

Abstract
Functional amino acids (FAAs) and α-aminoamides (AAAs) are two classes of antiepileptic drugs (AEDs) that exhibit pronounced anticonvulsant activities. We combined key structural pharmacophores present in FAAs and AAAs to generate a new series of compounds and document that select compounds exhibit activity superior to either the prototypical FAA (lacosamide) or the prototypical AAA (safinamide) in the maximal electroshock (MES) seizure model in rats. A representative compound, (R)-N-4′-((3″-fluoro)benzyloxy)benzyl 2-acetamido-3-methoxypropionamide ((R)-10), was tested in the MES (mice, ip), MES (rat, po), psychomotor 6 Hz (32 mA) (mice, ip), and hippocampal kindled (rat, ip) seizure tests providing excellent protection with ED50 values of 13, 14, ~10 mg/kg, and 12 mg/kg, respectively. In the rat sciatic nerve ligation model (ip), (R)-10 (12 mg/kg) provided an 11.2-fold attenuation of mechanical allodynia. In the mouse biphasic formalin pain model (ip), (R)-10 (15 mg/kg) reduced pain responses in the acute and the chronic inflammatory phases.
Epilepsy is a chronic disorder, characterized by recurrent, unprovoked seizures.1 A seizure is defined as a discrete clinical event arising from transient, hypersynchronous, abnormal neuronal behavior. Epilepsy, then, is not a disease but rather a syndrome arising from a group of nonspecific, dysfunctional events in the brain. The treatment mainstay for patients with epileptic disorders has been the long-term and consistent administration of anticonvulsant drugs.2,3 There are more than 40 pharmacologic therapies used for the treatment of epilepsy.4 Unfortunately, even when used optimally, these therapeutic interventions are ineffective for some 30% of patients. 5 Moreover, their use is associated, in more than 40% of patients, with untoward effects (e.g., drowsiness, dizziness, nausea, liver damage).6 The shortcomings of current regimens highlight the need for new, more effective agents.
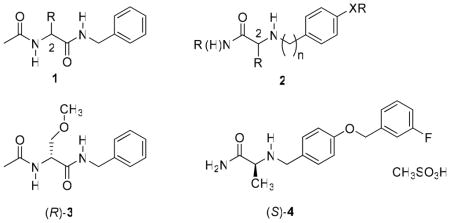
We have previously reported that functionalized amino acids (FAAs,a 1)7–18 exhibit excellent anticonvulsant activities in various animal seizure models. Whole-animal pharmacological studies for 1 showed a unique profile, which indicated a novel mechanism of action. 7–19 Similarly, studies have demonstrated that α-aminoamides (AAAs, 2) provide superb seizure protection. 20,21 Representative examples for each class of compounds have advanced through clinical trials. Lacosamide ((R)-3),16 the 1 prototype, is a first-in-class antiepileptic drug (AED) that was recently introduced in the United States and Europe for adjuvant treatment of partial-onset seizures in adults.22 Safinamide ((S)-4) is a leading representative for 2. (S)-4 exhibited excellent protection in seizure models, and positive responses have been reported in recent phase III human clinical trials for the treatment of Parkinson disorders.21,23,24
Recent electrophysiology studies using cultured rat cortical neurons demonstrated that (R)-3 selectively enhanced sodium channel slow inactivation in a time- and voltage-dependent manner, without affecting fast inactivation.25 Similarly, examination of (R)-3 with recombinant NaV 1.3 and 1.7 voltage-gated sodium channels expressed in HEK293 cells and of NaV1.8-type TTX-R currents from DRG neurons showed that (R)-3 selectively modulated the slow inactivation state in each of these sodium channel subtypes.26 (R)-3 is the only reported antiepileptic agent that selectively enhances slow inactivation without apparent interaction with fast inactivation gating.
Patch-clamp, whole-cell electrophysiological studies using hippocampal neurons demonstrated that (S)-4 inhibited tetrodotoxin-sensitive (TTX-S) fast Na+ currents in a concentration-dependent manner.21a The inhibition was voltage dependent, showing an IC50 of ~100 uM when currents were stimulated from a resting condition, while stronger inhibition (IC50 = 33 uM) was observed when sodium currents were stimulated from a −60 mV depolarized membrane potential. Together, these findings indicated that (S)-4 exhibited a higher affinity for the sodium channel’s inactivated state.24 Thus, the mechanisms associated with sodium channel inhibition for (R)-3 and (S)-4 are different.
At first glance, (R)-3 and (S)-4 appear structurally similar. Both have low molecular weights ((R)-3: MW =250; (S)-4: MW = 302 [free base]), each has a vicinal diamine backbone that contains a carbonyl (C=O) moiety, and each has one chiral center. In addition, both compounds contain an N-benzyl (PhCH2)–type substituent. Pharmacologically, both (R)-3 and (S)-4 exhibited excellent seizure protection16,19,27,28 in the maximal electroshock seizure (MES) animal model,29 and electrophysiology studies demonstrated that both modulate sodium currents.21a,25,26 Further inspection of (R)-3 and (S)-4 revealed stark differences in structure and function. First, (R)-3 is neutral and (S)-4 is basic. Second, the (R)-3 sequence of atoms attaches the N-benzyl moiety to an amide while in (S)-4 the substituted N-benzyl moiety attaches to an amine. Third, there is a different structure-activity relationship (SAR) in the MES test between the two classes of antiepileptic agents 1 and 2. For the 1 compounds, we observed a steady improvement in activity as the C(2) position was changed from H and CH2OH to CH3 to C6H5 to CH2OCH3.8,16 Correspondingly, for 2, activity in the MES test increased as the C(2) position was changed from C6H5 to CH2OH to H to CH3.20 Fourth, (R)-3 showed chiral specificity for function in the mouse (ip) (i.e., MES activity of (R)-3 vs (S)-3 is >22:1),16 but (S)-4 did not (i.e., MES activity of (S)-4 ≈ (R)-4).20,30 Fifth, (S)-4 administration provided protection against several chemoconvulsants27 while (R)-3 did not.19 Finally, (R)-3 enhanced the slow inactivation state of the voltage-gated sodium channel thereby selectively blocking the activity of chronically depolarized neurons25,26 while (S)-4 inhibited TTX-S fast sodium currents.21a
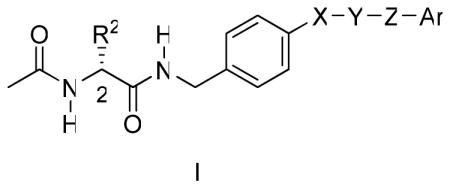
The structural differences between (R)-3 and (S)-4 likely account for their different modes of action and underscore that each compound has distinctive pharmacophores that contribute to drug function. In this study, we asked whether key structural units in 1 and 2 could be incorporated within a single compound to provide more effective anticonvulsant agents.31 Herein, we report the design, synthesis, and pharmacological evaluation of a series of compounds that conform to the general structure of I, and we document that they display excellent anticonvulsant activities.
RESULTS
Compound Design
In generating I, we used the generalized structure II for 1 and III for 2 (Figure 1). Both II and III include an “X-Y-Z” substituent at the N-benzyl 4′ site. “X-Y-Z” is a molecular unit 1–3 atoms long, or in the case I and III, it could also be a single bond. Our recent SAR study for the N-benzyl 4′ site in (R)-3 showed that the introduction of select substituents at this position provided compounds with excellent anticonvulsant properties.18 Similarly, the reported SAR for 2 demonstrated that significant anticonvulsant activity was observed with various linkers (“X-Y-Z”) at the N-benzyl position that bridged the two aromatic moieties in this agent.20 The area of pharmacophore overlap in I is portrayed in the box, and this overlap permitted the incorporation of key pharmacophores found in II (1) and III (2) within a unified structure (Figure 1). For convenience, we divided I into three sectors: A, B, and C. Sector A is the structural motif seen in II (1), sector B is the linker unit “X-Y-Z” reported in both II (1) and III (2), and sector C is the terminal aromatic ring (Ar) found in III (2).
Figure 1.

Overlay of Pharmacophores in I
Choice of Compounds
Tables 1–3 lists the I compounds evaluated in this study. We prepared 20 compounds (5–22) in which we varied the structural units in sectors A–C and the chirality of the C(2) center in sector A. Since the number of structural permutations was large, we maintained the structural pattern constancy for two sectors as we changed the other.
Table 1.
Novel Neurological Agents: Structure-Activity Relationship of Sector Aa
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd No |
R |
Stereo |
Mice (ip)b |
Rat (po)f |
|||||
| MES,c ED50 |
6Hz, ED50 |
Tox,d TD50 |
PIe |
MES,c ED50 |
Tox,g TD50 |
PIe |
|||
| 5 | H | - | >100, <300 [4.0] >30, <100 [2] |
>300 [0.5 and 4.0] | 31 [1.0] (18–53) |
>500 [1.0] | >16 | ||
| (R)-6 | (R)-Me | R | >30, <100 [0.5 and 4] | >300 [0.5] | 31 [4.0] (21–44) |
>500 [4.0] | >16 | ||
| (S)-6 | (S)-Me | S | >100,< 300 [0.5] > 300 [4.0] |
>300 [0.5] | > 30 [0.25 to 4.0] | > 30 [0.25 to 4.0] | |||
| (R)-7 | (R)-i-Pr | R | >300 [0.5 and 4] | >300 [0.5 and 4] | >30 | >30 | |||
| (R)-8 | (R)-t-Bu | R | >300 [0.5 and 4] | >300 [0.5 and 4] | |||||
| (R,S)-9 | R, S | 28 [0.5] (20–36) |
213 [2.0] (156–285) |
7.6 | >30 [4] | >30 [4] | |||
| (R)-10 | (R)-CH2OMe | R | 13 [0.25] (11–16) |
~10 [0.25] | 26 [0.5] (21–34) |
14 [0.5] (6.1–27) |
>500 [0.5] | >36 | |
| (S)-10 | (S)-CH2OMe | S | >300 | >300 | |||||
| (S)-4h | 8.0 (7.0–9.1) | 630 (560–700) | |||||||
| (R)-4h | 7.2 (5.9–8.9) | 580 (410–830) | |||||||
| (R)-3i | 4.5 [0.5] (3.7–5.5) |
27 [0.25] (26–28) |
3.9 [2.0] (2.9–6.2) |
>500 | |||||
| phenytoinj | 9.5 [2.0] (8.1–10) |
66 [2.0] (53–72) |
6.9 | 30 [4.0] (22–39) |
> 100 | ||||
| phenobarbitalj | 22 [1.0] (15–23) |
69 [0.5] (63–73) |
3.2 | 9.1 [5.0] (7.6–12) |
61 [0.5] (44–96) |
6.7 | |||
| valproatej | 270 [0.25] (250–340) |
430 [0.25] (370–450) |
1.6 | 490 [0.5] (350–730) |
280 [0.5] (190–350) |
0.6 | |||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
The compounds were administered orally. ED50 and TD50 values are in milligrams per kilogram.
Tox = behavioral toxicity.
Ref 30.
Ref 16.
Ref 32.
Table 3.
Novel Neurological Agents: Structure-Activity Relationship of Sector Ca
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd No |
R′ |
Mice (ip)b |
Rat (po)f |
|||||
| MES,c ED50 |
6Hz, ED50 |
Tox,d TD50 |
PIe |
MES,c ED50 |
Tox,g TD50 |
PIe |
||
| (R)-20 | - | <3 [0.5] | >10, <30 [0.5] | <10 [0.25 to 2.0] | >10 [0.25 to 2.0] | |||
| (R)-21 | 2-F | >3, <10 [0.5] | >30, <100 [0.5] | 11 [0.5] (7.9–13) |
>500 | >45 | ||
| (R)-10 | 3-F | 13 [0.25] (11–16) |
~10 [0.25] | 26 [0.5] (21–34) |
2 | 14 [0.5] (6.1–27) |
>500 [0.5] | >36 |
| (R)-22 | 4-F | >10, < 30 [0.5] | >30, < 100 [0.5] | 5.8 [0.5] (4.3–7.3) |
>500 | >86 | ||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
The compounds were administered orally. ED50 and TD50 values are in milligrams per kilogram.
