

Abstract
Background
Ecological, biochemical and genetic resemblance as well as clear differences of virulence between L. monocytogenes and L. innocua make this bacterial clade attractive as a model to examine evolution of pathogenicity. This study was attempted to examine the population structure of L. innocua and the microevolution in the L. innocua-L. monocytogenes clade via profiling of 37 internalin genes and multilocus sequence typing based on the sequences of 9 unlinked genes gyrB, sigB, dapE, hisJ, ribC, purM, gap, tuf and betL.
Results
L. innocua was genetically monophyletic compared to L. monocytogenes, and comprised four subgroups. Subgroups A and B correlated with internalin types 1 and 3 (except the strain 0063 belonging to subgroup C) and internalin types 2 and 4 respectively. The majority of L. innocua strains belonged to these two subgroups. Subgroup A harbored a whole set of L. monocytogenes-L. innocua common and L. innocua-specific internalin genes, and displayed higher recombination rates than those of subgroup B, including the relative frequency of occurrence of recombination versus mutation (ρ/θ) and the relative effect of recombination versus point mutation (r/m). Subgroup A also exhibited a significantly smaller exterior/interior branch length ratio than expected under the coalescent model, suggesting a recent expansion of its population size. The phylogram based on the analysis with correction for recombination revealed that the time to the most recent common ancestor (TMRCA) of L. innocua subgroups A and B were similar. Additionally, subgroup D, which correlated with internalin type 5, branched off from the other three subgroups. All L. innocua strains lacked seventeen virulence genes found in L. monocytogenes (except for the subgroup D strain L43 harboring inlJ and two subgroup B strains bearing bsh) and were nonpathogenic to mice.
Conclusions
L. innocua represents a young species descending from L. monocytogenes and comprises four subgroups: two major subgroups A and B, and one atypical subgroup D serving as a link between L. monocytogenes and L. innocua in the evolutionary chain. Although subgroups A and B appeared at approximately the same time, subgroup A seems to have experienced a recent expansion of the population size with higher recombination frequency and effect than those of subgroup B, and might represent the possible evolutionary direction towards adaptation to enviroments. The evolutionary history in the L. monocytogenes-L. innocua clade represents a rare example of evolution towards reduced virulence of pathogens.
Background
The genus Listeria encompasses six species, which could be divided into three major phylogenetic clusters: L. monocytogenes-L. innocua, L. ivanovii-L. seeligeri-L. welshimeri, and L. grayi [1]. L. monocytogenes is a well-recognized intracellular pathogen of humans and animals, with clinical features including meningitis, meningoencephalitis, septicemia, abortion, perinatal infections and gastroenteritis [2]. Three genetic lineages have been identified within L. monocytogenes strains, with lineage I including serovars 1/2b, 3b, 4b, 4d, 4e, 4ab and 7, lineage II covering serovars 1/2a, 3a, 1/2c and 3c, and lineage III comprising sublineages IIIA, IIIB and IIIC covering serovars 4a, 4c and atypical 4b [3-5]. Of the 13 L. monocytogenes serovars, four (4b, 1/2a, 1/2b and 1/2c) are responsible for over 98% of human listeriosis cases whereas other serovars (e.g., 4a and 4c) are seldom implicated in listeriosis [6,7]. L. innocua is of particular significance because it is most closely related to L. monocytogenes, and they usually co-exist in various environmental, food and clinical specimens [8]. Although these two species resemble each other ecologically, biochemically and genetically, L. innocua has no pathogenic inclination [1,9]. Therefore, the L. innocua-L. monocytogenes clade within the genus Listeria can be used as a model system to examine the evolution of pathogenicity.
Intriguingly, some L. monocytogenes strains tend to lose virulence factors that play critical roles in infection, which has been considered as a rare example of evolution towards reduced virulence of pathogens [4,10]. Certain L. monocytogenes lineage IIIA strains are presumed to have identifiable linkage between L. monocytogenes and L. innocua by possessing many genes common to L. monocytogenes [e.g., Listeria pathogenicity island I (LIPI-1), inlAB locus, bsh and hpt], and sharing many gene deletions similar to L. innocua (e.g., inlC, inlI, inlJ, internalin cluster between ascB and dapE, and arginine deiminase island lmo0036-lmo0041) [11-13]. Therefore, the population structure and biodiversity in L. innocua may, from the other side of the coin, provide clues for the evolutionary history in the L. monocytogenes-L. innocua clade. Unfortunately, comprehensive knowledge on the phylogenetic structure of L. innocua is still lacking.
Various strain typing methods have been developed and improved with a general shift from phenotype-based to genotype-based strategies [14]. Given its accuracy, reproducibility and increased speed of DNA sequencing, DNA sequence-based multilocus sequence typing (MLST) has gained more popularity [15], and provided an overview of the population structure of L. monocytogenes [4,16]. Internalin profiling seems to be instrumental to subtype L. monocytogenes strains into different serovars [17]. Moreover, internalin loci are also present in non-pathogenic species, including L. innocua, and seem to play broad roles not merely limited to attachment and invasion of host cells [18-20].
In this study, we attempted to delineate a phylogenetic framework based on internalin profiling and MLST analysis of a collection of L. innocua isolates from various food sources, and further to investigate microevolution in the L. innocua-L. monocytogenes clade.
Results
Biochemical patterns of L. innocua and L. monocytogenes strains
All L. innocua and L. monocytogenes strains displayed similar utilization patterns for xylose (negative), mannitol (negative) and glucose (positive), while hemolysis could distinguish these two species with L. monocytogenes showing β-hemolysis and all L. innocua strains being non-hemolytic. With regard to the rhamnose utilization pattern, three L. innocua strains 386, L19 and L103 (3/34, 8.8%) as well as L. monocytogenes sublineages IIIB and IIIC strains covering serovars 4a, 4b and 4c were atypically negative for rhamnose fermentation (Table 1).
Table 1.
