core of the fiber are more important to the mechanical properties of the gel than hydrogen bonds closer to the periphery,33 thus we expected that the closer the valines are to the hydrophobic alkyl tail, the stiffer the resulting gel is. PA 1, which has its valines closest to the center of the nanofiber, indeed formed a stiffer gel than PA 6, which had alanines on the interior of the fiber.
To understand the effects of placing additional valines and alanines into the peptide sequence, we compared the stiffness of PAs 2–5 as shown in Figure 1d. PA 2 has two fewer valines than PA 5 and is almost an order of magnitude less stiff. This indicates that adding two valines, an amino acid that has a strong preference for β-sheet formation, increases the storage modulus of these PA nanofiber gels. Comparing PA 2 to PA 4 shows that adding two alanines to the β-sheet region actually decreases the storage modulus of the gel, despite having a longer β-sheet region and more opportunities for hydrogen bonding. This effect is also observed in molecules with four valines and four alanines. Relative to PA 3, subtracting two alanines (PA 5) makes a stiffer gel while subtracting two valines (PA 4) substantially weakens the gels. These results suggest the presence of alanine may weaken the gel by reducing the strength of β-sheets within the PA fibers.
To further characterize the mechanical properties of the peptide amphiphiles studied, frequency sweeps, strain sweeps and time tests were done for all molecules (Figures S2–S7). Aside from changes in magnitude of the storage and loss moduli, rheological behavior remained relatively constant across the samples. In 5000s time tests, most of the increase in storage and loss moduli are seen in the first 500s and plateau after 1000s. The mechanical properties do not change significantly with respect to oscillation frequency, and show a slight linear decrease at lower frequencies that allow the nanofibers to slide past each other. During amplitude sweeps the mechanical properties stayed in the linear elastic region beyond 1% strain but saw significant decreases by the time a 10% strain was applied.
Electron Microscopy
All six PAs were imaged with transmission electron microscopy (TEM) using vitreous ice cryo-TEM. In this technique, a thin layer of PA solution is plunged into liquid ethane to rapidly freeze the sample in a thin sheet of vitreous ice. This preserves the morphology of structures in aqueous solution and avoids drying effects created by conventional TEM sample preparation. Cryo-TEM showed PA fibers that all nanofibers are typically 9–12 nm in diameter in solution and several microns or more in length, as shown in Figure 2. The rigid peptide amphiphile nanofibers are unbundled in all samples and had extended beyond the width of the image, making it difficult to quantify the length, however, there were no obvious differences among the cryoTEM samples.
Figure 2.

Transmission electron micrographs of (a) PA 1 (b) PA 2 (c) PA 3 (d) PA 4 (e) PA 5 and (f) PA 6 solutions cryo-frozen in vitreous ice.
FTIR
We used Fourier transform infrared spectroscopy (FTIR) to quantify the alignment of hydrogen bonding along the length of fibers and investigate its effect on gel mechanical properties. Previous FTIR studies of peptide amphiphiles have shown that the hydrogen bonding of β-sheets tends to be aligned along the long axis of fibers but with some amount of twisting or disruptions in the β-sheets.32,33,43 These studies were carried out on PA nanofiber monolayers using polarization modulation-infrared reflection absorption spectroscopy (PM-IRRAS), an FTIR method in which electric waves normal to a conductive surface are enhanced while the electromagnetic field parallel to the surface is eliminated. Transmission IR, on the other hand, probes the amide bending and stretching motions parallel and normal to the PA fibers on a surface, as depicted in Figure 3. Atomic force microscopy (AFM) was done to ensure that the IR samples were monolayers (Figure S8). The intensity of the amide I peak, which is predominantly a carbonyl stretching band aligned with the hydrogen bonding direction, will decrease in PM-IRRAS the more aligned the β-sheets are. This peak is normalized against the amide II peak, which is a mixture of a C-N stretch and a N-H bending modes, both of which have components parallel and normal to the substrate surface and should be equally attenuated in all PM-IRRAS samples, regardless of alignment. The amide II peak is typically found around 1550 cm−1 and is relatively insensitive to peptide secondary structure,32 while the amide I peak is found between 1610 cm−1 and 1690 cm−1, and its presence at 1630 cm−1 indicates β-sheet formation. In our all of the PA nanofibers used in this study the main Amide I peak is found around 1630 cm−1, with a minor peak at 1650 cm−1, indicative of turns or bends (Figure S9).44 If the hydrogen bonding is highly aligned down the fiber axis, the amide I peak should be greatly attenuated compared to the amide II as indicated by Figure 4a.