Tox = behavioral toxicity.
Ref 30.
Ref 16.
Ref 32.
For compounds listed in Table 1, sector A was varied while we restricted sectors B and C to an OCH2 and 3-(fluoro)phenyl moiety, respectively, to match (S)-4. Specifically, in this set of compounds we evaluated key structural features important to the 1 SAR. We showed that 1 compounds containing a small R substituent provided excellent seizure protection in the MES test and that anticonvulsant activity typically improved when a substituted heteroatom was introduced one atom removed from the C(2) center. Moreover, anticonvulsant activity for the 1 compounds principally resided in the D-configuration.9,10,11,16,18 Thus, we progressively increased the R substituent in 5–8 from hydrogen to methyl to isopropyl to tert-butyl and then included in our selection list 9 and 10, compounds that contained a substituted heteroatom positioned one atom removed from C(2). For compounds 6 and 10, we prepared the R- (D-configuration) and the S- (L-configuration) stereoisomers to determine whether the anticonvulsant activities for I mirrored those found for 1, in which a clear stereochemical preference was observed,9–11,16,18 or that for 2, in which both stereoisomers displayed comparable anticonvulsant activities.20,30 For all compounds listed in Table 1, except 5 and 9, we prepared I as a single stereoisomer. Compound 5 had no chiral center, and the synthetic route for 9 provided the racemic mixture.
For compounds 10–19 listed in Table 2, we varied sector B’s “X-Y-Z” linker region and employed the (R)-3 structural motif for sector A and the (S)-4 3-(fluoro)phenyl unit for sector C. Following the SAR reported for (S)-4,20 we incorporated one, two, and three atom linkers and varied the heteroatom content and degree of unsaturation of the linker. The choice of the “X-Y-Z” unit was also consistent with the SAR reported for (R)-3,18 in which we showed that incorporating select substituents (e.g., alkyl, substituted alkyl, vinyl, acetylenic, aryl) at the 4′ position of the N-benzyl ring gave compounds with superb seizure protection in the MES test29 in rodents with some analogs having activities comparable with (R)-3 and established AEDs.32
Table 2.
Novel Neurological Agents: Structure-Activity Relationship of Sector Ba
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd No |
-X-Y-Z- |
Mice (ip)b |
Rat (po)f |
|||||
| MES,c ED50 |
6Hz, ED50 |
Tox,d TD50 |
PIe |
MES,c ED50 |
Tox,g TD50 |
PIe |
||
| (R)-11 | - | >10, <30 [0.5] | <30 [0.5] | >100, <300 [0.5] | 2.4 [1.0] (1–3.9) |
>500 | >250 | |
| (R)-12 | -O- | <30 [0.5] | >30, <100 [0.5] | <10 [0.25–2.0] | >10 [0.25–4.0] | |||
| (R)-13 | -(CH2)2- | >10, <30 [0.5] | >30, <100 [0.5] | <30 [1] | >30 [1] | |||
| (R)-14 | -CH=CH- | >30, <100 [0.5] | < 30 [0.25] | >100, <300 [0.5] | ~30 [1.0, 4.0] | >30 [0.25, 4.0] | ||
| (R)-15 | -≡- | >30, <100 [1.0, 4.0] | >100, <300 [4.0] | 1.4 [4.0] (0.7–2.2) |
>63, <125 [4] | |||
| (R)-16 | -CH2O- | 5.9 [0.25] (4.3–7.3) |
10 [0.25] (9.1–13) |
1.8 | 19 [2] (13–25) |
>400 [0.5] | >21 | |
| (R)-17 | -N(H)CH2- | >10, <30 [0.5] | >30, <100 [0.5] | |||||
| (R)-10 | -OCH2- | 13 [0.25] (11–16) |
~10 [0.25] | 26 [0.5] (21–34) |
2 | 14 [0.5] (6.1–27) |
>500 [0.5] | >36 |
| (R)-18 | -CH2OCH2- | >30, <100 [0.5] | >30, <100 [0.5] | |||||
| (R)-19 | -OCH2CH2- | >30, <100 [0.5, 4.0] | >30, <100 [0.5] | |||||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
The compounds were administered orally. ED50 and TD50 values are in milligrams per kilogram.
Tox = behavioral toxicity.
Ref 30.
Ref 16.
Ref 32.
Finally, we evaluated the effect of the terminal aromatic ring by preparing compounds (R)-10, and (R)-20–(R)-22 (Table 3). Here, we varied sector C to include the unsubstituted aromatic compound (R)-20 and the three monofluorine–substituted regioisomers (R)-10, (R)-21, and (R)-22 and set sector A to match (R)-3 and sector B to match (S)-4. We also prepared compound 23, which contains the sector C structural motif found in many of our compounds.
Chemistry
Two similar routes were employed to prepare most I compounds in this study and depended only on the C(2)-R substituent. For most I derivatives that incorporated the (R)-3 framework in sector A, we used a recently reported procedure to prepare 4′-substituted N-benzylamide (R)-3 analogs18 beginning with either tert-Boc–protected (R)- or (S)-serine 24. The acid was coupled with the desired benzylamine 25 using the mixed anhydride method (isobutylchloroformate (IBCF), N-methyl morpholine (NMM)),33 unless otherwise indicated, to give the N-benzyl amide 26 without racemization of the C(2) chiral center (Scheme 1). Subsequent methylation of the serine hydroxy group (CH3I, Ag2O) gave ether 27. Deprotection of the tert-butoxycarbonyl group with acid followed by acetylation of the amine with acetyl chloride and triethylamine gave the desired product, I, in 43–80% yield. Using this method, we prepared (R)-10, (S)-10, and (R)-11–(R)-22.
Scheme 1.

General Procedure for the Preparation of (R)- and (S)-N-(4′-Substituted)benzyl 2-Acetamido-3-methoxypropionamide Derivatives I
A similar procedure was used to prepare compounds 5, (R)-6–(R)-8 and (S)-6 (Scheme 2). We began with a commercially available amino acid. Converting the amino acid to either the N-acetyl (28, (R)-29, (S)-29) or the N-tert-butoxycarbonyl ((R)-30, (R)-31) derivative permitted mixed anhydride coupling with benzylamine 32 to give the amides (5, (R)-6, (S)-6, (R)-33, (R)-34). For (R)-33 and (R)-34 that were protected as the tert-Boc derivatives, acid deprotection gave the amine, which was directly reacted with acetyl chloride and base to give (R)-7 and (R)-8, respectively.
Scheme 2.

General Procedure for the Preparation of (R)- and (S)-N-(4′-(3-fluorobenzyloxy)benzyl)benzyl 2-Acetamido-2-(substituted)-acetamide Derivatives 5–8
We developed a different route for the C(2) pyridyl derivative (R,S)-9 (Scheme 3). Beginning with commercially available ethyl 2-(pyridin-2yl)acetate (35), basic hydrolysis provided acid 36, which was coupled with 4-(((3′-fluoro)benzyloxy)phenyl)methanamine (32) using O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) to give 37. Treatment of 37 with NaNO2 in acetic acid yielded the oximes 38 as an ~1:1 mixture of syn- and anti-isomers. While oximes 38 could be separated by silica gel chromatography, the mixture was reduced to amine 39 with Zn dust in the presence of ammonium formate and then converted to racemic 9 with acetyl chloride and triethylamine.
Scheme 3.

Preparation of N-(4-(3-Fluorobenzyloxy)benzyl) 2-Acetamido-2-(pyridin-2-yl)acetamide ((R,S)-9)
The extended N-benzyl amide moiety within the I compounds was a key unit in our compounds. Thus, we used a series of methods to construct this moiety (Schemes 4–6). For many of the compounds, we reacted either 4-cyanophenol (40) with a substituted benzyl bromide (41–44) or a (cyano)aryl bromide (52, 53) with the substituted phenol (54) or aryl-substituted alcohol (55) under base conditions to give the ether (45–48, 56–58) (Scheme 4). Subsequent reduction (LiAlH4) of the nitrile in 45–48 and 56–58 gave the requisite benzylamine (32, 49–51, 59–61) for the mixed anhydride coupling reaction. We prepared nitrile 64 from 4-(hydroxy)benzonitrile (40) and 3-(fluoro)phenethanol (62) using Mitsunobu coupling conditions34 and then reduced nitrile 63 to benzylamine 64 with LiAlH4. Correspondingly, we reacted 4-(bromo)benzonitrile (52) with 3-(fluoro)benzylamine (65) under Buchwald-Hartwig coupling conditions38 to generate 66, which was reduced to amine 67 (Scheme 5). Finally, (R)-4-(iodo)benzyl 2-acetamido-3-methoxypropionamide18 ((R)-69) served as the starting material for I compounds (R)-11 and (R)-13–(R)-15 (Scheme 6). Coupling (R)-69 with 3-(fluoro)phenylboronic acid (70) under Suzuki coupling conditions36 gave (R)-11. When trans-2-((3′-fluoro)phenyl)vinylboronic acid (71) was substituted for 3-(fluoro)phenylboronic acid (70), we obtained (R)-14 with an embedded trans-double bond. Reduction of (R)-14 (10% Pd/C, H2) gave (R)-13. Sonogashira coupling37 of (R)-69 with 3-(fluoro)phenylacetylene (72) afforded (R)-15. For (R)-17 synthesis, we coupled amine 67 with (R)-2-acetamido-3-methoxypropanoic acid 17 ((R)-68) using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methoxymorpholinium chloride (DMTMM)38 (Scheme 5).
Scheme 4.

Benzylamine 32, 49–51, 59–61, and 64 Synthesis
Scheme 6.

Preparation of Compounds (R)-11, (R)-13, (R)-14 and (R)-15
Scheme 5.

Benzylamine 67 and Compound (R)-17 Synthesis
The enantiopurity of (R)-6–(R)-8, (R)-10–(R)-22, (S)-6, and (S)-10 was assessed by the detection of a single acetyl methyl signal in the 1H NMR spectrum for each compound when a saturated solution of (R)-(−)-mandelic acid was added.39 In the cases of (R)-10-(R)-22, we also observed a single O-methyl peak upon addition of (R)-(−)-mandelic acid.
We report, in the Experimental Section, the details (synthetic procedure, characterization) of the final step for all the compounds evaluated in the seizure models. In Supporting Information, we provide a synthetic scheme for each compound tested and the experimental procedures used and physical and full spectroscopic properties for all the synthetic compounds prepared in this study.