MLST and mouse assay of L. innocua and L. monocytogenes strains
| Strain | Source | Subgroup/ lineage (serovar) |
rhamnose | Allele designation | Virulence gene | Mouse assay | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gyrB | dapE | hisJ | sigB | ribC | purM | betL | gap | tuf | STa | bsh | inlJ | CFU | Relative virulence (%) | ||||||
| L. innocua | |||||||||||||||||||
| ATCC33090 | reference | A | + | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | -- | -- | 3.5 × 107 | 0 | ||
| 90001 | reference | B | + | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | -- | -- | 2.7 × 107 | 0 | ||
| 1603 | reference | B | + | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 2 | 3 | + | -- | 2.0 × 107 | 0 | ||
| AB2497 | reference | A | + | 1 | 1 | 4 | 1 | 1 | 1 | 1 | 1 | 1 | 4 | -- | -- | 3.3 × 107 | 0 | ||
| CLIP11262 | reference | A | + | 1 | 4 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 5 | -- | -- | ND | ND | ||
| 0063 | meat | C | + | 4 | 5 | 6 | 4 | 4 | 5 | 4 | 1 | 1 | 6 | -- | -- | 5.3 × 107 | 0 | ||
| 0065 | meat | A | + | 1 | 6 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 7 | -- | -- | 1.5 × 107 | 0 | ||
| 0068 | meat | B | + | 2 | 2 | 5 | 2 | 5 | 2 | 5 | 1 | 2 | 8 | -- | -- | 1.8 × 107 | 0 | ||
| 0072 | meat | A | + | 1 | 7 | 5 | 5 | 1 | 4 | 3 | 2 | 1 | 9 | -- | -- | 2.1 × 107 | 0 | ||
| 0082 | meat | A | + | 1 | 7 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 10 | -- | -- | 2.3 × 107 | 0 | ||
| 0083 | meat | A | + | 1 | 8 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 11 | -- | -- | 1.7 × 107 | 0 | ||
| 0173 | meat | A | + | 4 | 9 | 1 | 6 | 6 | 1 | 6 | 1 | 1 | 12 | -- | -- | 2.2 × 107 | 0 | ||
| 0197 | meat | A | + | 4 | 10 | 1 | 6 | 6 | 1 | 6 | 1 | 1 | 13 | -- | -- | 1.9 × 107 | 0 | ||
| 01174 | meat | A | + | 5 | 11 | 7 | 7 | 7 | 4 | 1 | 1 | 1 | 14 | -- | -- | 1.3 × 107 | 0 | ||
| 01178 | meat | A | + | 1 | 12 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 15 | -- | -- | 3.3 × 107 | 0 | ||
| 01182 | meat | A | + | 1 | 13 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 16 | -- | -- | 2.3 × 107 | 0 | ||
| 317 | milk | A | + | 1 | 1 | 5 | 1 | 1 | 1 | 7 | 1 | 1 | 17 | -- | -- | 3.3 × 107 | 0 | ||
| 337 | milk | B | + | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 2 | 3 | -- | -- | 2.0 × 107 | 0 | ||
| 376 | milk | A | + | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | -- | -- | 2.5 × 107 | 0 | ||
| 380 | milk | B | + | 3 | 3 | 3 | 3 | 8 | 3 | 2 | 1 | 2 | 18 | -- | -- | 2.1 × 107 | 0 | ||
| 386 | milk | B | -- | 5 | 14 | 8 | 8 | 9 | 4 | 8 | 1 | 1 | 19 | + | -- | 3.0 × 107 | 0 | ||
| 438 | milk | B | + | 6 | 15 | 9 | 9 | 10 | 6 | 3 | 3 | 1 | 20 | -- | -- | 1.6 × 107 | 0 | ||
| 693 | milk | A | + | 1 | 12 | 7 | 1 | 1 | 4 | 3 | 1 | 1 | 21 | -- | -- | 2.3 × 107 | 0 | ||
| 694 | milk | A | + | 5 | 11 | 1 | 7 | 11 | 4 | 9 | 1 | 1 | 22 | -- | -- | 4.7 × 107 | 0 | ||
| ZS14 | seafood | A | + | 1 | 12 | 5 | 1 | 1 | 7 | 10 | 1 | 1 | 23 | -- | -- | 4.3 × 107 | 0 | ||
| ZXF | seafood | B | + | 3 | 3 | 3 | 3 | 12 | 3 | 2 | 1 | 2 | 24 | -- | -- | 3.3 × 107 | 0 | ||
| 1571 | seafood | A | + | 1 | 16 | 5 | 1 | 1 | 4 | 3 | 1 | 1 | 25 | -- | -- | 2.0 × 107 | 0 | ||
| L19 | seafood | B | -- | 7 | 17 | 10 | 10 | 13 | 8 | 11 | 4 | 2 | 26 | -- | -- | 1.5 × 107 | 0 | ||
| L43 | seafood | D | + | 8 | 18 | 11 | 11 | 14 | 9 | 12 | 5 | 3 | 27 | -- | + | 1.7 × 107 | 0 | ||
| NB2 | seafood | B | + | 3 | 3 | 3 | 3 | 15 | 3 | 2 | 1 | 2 | 28 | -- | -- | 3.5 × 107 | 0 | ||
| NB3 | seafood | B | + | 3 | 3 | 3 | 3 | 15 | 3 | 2 | 1 | 2 | 28 | -- | -- | 3.7 × 107 | 0 | ||
| NB24 | seafood | B | + | 5 | 19 | 12 | 12 | 16 | 2 | 13 | 1 | 1 | 29 | -- | -- | 2.9 × 107 | 0 | ||
| L87 | pork | B | + | 5 | 19 | 12 | 7 | 16 | 10 | 13 | 1 | 1 | 30 | -- | -- | 1.3 × 107 | 0 | ||
| L103 | chicken | A | -- | 1 | 12 | 5 | 1 | 1 | 11 | 14 | 1 | 1 | 31 | -- | -- | 4.0 × 107 | 0 | ||
| L. monocytogenes | |||||||||||||||||||
| SH3 | pork | I (1/2b) | + | 9 | 20 | 13 | 13 | 17 | 12 | 15 | 6 | 4 | 32 | + | + | 4.3 × 107 | 100 | ||
| NB26 | seafood | I (1/2b) | + | 10 | 21 | 14 | 14 | 18 | 13 | 16 | 6 | 4 | 33 | + | + | 6.5 × 107 | 100 | ||
| NB27 | seafood | I (1/2b) | + | 11 | 22 | 15 | 14 | 19 | 14 | 17 | 7 | 4 | 34 | + | + | 5.5 × 107 | 80 | ||
| M1 | milk | I (1/2b) | + | 11 | 23 | 13 | 14 | 19 | 15 | 18 | 8 | 4 | 35 | + | + | 3.0 × 107 | 100 | ||
| ScottA | reference | I (4b) | + | 11 | 24 | 15 | 15 | 19 | 16 | 19 | 8 | 4 | 36 | + | + | 3.3 × 107 | 100 | ||
| NB4 | seafood | I (4b) | + | 11 | 25 | 16 | 15 | 19 | 14 | 20 | 6 | 4 | 37 | + | + | 2.1 × 107 | 100 | ||
| NB6 | seafood | I (4b) | + | 11 | 26 | 17 | 16 | 20 | 16 | 21 | 6 | 4 | 38 | + | + | 3.3 × 107 | 100 | ||
| NB7 | seafood | I (4b) | + | 11 | 26 | 17 | 16 | 20 | 16 | 22 | 6 | 4 | 39 | + | + | 4.6 × 107 | 100 | ||
| NB25 | seafood | I (4b) | + | 11 | 27 | 18 | 15 | 19 | 17 | 19 | 6 | 4 | 40 | + | + | 4.6 × 107 | 100 | ||
| 90SB1 | animal | I (4b) | + | 11 | 27 | 16 | 15 | 19 | 17 | 19 | 6 | 4 | 41 | + | + | 5.5 × 107 | 100 | ||
| EGDe | reference | II (1/2a) | + | 12 | 28 | 19 | 17 | 21 | 18 | 20 | 9 | 5 | 42 | + | + | 5.5 × 107 | 100 | ||
| 10403S | reference | II (1/2a) | + | 13 | 29 | 20 | 17 | 22 | 19 | 20 | 10 | 5 | 43 | + | + | 5.0 × 107 | 100 | ||
| SH2 | vegetable | II (1/2a) | + | 13 | 29 | 21 | 17 | 23 | 20 | 20 | 9 | 5 | 44 | + | + | 4.3 × 107 | 100 | ||
| SH4 | chicken | II (1/2a) | + | 13 | 29 | 20 | 17 | 22 | 19 | 20 | 11 | 5 | 45 | + | + | 5.0 × 107 | 100 | ||
| NB5 | seafood | II (1/2a) | + | 13 | 29 | 22 | 17 | 24 | 21 | 23 | 12 | 5 | 46 | + | + | 4.5 × 107 | 100 | ||
| NB21 | seafood | II (1/2a) | + | 13 | 29 | 23 | 17 | 25 | 22 | 23 | 13 | 5 | 47 | + | + | 3.9 × 107 | 80 | ||
| P3 | pork | II (1/2a) | + | 13 | 23 | 24 | 18 | 26 | 23 | 24 | 13 | 5 | 48 | + | + | 4.0 × 107 | 100 | ||
| NB28 | seafood | II (1/2c) | + | 12 | 23 | 19 | 17 | 21 | 18 | 20 | 14 | 6 | 49 | + | + | 4.1 × 107 | 100 | ||
| V1 | vegetable | II (1/2c) | + | 12 | 23 | 19 | 17 | 21 | 18 | 20 | 9 | 6 | 50 | + | + | 3.0 × 107 | 100 | ||
| P19 | chicken | II (1/2c) | + | 12 | 30 | 19 | 17 | 21 | 18 | 20 | 9 | 6 | 51 | + | + | 5.0 × 107 | 100 | ||
| 54006 | reference | IIIA (4a) | + | 14 | 31 | 25 | 19 | 27 | 24 | 3 | 15 | 2 | 52 | + | -- | 1.3 × 107 | 0 | ||
| F2-695 | reference | IIIA(4a) | + | 15 | 32 | 26 | 20 | 28 | 25 | 25 | 15 | 7 | 53 | + | + | 1.2 × 107 | 40 | ||