Figure 3.
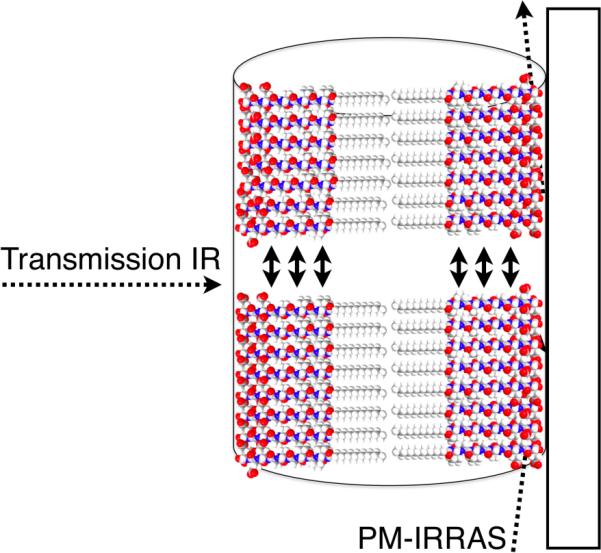
Schematic of FTIR setup in transmission, where the IR beam is nearly perpendicular to the surface and PM-IRRAS, where the IR beam is almost parallel to the substrate surface. Two β-sheet in the fiber are shown, with arrows indicating hydrogen bonds.
Figure 4.

(a) Transmission and PM-IRRAS spectra for the amide region of C16 H31O - V4A4E3 (PA 4). (b) The hydrogen bond alignment quantified from the reduction of the Amide I absorbances (1630 cm−1) from transmission IR to PM-IRRAAS normalized to amibe II absorbance (1550 cm−) for PAs 1–6.
The ratio between the amide I peak in PM-IRRAS to the amide I peak in transmission was used to quantify the hydrogen bond alignment for each of the PAs studied (see Figure 4b). Even in perfectly linear β-sheets there will still be a small signal due to surface roughness and both the N-H bending and C-N stretching components of the amide I, but they are minor components of the 1630 cm−1 peak and the decrease in PM-IRRAS intensity is due to the carbonyl stretching being in the plane of the substrate surface. As shown in Figure 4b, the attenuation of the amide I peak, which corresponds to less twisted β-sheets, is greater in PAs 1, 3 and 5 and smaller in PAs 4, 6 and 7. Comparing the FTIR results with the rheology studies strongy indicates that PAs that form stiffer gels tend to have their internal hydrogen bonds aligned with the fiber axis relative to their weaker gels.