Pharmacological Activity
Compounds 5, (R)-6–(R)-8, (R,S)-9, (R)-10–(R)-22, (S)-6, and (S)-10 were tested for anticonvulsant activity at the Anticonvulsant Screening Program (ASP), which is part of the National Institute of Neurological Disorders and Stroke (NINDS) at the U.S. National Institutes of Health. Screening was performed using the procedures described by Stables and Kupferberg.40 The pharmacological data from the MES test 29 are summarized in Tables 1–3 and similar results obtained for (R)-3,16 (R)-4,30 (S)-4,30 and the clinical antiepileptic drugs (AEDs) phenytoin,32 valproate,32 and phenobarbital32 are given in Table 1. All compounds were administered intraperitoneally (ip) to mice and orally (po) to rats. Tables 1–3 lists the values that were determined to be protective in blocking hind limb extension induced in the electrically induced MES seizure model from the rodent identification studies. For compounds that showed significant activity, we report the 50% effective dose (ED50) values obtained in quantitative screening evaluations. Also provided are the median doses for 50% neurological impairment (TD50) in mice, using the rotorod test,41 and the behavioral toxicity effects observed in rats.42 TD50 values were determined for those compounds that exhibited significant activity in the MES test. The protective index (PI = TD50/ED50) for each of these analogs is also listed. Select compounds were also evaluated in the psychomotor 6 Hz (32 mA) seizure models (mice, ip).43 When the I derivatives were evaluated in the subcutaneous Metrazol (scMet) seizure model44 none provided protection at doses up to 300 mg/kg at two time points (0.5 and 4 h) (data not shown). The absence of seizure protection in this assay is a hallmark of FAA activity 7–17 and contrasted with the data reported for (S)-4.27
The SAR data for I provided distinctive trends. In Table 1, we varied the C(2) R group in sector A while maintaining the (S)-4 structural components found in sectors B and C. Using the mice (ip) data, we observed that as the size of the C(2) alkyl group increased from methyl ((R)-6, MES ED50 = >30, <100 mg/kg) to isopropyl ((R)-7, MES ED50 = >300 mg/kg) to tert-butyl ((R)-8, MES ED50 = >300 mg/kg), the anticonvulsant activity decreased. We also found that MES seizure protection significantly increased upon the inclusion of a C(2)-substituted heteroatom group one atom removed from the C(2) center ((R,S)-9, MES ED50 = 28 mg/kg; (R)-10, MES ED50 = 13 mg/kg versus (R)-6, MES ED50 = >30, <100 mg/kg; (R)-7, MES ED50 = >300 mg/kg; (R)-8, MES ED50 = >300 mg/kg). Similar SAR patterns were observed for 1 compounds.7,11–14 Furthermore, for both 6 and 10, we tested the (R)- and (S)-enantiomers and found that the principal activity resided in the (R)-stereoisomer (D-configuration) (MES ED50 (mice, ip): (R)-6, >30, <100 mg/kg vs (S)-6, >100, <300 mg/kg, and (R)-10, 13 mg/kg vs (S)-10, >300 mg/kg). Both the C(2) SAR pattern and the stereochemical preference for the (R)-enantiomer for seizure protection strongly indicated that anticonvulsant activity in the MES test for I compounds resembled the activity in 1 compounds. (R)-10 was the most active among the sector A compounds, exhibiting an MES ED50 = 13 mg/kg. This placed (R)-10 midway between phenytoin and phenobarbital in activity32 and approximately three times less active than (R)-3.16 The protective index (PI = TD50/ED50) in mice (ip) for (R)-10 was 2, which was lower than (R)-3 (PI = 5.2).16 When sector A I compounds were evaluated in the rat (po), the effect of C(2) R substitution on MES seizure protection was less noticeable. In this model, we found that (R)-10 (MES ED50 = 14 mg/kg) was more active than either 5 (MES ED50 = 31 mg/kg) or (R)-6 (MES ED50 = 31 mg/kg) but that the glycine derivative 5 and the alanine analog (R)-6 showed equal seizure protection. Interestingly, none of the three compounds displayed behavioral toxicity, even at doses up to 500 mg/kg. The activity observed for (R)-10 in the rat (po) exceeded that of phenytoin and phenobarbital.32
Further SAR information was gathered by varying the sector B linker in I (Table 2). For these compounds, we maintained the (R)-3 framework for sector A and the (S)-4 (3-fluoro)phenyl substituent for sector C. We observed differences between the mice (ip) and rat (po) data sets. In the mice, anticonvulsant activity in the MES test decreased as the linker changed from CH2O ((R)-16) [MES ED50 = 5.9 mg/kg] to OCH2 ((R)-10) [MES ED50 = 13 mg/kg] to no linker ((R)-11), O ((R)-12), CH2CH2 ((R)-13), N(H)CH2 ((R)-17) [MES ED50 = >10, <30 mg/kg] to C(H)=C(H) ((R)-14), C≡C ((R)-15), CH2OCH2 ((R)-18), OCH2CH2 ((R)-19) [MES ED50 = >30, <100 mg/kg]. Correspondingly, in the rat (po), we observed that the anticonvulsant activity decreased by going from C≡C ((R)-15) [MES ED50 = 1.4 mg/kg], no linker ((R)-11) [MES ED50 = 2.4 mg/kg], to O ((R)-12) [MES ED50 = <10 mg/kg], OCH2 ((R)-10) [MES ED50 = 14 mg/kg], CH2O ((R)-16) [MES ED50 = 19 mg/kg], CH2CH2 ((R)-13) [MES ED50 = <30 mg/kg] to C(H)=C(H) ((R)-14) [MES ED50 = ~30 mg/kg]. We were interested to find that (R)-15 and (R)-11 exhibited superb seizure protection in the MES test (rat, po) with ED50 values of 1.4 mg/kg and 2.4 mg/kg, respectively. Moreover, (R)-11 displayed no evidence of behavioral toxicity at 500 mg/kg. Earlier, we reported the anticonvulsant activity of (R)-N-(biphenyl-4-yl)methyl 2-acetamido-3-methoxypropionamide ((R)-73).18 (R)-73, like (R)-11, exhibited outstanding seizure protection in the MES test (rat, po: MES ED50 = 2.0 mg/kg; TD50 = 49 mg/kg) but, unlike (R)-11, was found to have noticeable neurotoxicity. The precise role of the 3′-fluoro group in (R)-11 in modulating behavioral neurotoxicity is unclear but is under investigation. Finally, for (R)-15 we not only observed excellent anticonvulsant protection but also the duration of seizure protection extended for the entire 4 h testing period. The activities of (R)-11 and (R)-15 exceeded (R)-3 (ED50 = 3.8 mg/kg)16 and other AEDs.32
Sector C’s terminal aryl group was the last structural unit evaluated (Table 3). For this series of compounds, we maintained the (R)-3 framework for sector A, and the (S)-4 OCH2 unit for sector B since both moieties provided I compounds with excellent activity (Tables 1, 2). We chose to evaluate the unsubstituted aryl compound, (R)-20 and the three mono-fluorophenyl derivatives, (R)-10, (R)-21, and (R)-22. For the three fluorinated compounds, we observed in mice (ip) that the 2″-fluoro derivative (R)-21 was the most active (MES ED50 = >3, <10 mg/kg). Correspondingly, in the rat (po) model, the 4″-fluoro isomer (R)-22 was the most potent (MES ED50 = 5.8 mg/kg) followed by the 2″-fluoro ((R)-21, MES ED50 = 11 mg/kg) and the 3″-fluoro ((R)-10, MES ED50 = 14 mg/kg) analogs. Removing the fluoro substituent to give unsubstituted (R)-20 increased seizure protection (MES ED50 = <3 mg/kg) in mice (ip), making this agent among the most potent I compounds tested under these conditions. Collectively, these findings demonstrated that the terminal aryl ring in I can influence anticonvulsant activities, thus warranting our further SAR exploration of this unit. We asked whether the extended N-aryl substituent in I directly contributed to the observed anticonvulsant activity. Accordingly, we evaluated 23 in the MES test (mice, ip) and observed modest anticonvulsant activity (MES ED50 = >30, <100 mg/kg).
The excellent activity for (R)-10 warranted its further pharmacological evaluation in other models. In the psychomotor 6 Hz (32 mA) seizure test,43 (R)-10 exhibited an ED50 = ~10 mg/kg in mice (ip) (Table 1), a value comparable with that observed for (R)-3 (ED50 = 10 mg/kg).19 In the sensitive rat (ip) hippocampal kindled seizure test,44,45 its ED50 value was 12 mg/kg. This closely matched the value for (R)-3 (ED50 = 14 mg/kg) in similar screening.12 The hippocampal kindled seizure assay is a model of partial complex seizures or temporal lobe seizures, which are the most common and drug-resistant type of adult focal epilepsy.45a,46
Further evaluations of (R)-10 were undertaken using the sciatic nerve ligation model,47 which is used to predict potential efficacy against chronic neuropathic pain in humans. Recent pharmacological and clinical studies have documented that certain anticonvulsant compounds are effective in several different models of inflammatory and neuropathic pain.48 Thus, it is not surprising that these antiepileptic agents are also used to manage neuropathic pain. 48,49 Neuropathic pain results from excessive neuronal activity and damage resulting in dysfunction of neuronal pathways within both the peripheral and the central nervous systems.48,49 The sciatic ligation model was used to assess the efficacy of (R)-10 in rats. Using this test, we showed that administration of (R)-10 (12 mg/kg; rats, ip) provided a 11.2-fold attenuation of mechanical allodynia at 1 h.
The formalin test, a chemically induced biphasic pain model, was also employed.50 There are two phases of response measured in the test, early (acute) and late (chronic inflammatory). The acute phase results in a behavioral licking response, which is believed to be mediated by chemical activation of local C-fibres.50 The late phase is likely due to the development of peripheral inflammation and central sensitization of dorsal horn neurons. Thus, this model is believed to provide preliminary information about the utility of the test candidate for the treatment of acute and chronic inflammatory pain. When evaluated in the formalin model, (R)-10 (15 mg/kg, mice, ip) significantly reduced the pain response in both the acute (35% of control; p <0.01) and the late (44% of control; p <0.01) phases. These results compared favorably with (R)-3, where comparable reduction in pain was observed only in the late phase when 16 mg/kg was administered.51
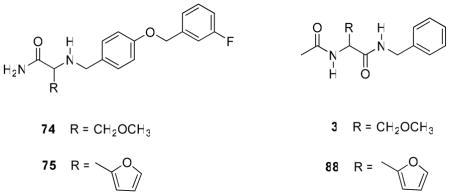
In 5–22, we constructed compounds with the principal structural motif seen in 1 and then extended the N-benzyl amide moiety to resemble 2. We briefly explored the reverse, where we began with the 2 core structure and then incorporated at C(2) 1 units shown to have excellent anticonvulsant activities. Accordingly, we prepared racemic 74 and 75 using the synthetic procedures shown in Schemes 7 and 8, respectively. Compound 74 contained the (R)-3 (C)2-methoxymethylene unit, and compound 75 had a 2-furanyl moiety. In the 1 series, racemic 3 and 88 displayed MES ED50 values (mice, ip) of 8.3 and 10 mg/kg, respectively (Table 4). When tested, (R,S)-74 and (R,S)-75 displayed good-to-moderate anticonvulsant activity in mice (ip) ((R,S)-74, MES ED50 = 30–100 mg/kg; (R,S)-75, MES ED50 = 10–30 mg/kg), values that were higher (lower activity) than observed for (R,S)-3 and (R,S)-88 in this test (MES ED50 = 8.3–10 mg/kg).11,16 Interestingly, we observed little or no activity in the scMet seizure model for (R,S)-74 (scMet ED50 = >300 mg/kg) and (R,S)-75 (scMet ED50 = 100–300 mg/kg). This finding contrasted with the excellent activity reported for (S)-4 in this model (scMet ED50 = 27 mg/kg).27 We concluded that the SAR guidelines for 17–16 did not help improve the pharmacological activity of 2.
Scheme 7.

Preparation of 74
Scheme 8.

Preparation of 75
Table 4.