| F2-086 | reference | IIIB (4a) | -- | 16 | 33 | 27 | 21 | 29 | 26 | 26 | 16 | 8 | 54 | + | -- | 1.7 × 107 | 100 | ||
| F2-407 | reference | IIIB (4a) | -- | 17 | 34 | 28 | 21 | 30 | 26 | 27 | 16 | 9 | 55 | + | -- | 1.5 × 107 | 100 | ||
| F2-270 | reference | IIIB (4a) | -- | 18 | 35 | 29 | 21 | 31 | 27 | 28 | 16 | 8 | 56 | + | -- | 2.2 × 107 | 100 | ||
| F2-208 | reference | IIIC (4a) | -- | 19 | 36 | 30 | 22 | 32 | 28 | 29 | 16 | 10 | 57 | + | -- | 3.5 × 107 | 100 | ||
| F2-525 | reference | IIIA (4b) | + | 20 | 37 | 31 | 23 | 33 | 29 | 30 | 17 | 11 | 58 | + | + | 2.8 × 107 | 100 | ||
| J1-158 | reference | IIIB (4b) | -- | 21 | 34 | 28 | 21 | 29 | 30 | 31 | 16 | 8 | 59 | + | -- | 2.2 × 107 | 40 | ||
| J2-071 | reference | IIIA (4c) | + | 22 | 38 | 26 | 20 | 28 | 31 | 25 | 15 | 12 | 60 | + | + | 1.5 × 107 | 100 | ||
| W1-111 | reference | IIIC (4c) | -- | 23 | 39 | 32 | 24 | 34 | 32 | 32 | 18 | 2 | 61 | + | -- | 2.8 × 107 | 80 | ||
| L. welshimeri | |||||||||||||||||||
| SLCC5334 | reference | ND | -- | 24 | 40 | 33 | 25 | 35 | 33 | 33 | 19 | 13 | 62 | -- | -- | ND | ND | ||
| C15 | reference | ND | -- | 25 | 41 | 34 | 26 | 36 | 34 | 34 | 20 | 14 | 63 | -- | -- | 5.5 × 107 | 0 | ||
| AB1626 | reference | ND | -- | 25 | 41 | 35 | 27 | 36 | 34 | 34 | 20 | 14 | 64 | -- | -- | ND | ND | ||
| AB2500 | reference | ND | -- | 24 | 40 | 36 | 25 | 35 | 33 | 33 | 19 | 13 | 65 | -- | -- | ND | ND | ||
ND, not done. a ST, sequence type.
Internalin profiling indicates five internalin types (ITs) of L. innocua
Upon examination of 14 L. monocytogenes-L. innocua common, 4 L. innocua-specific and 19 L. monocytogenes-specific internalin genes, L. innocua strains harbored 15 to 18 internalin genes, with three L. monocytogenes-L. innocua common and one L. innocua-specific internalin genes absent individually or in combination in certain L. innocua strains (Table S1; Additional file 1). Eighteen L. monocytogenes-specific internalin genes were absent in all L. innocua strains except for L43 having inlJ (Table 1). These L. innocua strains could be separated into five internalin types (ITs), with IT1 containing a whole set of L. monocytogenes-L. innocua common and L. innocua-specific internalin genes, IT2 lacking lin1204, IT3 lacking lin1204 and lin2539, IT4 lacking lin0661, lin0354 and lin2539, and IT5 lacking lin1204 but bearing inlJ. The majority of L. innocua strains fell into IT1 (17/34, 50%) and IT2 (12/34, 35.4%). Among the remainders, three belonged to IT3 (8.8%), one to IT4 (2.9%) and one to IT5 (2.9%). In addition, all L. monocytogenes strains contained L. monocytogenes-L. innocua common internalin genes ranging from 6 to 13, and lacked all L. innocua-specific internalin genes (Table 2).
Table 2.
Internalin profiling of L. innocua and L. monocytogenes strains
| IT | No. of internalin genes | Characteristics | No. (%) of strains | No. (%) of strains belonging to each subgroup | ||||
|---|---|---|---|---|---|---|---|---|
| common and L. innocua -specific |
L. monocytogenes -specific |
A | B | C | D | |||
| 1 | 18 | 0 | whole set of common and L. innocua-specific internalin genes | 17 (50.0%) | 17 | 0 | 0 | 0 |
| 2 | 17 | 0 | lin1204 negative | 12 (35.4%) | 0 | 12 | 0 | 0 |
| 3 | 16 | 0 | Lin1204, lin2539 negative | 3 (8.8%) | 2 | 0 | 1 | 0 |
| 4 | 15 | 0 | lin0661, lin0354, lin2539 negative | 1 (2.9%) | 0 | 1 | 0 | 0 |
| 5 | 17 | 1 | lin1204 negative, and inlJ positive | 1 (2.9%) | 0 | 0 | 0 | 1 |
| Total | 18 | 19 | -- | 34 | 19 (55.9%) | 13 (38.3%) | 1 (2.9%) | 1 (2.9%) |
MLST correlates with internalin profiling of L. innocus strains
Sixty-four strains in the L. monocytogenes-L. innocua clade were classified into 61 unique sequence types (ST) in the MLST scheme with a high discrimination index (DI = 0.99, 0.76 to 0.98 per gene). The concatenated sequence data showed that L. innocua was genetically monophyletic as compared to L. monocytogenes, with 34 L. innocua and 30 L. monocytogenes strains bearing 391 (6.69%) and 820 (14.03%) polymorphisms respectively. The average nucleotide diversity π of L. innocua was lower than that of L. monocytogenes (1.06% vs 4.38%). However, the nonsynonymous/synonymous mutation rate of L. innocua was higher than that of L. monocytogenes (0.0865 vs 0.0500) (Table 3).
Table 3.
Polymorphisms at nine genes in the L. innocua-L. monocytogenes clade
| Gene | No. strains | Size (bp) | No. alleles | No. (%) polymorphic sites | D.I. | Ks | Ka | Ka/Ks | π |
|---|---|---|---|---|---|---|---|---|---|
| gyrB | 64 | 657 | 23 | 74 (11.26%) | 0.91 | 0.1991 | 0.0010 | 0.0050 | 0.0384 |
| dapE | 64 | 669 | 39 | 146 (21.82%) | 0.98 | 0.2337 | 0.0152 | 0.0650 | 0.0564 |
| hisJ | 64 | 714 | 32 | 187 (26.19%) | 0.95 | 0.3999 | 0.0380 | 0.0951 | 0.1000 |
| sigB | 64 | 642 | 24 | 83 (12.93%) | 0.92 | 0.2588 | 0.0054 | 0.0207 | 0.0518 |
| ribC | 64 | 633 | 34 | 163 (25.75%) | 0.93 | 0.6002 | 0.0209 | 0.0348 | 0.1093 |
| purM | 64 | 693 | 31 | 170 (24.53%) | 0.94 | 0.3955 | 0.0194 | 0.0490 | 0.0853 |
| betL | 64 | 534 | 32 | 171 (32.02%) | 0.94 | 0.7918 | 0.0312 | 0.0394 | 0.1325 |
| gap | 64 | 621 | 18 | 28 (4.51%) | 0.76 | 0.0240 | 0.0013 | 0.0547 | 0.0067 |
| tuf | 64 | 681 | 11 | 14 (2.06%) | 0.80 | 0.0182 | 0.0021 | 0.1160 | 0.0058 |
| Concatenated | 64 | 5,844 | 61 | 1036 (17.73%) | 0.99 | 0.2621 | 0.0147 | 0.0559 | 0.0623 |
| Concatenated, L. innocua | 34 | 5,844 | 31 | 391 (6.69%) | 0.99 | 0.0365 | 0.0032 | 0.0865 | 0.0106 |
| Concatenated, subgroupA | 19 | 5,844 | 18 | 90 (1.54%) | 0.99 | 0.0142 | 0.0018 | 0.1241 | 0.0046 |
| Concatenated, subgroup B | 13 | 5,844 | 11 | 135 (2.31%) | 0.97 | 0.0280 | 0.0018 | 0.0628 | 0.0077 |
| Concatenated, subgroup C | 1 | 5,844 | 1 | -- | -- | 0.4659 | 0.0241 | 0.0517 | -- |
| Concatenated, subgroup D | 1 | 5,844 | 1 | -- | -- | 0.4867 | 0.0225 | 0.0461 | -- |
| Concatenated, L. monocytogenes | 30 | 5,844 | 30 | 820 (14.03%) | 1.00 | 0.1781 | 0.0089 | 0.0500 | 0.0438 |
| Concatenated, lineage I | 10 | 5,844 | 4 | 84 (1.44%) | 1.00 | 0.0174 | 0.0019 | 0.1112 | 0.0055 |
| Concatenated, lineage II | 10 | 5,844 | 6 | 250 (4.28%) | 1.00 | 0.0493 | 0.0027 | 0.0537 | 0.0131 |
| Concatenated, lineage III | 10 | 5,844 | 10 | 522 (8.93%) | 1.00 | 0.1459 | 0.0084 | 0.0575 | 0.0374 |
D.I.: discrimination index; Ks: number of synonymous changes per synonymous site; Ka: number of non-synonymous changes per non-synonymous site; π: nucleotide diversity.