Circular Dichroism
To further understand the effect of PA sequence on mechanical properties, circular dichroism (CD) was used to study the peptide secondary structure. Previous work on PAs has shown that non-spherical assembly of molecules correlates with a β-sheet signal in CD, and the hydrogen bonded β-sheet structure combined with bioactive domains is likely the reason why most PAs with our original design form cylindrical nanofibers and not spherical micelles.33 Supramolecular assemblies of PAs can have heterogeneous secondary structures, and this can explain differences in mechanical properties among sequences.33 Of the common secondary structures, the β-sheet is the only one that is likely to have significant intermolecular hydrogen bonding and thus has the most potential to impact on mechanical properties of the nanofibers and the networks they form. In our CD studies we found that all PAs had spectra that were predominantly those expected for β-sheets, and this was supported by our FTIR data (Figure S9), thus changes in mechanical properties are not due to difference in the amounts of the different secondary structures. However, CD spectra were red-shifted relative to those in typical β-sheets with a maximum at 195 nm and a minimum at 216 nm45 (see Figure 5a). The red-shifted β-sheet signal has been shown to be associated with a twisted structure as opposed to the standard planar β-sheet,33,46 with more twisting increasing the red-shift. In natural proteins47 and model peptides it is found that β-sheets have a varying degree of twist which increases the entropy of the β-sheet53. However, by twisting the β-sheet the hydrogen bonds on the periphery of the β-sheet increase in length and are weakened.47,48 There are also differences in the intensity of the CD signals of the peptide amphiphiles studied, with the PA sequence which forms the stiffest gel having the strongest signal, while that which forms the most compliant gel having the weakest signal. Since peptide amphiphiles are supramolecular aggregates, scattering of circularly polarized light is possible, but another explanation for the differences in intensity is that varying amounts of disorder in the β-sheets are contributing to the differences in signal. Having shorter, disordered β-sheets inside a PA nanofiber would likely influence the CD signal and the mechanical properties of the PA nanofiber. It should be noted that more twisted β-sheets would have larger hydrogen bonding distances, which increases the likelihood of imperfections, so increases in β-sheet twisting would likely increase disorder. Comparing the circular dichroism (Figure 5) with the rheology (Figure 2), the PAs with the highest storage modulus are the least red-shifted, suggesting that increasing the amount of twist and disorder in β-sheets decreases the mechanical properties of the PA gel. PAs 4, 6 and 7 were more red-shifted than PAs 3 and 5, as shown in Figure 5b. These findings support a model that more twisted and disordered β-sheets leads to a weaker PA fiber thus reducing the stiffness of the gels they form. There are other interactions, such as interfiber bonding and entanglements that are likely to be important in determining mechanical properties of PA gels. There were no obvious differences in nanofiber length observed in cryoTEM and all molecules formed one-dimensional nanostructures that exceeded measurable lengths. However, it is possible that changes in β-sheet sequence could change not only the mechanical properties of the individual fiber, but also the length of the fiber and their entanglement. The fiber-fiber interactions should be similar due to the conserved three glutamic acid termini across the library of molecules studied, but any difference in entanglement densities or bundling propensity due to fiber length or rigidity in the gel state would have an impact on gel mechanical properties. In order to determine the contribution that fiber length and entanglement make to mechanical properties, it would be necessary to measure the mechanical properties of the individual nanostructures. We explore below reasons why β-sheet twisting would affect mechanical properties of gels.
Figure 5.
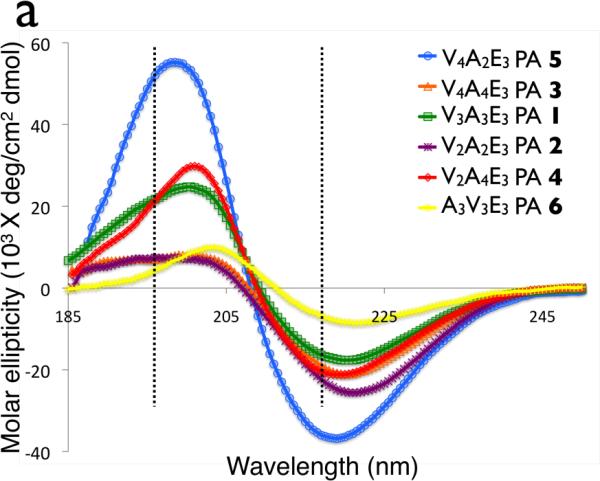
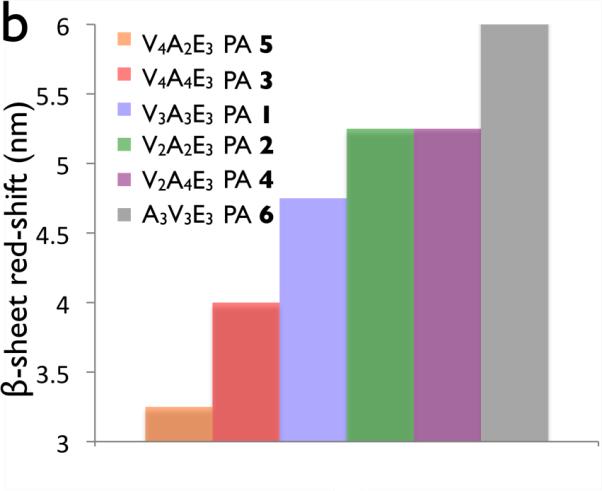
(a)Cricular dichroism of palmitoyl-(val)x-(Ala)y-(Glu)3 peptide amphiphiles. Dashed lines indicate 195nm and 216 nm, the canonical maximum and minimum of a β-sheet. (b) Red-shifting of the β-sheet for PAs 1–6.