Pharmacological Data for α-Aminoamide Derivatives 74 and 75 and their Functionalized Amino Acid Counterparts 3 and 88a
 | ||||
|---|---|---|---|---|
| No. |
R4 |
Mice (ip)b |
||
| MES,c ED50 |
Tox,d TD50 |
PI e |
||
| (R,S)-74 | >30, <100 [0.5] | >100, <300 [0.5] | ||
| (R,S)-75 | >10, <30 [0.5] | >100, <300 [0.5] | ||
| (R,S)-3f | 8.3 [0.5] (7.9–9.8) |
43 [0.25] (38–47) |
5.2 | |
| (R,S)-88g | 10 (9.1–12) | ~40 | ~4.0 | |
| (S)-4h | 8.0 (7.0–9.1) | 630 (560–700) | 78 | |
| (R)-4h | 7.2 (5.9–8.9) | 580 (410–830) | 88 | |
| phenytoini | 9.5 [2] (8.1–10) |
66 [2] (53–72) |
6.9 | |
| phenobarbitalli | 22 [1] (15–23) |
69 [0.5] (63–73) |
3.2 | |
| valproatei | 270 [0.25] (250–340) |
430 [0.25] (370–450) |
1.6 | |
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
Ref 16.
Ref 11.
Ref 30.
Ref 32.
DISCUSSION
In this study, we combined key pharmacophores found in 1 and 2 to provide I, anticipating that I might exhibit potent anticonvulsant activity. The structural design for I permitted the near seamless overlap of the pharmacophores. Moreover, the structural economy gained from this overlap permitted the design of a compact agent that conforms to Lipinski’s pharmacokinetic rules for low molecular weight, oral therapeutic agents.52 We expected several additional benefits from this design. First, the overlap eliminated the need for extraneous bridging units, which could confound receptor binding and provide unwanted sites for metabolism and toxicity. Second, we anticipated that I, like 1 and 2, 22,24 would exhibit favorable CNS biodistribution.
We found that I did not prevent scMet-induced seizures in mice and that it showed a stereochemical preference for function, which corresponds to the D-amino acid, in the MES-induced seizure test in rodents. Moreover, anticonvulsant activity increased with placement of a heteroatom one atom removed from the C(2) atom in I (Table 1) These whole animal pharmacological responses are similar to those observed for 1 ((R)-3)7–18 but not 2 ((S)-4).20,30 Table 2 shows that the most potent I derivatives in the MES test were those in which “X-Y-Z” was a single bond ((R)-11), a rigid, linear unit ((R)-15), or a short moiety (i.e., O ((R)-12); “X-Y” [where X and Y are C(H), N(H), O, and CH2] ((R)-10, (R)-13, (R)-14, (R)-16, (R)-17). Of these, (R)-11 and (R)-15 were the most potent anticonvulsants when tested in the rat (po) (MES ED50 = 1.4–2.4 mg/kg).
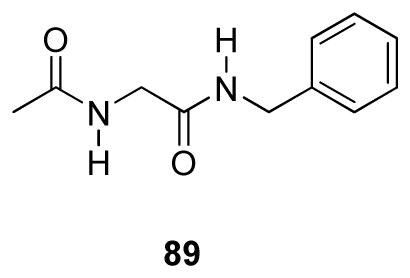
The excellent anticonvulsant activity for several I compounds demonstrated that seizure protection could be improved over their 1 counterpart by incorporating an extended N-aryl substituent similar to that found in 2 ((S)-4). For example, in the MES test (mice, ip), the glycine FAA 89 exhibited weak anticonvulsant activity (MES ED50 = >100, <300 mg/kg),8 but glycine 5 exhibited a MES ED50 = >30, <100 mg/kg, mice (ip). When 5 was tested in the rat (po), the MES ED50 = 31 mg/kg, and we observed no apparent neurological toxicity at doses of 500 mg/kg. Similarly, we found superb anticonvulsant activity for (R)-11 (MES ED50 = 2.4 mg/kg) and (R)-15 (MES ED50 = 1.4 mg/kg) in the rat (po), which surpassed that of (R)-3 (MES ED50 = 3.9 mg/kg). We evaluated 23 that contained the same extended N-benzyl substituent that was incorporated in 5 and (R)-6–(R)-10 and observed only modest anticonvulsant activity in mice (ip) (MES ED50 = >30, <100 mg/kg), indicating that this group alone was not responsible for the observed seizure protection. Finally, we found that several I compounds ((R)-6, (R)-15) exhibited extended duration of action in the MES model in mice (ip) and rat (po).
We tested (R)-N-4′-((3″-fluoro)benzyloxy)benzyl 2-acetamido-3-methoxypropionamide ((R)-10) in other neurological models. We first evaluated (R)-10 in two additional seizure models. In the psychomotor 6 Hz (32 mA) test in mice (ip) 43 and the rat (ip) hippocampal kindled seizure test,45 (R)-10 provided excellent protection, giving an ED50 of ~10 and 12 mg/kg, respectively. Since several clinically available AEDs are used to treat pain disorders,48,49 we evaluated (R)-10 in two animal pain models. When (R)-10 was tested at 12 mg/kg in the sciatic nerve ligation model47 in rats (ip) we observed an 11.2-fold attenuation of mechanical allodynia at 1 h. In the biphasic, chemically induced formalin pain model in mice,50 ip administration of (R)-10 (15 mg/kg) reduced pain response in both the acute (35% of control) and the late (44% of control) phases. These findings document the potential value of I to treat a range of neurological conditions.
CONCLUSIONS
Our findings demonstrate that the incorporation of key pharmacophores in 1 and 2 to provide I gave compounds of significant neurological interest. Using animal tests, we observed excellent seizure protection and pronounced pain reduction. The pharmacological basis for I anticonvulsant activity and pain protection has not been determined. Anticonvulsants, like many neurological agents,53 typically exert their activity through multiple pathways.54 Current studies are directed at optimizing sectors A–C in I and evaluating these compounds in electrophysiology, radioligand displacement, and functional assays to provide information about the mode(s) of action of these novel hybrids.
EXPERIMENTAL SECTION
General Methods
The general methods used in this study were identical to those previously reported18 and are summarized in the Supporting Information Section. All compounds were checked by TLC, 1H and 13C NMR, MS, and elemental analyses. The analytical results are within +/− 0.40% of the theoretical value. The TLC, NMR and the analytical data confirmed the purity of the products was ≥95%.
Preparation of N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamidoacetamide (5)
A THF solution (120 mL) of 28 (1.50 g, 12.8 mmol) was stirred and cooled at −78 °C under Ar and then 4-methylmorpholine (NMM) (1.7 mL, 15.4 mmol) was added dropwise. After 2 min of stirring at this temperature, isobutylchloroformate (IBCF) (2.0 mL, 15.4 mmol) was added dropwise leading to the precipitation of a white solid. The reaction was allowed to proceed for additional 2 min and 32 (3.26 g, 14.1 mmol) was added portionwise at −78 °C. The mixture was allowed to stir at room temperature (2 h), and then the white solid filtered and the organic layer concentrated in vacuo. The solid was purified by flash column chromatography on silica gel with methanol/EtOAc (0/10 -> 5/5) as the eluant to obtain 5 as white solid (1.30 g, 31%): Rf = 0.11 (EtOAc); mp 155–156 °C; 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.92 (d, J = 5.1 Hz, 3H), 4.38 (d, J = 5.7 Hz, 2H), 5.05 (s, 1H), 6.24–6.35 (br m, 2H), 6.92 (m, 2H), 6.97–7.04 (m, 1H), 7.11–7.21 (m, 4H), 7.30–7.38 (m, 1H); MS (M+H+)(ESI+) 331.1 [M + H+] (calcd for C18H19FN2O3H+ 331.1); Anal. (C18H19FN2O3•0.35 H2O): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamido-propionamide ((R)-6)
Employing the procedure to prepare 5 and using THF (100 mL), (R)-29 (1.50 g, 11.4 mmol), NMM (1.5 mL, 13.7 mmol), IBCF (1.8 mL, 13.7 mmol), and 32 (2.90 g, 12.6 mmol) gave (R)-6 that was purified by recrystallization (EtOAc) as a white solid (905 mg, 22%): Rf = 0.16 (EtOAc); mp 158 °C; [α]26.2D +27.3° (c 1, CHCl3); 1H NMR (CDCl3) δ 1.37 (d, J = 6.9 Hz, 3H), 1.94 (s, 3H), 4.32 (d, J = 5.7 Hz, 2H), 4.46–4.56 (m, 1H), 5.02 (s, 2H), 6.41 (d, J = 7.5 Hz, 1H), 6.79–6.85 (br m, 1H), 6.87–6.92 (m, 2H), 7.00 (td, J = 2.4, 8.4 Hz, 1H), 7.10–7.20 (m, 4H), 7.30–7.37 (m, 1H); MS (M+H+)(ESI+) 345.2 [M + H +] (calcd for C19H21FN2O3H+ 344.2); Anal. (C19H21FN2O3): C, H, F, N.
Preparation of (S)-N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamidopropionamide ((S)-6)
Employing the procedure of (R)-6 and using (S)-29 (1.50 g, 11.4 mmol), NMM (1.5 mL, 13.7 mmol), IBCF, 32 (2.90 g, 12.6 mmol) gave after workup and recrystallization (EtOAc) a white solid (1.50 g, 38%): Rf = 0.16 (EtOAc); mp 153–154 °C; [α]26.2D -28.1° (c 1, CHCl3); 1H NMR (CDCl3) δ 1.38 (d, J = 7.2 Hz, 3H), 1.94 (s, 3H), 4.32 (d, J = 5.7 Hz, 2H), 4.46–4.57 (m, 1H), 5.02 (s, 2H), 6.38–6.46 (br d, 1H), 6.78–6.85 (br m, 1H), 6.89 (d, J = 8.4 Hz, 2H), 6.98–7.04 (m, 1H), 7.10–7.20 (m, 4H), 7.30–7.37 (m, 1H); MS (M+H+)(ESI+) 345.2 [M + H+] (calcd for C19H21FN2O3H+ 345.2); Anal. (C19H21FN2O3): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamido-3-methylbutanamide ((R)-7)
Trifluoroacetic acid (2 mL) was added to a CH2Cl2 (10 mL) solution of solution (R)-33 (1.00 g, 2.3 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo, dried (30 min), and CH2Cl2 (20 mL) and a saturated aqueous Na2CO3 solution (20 mL) were added. The layers were separated and the aqueous layer was washed with CH2Cl2 (2 × 20 mL). The organic layers were combined and concentrated under vacuum.
The residue was dissolved in CH2Cl2 (20 mL) and Et3N (0.49 mL, 3.5 mmol) and AcCl (200 μL, 2.8 mmol) were successively added at 0 °C. The mixture was stirred at room temperature (3 h), aqueous 10% citric acid (60 mL) was added, and the organic layer was separated. The aqueous layer was washed with CH2Cl2 (2 x 30 mL). All the organic layers were combined, washed with aqueous saturated NaHCO3 (30 mL), and H2O (30 mL), dried (MgSO4), and concentrated in vacuo. The solid was recrystallized with EtOAc to obtain (R)-7 (585 mg, 68%) as a white solid: Rf = 0.26 (EtOAc); mp 199–200 °C; [α]25.2D +25.6° (c 0.5, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 0.81 (d, J = 3.0 Hz, 3H), 0.83 (d, J = 3.0 Hz, 3H), 1.87 (s, 3H), 1.90–1.99 (m, 1H), 4.12–4.43 (dd, J = 6.4, 8.8 Hz, 1H), 4.19 (d, J = 5.8 Hz, 2H), 5.11 (s, 2H), 6.95 (d, J = 8.4 Hz, 2H), 7.11–7.19 (m, 3H), 7.24–7.28 (m, 2H), 7.39–7.46 (m, 1H), 7.87 (d, J = 8.8 Hz, 1H), 8.38 (t, J = 5.8 Hz, 1H); HRMS (M+Na+)(ESI+) 395.1747 [M + Na+] (calcd for C21H25FN2O3Na+ 395.1747); Anal. (C21H25FN2O3): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamido-3,3-dimethylbutanamide ((R)-8)
Employing a procedure similar to (R)-7 and using (R)-N-4′-((3″-fluoro)benzyloxy)benzyl 2-amino-3,3-dimethylbutanamide (1.20 g, 3.3 mmol), CH2Cl2 (40 mL), Et3N (0.93 mL, 6.6 mmol), and AcCl (0.34 mL, 4.8 mmol) gave after workup and purification by flash column chromatography on silica gel with EtOAc/hexanes (8/2 to 10/0) as the eluant (R)-8 as a white solid (1.03 g, 81%): Rf = 0.56 (EtOAc); mp 64–66 °C; [α]27.0D -15.6° (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.99 (s, 9H), 1.89 (s, 3H), 4.42 (1/2 ABq, J = 5.2, 14.4 Hz, 1H), 4.34–4.40 (m, CH, 1H), 5.02 (s, 2H), 6.41 (br d, J = 8.8 Hz, 1H), 6.88 (d, J = 8.8 Hz, 2H), 6.85–6.96 (br m, 1H), 7.00 (t, J = 8.4 Hz, 1H), 7.11–7.18 (m, 4H), 7.30–7.36 (m, 1H); HRMS (M+Na+)(ESI+) 409.1903 [M + Na+] (calcd for C22H27FN2O3Na+ 409.1903); Anal. (C22H27FN2O3): C, H, F, N.