With L. welshimeri as the outgroup species, the phylogenetic tree revealed nine major branches of the L. monocytogenes-L. innocua clade, four corresponding to the recognized L. monocytogenes lineages I, II, IIIA/C and IIIB, one harboring the low-virulent L. monocytogenes lineage IIIA strains reported in our previous study [11], and the other four beloning to L. innocua (Figure 1). The majority of L. innocua strains were placed in two branches: one contained 19 strains (55.9%) representing STs 1, 4, 5, 7, 9-17, 21-23, 25 and 31, and the other harbored 13 strains (38.2%) representing STs 2, 3, 6, 8, 18-20, 24, 26 and 28-30. Remarkably, L. innocua strain L43 (ST27) showed the least genetic distance to the main cluster of L. monocytogenes. This strain seems to serve as the evolutionary intermediate between L. monocytogenes and L. innocua main clusters together with the low-virulent L. monocytogenes lineage IIIA strain 54006. Additionally, L. innocua strain 0063 (ST6) was present on the halfway between the L. innocua main cluster and strain L43 (Figure 1).
Figure 1.

Neighbor-joining cladogram of 34 L. innocua and 30 L. monocytogenes strains by the concatenated data set gyrB-dapE-hisJ-sigB-ribC-purM-betL-gap-tuf with L. welshimeri as outgroup species. Leaves are labeled with sequence type (ST) designations. The numbers I, II, IIIA, IIIB, IIIC, A, B, C and D, on the branches represent L. monocytogenes lineages I, II, IIIA, IIIB, and IIIC, and L. innocua subgroups A, B, C and D respectively. IIIA* represents the low-virulent L. monocytogenes sublineages IIIA (seovar 4a) strain [11].
Based on the MLST scheme and internalin profiling, L. innocua could be divided into at least four subgroups. Two main subgroups A and B located in the two major branches of the phylogenetic tree, which correlated with IT1 and IT3 (except strain 0063) (19/34, 55.9%), and IT2 and IT4 (13/34, 38.2%) respectively. In addition, one IT3 strain 0063 and one IT5 strain L43 present in two individual branches formed subgroups C and D respectively (Table 2).
Phylogeny and population history of L. innocua
As aforementioned, L. innocua was genetically monophyletic (π = 1.06%) as compared to L. monocytogenes (π = 4.38%). When sequence data were analyzed after stratification by subgroups, the number of polymorphisms and genetic diversity within each subpopulations were reduced (Table 3), suggesting a barrier for genetic exchange between these L. innocua subgroups. Such barrier was also observed between L. monocytogenes lineages (Table 3), consistent with one previous report [21].
Tajima's D test revealed that L. innocua and L. monocytogenes did not evolve under neutrality. A marginal positive value of Tajima's D observed for ribC in L. monocytogenes (1.9963, 0.05 < p < 0.10) became smaller or negative when analyses were performed for separate lineages, suggesting a divided population structure. Similarly, significant or marginal positive Tajima's D values were observed for gyrB (2.0401, p < 0.05) in L. monocytogenes lineage II, and for sigB (2.0426, p < 0.05) and gap (1.7746, 0.05 < p < 0.10) in lineage III, supporting that lineages II and III represented diverse populations as compared to lineage I (Table 4). On the other hand, gyrB (-2.2650, p < 0.01), betL (-2.5954, p < 0.001) and gap (-2.4190, p < 0.01) showed significant negative Tajima's D values in L. innocua, indicative of a bottleneck or selective sweep [22,23]. Also, Tajima's D were marginal negative for betL in L. innocua subgroup A (-1.7315, 0.05 < p < 0.10) and gap in subgroup B (-1.6523, 0.05 < p < 0.10) (Table 4).
Table 4.
Tajima's D test for the L. innocua-L. monocytogenes clade
| Gene | L. innocua | L. monocytogenes | |||||
|---|---|---|---|---|---|---|---|
| A | B | all | I | II | III | all | |
| gyrB | -0.3479 | 0.3871 | -2.2650** | -1.6671# | 2.0401* | 0.0136 | 0.7361 |
| dapE | 0.7970 | 1.1138 | -1.0723 | -0.0394 | -0.4958 | 0.9003 | -0.3265 |
| hisJ | 1.2046 | 0.1750 | 0.2478 | -0.1104 | -0.6528 | 0.0336 | 1.4256 |
| sigB | -0.1097 | 0.5901 | 0.2092 | 0.5444 | -1.1117 | 2.0426* | 1.2456 |
| ribC | 0.0511 | 0.2773 | 0.2987 | 1.5368 | -1.5344 | 0.4909 | 1.9963# |
| purM | 0.5044 | 0.2217 | -1.4464 | 0.0235 | -0.2856 | 0.9867 | 0.4698 |
| betL | -1.7315# | -1.5047 | -2.5954*** | -0.2912 | -0.1839 | 0.5179 | 0.0554 |
| gap | -1.1648 | -1.6523# | -2.4190** | -0.6910 | -0.8223 | 1.7746# | 0.2481 |
| tuf | N/Aa | 0.9505 | -0.0101 | N/A | 0.8198 | 0.5380 | 0.4709 |
| Concatenated | 0.1719 | 0.1492 | 0.3847 | 0.3655 | -0.7070 | 0. 7379 | 0.7452 |
#, 0.05 < p < 0.10; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
a. No polymorphisms in the data, resulting in inability to compute Tajima's test.
The exterior/interior branch length ratio test demonstrated that L. innocua and its subgroup A as well as L. monocytogenes and its lineage I showed a significantly smaller exterior/interior branch length ratio (p < 0.05) than expected under the coalescent model (Figure 2). This suggests that the contemporary L. innocua population experienced a recent expansion of its population size, consistent with a population bottleneck. Specifically, L. innocua subgroup A underwent expansion of the population size (p = 0.027), while subgroup II did not (p = 0.176) (Figure 2).
Figure 2.

Population history in L. innocua-L. monocytogenes clade inferred by the distribution of the exterior/interior branch length ratio of trees resulting from ClonalFrame analysis as compared to trees simulated under the coalescent model. L. innocua spp. (A) and its group subgroup I (B), and L. monocytogenes spp. (D) and its lineage I (E) show a significantly smaller exterior/interior branch length ratio (p < 0.05) than expected under the coalescent model, while L. innocua subgroup II (C) and L. monocytogenes lineages II (F) and III (G) do not.
The rate of recombination within bacterical species can differ widely from one species to another. In the L. innocua-L. monocytogenes clade, both the relative frequency of occurrence of recombination versus mutation (ρ/θ) and the relative effect of recombination versus point mutation (r/m) were about two to three times higher in L. innocua than in L. monocytogenes (Table 5). L. innocua subgroup A exhibited significantly higher frequency (ρ/θ = 3.7697) and effect (r/m = 12.0359) of recombination than subgroup B (ρ/θ = 0.2818; r/m = 4.8132), consistent with a definite population expansion of subgroup A as aforementioned. However, the higher recombination rate of L. innocua subgroup A did not seem to contribute to nucleotide diversity (π for subgroups A and B are 0.46% and 0.77% respectively) (Table 3 and Table 5). On the other hand, both the frequency and effect of recombination in L. monocytogenes lineage II were higher than those in lineages I and III (Table 5).