Figure 6 shows a schematic representation of the geometry in a twisted and an untwisted β-sheet. As shown in Figure 6a, in a perfectly planar untwisted β-sheet each PA molecule lines up perfectly with the one next to it down the long axis of the. In this model the hydrogen bond length should remain constant throughout the length of two adjacent PA molecules. Twisting of the β-sheet causes a small rotation between each PA molecule in the β-sheet down the long axis of the fiber, which will increase the hydrogen bond length between amino acids closer to the periphery of the fiber and likely increase disorder, as shown in Figure 6b.48 The more twisted a β-sheet is, the weaker the bonds are between two PA molecules on the periphery of the fiber. In addition the amino acids at the periphery of the fiber can adopt secondary structures other than β-sheets,33 which would further weaken bonding among PA molecules. All of the PAs gave rise to CD spectra with the signature of β-sheets, but the number of α-helix forming amino acids may be important in determining the amount of twist in the β-sheets. Even if alanine and glutamic acid are not able to form α-helices, their preferred secondary structure, they may be able to twist the β-sheet and increase the amount of disorder in the β-sheet, which would diminish the mechanical properties of the fibers and and the stiffness of gels.
Figure 6.

Schematic representation of a single β –sheet in (a) linear and (b) twisted/disordered geometry in a peptide amphiphile nanofiber.
In model systems consisting of peptides without hydrophobic alkyl tails, the twisting β-sheet is centered around the middle of the peptide sequence.49 However, due to the unique geometry of the peptide amphiphile in which the alkyl tails are in the center of a cylindrical micelle, the center of a β-sheet's twist is the center of the nanofiber, and not the center of the β-sheet. When a V3A3E3 peptide is compare to the V3A3E3 PA (PA 1), it is seen that the β-sheet in the peptide is more twisted than in the PA (Figure S10). In a twisted β-sheet, the hydrogen bonding distance increases as the angle between two peptides increases and the further out you go from the center of twist. In the same vein, in a PA nanofiber, a longer alkyl segment will increase the distance between the β-sheet and the center of the β-sheet's twist, which either leads to an increased hydrogen bonding distance or, more likely, a decreased angle between peptide groups (ie. The β-sheets are less twisted). Because of this, one would expect the addition of the alkyl tail to decrease the amount of twist over a cylindrical micelle without the alkyl segment and, to an even larger extent, a peptide nanofiber where the center of twist is in the center of the peptide sequence.
The two main types of β-sheets are parallel β-sheets, where each β-strand in the β-sheet has the same peptide C-N direction, and anti-parallel β-sheets where the β-strands peptides have alternating C-N directions. In our system the alkyl tails are contained within the hydrophobic core and are attached to the N-terminus of our peptides. Since every peptide has the same C-N direction, it is likely that our fibers contain only parallel β-sheets. However, we were unable to conclusively rule out a contribution from anti-parallel β-sheets in the CD and FTIR spectra obtained.
Conclusions
We found that systematic variations in the β-sheet sequences of peptide amphiphiles are very effective in tailoring the stiffness of gels formed by their nanofiber networks. Both the number and fraction of valine residues are especially effective at raising mechanical stiffness whereas alanines tend to reduce it. This work also shows that high gel stiffness correlates with alignment of hydrogen bonds along the fiber axis as well as the placement of β-sheet forming residues near the hydrophobic core of nanofibers. On the other hand, supramolecular design of softer gels can be achieved with sequences that promote twisting of β-sheets given their ability to weaken their peripheric hydrogen bonds. The supramolecular elements linked here to gel stiffness of peptide amphiphile nanofiber networks could be useful in the design of systems that control the response of cells to artificial matrices based on mechano-biology.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (NIH)/NIBIB Award No. 5R01EB003806-04, and NIH/NIDCR award 5 R01 DE015920-. The AFM and FTIR experiments were performed in the NIFTI and KECKII facilities, respectively, of the Northwestern University Atomic and Nanoscale Characterization Experimental Center (NUANCE) at Northwestern University. NUANCE is supported by NSF-NSEC, NSF-MRSEC, Keck Foundation, the State of Illinois, and Northwestern University. We are grateful for the use of instruments in the Keck Biophysics facility, the Biological Imaging Facility, the Institute for BioNanotechnology in Medicine, and the Analytical Services Laboratory at Northwestern University. We are also grateful to Prof. Wes Burghardt for the use of rheological equipment and J. Colee for help with statistical anlysis. Finally, we are thankful to L. Chow, J. Hulvat, A. Cheetham, H. Jiang and K. Niece of the authors' laboratory for useful discussions.