Preparation of N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamido-2-(pyridin-2-yl)acetamide ((R,S)-9)
To a solution of 38 (1.81 g, 4.77 mmol, 1 equiv) in MeOH (95 mL) was added ammonium formate (1.21 g, 19.08 mmol, 4 equiv) as a solid and then the reaction mixture was stirred at room temperature (5 min). Zn dust (Sigma-Aldrich <10 micron, 1.20 g, 19.08 mmol, 4 equiv) was added and the reaction heated at reflux (6 h), and then maintained at room temperature (16 h). The reaction mixture was filtered through Celite®. The filtrate was concentrated and the residue was dissolved in CH2Cl2 (100 mL). The CH2Cl2 layer was washed with a brine (2 × 100 mL), dried (Na2SO4), and concentrated in vacuo. The crude 39 was used without further purification for the next step: Rf = 0.00 (EtOAc).
Compound 39 (4.77 mmol, 1 equiv) was dissolved in CH2Cl2 (100 mL) and then triethylamine (0.8 mL, 5.72 mmol, 1.2 equiv) and AcCl (0.4 mL, 5.72 mmol, 1.2 equiv) were carefully added at 0 °C and the resulting solution was stirred at room temperature (2 h). An aqueous saturated NaHCO3 solution (100 mL) was added and the organic layer was extracted with CH2Cl2 (3 x 100 mL). The organic layers were combined, dried (Na2SO4), and concentrated in vacuo. The residue was purified by chromatography on silica gel with EtOAc/hexanes (7/3 to 10/0) as the eluent. The residue was recrystallized (EtOAc) to obtain (R,S)-9 as a white solid (935 mg, 48%): Rf = 0.47 (EtOAc); mp 154–155 ºC; 1H NMR (CDCl3) δ 2.14 (s, 3H), 4.29–4.41 (m, 2H), 5.03 (s, 2H), 5.56 (d, J = 6.0 Hz, 1H), 6.87 (d, J = 9.0 Hz, 2H), 6.98–7.43 (m, 10H), 7.70 (dt, J = 1.6, 7.8 Hz, 1H), 8.50–8.52 (m, 1H); Anal. (C23H22FN3O3): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-10)
A saturated HCl solution in dioxane (1 mmol/2 mL, 21.75 mL) was added to (R)-N-4′-((3″-fluoro)benzyloxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (4.70 g, 10.9 mmol) at 0 °C and the solution was stirred at room temperature (4 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue, CH2Cl2 (40 mL), Et3N (4.47 mL, 32.6 mmol), and AcCl (1.16 mL, 16.30 mmol) gave after workup and recrystallization (EtOAc) (R)-10 (2.60 g, 65%) as a white solid: Rf = 0.29 (7/3 hexanes/EtOAc); mp 152 °C; [α]24.5D -18.9° (c 1, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.37 (s, 3H), 3.43 (dd, J = 7.2, 9.0 Hz, 1H), 3.79 (dd, J = 3.9, 9.0 Hz, 1H), 4.40 (d, J = 5.7 Hz, 2H), 4.49–4.55 (m, 1H), 5.05 (s, 2H), 6.43 (br d, J = 7.2 Hz, 1H), 6.64–6.83 (br m, 1H), 6.89–7.05 (m, 3H), 7.10–7.22 (m, 4H), 7.31–7.38 (m, 1H); HRMS (M+H+)(ESI+) 375.1720 [M + H+] (calcd for C20H23FN2O4H+ 375.1720); Anal. (C20H23FN2O4): C, H, F, N.
Preparation of (S)-N-4′-((3″-Fluoro)benzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((S)-10)
A saturated HCl solution in dioxane (1 mmol/2 mL, 20.8 mL) was added to (S)-N-4′-((3″-fluoro)benzyloxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (4.50 g, 10.4 mmol) at 0 °C and the solution was stirred at room temperature (4 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue, CH2Cl2 (40 mL), Et3N (4.3 mL, 31.2 mmol), and AcCl (1.1 mL, 15.6 mmol) gave after recrystallization (EtOAc) (S)-10 (3.10 g, 80%) as a white solid: Rf = 0.29 (7/3 hexanes/EtOAc); mp 149–150 °C; [α]24.5D +18.8° (c 1, CHCl3); 1H NMR (CDCl3) δ 2.02 (s, 3H), 3.36 (s, 3H), 3.43 (dd, J = 7.5, 9.1 Hz, 1H), 3.79 (dd, J = 4.2, 9.1 Hz, 1H), 4.40 (d, J = 5.7 Hz, 2H), 4.50–4.55 (m, 1H), 5.05 (s, 2H), 6.47 (br d, J = 6.0 Hz, 1H), 6.70–6.79 (br m, 1H), 6.90–7.05 (m, 3H), 7.10–7.22 (m, 4H), 7.31–7.38 (m, 1H); HRMS (M+H+)(ESI+) 375.1720 [M + H+] (calcd for C20H23FN2O4H+ 375.1720); Anal. (C20H23FN2O4): C, H, F, N.
Preparation of (R)-N-(3″-Fluorobiphenyl-4-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-11)
To a flame-dried Schlenck tube, under Ar, containing a dioxane (22.5 mL) solution of (R)-6918 (1.50 g, 4.0 mmol), palladiumtetrakis(triphenylphosphine) (464 mg, 0.402) and 3-fluorophenylboronic acid (70) (670 mg, 4.80 mmol) was added an aqueous solution (9 mL) of Cs2CO3 (2.60 g, 8.0 mmol). The mixture was stirred at reflux (16 h). Then MeOH and silica gel were added, and the volatiles were concentrated in vacuo. The residue was purified by flash chromatography on silica gel with EtOAc/MeOH (10/0 to 9/1) as the eluant to obtain (R)-11 (0.95 g, 60%) as a yellowish solid. To remove traces of palladium impurities, the solid was treated with 6.00 g of resin scavenger (SPM32, PhosPhonics) in CH2Cl2. The mixture was stirred at room temperature (2 h), filtered, and the filtrate evaporated under vacuum to obtain 800 mg (58%) of (R)-11 as a white solid: Rf = 0.22 (EtOAc); mp 170–172 °C; [α]25.3D = −8.1° (c 0.5, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.39 (s, 3H), 3.47 (d, J = 7.5, 9.3 Hz, 1H), 3.81 (d, J = 3.9, 9.3 Hz, 1H), 4.45–4.55 (m, 2H), 4.56–4.63 (m, 1H), 6.53 (br d, J = 6.6 Hz, 1H), 6.93–7.07 (m, 2H), 7.23–7.51 (m, 5H), 7.53 (d, J = 8.1 Hz, 2H); HRMS (M+Cs+)(ESI+) 477.0591 [M + Cs+] (calcd for C19H21FN2O3Cs+ 477.0587); Anal. (C19H21FN2O3): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)phenoxy)benzyl 2-N-Acetamido-3-methoxypropionamide ((R)-12)
A saturated HCl solution in dioxane (1 mmol/2 mL, 16.7 mL) was added to an Et2O (8 mL) solution of (R)-N-4′-((3″-fluoro)phenoxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (3.50 g, 8.4 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue, CH2Cl2 (40 mL), Et3N (3.52 mL, 25.1 mmol), and AcCl (0.91 mL, 12.5 mmol) gave after workup and purification by flash column chromatography on silica gel with EtOAc as the eluant (R)-12 as a white solid (1.30 g, 43%): Rf = 0.45 (EtOAc); mp 125–126 °C; [α]25.3D –14.8° (c 1, CHCl3); 1H NMR (300 MHz, CDCl3) δ 2.04 (s, 3H), 3.39 (s, 3H), 3.45 (dd, J = 7.5, 9.3 Hz, 1H), 3.81 (dd, J = 4.2, 9.3 Hz, 1H), 4.45 (d, J = 6.0 Hz, 2H), 4.53–4.59 (m, 1H), 6.48 (br d, J = 6.0 Hz, 1H), 6.68 (dt, J = 2.4, 10.2 Hz, 1H), 6.74–6.89 (m, 3H), 6.99 (d, J = 9.0 Hz, 2H), 7.21–7.34 (m, 3H); HRMS (M+H+)(ESI+) 361.1564 [M + H+] (calcd for C19H21FN2O4H+ 361.1563); Anal. (C19H21FN2O4): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)phenethyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-13)
Pd/C (18 mg) was added to an EtOH solution of (R)-14 (180 mg, 0.49 mmol), and the mixture was stirred at room temperature under H2 (1 atm) (36 h). The reaction mixture was filtered through a pad of Celite®, and the pad was washed successively with EtOH and CH2Cl2. The filtrate was concentrated under vacuum to obtain (R)-13 (170 mg, 94%) as a white solid: Rf = 0.29 (EtOAc); mp 134–136 °C; [α]24.4D = −12.3° (c 0.48, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.03 (s, 3H), 2.90 (s, 4H), 3.38 (s, 3H), 3.43 (dd, J = 7.6, 9.2 Hz, 1H), 3.81 (dd, J = 4.0, 9.2 Hz, 1H), 4.40–4.47 (m, 2H), 4.50–4.55 (m, 1H), 6.41–6.47 (br m, 1H), 6.68–6.75 (br m, 1H), 6.84–6.94 (m, 3H), 7.11–7.25 (m, 5H);HRMS (M+H+)(ESI+) 373.1927 [M + H+] (calcd for C21H25FN2O3H+ 373.1927); Anal. (C21H25FN2O3•0.32H2O): C, H, N.
Preparation of (2-R,E)-N-4′-((3″-Fluoro)styryl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-14)
To a flame-dried Schlenck tube, under Ar, containing a dioxane (22.5 mL) solution of (R)-6918 (1.50 g, 4.0 mmol), palladiumtetrakis(triphenylphosphine) (464 mg, 0.402) and trans-2-((3-fluoro)phenyl)vinylboronic acid (71) (800 mg, 4.82 mmol) was added an aqueous solution (9 mL) of Cs2CO3 (2.60 g, 8.0 mmol). The mixture was stirred at reflux (16 h). Then, MeOH and silica gel were added. The volatiles were concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc/MeOH (10/0 to 9/1) as the eluant to obtain (R)-14 (0.90 g, 60%) as a yellowish solid. To remove traces of palladium impurities, the solid was treated with 6.00 g of resin scavenger (SPM32, PhosPhonics) in CH2Cl2. The mixture was stirred at room temperature (2 h), and filtered, the filtrate evaporated under vacuum to obtain 560 mg (37%) of (R)-14 as a white solid: Rf = 0.53 (EtOAc); mp 206–208 °C; [α]27D = −20.6° (c 0.5, CHCl3); 1H NMR (CDCl3) δ 2.04 (s, 3H), 3.40 (s, 3H), 3.40–3.48 (m, 1H), 3.83 (d, J = 3.9, 8.7 Hz, 1H), 4.47–4.56 (m, 3H), 6.41–6.49 (br d, 1H), 6.75–7.02 (br t, 1H), 6.92–7.01 (m), 7.07 (d, J = 2.7 Hz), 7.18–7.35 (m), 7.47 (d, J = 8.4 Hz) (10H); LRMS (M+Na+)(ESI+) 393.1 [M + Na+] (calcd for C21H23FN2O3Na+ 393.1); Anal. (C21H23FN2O3): C, H, F, N.