Table 5.
Recombination rates in the L. innocua-L. monocytogenes clade and other bacteria
| r/ma | ρ/θb | Reference | |
|---|---|---|---|
| L. innocua | 3.144 (2.234-4.071) | 0.535 (0.396-0.764) | This study |
| L. innocua subgroup A | 12.036 (5.404-20.716) | 3.770 (2.021-6.188) | This study |
| L. innocua subgroup B | 4.813 (1.431-20.455) | 0.282 (0.095-1.124) | This study |
| L. monocytogenes | 1.847 (1.293-2.641) | 0.179 (0.135-0.258) | This study |
| L. monocytogenes lineage I | 5.752 (1.413-18.660) | 0.055 (0.023-0.118) | This study |
| L. monocytogenes lineage II | 7.610 (5.096-11.065) | 0.518 (0.244-0.801) | This study |
| L. monocytogenes lineage III | 1.869 (0.720-5.117) | 0.195 (0.066-0.661) | This study |
| L. innocua-L. monocytogenes clade | 2.783 (2.326-3.307) | 0.334 (0.284-0.395) | This study |
| Bacillus anthracis-Bacillus cereus clade | ND | 0.2-0.5 | Didelot et al. 2007 |
| Clostridium perfringens | ND | 3.2 | Rooney et al. 2006 |
| Neisseria meningitis | ND | 1.1 | Jolley et al. 2005 |
| Staphylococcus aureus | ND | 0.11 | Fraser et al. 2005 |
| Streptococcus pneumoniae | ND | 2.1 | Fraser et al. 2005 |
ND, not done.
a. The ratio of probabilities that a given site is altered through recombination and mutation, representing a measure of how important the effect of recombination is in the diversification of the sample relative to mutation.
b. The ratio of rates at which recombination and mutation occur, representing a measure of how often recombination events happen relative to mutations.
The phylogram based on the analysis with correction for recombination revealed that the time to the most recent common ancestor (TMRCA) of L. innocua subgroups A and B was similar (Figure 3), suggesting that these two subgroups appeared at approximately the same time. In addition, our study also showed the TMRCA of L. monocytogenes lineages I and II were similar, consistent with a recent report [24].
Figure 3.

A 95% majority-rule consensus tree based on ClonalFrame output with correction for recombination. The X-axis represents the estimated time to the most recent common ancestors (TMRCA) of the L. innocua-L. monocytogenes clade. Blue dash line shows the estimated time to the most recent common ancestors of L. innocua subgroups I and II.
Distribution of L. innocua isolates among different sources
Of the 29 L. innocua food isolates, 13 were obtained from meat, 8 from milk and 8 from seafoods. The majority of meat isolates (10/13, 76.9%) belonged to subgroup A, while most seafood isolates (5/8, 62.5%) belonged to subgroup B. There were significant associations between subgroups and source of isolation (p < 0.05).
L. innocua isolates lack virulence genes found in L. monocytogenes, and were nonpathogenic to mice
All L. innocua strains lacked 17 virulence genes examined, with the exception of the subgroup D strain (L43) harboring inlJ (87.5%-93.6% nucleotide identities to L. monocytogenes reference strains EGDe and F2365) and two subgroup B strains (1603 and 386) bearing bsh (97.7%-99.4% nucleotide identities to EGDe and F2365). All of these L. innocua strains were nonpathogenic to ICR mice (Table 1).
Discussion
The ecological, biochemical and genetical resemblance as well as the clear differences of virulence between L. monocytogenes and L. innocua make this bacterial clade attractive as models to examine the evolution of pathogenicity in Listeria genus. L. monocytogenes causes life-threatening infections in animals and human populations, and exhibits a diversity of strains with different pathogenicity [25]. L. innocua has once been postulated as the nonpathogenic variant of L. monocytogenes, and holds the key to understanding the evolutionary history of the L. monocytogenes-L. innocua clade. However, information on the phylogenetic structure and microevolution of L. innocua is still lacking. Thus, we characterized L. innocua strains in our laboratory stock from phylogentic perspectives.
Profiling of 37 internalin genes grouped the L. innocua strains into five internalin types, IT1 to IT5, with IT1 and IT2 as the major types (Table 2). The MLST scheme identified two major phylogenetic branches containing the majority of sequence types (29/31, 93.5%), and other two bearing one strain each (Fig 1). Consequently, L. innocua consists of at least four distinct subgroups A, B, C and D. Subgroup A correlates with one of the major branches including all the IT1 and IT3 strains with the exception of one IT3 strain 0063 belonging to subgroup C, while subgroup B correlates with the other major branch covering all the IT2 and IT4 strains (Table 2B). Therefore, it is inferred that a certain L. innocua subgroup possibly contains several serovars and exhibits different internalin patterns, which is similar to the fact that each lineage of L. monocytogenes contains several serovars and exhibits more than one internalin patterns, as exemplified by the internalin island between ascB and dapE in our previous report [17].
The majority of L. monocytogenes lineage I strains harbor inlC2DE, and a small number of 1/2b strains carry inlGC2DE instead. Within L. monocytogenes lineage II strains, the majority of 1/2a and 1/2c strains harbor inlGC2DE and inlGHE respectively. In addition, L. monocytogenes lineage III strains show the greatest level of diversity [8,17]. The L. innocua subgroup A strains either contain a whole set of L. monocytogenes-L. innocua common and L. innocua-specific internalin genes, or lack lin1204 and lin2539, and the L. innocua subgroup B strains either lack lin1204 or lack lin0661, lin0354 and lin2539 instead. Besides, the subgroup D strain L43, which shows the least genetic distance to L. monocytogenes, lacks lin1204 but bears L. monocytogenes-specific inlJ in the counterpart region in L. monocytogenes genomes (Table 2). We propose that certain internalin genes such as lin0354, lin0661, lin1204 and lin2539 could be potential genetic markers for subgroups of L. innocua.
The phylogenetic tree revealed nine major branches of the L. innocua-L. monocytogenes clade, five belonged to L. monocytogenes representing lineages I, II, and III, consistent with previous reports [11,24,26], and the other four represented L. innocua subgroups A, B, C and D (Fig 1). Overall, L. innocua is genetically monophyletic compared to L. monocytogenes, and the nucleotide diversity of the L. innocua species is similar to that of L. monocytogenes lineage I but less than those of L. monocytogenes lineages II and III. In evolutionary terms, younger bacterial species has lower level of genetic diversity [15]. The results from this study offer additional evidence that L. innocua possibly represents a relatively young species as compared to its closest related pathogenic species L. monocytogenes.
Previous studies suggest that L. monocytogenes represents one of the bacterial species with the lowest rate of recombination [4,27]. In this study, strains in the L. innocua-L. monocytogenes clade exhibit similar value of ρ/θ to those of the Bacillus anthracis-Bacillus cereus clade [28] and slightly higher than those of Staphylococcus aureus [29], but still considerably lower than those of pathogens such as Clostridium perfringens [30], Neisseria meningitis [31] and Streptococcus pneumoniae [29]. Both the relative frequency of occurrence of recombination versus mutation (ρ/θ) and the relative effect of recombination versus point mutation (r/m) of L. innocua were higher than those of L. monocytogenes. More strikingly, recombination rates of L. innocua subgroup A were particularly high (Table 5). Wirth et al. [32] proposed from the data for Escherichia coli that epidemic and virulent bacteria face an increased selective pressure for rapid diversification in response to host immune defenses, resulting in higher recombination rates. L. monocytogenes is an opportunistic pathogen with wide host ranges as well as a saprotroph found in different environments [2,33]. Though lineage I strains were responsible for almost all major human listeriosis outbreaks and the majority of sporadic cases [6], those of lineage II exhibited higher recombination rate according to our observation and the findings by Bakker et al. [24]. Bakker et al. [24] proposed that higher recombination in lineage II was not due to selective forces involved in its virulence. Recombination may be critical for lineage II to successfully compete and survive in a board range of different environments. Lineage II strains are more commonly found at higher levels than lineage I strains in natural environments including foods [24,34]. Similarly, we postulate that the nonpathogenic species L. innocua descending from its pathogenic ancestor has better adaptability to contemporary environmental niches. Removal of some gene loci related to virulence (e.g., LIPI-1, inlAB and bsh) in Listeria could be regarded as adaptive gene loss, which favors its survival in environmental niches as a saprotroph [9,11].