Footnotes
Supporting Information Available: Details of synthesis and purification, rheological studies (strain sweeps, frequency sweeps and time tests for all molecules), transmission FTIR, circular dichroism and statistical information. This material is available free of charge via the Internet at http://pubs.ac.org.
References
- (1).Lutolf MP, Hubbell JA. Nat. Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- (2).Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- (3).Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7791–7796. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- (5).Ellis-Behnke RG, Liang YX, You SW, Tay DKC, Zhang SG, So KF, Schneider GE. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5054–5059. doi: 10.1073/pnas.0600559103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, Stupp SI, Kessler JA. Journal of Neuroscience. 2008;28:3814–3823. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rajangam K, Behanna HA, Hui MJ, Han XQ, Hulvat JF, Lomasney JW, Stupp SI. Nano Letters. 2006;6:2086–2090. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- (8).Aggeli A, Bell M, Boden N, Keen JN, McLeish TCB, Nyrkova I, Radford SE, Semenov A. Journal of Materials Chemistry. 1997;7:1135–1145. [Google Scholar]
- (9).Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TCB, Pitkeathly M, Radford SE. Nature. 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- (10).Deechongkit S, Powers ET, You SL, Kelly JW. Journal of the American Chemical Society. 2005;127:8562–8570. doi: 10.1021/ja050558c. [DOI] [PubMed] [Google Scholar]
- (11).Zhang SG, Holmes T, Lockshin C, Rich A. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:3334–3338. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang SG. Nat. Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- (13).Santoso S, Hwang W, Hartman H, Zhang SG. Nano Letters. 2002;2:687–691. [Google Scholar]
- (14).Fields GB, Lauer JL, Dori Y, Forns P, Yu YC, Tirrell M. Biopolymers. 1998;47:143–151. doi: 10.1002/(SICI)1097-0282(1998)47:2<143::AID-BIP3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (15).Murakami Y, Nakano A, Yoshimatsu A, Uchitomi K, Matsuda Y. Journal of the American Chemical Society. 1984;106:3613–3623. [Google Scholar]
- (16).Neumann R, Ringsdorf H. Journal of the American Chemical Society. 1986;108:487–490. doi: 10.1021/ja00263a021. [DOI] [PubMed] [Google Scholar]
- (17).Kraehenbuehl TP, Zammaretti P, Van der Vlies AJ, Schoenmakers RG, Lutolf MP, Jaconi ME, Hubbell JA. Biomaterials. 2008;29:2757–2766. doi: 10.1016/j.biomaterials.2008.03.016. [DOI] [PubMed] [Google Scholar]
- (18).Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- (19).Engler AJ, Griffin MA, Sen S, Bonnetnann CG, Sweeney HL, Discher DEJ. Cell Biol. 2004;166:877–887. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ryadnov MG, Woolfson DN. Nat. Mater. 2003;2:329–332. doi: 10.1038/nmat885. [DOI] [PubMed] [Google Scholar]
- (21).Caplan MR, Schwartzfarb EM, Zhang SG, Kamm RD, Lauffenburger DAJ. Biomater. Sci.-Polym. Ed. 2002;13:225–236. doi: 10.1163/156856202320176493. [DOI] [PubMed] [Google Scholar]
- (22).Caplan MR, Schwartzfarb EM, Zhang SG, Kamm RD, Lauffenburger DA. Biomaterials. 2002;23:219–227. doi: 10.1016/s0142-9612(01)00099-0. [DOI] [PubMed] [Google Scholar]
- (23).Hartgerink JD, Beniash E, Stupp SI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yu Y-C, Pakalns T, Dori Y, McCarthy JB, Tirrell M, Fields GB. Methods in Enzymology. 1997;289:571–582. doi: 10.1016/s0076-6879(97)89065-9. [DOI] [PubMed] [Google Scholar]