Preparation of (R)-N-4′-(((3″-Fluoro)phenyl)ethynyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-15)
To an anhydrous THF (70 mL) solution of (R)-6918 (2.60 g, 7.0 mmol) were sequentially added triethylamine (0.95 mL, 14.0 mmol), 3-(fluoro)phenylacetylene (72) (1.20 ml, 10.37 mmol), dichlorobis(triphenylphosphine)palladium (II) (491 mg, 0.70 mmol), and CuI (200 mg, 0.1.05 mmol) under Ar. The mixture was stirred at room temperature (16 h), and then MeOH and silica gel were added. The volatiles were concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc/MeOH (9/1) as the eluant to obtain (R)-15 (2.40 g, 93%) as a yellowish solid. To remove traces of palladium impurities, the solid was treated with 21.00 g of resin scavenger (SPM32, PhosPhonics) in CH2Cl2. The mixture was stirred at room temperature (2 h), and filtered, the filtrate evaporated under vacuum. The solid was recrystallized with EtOAc to obtain 1.20 g (46%) of (R)-15 as a white solid: Rf = 0.26 (EtOAc); mp 200–202 °C; [α]24D = −2.6° (c 0.5, CHCl3); 1H NMR (DMSO-d6) δ 1.88 (s, 3H), 3.27 (s, 3H), 3.48–3.57 (m, 2H), 4.33 (d, J = 6.1 Hz, 2H), 4.45–4.53 (m, 1H), 7.25–7.32 (m, 3H), 7.38–7.53 (m, 5H), 8.13 (d, J = 6.3 Hz, 1H), 8.56 (br t, J = 6.1 Hz, 1H); HRMS (M+H+)(ESI+) 369.1614 [M + H+] (calcd for C21H21FN2O3H+ 369.1614); Anal. (C21H21FN2O3): C, H, F, N.
Preparation of (R)-N-4′-(((3″-Fluoro)phenoxy)methyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-16)
A saturated HCl solution in dioxane (1 mmol/2 mL, 10.2 mL) was added to (R)-N-4′-(((3″-fluoro)phenoxy)methyl)benzyl 2 -N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (2.20 g, 5.1 mmol) at 0 °C and the solution was stirred at room temperature (2 h). The reaction solution was concentrated in vacuo and dried (30 min) to provide (R)-2-amino-N-4′-(((3″-fluoro)phenoxy)methyl)benzyl-3-methoxypropionamide hydrochloride as a white solid (1.80 g, quant.).
Employing a procedure similar to (R)-7, and using triethylamine (1.5 mL, 5.2 mmol), acetyl chloride (380 μL, 10.7 mmol), CH2Cl2 (20 mL), and (R)-N-4′-(((3″-fluoro)phenoxy)methyl)benzyl 2-amino-3-methoxypropionamide hydrochloride (1.30 g, 3.5 mmol) gave after workup and recrystallization (EtOAc) (R)-16 (900 mg, 68%) as a white solid: Rf = 0.18 (EtOAc); mp 140–142 °C; [α]26.9D -21.0° (c 1, CHCl3); 1H NMR (CDCl3) δ 2.04 (s, 3H), 3.41 (s, 3H), 3.43 (dd, J = 7.5, 9.2 Hz, 1H), 3.82 (dd, J = 3.9, 9.2 Hz, 1H), 4.47–4.59 (m, 3H), 5.03 (s, 2H), 6.38–6.43 (br d, 1H), 6.64–6.78 (m, 4H), 7.14–7.30 (m, 3H), 7.40 (d, J = 8.4 Hz, 2H); LRMS (M+Na+)(ESI+) 397.1 [M + Na+] (calcd for C20H23FN2O4H+ 397.1); Anal. (C20H23FN2O4): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)benzylamino)benzyl 2-Acetamido-3-methoxypropionamide ((R)-17)
Compound 67. HCl (293 mg, 1.1 mmol) was added to a THF (10 mL) solution of the (R)-68 (161 mg, 1.0 mmol) and the mixture was stirred at room temperature (5 min) and then NMM (121 μL, 1.1 mmol) was added. The mixture was stirred at room temperature (5 min) and DMTMM (332 mg, 1.2 mmol) was added, and the mixture was stirred at room temperature (16 h). The white precipitate was filtered and the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc/hexanes (5/5) to EtOAc/acetone (5/5) as the eluant to obtain (R)-17 as a yellow solid (140 mg, 35%): Rf = 0.37 (EtOAc); mp 78–81 °C; [α]26.9D −15.0° (c 0.5, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.36 (s, 3H), 3.40 (dd, J = 7.2, 9.0 Hz, 1H), 3.82 (dd, J = 4.2, 9.0 Hz, 1H), 4.12–4.19 (br m, 1H), 4.31–4.37 (m, 4H), 4.46–4.52 (m, 1H), 6.38–6.45 (br m, 1H), 6.57 (d, J = 9.0 Hz, 3H), 6.91–6.89 (m, 1H), 7.05–7.15 (m, 4H), 7.27–7.34 (m, 1H); HRMS (M+H+)(ESI+) 374.1880 [M + H+] (calcd for C20H24FN3O3H+ 374.1879).
Preparation of (R)-N-4′-(((3″-Fluoro)benzyloxy)methyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-18)
A saturated HCl solution in dioxane (1 mmol/2 mL, 1.2 mL) was added to (R)-N-4′-(((3″-fluoro)benzyloxy)methyl)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (1.10 g, 5.8 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue, CH2Cl2 (20 mL), Et3N (1.40 mL, 9.8 mmol), and AcCl (356 μL, 4.9 mmol) gave after workup and recrystallization (EtOAc) (R)-18 (450 mg, 47%) as a white solid: Rf = 0.26 (EtOAc); mp 140–142 °C; [α]25.2D −21.0° (c 0.5, CHCl3); 1H NMR (CDCl3) δ 2.04 (s, 3H), 3.39 (s, 3H), 3.43 (dd, J = 7.8, 9.0 Hz, 1H), 3.82 (dd, J = 3.9, 9.0 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 4.48–4.56 (m, 1H), 4.54 (s, 2H), 4.55 (s, 2H), 6.42 (br d, J = 6.6 Hz, 1H), 6.71–6.79 (br t, 1H), 6.96–7.15 (m, 3H), 7.24–7.35 (m, 5H); Anal. (C21H25FN2O4): C, H, F, N.
Preparation of (R)-N-4′-((3″-Fluoro)phenethoxy)benzyl 2-N-Acetamido-3-methoxypropionamide ((R)-19)
A saturated HCl solution in dioxane (1 mmol/2 mL, 10.0 mL) was added to an Et2O (5 mL) solution of (R)-N-4′-((3″-fluoro)phenethoxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (2.20 g, 5.0 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue, CH2Cl2 (30 mL), Et3N (2.1 mL, 15.0 mmol), and AcCl (0.54 mL, 7.5 mmol) gave after workup and recrystallization (EtOAc) (R)-19 as a white solid (1.30 g, 66%): Rf = 0.28 (EtOAc); mp 147–148 °C; [α]25.2D −16.6° (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.02 (s, 3H), 3.08 (t, J = 7.1 Hz, 2H), 3.36 (s, 3H), 3.39–3.44 (m, 1H), 3.79 (dd, J = 4.8, 9.6 Hz, 1H), 4.15 (t, J = 7.1 Hz, 2H), 4.33–4.44 (m, 2H), 4.49–4.54 (m, 1H), 6.44 (br d, J = 6.4 Hz, 1H), 6.65–6.73 (br t, 1H), 6.84 (d, J = 8.0 Hz, 2H), 6.90–7.06 (m, 3H), 7.16 (d, J = 8.0 Hz, 2H), 7.23–7.29 (m, 1H); HRMS (M+Na+)(ESI+) 411.1696 [M + H+] (calcd for C21H25FN2O4Na+ 411.1697); Anal. (C21H25FN2O4): C, H, F, N.
Preparation of (R)-N-4′-(Benzyloxy)benzyl 2-N-Acetamido-3-methoxypropionamide ((R)-20)
A saturated HCl solution in dioxane (1 mmol/2 mL, 24.1 mL) was added to an Et2O (10 mL) solution of (R)-N-4′-(benzyloxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (5.00g, 12.1 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue, CH2Cl2 (60 mL), Et3N (5.1 mL, 36.3 mmol), and AcCl (1.4 mL, 18.8 mmol) gave after workup and recrystallization (EtOAc) (R)-20 as a white solid (2.60 g, 60%): Rf = 0.28 (EtOAc); mp 149 °C; [α]25.1D −26.8° (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.02 (s, 3H), 3.36 (s, 3H), 3.39–3.44 (br m, 1H), 4.02–3.79 (dd, J = 4.2, 9.4 Hz, 1H), 4.36–4.44 (m, 2H), 4.48–4.55 (m, 1H), 5.05 (s, 2H), 6.42 (br d, J = 6.0 Hz, 1H), 6.64–6.71 (br m, 1H), 6.93 (d, J = 7.8 Hz, 2H), 7.18 (d, J = 7.8 Hz, 2H), 7.29–7.44 (m, 5H); HRMS (M+Na+)(ESI+) 379.1634 [M + Na+] (calcd for C20H24N2O4Na+ 379.1634); Anal. (C20H24N2O4): C, H, N.
Preparation of (R)-N-4′-((2″-Fluoro)benzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-21)
A saturated HCl solution in dioxane (1 mmol/2 mL, 11.57 mL) was added to (R)-N-4′-((2″-fluoro)benzyloxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (2.50 g, 5.8 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue (1.70 g, 5.1 mmol), CH2Cl2 (20 mL), Et3N (2.10 mL, 15.3 mmol), and AcCl (550 μL, 7.6 mmol) gave after workup and recrystallization (EtOAc) (R)-21 (1.25 g, 65%) as a white solid: Rf = 0.28 (EtOAc); mp 173–174 °C; [α]24.6D −20.7° (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.03 (s, 3H), 3.37 (s, 3H), 3.42 (dd, J = 7.6, 9.0 Hz, 1H), 3.79 (dd, J = 4.0, 9.0 Hz, 1H), 4.34–4.44 (m, 2H), 4.49–4.54 (m, 1H), 5.12 (s, 2H), 6.43 (br d, J = 6.4 Hz, 1H), 6.66–6.72 (br t, 1H), 6.94 (d, J = 8.0 Hz, 2H), 7.06–7.21 (m, 4H), 7.28–7.34 (m, 1H), 7.49 (td, J = 1.6, 7.6 Hz, 1H); HRMS (M+H+)(ESI+) 375.1720 [M + H+] (calcd for C20H23FN2O4H+ 375.1720); Anal. (C20H23FN2O4): C, H, F, N.
Preparation of (R)-N-4′-((4″-Fluoro)benzyloxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-22)
A saturated HCl solution in dioxane (1 mmol/2 mL, 11.57 mL) was added to (R)-N-4′-((4″-fluoro)benzyloxy)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (2.50 g, 5.8 mmol) at 0 °C and the solution was stirred at room temperature (16 h). The reaction solution was concentrated in vacuo and dried (30 min).