L. innocua subgroups A and B strains have similar TMRCA and exhibit similar genetic distances to L. monocytogenes, suggesting that these two subgroups appeared at approximately the same time (Fig 2). However, subgroup A experienced a recent expansion of the population size, consistent with the higher recombination frequency (r/m) and effect (ρ/θ) of subgroup A as compared to those of subgroup B. This further implies that these two subgroups have distinct inclinations and adaptive abilities to environments and occupy different habitats, while subgroup A might face increased selective pressures resulting in higher recombination rates. Additional support for this indication is that the majority of subgroup A isolates (belonging IT1) contain a whole set of L. monocytogenes-L. innocua common and L. innocua-specific internalin genes which may play broad roles in enhancing the adaption to various environments. Hence, the L. innocua subgroup A strains might represent the possible evolutionary direction towards adaptation. Interestingly, the higher recombination rate of L. innocua subgroup A did not seem to contribute to nucleotide diversity. One possible explanation is that members of subgroup A recombines only with other members of subgroup A, so that recombination does not introduce novel polymorphisms into subgroup A and therefore does not increase the genetic diversity.
Previous reports indicate that horizontal gene transfer might have occurred earlier to form a more ancestral L. monocytogenes strain, which would then give rise to L. innocua through gene deletion events possibly via low-virulent L. monocytogenes lineage IIIA strains [11,13]. In this study, L. innocua subgroup D strain L43 exhibits the least genetic distances to L. monocytogenes (Fig 1), and constituted another evolutionary intermediates between L. monocytogenes and L. innocua main clusters. Therefore, L. innocua strain L43 and L monocytogenes strain 54006 [11] might serve as intermediate linkage strains in deciphering the evolution of the L. innocua-L. monocytogenes clade.
The strain L43 seems to share a "hybrid" genetic background derived from L. innocua and L. monocytogenes by the MLST data and its carriage of L. monocytogenes-specific virulence gene inlJ. InlJ is a sortase-anchored adhesin specifically expressed in vivo [35], but its function in atypical L. innocua strains requires further investigation. Another atypical L. innocua strain PRL/NW 15B95 has been characterized as having the entire LIPI-1 embedded into an otherwise typical L. innocua genetic background [9]. However, we did not see its presence in the strain L43. PRL/NW 15B95 falls into the main L. innocua cluster based on sequencing of 16S-23S intergenic regions, 16S rRNA and iap genes, and has possibly acquired LIPI-1 by a later transposition event, based on the finding of a 16 bp Tn1545 integration consensus sequence flanking the virulence island [9]. Thus, unlike L43, PRL/NW 15B95 does not constitute an evolutional intermediate between L. monocytogenes and L. innocua. Complementary transfer of only some of the virulence genes such as LIPI-1 did not change the avirulent character of PRL/NW 15B95 [9]. In this study, all L. innocua strains were nonpathogenic in mice models (Table 1).
Conclusion
This study reveals that L. innocua is a relatively young species descending from L. monocytogenes. The evolutionary history in the L. monocytogenes-L. innocua clade represents a rare example of evolution towards reduced virulence of pathogens. L. innocua is genetically monophyletic and comprises four subgroups based on internalin profiling and MLST scheme. The majority of L. innocua strains belong to two major subgroups A and B, and one atypical subgroup might serve as a link between L. monocytogenes and L. innocua main cluster in the evolutionary chain. While subgroups A and B appeared at approximately the same time, the subgroup A strains seem to represent the possible evolutionary direction towards adaptation to enviroments. It is believed that the phylogenetic structure and evolutionary history of L. innocua will be much clearer if a larger strain collection and the whole genome sequences of more representative strains become available.
Methods
Bacterial strains
A total of 68 Listeria strains were examined in this study (Table 1). These included 30 L. monocytogenes strains representing three lineages, 34 L. innocua strains, 5 from reference collections, 13 from meat, 8 from milk and 8 from seafoods, and 4 L. welshimeri strains. Listeria strains were retrieved from glycerol stocks maintained at -80°C, and cultured in brain heart infusion broth (BHI; Oxoid, Hampshire, England) at 37°C.
Carbohydrate fermentation and hemolytic reactions
The recommended biochemical patterns for differentiating Listeria spp. included L-rhamnose, D-xylose, D-mannitol and glucose utilization and hemolytic reactivity, and were tested by using conventional procedures [36,37].
DNA manipulations
Genomic DNA was extracted using a protocol reported previously [12]. Oligonucleotide primers were synthesized by Invitrogen Biotechnology (Shanghai, China) (Table 6 and Additional file 1; table S2), and Taq DNA polymerase (TaKaRa Biotech Co. Ltd., Dalian, China) was used for PCR amplification. PCR was conducted using a PT-200 thermal cycler (MJ Research Inc. MA, Boston, USA), with annealing temperatures depending on specific primer pairs (Table 6 and Additional file 1; table S2), and the duration of extension depending on the expected length of amplicon (1 min per kb, at 72°C). For DNA sequencing analysis, PCR fragments were purified with the AxyPrep DNA Gel Extraction Kit (Axygen Inc., USA) and their sequences determined by dideoxy method on ABI-PRISM 377 DNA sequencer.
Table 6.
Primers used for MLST
| Locus | Putative function | Locationa | Forward primer | Reverse primer | Length (bp) |
|---|---|---|---|---|---|
| gyrB | DNA gyrase subunit B | 6,031-7,971 | TGGTGCATCGGTAGTTAATGC | CAACATCTGGGTTTTCCATCAT | 657 |
| dapE | Succinyl diaminopimelate desuccinylase | 301,402-302,538 | GTAAATATTGATTCGACTAATG | CACTAGCACTTGTTTCACTG | 669 |
| hisJ | Histidinol phosphate phosphatase | 606,408-607,235 | TCCACATGGTACGCATGAT | GGACATGTCAAAATGAAAGATC | 714 |
| sigB | Stess responsive alternative sigma factor B | 924,734-925,513 | CCAAAAGTATCTCAACCTGAT | CATGCATTTGTGATATATCGA | 642 |
| ribC | Riboflavin kinaseand FAD synthase | 1,364,536-1,365,480 | AAGACGATATACTTACATCAT | GTCTTTTTCTAACTGAGCA | 633 |
| purM | Phosphoribosyl aminoimidazole synthase | 1,893,107-1,894,153 | CAAGCTCCACTTTGACAGCTAA | TAAAGCAGGCGTGGACGTA | 693 |
| betL | Glycine betaine transporter | 2,216,882-2,218,405 | ACAGAACATTATCCAAATGAGTT | ACGTTGTGATTTTTTCGGTC | 534 |
| gap | Glyceraldehyde 3-phosphate dehydrogenase | 2,578,558-2,579,584 | CTGGATCAGAAGCTGCTTCCA | GTCGTATTCAAAATGTGGAAGGA | 621 |
| tuf | Translation elongation factor | 2,816,958-2,818,145 | CATTTCTACTCCAGTTACTACT | GCTCTAAACCCCATGTTA | 681 |
| Subtotal | 5,844 |
a, Positions correspond to complete genome sequence of L. innocua strain CLIP11262 (AL592022).
Internalin profiling
By sequence comparison of L. monocytogenes strains F2365, H7858 (serovar 4b), EGDe and F6854 (serovar 1/2a) and L. innocua strain CLIP11262, we investigated the presence or absence of 14 L. monocytogenes-L. innocua-common and 4 L. innocua-specific internalin genes as well as 19 L. monocytogenes-specific internalin genes by PCR with specific primers outlined in Additional file 1; table S1. Due to the conserved repeats present in internalin multigene family [19], primers were designed based on the distinguishable regions through sequence comparison. As inlH and inlC2 shared highly identical nucleotide sequences, a common primer set was employed [17].