- (25).Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI. Biomaterials. 2007;28:4608–4618. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- (26).Behanna HA, Donners J, Gordon AC, Stupp SI. Journal of the American Chemical Society. 2005;127:1193–1200. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- (27).Niece KL, Hartgerink JD, Donners JJJM, Stupp SI. Journal of the American Chemical Society. 2003;125:7146–7147. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- (28).Hsu L, Cvetanovich GL, Stupp SI. Journal of the American Chemical Society. 2008;130:3892–3899. doi: 10.1021/ja076553s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Biesalski MA, Knaebel A, Tu R, Tirrell M. Biomaterials. 2006;27:1259–1269. doi: 10.1016/j.biomaterials.2005.08.002. [DOI] [PubMed] [Google Scholar]
- (30).Biesalski M, Tu R, Tirrell MV. Langmuir. 2005;21:5663–5666. doi: 10.1021/la0504558. [DOI] [PubMed] [Google Scholar]
- (31).Tovar JD, Claussen RC, Stupp SI. Journal of the American Chemical Society. 2005;127:7337–7345. doi: 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]
- (32).Jiang HZ, Guler MO, Stupp SI. Soft Matter. 2007;3:454–462. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- (33).Paramonov SE, Jun HW, Hartgerink JD. Journal of the American Chemical Society. 2006;128:7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- (34).Discher DE, Janmey P, Wang YL. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- (35).Guo WH, Frey MT, Burnham NA, Wang YL. Biophysical Journal. 2006;90:2213–2220. doi: 10.1529/biophysj.105.070144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Flanagan LA, Ju YE, Marg B, Osterfield M, Janmey PA. Neuroreport. 2002;13:2411–2415. doi: 10.1097/01.wnr.0000048003.96487.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Jung JP, Jones JL, Cronier SA, Collier JH. Biomaterials. 2008;29:2143–2151. doi: 10.1016/j.biomaterials.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Georges PC, Miller WJ, Meaney DF, Sawyer ES, Janmey PA. Biophysical Journal. 2006;90:3012–3018. doi: 10.1529/biophysj.105.073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- (40).Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812–1816. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- (41).Levitt M. Biochemistry. 1978;17:4277–4284. doi: 10.1021/bi00613a026. [DOI] [PubMed] [Google Scholar]
- (42).Kim CWA, Berg JM. Nature. 1993;362:267–270. doi: 10.1038/362267a0. [DOI] [PubMed] [Google Scholar]
- (43).Hung AM, Stupp SI. Langmuir. 2009;25:7084–7089. doi: 10.1021/la900149v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kubelka J, Keiderling TA. Journal of the American Chemical Society. 2001;123:12048–12058. doi: 10.1021/ja0116627. [DOI] [PubMed] [Google Scholar]
- (45).Sreerama R. a. W., R.W. In: Circular Dichroism: Principles and Applications. Berova N, Nakanishi K, Woody RW, editors. Wiley-VCH; New York: 2000. pp. 601–620. [Google Scholar]
- (46).Manning MC, Illangasekare M, Woody RW. Biophys. Chem. 1988;31:77–86. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]
- (47).Salemme FR. Prog. Biophys. Mol. Biol. 1983;42:95–133. doi: 10.1016/0079-6107(83)90005-6. [DOI] [PubMed] [Google Scholar]
- (48).Salemme FR, Weatherford DWJ. Mol. Biol. 1981;146:101–117. doi: 10.1016/0022-2836(81)90368-5. [DOI] [PubMed] [Google Scholar]
- (49).Aggeli A, Nyrkova IA, Bell M, Harding R, Carrick L, McLeish TCB, Semenov AN, Boden N. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11857–11862. doi: 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.