Employing a procedure similar to (R)-7, and using the residue (1.85 g, 5.6 mmol), CH2Cl2 (30 mL), Et3N (2.36 mL, 16.8 mmol), and AcCl (608 μL, 8.4 mmol) gave after workup and recrystallization (EtOAc) (R)-22 (1.28 g, 61%) as a white solid: Rf = 0.22 (EtOAc); mp 166–167 °C; [α]25.2D −19.4° (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.03 (s, 3H), 3.37 (s, 3H), 3.42 (dd, J = 7.6, 9.4 Hz, 1H), 3.79 (dd, J = 4.0, 9.4 Hz, 1H), 4.40 (d, J = 5.2 Hz, 2H), 4.49–4.54 (m, 1H), 5.01 (s, 2H), 6.40 (br d, J = 5.6 Hz, 1H), 6.62–6.69 (br t, 1H), 6.92 (d, J = 8.8 Hz, 2H), 7.07 (t, J = 8.8 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.37–7.41 (m, 2H); HRMS (M+H+)(ESI+) 375.1720 [M + H+] (calcd for C20H23FN2O4H+ 375.1720); Anal. (C20H23FN2O4): C, H, F, N.
Preparation of N-4′-((3″-Fluoro)benzyloxy)benzyl Acetamide (23)
Employing a procedure similar to (R)-7, and using 4-((3′-fluoro)benzyloxy)benzylamine (32) (1.00 g, 4.3 mmol), CH2Cl2 (40 mL), Et3N (728 μL, 5.2 mmol), and AcCl (376 μL, 5.2 mmol) gave after workup and trituration (Et2O) 23 (810 mg, 69%) as a white solid: Rf = 0.39 (EtOAc); mp 131–132 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.84 (s, 3H), 4.16 (d, J = 5.6 Hz, 2H), 5.11 (s, 2H), 6.95 (d, J = 8.8 Hz, 2H), 7.11–7.18 (m, 3H), 7.24–7.29 (m, 2H), 7.40–7.45 (m, 1H), 8.21–8.24 (br t, 1H); HRMS (M+Na+)(ESI+) 296.1063 [M + Na+] (calcd for C16H16FNO2Na+ 296.1062); Anal. (C16H16FNO2): C, H, F, N.
Preparation of 2-(4′-((3″-Fluoro)benzyloxy)benzyl)amino-3-methoxypropionamide (74)
A solution of 79 (1.50 g, 4.32 mmol) in NH3 (7 N in MeOH, 150 mL) was stirred at room temperature in a sealed tube (7 d). The solution was concentrated in vacuo, and the residue was recrystallized (EtOAc) to obtain 74 as a white solid (350 mg, 24%): Rf = 0.25 (EtOAc); mp 84–85 °C; 1H NMR (300 MHz, CDCl3) δ 3.31–3.36 (m, 4H), 3.60 (d, J = 5.7 Hz, 2H), 3.71 (½ ABq, J = 12.9 Hz, 1H), 3.78 (½ ABq, J = 12.9 Hz, 1H), 5.06 (s, 2H), 5.40–5.44 (br s, 1H), 7.10 (d, J = 9.0 Hz, 2H), 6.96–7.05 (br dt, 1H), 7.13–7.25 (m, 4H), 7.31–7.38 (m, 1H); Mr (+ESI) 355.16 [M+Na]+ (calcd for C18H21FN2O3Na+ [M+355.14]+). Anal. (C18H21FN2O3): C, H, F, N.
Preparation of 2-(4′-((3″-Fluoro)benzyloxy)benzyl)amino-2-(furan-2-yl)acetamide Oxalate (75)
A solution of 87 (880 mg, 0.05 mmol) in NH3 (7 N in MeOH, 88 mL) was stirred at 4 °C (16 h). The solution was concentrated in vacuo, and the residue was dissolved in THF (2.3 mL) to obtain a 1 N solution. To this solution, oxalic acid (2 N in THF, 4.6 mL) was added. After standing at room temperature (16 h) the precipitate was collected, dried, and recrystallized with i-PrOH. The white solid was recrystallized (absolute EtOH) to obtain 75 as a white solid (520 mg, 51%): Rf = 0.15 (EtOAc); mp 194–195 °C; 1H NMR (DMSO-d6) δ 3.75 (s, 2H), 4.54 (s, 1H), 5.14 (s, 2H), 6.44–6.45 (m, 1H), 6.48–6.50 (m, 1H), 7.51 (d, J = 9.0 Hz, 2H), 7.03–7.19 (m, 1H), 7.26–7.31 (m, 3H), 7.41–7.51 (m, 2H), 7.71–7.73 (m, 2H); Mr (+ESI) 355.13 [M+H]+ (calcd for C22H21FN2O7H+ 355.15 [M+H]+). Anal. (C22H21FN2O7): C, H, F, N.
Pharmacology
Compounds were screened under the auspices of the National Institutes of Health’s Anticonvulsant Screening Program. Experiments were performed in male rodents [albino Carworth Farms No. 1 mice (intraperitoneal route, ip), albino Spague-Dawley rats (oral route, po)]. Housing, handling, and feeding were in accordance with recommendations contained in the ‘Guide for the Care and Use of laboratory Animals’. Anticonvulsant activity was established using the MES test,29 6 Hz,42 hippocampal kindled seizure,45 and the scMet test,44 according to previously reported methods.16
Formalin Test50a
The formalin test involved injection of 0.5% formalin into the mouse hind paw. Injection leads to a biphasic behavioral response characterized by licking of the affected paw. The number of licks is measured as a proxy for perceived pain. The first phase is termed the “acute” phase, and the second phase is termed the “inflammatory” phase. Each trial involved 16 animals, 8 controls given an ip injection of vehicle and 8 given the test compound at a specified dose. The amount of time that each animal spends licking is monitored at 2 min intervals, and monitoring continues for 45 min. Plots of time licking versus time provides a biphasic response and permits the area under the curve (AUC) to be determined for each animal for the acute and inflammatory phases. The AUC for each compound-treated animal is compared to the average result from the control group, yielding an average percent of control (reported with the SEM and p value).
Partial Sciatic Ligation Model47
Rats are anesthetized with sodium pentobarbital and the depth of anesthesia monitored by their response to a tail pinch and observation of the depth of respiration. After surgical exposure of the sciatic nerve, the nerve is slightly elevated, and approximately one-third to one-half of the nerve is tied off. Typically, the surgical procedure is done on the right side while a sham surgery is performed on the left hind leg where the sciatic nerve is only exposed. After recovery (7 d), the animals are tested for the development of mechanical allodynia. The animals are placed in a bottomless plexiglass box placed on a wire mesh (1/4″) platform. After a 30–60 min acclimation period, a baseline mechanical sensitivity is determined by applying a series of calibrated Von Frey fibres perpendicularly to the plantar surface of each hind paw and holding it in place for 6 sec with enough force to slightly bend the fibre. After a positive response (withdrawal of the foot) is observed a weaker fibre is applied until a 50% threshold for withdrawal can be determined. The allodynic threshold is then re-determined after ip administration of the test compound. Testing is conducted at the time-to-peak effect of the compound in the MES test.
Supplementary Material
Acknowledgments
The authors thank the NINDS and the ASP at the National Institutes of Health with Drs. Tracy Chen and Jeffrey Jiang, for kindly performing the pharmacological studies via the ASP’s contract site at the University of Utah with Drs. H. Wolfe, H.S. White, and K. Wilcox. The project was supported by Award Number UL1RR025747 from the National Center for Research Resources and grant R01NS054112 (H.K.) from the National Institute of Neurological Disorders and Stroke. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, National Institute of Neurological Disorders and Stroke or the National Institutes of Health. Harold Kohn has a royalty-stake position in (R)-3.
Footnotes
Abbreviations: FAA, functionalized amino acids; AAA, α-aminoamides; AED, antiepileptic drug; TTX-S, tetrodotoxin-sensitive; MES, maximal electroshock seizure; SAR, structure-activity relationship; IBCF, isobutylchloroformate; NMM, N-methyl morpholine; TBTU, O-(benzotriazol-1-yl), N,N,N′,N′-tetramethyluronium tetrafluoroborate; DMTMM, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methoxymorpholinium chloride; ASP, Anticonvulsant Screening Program; NINDS, National Institute of Neurological Disorders and Stroke; scMet, subcutaneous Metrazol.