Multilocus sequence typing (MLST)
The MLST scheme was based on the sequence analysis of 9 unlinked genes, including 7 housekeeping genes gyrB, dapE, hisJ, ribC, purM, gap and tuf, and 2 stress-response genes sigB and betL. Sequences generated in this study have been deposited in GenBank within the accession numbers FJ774089 to FJ774121 (gyrB), FJ774145 to FJ774177 (sigB), FJ774274 to FJ774282, FJ774257 to FJ774273, FJ774283 to FJ774293, FJ774295 to FJ774297, FJ774299 to FJ4300 (gap), FJ774313 to FJ774344, FJ774368 (hisJ), FJ774369 to FJ774400, FJ774424 (purM), FJ774425 to FJ774457 (ribC), FJ774481 to FJ774513 (dapE), FJ774537 to FJ774568 (tuf), and FJ774593 to FJ774625 (betL).
Detection of virulence genes
Five categories of virulence genes found in L. monocytogenes were assessed by using primers listed in Additional file 1; table S2, including (i) stress response genes conferring tolerance to harsh conditions within the host (e.g. bsh, arcB, arcD, lmo0038 and arcC); (ii) internalin genes responsible for adhesion and invasion of host cells (e.g. inlA, inlB, inlC, inlF and inlJ); (iii) genes involved in escape from vacuole and intracellular multiplication (e.g. plcA, hly, mpl, plcB and hpt); (iv) the gene associated with intracellular and intercellular spread (e.g. actA); and (v) regulatory genes (e.g. prfA).
Mouse infection
The virulence potential of 33 L. innocua strains and 30 L. monocytogenes isolates was assessed in ICR mice by a previously reported protocol [38]. The animal experiment was approved by the Laboratory Animal Management Committee of Zhejiang University, and the mice were handled under strict ethical conditions. Briefly, 5 female ICR mice at 20-22 g (Zhejiang College of Traditional Chinese Medicine, Hangzhou, China) were inoculated intraperitoneally with ~108 CFU each strain in a 0.1 ml-volume. Mice in the control group were injected with 0.1 ml PBS. The mice were observed daily and mortalities recorded until all of the mice inoculated with the virulent EGDe strain died. Relative virulence (%) was calculated by dividing the number of dead mice with the total number of mice tested. On the 15th day post- inoculation, all surviving mice were euthanized.
Data analysis
For each MLST locus, an allele number was given to each distinct sequence variant, and a distinct sequence type (ST) number was given to each distinct combination of alleles of the 9 genes. MEGA 4.0 was used to construct a neighbor-joining tree of L. innocua and L. monocytogenes isolates using the number of nucleotide differences in the concatenated sequences of 9 loci with 1,000 bootstrap tests [39]. L. welshimeri was used as outgroup species. DNAsp v4.10.9 [40] was used to calculate the number of alleles, number of polymorphic sites, nucleotide diversity (π, mean pairwise nucleotide difference per site), Tajima's D (for testing the hypothesis that all mutations are selectively neutral [41]), numbers of synonymous mutations and nonsynonymous mutations and the rate of nonsynonymous to synonymous changes with a Jukes-Cantor correction. Discrimination index (D.I.) values of selected genes were calculated according to the method previously described [42] on the basis of allelic types [j], numbers of strains belonging to each type [nj], and the total numbers of strains analyzed [N] with the following equation (higher D.I. values indicate better discriminatory power):
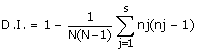 |
ClonalFrame v1.1 was employed to show the evolution of ρ/θ and r/m as chain run. These two complementary measures were used to assess the relative contribution of recombination and mutation in the creation of the sample population from a common ancestor. Specifically, ρ/θ is the ratio of rates at which recombination and mutation occur, representing a measure of how often recombination happen relative to mutation [43], while r/m is the ratio of probabilities that a given site is altered through recombination and mutation, representing a measure of how important the effect of recombination is in the diversification of the sample population relative to mutation [44].
To infer the population history in the L. innocua-L. monocytogenes clade, ClonalFrame GUI, which treated each gene as an independent unit in the input file, was used to calculate the ratio of the sum of the external branches (the ones that connect a leaf of the tree) to the sum of the internal branches (the ones that connect two internal nodes of the tree) [45]. The distribution of these ratios was then compared to the distribution of the external/internal branch length ratio as expected under the coalescent model [46]. If the external/internal branch length ratio is significantly smaller than expected, it means that the inferred genealogy is unexpectedly "star-like", which is consistent with an expansion of the population size.
The chi-squared test was used to test significant associations between L. innocua subgroup and isolate source.
Authors' contributions
JC conceived and designed the study, performed and interpreted the phylogenetic and statistical analyses, participated in the collection of the sequence data and animal assays, and drafted the manuscript. QC performed the PCR amplification and participated in the collection of the sequence data. LJ participated in evaluation of the results and in revision of the manuscript. CC and FB participated in the PCR amplification, biochemical tests and animal assays. JW and FM participated in the analysis of sequence data. WF supervised the project, participated in the design of the study and data interpretation, and helped draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table S1 and S2: Internalin Types (ITs) based on 14 L. monocytogenes-L. innocua-common and 4 L. innocua-specific (in grey shading) internalin genes.
Contributor Information
Jianshun Chen, Email: jianshunchan@gmail.com.
Qiaomiao Chen, Email: bonny09222004@yahoo.com.cn.
Lingli Jiang, Email: lljiang@zju.edu.cn.
Changyong Cheng, Email: jackenmickey@qq.com.
Fan Bai, Email: rachelbai726@126.com.
Jun Wang, Email: junwang@zju.edu.cn.
Fan Mo, Email: mofan.hz@gmail.com.
Weihuan Fang, Email: whfang@zju.edu.cn.
Acknowledgements
This study was supported by grants from National Natural Science Foundation (Contract No. 30870068). We thank Dr. Martin Wiedmann for kindly providing L. monocytogenes lineage III strains, and Drs. Dongyou Liu and John Bowman for helpful discussion on the subtyping of Listeria strains.
References
- Schmid MW, Ng EYW, Lampidis R, Emmerth M, Walcher M, Kreft J, Goebel W, Wanger M, Schleifer K. Evolutionary history of the genus Listeria and its virulence genes. Syst Appl Microbiol. 2005;28:1–18. doi: 10.1016/j.syapm.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TJ, Gorski L, Borucki MK, Mandrell RE, Hutchins J, Pupedis K. Intraspecific phylogeny and lineage group identification based on the prfA virulence gene cluster of Listeria monocytogenes. J Bacteriol. 2004;186:4994–5002. doi: 10.1128/JB.186.15.4994-5002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Monnier AL, Brisse S. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008;4:1–14. doi: 10.1371/journal.ppat.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Lawrence ML, Wiedmann M, Gorski L, Mandrell RE, Ainsworth AJ, Austin FW. Listeria monocytogenes subgroups IIIA, IIIB and IIIC delineate genetically distinct populations with varied virulence potential. J Clin Microbiol. 2006;44:4229–4233. doi: 10.1128/JCM.01032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan B, Gerner-Smidt P. The epidemiology of human listeriosis. Microbes Infect. 2007;9:1236–1243. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Goulet V, Jacquet C, Martin P, Vaillant V, Laurent E, Valk Hd. Surveillance of human listeriosis in France, 2001-2003. Euro Surveill. 2006;11:79–81. [PubMed] [Google Scholar]
- Chen J, Chen Q, Jiang J, Hu H, Ye J, Fang W. Serovar 4b complex predominates among Listeria monocytogenes isolates from imported aquatic products in China . Foodborne Pathog Dis . 2009;7:31–41. doi: 10.1089/fpd.2009.0353. [DOI] [PubMed] [Google Scholar]
- Johnson J, Jinneman K, Stelma G, Smith BG, Lye D, Messer J, Ulaszek J, Evsen L, Gendel S, Bennett RW, Swaminathan B, Pruckler J, Steigerwalt A, Kathariou S, Yildirim S, Volokhov D, Rasooly A, Chizhikov V, Wiedmann M, Fortes E, Duvall RE, Hitchins AD. Natural atypical Listeria innocua strains with Listeria monocytogenes pathogenicity Island 1 genes. Appl Environ Microbiol. 2004;70:4256–4266. doi: 10.1128/AEM.70.7.4256-4266.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale KK, Ivy RA, Ho AJ, Fortes ED, Njaa BL, Peters RM, Wiedmann M. inlA premature stop codons are common among Listeria monocytogenes isolates from foods and yield virulence-attenuated strains that confer protection against fully virulent strains. Appl Environ Microbiol. 2008;74:6570–6583. doi: 10.1128/AEM.00997-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Jiang L, Chen X, Luo X, Chen Y, Yu Y, Tian G, Liu D, Fang W. Listeria monocytogenes serovar 4a is a possible evolutionary intermediate between L. monocytogenes serovars 1/2a and 4b and L. innocua. J Microbiol Biotechnol. 2009;19:238–249. [PubMed] [Google Scholar]
- Chen J, Jiang L, Chen Q, Zhao H, Luo X, Chen X, Fang W. lmo0038 is involved in acid and heat stress responses and specific for Listeri monocytogenes lineages I and II, and Listeri ivanovii. Foodborne Pathog Dis. 2009;6:365–376. doi: 10.1089/fpd.2008.0207. [DOI] [PubMed] [Google Scholar]
- Doumith M, Cazalet C, Simoes N, Frangeul L, Jacquet C, Kunst F, Martin P, Cossart P, Glaser P, Buchrieser C. New aspects regarding evolution and virulence of Listeria monocytogenes revealed by comparative genomics and DNA arrays. Infect Immun. 2004;72:1072–1083. doi: 10.1128/IAI.72.2.1072-1083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. Identification, subtyping and virulence determination of Listeria monocytogenes, an important foodborne pathogen. J Med Microbiol. 2006;55:645–659. doi: 10.1099/jmm.0.46495-0. [DOI] [PubMed] [Google Scholar]
- Urwin R, Maiden MCJ. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;11:479–487. doi: 10.1016/j.tim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Meinersmann R, Phillips R, Wiedmann M, Berrang M. Multilocus sequence typing of Listeria monocytogenes by use of hypervariable genes reveals clonal and recombination histories of three lineages. Appl Environ Microbiol. 2004;70:2193–2203. doi: 10.1128/AEM.70.4.2193-2203.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Luo X, Jiang L, Jin P, Wei W, Liu D, Fang W. Molecular characteristics and virulence potential of Listeria monocytogenes isolates from Chinese food systems. Food Microbiol. 2009;26:103–111. doi: 10.1016/j.fm.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakratory T, Charbit A, Chetouani F, Couve E, Daruvar Ad, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Portillo FG, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, Pablos Bd, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- Bierne H, Sabet C, Personnic N, Cossart P. Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect. 2007;9:1156–1166. doi: 10.1016/j.micinf.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Milillo SR, Wiedmann M. Contributions of six lineage-specific internalin-like genes to invasion efficiency of Listeria monocytogenes. Foodborne Pathog Dis. 2008;6:57–70. doi: 10.1089/fpd.2008.0140. [DOI] [PubMed] [Google Scholar]
- Roberts A, Nightingale K, Jeffers G, Fortes E, Kongo JM, Wiedmann M. Genetic and phenotypic characterization of Listeria monocytogenes lineage III. Microbiology. 2006;152:685–693. doi: 10.1099/mic.0.28503-0. [DOI] [PubMed] [Google Scholar]
- Nielsen R. Statistical tests of selective neutrality in the age of genomics. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- Simonsen K, Churchill G, Aquadro C. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker HC, Didelot X, Fortes ED, Nightingale KK, Wiedmann M. Lineage specific rates and microevolution in Listeria monocytogenes. BMC Evol Biol. 2008;8:277. doi: 10.1186/1471-2148-8-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedmann M, Bruce JL, Keatine C, Johnson AE, McDonough PL, Batt CA. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect Immun. 1997;65:2707–2716. doi: 10.1128/iai.65.7.2707-2716.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi RH, Sun Q, Wiedmann M. Genome-wide analyses reveal lineage specifi contributions of positive selection and recombination to the evolution of Listeria monocytogenes. BMC Evol Biol. 2008;8:233. doi: 10.1186/1471-2148-8-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo C, Arreaza L, Alcala B, Fuente L, Vazquez JA. Development of a multilocus sequence typing method for analysis of Listeria monocytogenes clone. J Clin Microbiol. 2003;41:757–762. doi: 10.1128/JCM.41.2.757-762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C, Hanage W, Spratt B. Neutral microepidemic evolution of bacterial pathogens. Proc Natl Acad Sci USA. 2005;102:1968–1073. doi: 10.1073/pnas.0406993102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney AP, Swezey JL, Friedman R, Hecht DW, Maddox CW. Analysis of core housekeeping and virulence genes reveals cryptic lineages of Clostridium perfringens that are associated with distinct disease presentations. Genetics. 2006;172:2081–2203. doi: 10.1534/genetics.105.054601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Wilson DJ, Kriz P, McVean G, Maiden MC. The influence of mutation, recombination, population history, and selection on patterns of genetic diversity in Neisseria meningitis. Mol Biol Evol. 2005;22:562–569. doi: 10.1093/molbev/msi041. [DOI] [PubMed] [Google Scholar]
- Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler L, Karch H, Reeves P, Maiden M, Ochman H, Achtman M. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006;60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori MA, Soubigou G, Régnault B, Coppée JY, Lecuit M, Johansson J, Cossart P. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- Chen Y, Ross WH, Gray MJ, Wiedmann M, Whiting RC, Scott VN. Attributing risk to Listeria monocytogenes subgroups: does response in relation to genetic lineages. J Food Prot. 2006;69:335–344. doi: 10.4315/0362-028x-69.2.335. [DOI] [PubMed] [Google Scholar]
- Sabet C, Lecuit M, Cabanes D, Cossart P, Bierne H. LPXTG protein InlJ, a newly identified internalin involved in Listeria monocytogenes virulence. Infect Immun. 2008;73:6912–6922. doi: 10.1128/IAI.73.10.6912-6922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLauchlin J. The identification of Listeria species. Int J Food Microbiol. 1997;38:77–81. doi: 10.1016/S0168-1605(97)00086-X. [DOI] [PubMed] [Google Scholar]
- Geoffroy C, Gaillard JL, Alouf JE, Berche P. Production of thiol-dependent haemolysins by Listeria monocytogenes and related species. J Gen Microbiol. 1989;135:481–487. doi: 10.1099/00221287-135-3-481. [DOI] [PubMed] [Google Scholar]
- Liu D. Listeria monocytogenes: comparative interpretation of mouse virulence assay. FEMS Microbiol Lett. 2004;233:159–164. doi: 10.1016/j.femsle.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007. pp. 1596–1599. [DOI] [PubMed]
- Rozas J, Sánchez-DelBarrio J, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Jayarao BM, Knabel SJ. Multi-virulence-locus sequence typing of Listeria monocytogenes. Appl Environ Microbiol. 2004;70:913–920. doi: 10.1128/AEM.70.2.913-920.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkman R, Bridges M. Molecular evolution of the Escherichia coli chromosome. III. Clonal Frames. Genetics. 1990;126:505–517. doi: 10.1093/genetics/126.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman DS, Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- Fiala KI, Sokal RR. Factors determining the accuracy of cladogram estimation-evaluation using computer-simulation. Evolution. 1985;39:609–622. doi: 10.2307/2408656. [DOI] [PubMed] [Google Scholar]
- Kingman JFC. The Coalescent. Stochastic Processes and their applications. 1982;13:235–248. doi: 10.1016/0304-4149(82)90011-4. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 and S2: Internalin Types (ITs) based on 14 L. monocytogenes-L. innocua-common and 4 L. innocua-specific (in grey shading) internalin genes.