Supporting Information Available: Synthetic procedures for the intermediates leading to the preparation of 5,9, (R)-6–8, (R)-10–22, (S)-6, (S)-10, 74, and 75, elemental analyses, 1H and 13C NMR spectra of compounds 5, (R)-6–8, 9, (R)-10–22, (S)-6, (S)-10, 74, and 75 evaluated in this study. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Evans JH. Post-traumatic epilepsy. Neurology. 1962;12:665–674. doi: 10.1212/wnl.12.10.665. [DOI] [PubMed] [Google Scholar]; (b) Lindsay JM. Genetics and epilepsy. Epilepsia. 1971;12:47–54. doi: 10.1111/j.1528-1157.1971.tb03914.x. [DOI] [PubMed] [Google Scholar]
- 2.(a) Rogawski MA, Porter RJ. Antiepileptic drugs: Pharmacological mechanisms and clinical efficacy with consideration of promising development stage compounds. Pharmacol Reviews. 1997;42:223–286. [PubMed] [Google Scholar]; (b) Aiken SP, Brown WM. Treatment of epilepsy: Existing therapies and future developments. Frontiers in Bioscience. 2000;5:124–152. doi: 10.2741/aiken. [DOI] [PubMed] [Google Scholar]
- 3.(a) Brodie MJ, Dichter MA. Antiepileptic drugs. N Engl J Med. 1996;334:168–175. doi: 10.1056/NEJM199601183340308. [DOI] [PubMed] [Google Scholar]; (b) Dichter MA, Brodie MJ. New antiepileptic drugs. New England J Med. 1996;334:1583–1590. doi: 10.1056/NEJM199606133342407. [DOI] [PubMed] [Google Scholar]
- 4.McNamara JO. Pharmacotherapies of the epilepsies. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11. Chapt 19. McGraw-Hill; New York: 2006. pp. 501–525. [Google Scholar]
- 5.(a) McCorry D, Chadwick D, Marson A. Current drug treatment of epilepsy in adults. Lancet Neurol. 2004;3:729–735. doi: 10.1016/S1474-4422(04)00935-4. [DOI] [PubMed] [Google Scholar]; (b) Duncan JS. The promise of new antiepileptic drugs. Brit J Clin Pharmacol. 2002;53:123–131. doi: 10.1046/j.0306-5251.2001.01540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellock JM, Willmore LJ. A rational guide to monitoring in patients receiving anticonvulsants. Neurology. 1991;41:961–964. doi: 10.1212/wnl.41.7.961. [DOI] [PubMed] [Google Scholar]
- 7.Cortes S, Liao ZK, Watson D, Kohn H. Effect of structural modification of the hydantoin ring on anticonvulsant activity. J Med Chem. 1985;28:601–606. doi: 10.1021/jm50001a012. [DOI] [PubMed] [Google Scholar]
- 8.Conley JD, Kohn H. Functionalized D,L-amino acid derivatives. Potent new agents for the treatment to epilepsy. J Med Chem. 1987;30:567–574. doi: 10.1021/jm00386a021. [DOI] [PubMed] [Google Scholar]
- 9.Kohn H, Conley JD. New antiepileptic agents. Chem Br. 1988;24:231–234. [Google Scholar]
- 10.Kohn H, Conley JD, Leander JD. Marked stereospecificity in a new class of anticonvulsants. Brain Res. 1988;457:371–375. doi: 10.1016/0006-8993(88)90709-3. [DOI] [PubMed] [Google Scholar]
- 11.Kohn H, Sawhney KN, LeGall P, Conley JD, Robertson DW, Leander JD. Preparation and anticonvulsant activity of a series of functionalized α-aromatic and α-heteroaromatic amino acids. J Med Chem. 1990;33:919–926. doi: 10.1021/jm00165a006. [DOI] [PubMed] [Google Scholar]
- 12.Kohn H, Sawhney KN, LeGall P, Robertson DW, Leander JD. Preparation and anticonvulsant activity of a series of functionalized α-heteroatom-substituted amino acids. J Med Chem. 1991;34:2444–2452. doi: 10.1021/jm00112a020. [DOI] [PubMed] [Google Scholar]
- 13.Kohn H, Sawhney KN, Bardel P, Robertson DW, Leander JD. Synthesis and anticonvulsant activities of α-heterocyclic α-acetamido-N-benzylacetamide derivatives. J Med Chem. 1993;36:3350–3360. doi: 10.1021/jm00074a016. [DOI] [PubMed] [Google Scholar]
- 14.Bardel P, Bolanos A, Kohn H. Synthesis and anticonvulsant activities of α-acetamido-N-benzylacetamide derivatives containing an electron-deficient α-heteroaromatic substituent. J Med Chem. 1994;37:4567–4571. doi: 10.1021/jm00052a017. [DOI] [PubMed] [Google Scholar]
- 15.Kohn H, Sawhney KN, Robertson DW, Leander JD. Anticonvulsant properties of N-substituted α,α-diamino acid derivatives. J Pharm Sci. 1994;83:689–691. doi: 10.1002/jps.2600830519. [DOI] [PubMed] [Google Scholar]
- 16.Choi D, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl-2-acetamidopropionamide derivatives. J Med Chem. 1996;39:1907–1916. doi: 10.1021/jm9508705. [DOI] [PubMed] [Google Scholar]
- 17.Morieux P, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamide derivatives. Bioorg Med Chem. 2008;16:8968–8975. doi: 10.1016/j.bmc.2008.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salomé C, Salomé-Grosjean E, Park KD, Morieux P, Swendiman R, DeMarco E, Stables JP, Kohn H. Synthesis and anticonvulsant activities of (R)-N-(4′-substituted)benzyl 2-acetamido-3-methoxypropionamides. J Med Chem. 2010;53:1288–1305. doi: 10.1021/jm901563p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoehr T, Kupferberg HJ, Stables JP, Choi D, Harris RH, Kohn H, Walton N, White HS. Lacosamide, a novel anticonvulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epil Res. 2007;74:147–154. doi: 10.1016/j.eplepsyres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Pevarello P, Bonsignori A, Dostert P, Heidempergher F, Pinciroli V, Colombo M, McArthur RA, Salvati P, Post C, Fariello RG, Varasi M. Synthesis and anticonvulsant activity of a new class of 2-[(arylalkyl)amino]alkanamide derivatives. J Med Chem. 1998;41:579–590. doi: 10.1021/jm970599m. [DOI] [PubMed] [Google Scholar]
- 21.Salvati P, Maj R, Caccia C, Cervini MA, Fornaretto MG, Lamberti E, Pevarello P, Skeen GA, White HS, Wolf HH, Faravelli L, Mazzanti M, Mancinelli M, Varasi M, Fariello RG. Biochemical and electrophysiological studies on the mechanism of action of PNU-151774E, a novel antiepileptic compound. J Pharmacol Exp Ther. 1999;288:1151–1159. [PubMed] [Google Scholar]
- 22.Perucca E, Yasothan U, Clincke G, Kirkpatrick P. Lacosamide. Nat Rev Drug Disc. 2008;7:973–974. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]
- 23.Caccia C, Maj R, Calabresi M, Maestroni S, Faravelli L, Curatolo L, Salvati P, Fariello RG. Safinamide: From molecular targets to a new anti-Parkinson drug. Neurology. 2006;67(7, Suppl 2):S18–S23. doi: 10.1212/wnl.67.7_suppl_2.s18. [DOI] [PubMed] [Google Scholar]; (b) http://www.merckserono.com/corp.merckserono/en/images/20090203_en_tcm112_35396.pdf
- 24.Fariello RG. Safinamide. Neurotherap. 2007;4:110–116. doi: 10.1016/j.nurt.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Errington AC, Stoehr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol. 2008;73:157–169. doi: 10.1124/mol.107.039867. [DOI] [PubMed] [Google Scholar]
- 26.Sheets PL, Heers C, Stoehr T, Cummins TR. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J Pharmacol Exp Ther. 2008;326:89–99. doi: 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- 27.Fariello RG, McArtur RA, Bonsignori A, Cervini MA, Maj R, Marrari P, Pevarello P, Wolf HH, Woodhead JW, White HS, Varasi M, Salvati P, Post C. Preclinical evaluation of PNU-151774E as a novel anticonvulsant. J Pharmacol Exp Therap. 1998;285:397–403. [PubMed] [Google Scholar]
- 28.Maj R, Fariello R, Pevarello P, Varasi M, McArthur RA, Salvati P. Anticonvulsant activity of PNU-151774E in the amygdala kindled model of complex partial seizures. Epilepsia. 1999;40:1523–1528. doi: 10.1111/j.1528-1157.1999.tb02035.x. [DOI] [PubMed] [Google Scholar]
- 29.Levy RH, Mattson R, Meldrum B. Antiepileptic Drugs. 4. Chapter 6 Raven Press; New York: 1995. [Google Scholar]
- 30.Pavarello P, Bonsignori A, Caccia C, Amici R, McArthur RA, Fariello RG, Salvatti P, Varasi M. Sodium channel activity and sigma binding of 2-aminopropanamide anticonvulsants. Bioorg Med Chem Lett. 1999;9:2521–2524. doi: 10.1016/s0960-894x(99)00415-1. [DOI] [PubMed] [Google Scholar]
- 31.For related examples that use hydrid (dual) agents, see: Barbachyn MR. In: Annual Reports in Medicinal Chemistry. Macor JE, editor. Vol. 43. Elsevier; London: 2008. pp. 281–292. Chapt. 17.Yogeeswari P, Ragevendran JV, Sriram D, Nageswari Y, Kavya R, Sreevatsan N, Vanitha K, Stables J. Discovery of 4-aminobutyric acid derivatives possessing anticonvulsant and antinociceptive activities: a hybrid pharmacophore approach. J Med Chem. 2007;50:2459–2467. doi: 10.1021/jm061431g.
- 32.Porter RJ, Cereghino JJ, Gladding GD, Hessie BJ, Kupferberg HJ, Scoville B, White BG. Antiepileptic drug development program. Cleveland Clin Q. 1984;51:293–305. doi: 10.3949/ccjm.51.2.293. [DOI] [PubMed] [Google Scholar]
- 33.Anderson GW, Zimmerman JE, Callahan FM. A reinvestigation of the mixed carbonic anhydride method of peptide synthesis. J Am Chem Soc. 1967;87:5012–5017. doi: 10.1021/ja00995a032. [DOI] [PubMed] [Google Scholar]
- 34.Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Mitsunobu and related reactions: advances and applications. Chem Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 35.Maes BUW, Tapolcsanyi P, Meyers C, Matyus P. Palladium-catalyzed reactions on 1,2-diazines. Curr Org Chem. 2006;10:377–417. [Google Scholar]
- 36.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 37.Chinchilla R, Najera C. The Sonogahira reaction: a booming methodology is synthetic organic chemistry. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 38.Kunishima M, Kawachi C, Monta J, Terao K, Iwasaki F, Tani S. 2-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: An efficient condensing agent leading to the formation of amides and esters. Tetrahedron. 1999;55:13159–13170. [Google Scholar]
- 39.For comparable procedures for resolving stereoisomers, see the following: Weisman GR. In: Asymmetric Synthesis-Analytical Methods. Morrison JD, editor. Vol. 1. Academic Press; New York: 1983. pp. 153–171.Parker D, Taylor RJ. Direct 1H NMR assay of the enantiomeric composition of amines and β-amino alcohols using O-acetyl mandelic acid as a chiral solvating agent. Tetrahedron. 1987;43:5431–5456.
- 40.Stables JP, Kupferberg HG. In: Molecular and Cellular Targets for Antiepileptic Drugs. Avanzini G, Tanganelli P, Avoli M, editors. John Libbey; London: 1977. pp. 191–198. [Google Scholar]
- 41.Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- 42.White HS, Woodhead JH, Wilcox KS, Stables JP, Kupferberg HJ, Wolf HH. General Principles: Discovery and Preclinical Development of Antiepileptic Drugs. In: Levy RH, Mattson RH, Meldrum BS, Perruca E, editors. Antiepileptic Drugs. 5. Lippincott, Williams and Wilkins; Philadelphia, PA: 2002. pp. 36–48. [Google Scholar]
- 43.Barton ME, Klein BD, Wolff HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epil Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 44.Swinyard EA. Laboratory evaluation of antiepileptic drugs: review of laboratory methods. Epilepsia. 1969;10:107–119. doi: 10.1111/j.1528-1157.1969.tb03838.x. [DOI] [PubMed] [Google Scholar]
- 45.Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]; (b) Lothman EW, Williamson JM. Closely spaced recurrent hippocampal seizures elicit two types of heightened epileptogenesis: a rapidly developing, transient kindling and a slowly developing, enduring kindling. Brain Res. 1994;649:71–84. doi: 10.1016/0006-8993(94)91050-2. [DOI] [PubMed] [Google Scholar]
- 46.(a) McNamara JO, Byrne MC, Dasheiff RM, Fitz JG. The kindling model of epilepsy: A review. Prog Neurobiol. 1980;15:139–159. doi: 10.1016/0301-0082(80)90006-4. [DOI] [PubMed] [Google Scholar]; (b) Loscher W, Schmidt D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epil Res. 1988;2:145–181. doi: 10.1016/0920-1211(88)90054-x. [DOI] [PubMed] [Google Scholar]
- 47.Seltzer Z, Duner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- 48.Dickinson AH, Matthews EA, Suzuki R. Neurobiology of neuropathic pain: Mode of action of anticonvulsants. Europ J Pain. 2002;6(Suppl A):51–60. doi: 10.1053/eujp.2001.0323. [DOI] [PubMed] [Google Scholar]
- 49.Dickinson T, Lee K, Spanswick D, Munro FE. Leading the charge–pioneering treatments in the fight against neuropathic pain. Trends Pharmacol Sci. 2003;24:555–557. doi: 10.1016/j.tips.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 50.(a) Hunskaar S, Fasmer OB, Hole K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods. 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]; (b) Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- 51.Stoehr T, Krause E, Selve N. Lacosamide displays potent antinociceptive effects in animal models for inflammatory pain. Europ J Pain. 2006;10:241–249. doi: 10.1016/j.ejpain.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and developmental settings. Adv Drug Del Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 53.(a) Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Dis. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]; (b) Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 54.(a) Macdonald RL. Antiepileptic drug actions. Epilepsia. 1989;30:S19–S28. doi: 10.1111/j.1528-1157.1989.tb05810.x. [DOI] [PubMed] [Google Scholar]; (b) Stahl SM. Psychopharmacology of anticonvulsants: Do all anticonvulsants have the same mechanism of action? J Clin Psych. 2004;65:149–150. doi: 10.4088/jcp.v65n0201. [DOI] [PubMed] [Google Scholar]; (c) Errington AC, Stoehr T, Lees G. Voltage gated ion channels: Targets for anticonvulsant drugs. Curr Targ Med Chem. 2005;5:15–30. doi: 10.2174/1568026053386872. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.