

Abstract
Cognitive deficits such as impaired decision-making can be a consequence of persistent pain. Normal functions of the intact amygdala and prefrontal cortex are required for emotion-based decision-making that relies on the ability to assess risk, attribute value, and identify advantageous strategies. We tested the hypothesis that pain-related cognitive deficits result from amygdala-driven impairment of medial prefrontal cortical (mPFC) function. To do this, we used electrophysiological single-unit recordings in vivo, patch clamp in brain slices, and various behavioral assays to show that increased neuronal activity in the amygdala in an animal model of arthritis pain was accompanied by decreased mPFC activation and impaired decision-making. Furthermore, pharmacologic inhibition (with a corticotropin-releasing factor 1 receptor antagonist) of pain-related hyperactivity in the basolateral amygdala (BLA), but not central amygdala (CeA), reversed deactivation of mPFC pyramidal cells and improved decision-making deficits. Pain-related cortical deactivation resulted from a shift of balance between inhibitory and excitatory synaptic transmission. Direct excitatory transmission to mPFC pyramidal cells did not change in the pain model, whereas polysynaptic inhibitory transmission increased. GABAergic transmission was reduced by non-NMDA receptor antagonists, suggesting that synaptic inhibition was glutamate driven. The results are consistent with a model of BLA-driven feedforward inhibition of mPFC neurons. In contrast to the differential effects of BLA versus CeA hyperactivity on cortical-cognitive functions, both amygdala nuclei modulate emotional-affective pain behavior. Thus, this study shows that the amygdala contributes not only to emotional-affective but also cognitive effects of pain. The novel amygdalo-cortical pain mechanism has important implications for our understanding of amygdala functions and amygdalo-cortical interactions.
Introduction
Decision-making represents a critical cognitive function that requires the integration of sensory, affective, and cognitive processes (Bechara et al., 1999; Paulus, 2007; Seymour and Dolan, 2008). A strong emotional stimulus, pain, can impair decision-making in humans (Apkarian et al., 2004b) and animals (Pais-Vieira et al., 2009). The underlying mechanism remains to be determined, but recent evidence suggests that dysfunction of prefrontal cortical (PFC) areas is involved (Apkarian et al., 2004a; Metz et al., 2009). The PFC, particularly its medial and orbital regions, plays a key role in value-based decision-making that avoids risky choices and guides advantageous goal-directed behaviors in animals and humans (Bechara et al., 1999; Vertes, 2006; Stalnaker et al., 2007a; Kouneiher et al., 2009). Patients with PFC lesions (Bechara et al., 1999) show similar deficits in an emotion-based decision-making task as pain patients (Apkarian et al., 2004b). They perseverate in making disadvantageous choices that yield high immediate monetary gains at the risk of higher future losses in a gambling task.
Advantageous decision-making in the gambling task requires also the intact amygdala (Bechara et al., 1999, 2003), a key player in emotions and affective disorders (Maren and Quirk, 2004; Phelps and LeDoux, 2005; Seymour and Dolan, 2008; Ehrlich et al., 2009). The amygdala consists of several functionally distinct nuclei, including the lateral (LA), basolateral (BLA), and central (CeA) nuclei (Sah et al., 2003). Associative processing in the LA–BLA network attaches emotional significance to sensory stimuli. Affect-related information is then transmitted to the CeA to regulate behavioral responses through interactions with hypothalamus and brainstem (Maren and Quirk, 2004; Phelps and LeDoux, 2005; Ehrlich et al., 2009). The amygdala can also modulate cortical functions and interactions between BLA and medial PFC (mPFC) are important for emotional associative learning and behavior (Garcia et al., 1999; Holland and Gallagher, 2004; McGaugh, 2004; Laviolette and Grace, 2006; Herry et al., 2008; Roozendaal et al., 2009).
Recent work from our group and others showed that pain-related plasticity in the CeA contributes critically to the emotional-affective component of pain (Neugebauer et al., 2004, 2009; Carrasquillo and Gereau, 2007; Ikeda et al., 2007; Myers and Greenwood-Van Meerveld, 2007). Less is known about the role of the BLA in pain. Although the BLA receives polymodal, including nociceptive, information (Neugebauer et al., 2009) and has been implicated in top-down pain modulation (McGaraughty and Heinricher, 2002; Jasmin et al., 2003), processing of pain-related information in the BLA and upstream interactions with cortical areas in pain remain to be determined. Based on the LA–BLA–CeA circuitry model of amygdala function in aversive behavior (Sah et al., 2003; Maren and Quirk, 2004; Phelps and LeDoux, 2005; Ehrlich et al., 2009), we hypothesized that pain leads to hyperactivity in the BLA, as in the CeA (Neugebauer et al., 2009), and that both nuclei contribute to emotional-affective aspects of pain. Different from the CeA, however, the BLA is a major source of input to the mPFC (Kita and Kitai, 1990; McDonald, 1991; Bacon et al., 1996; Gabbott et al., 2006). Therefore, we further hypothesized that pain-related plasticity in the BLA, but not CeA, impairs mPFC function and produces cognitive decision-making deficits.
Materials and Methods
Arthritis pain model
A localized mono-arthritis was induced in the left knee joint by intra-articular injections of kaolin (4%, 80–100 μl) and carrageenan (2%, 80–100 μl) through the patellar ligament (Neugebauer et al., 2007). This treatment paradigm reliably leads to inflammation and swelling of the knee within 1–3 h, reaches a maximum plateau at 5–6 h, and persists for days (Neugebauer et al., 2007). The 5–6 h time point was selected for testing drug effects, behavioral assays, and obtaining brain slices for electrophysiology. Details of the kaolin–carrageenan arthritis pain model have been given previously (Neugebauer et al., 2007).
Electrophysiology in vivo
Animal preparation.
Extracellular single-unit recordings were made in pentobarbital-anesthetized adult rats (250–350 g) as described previously in detail (Neugebauer and Li, 2003; Ji and Neugebauer, 2007). Animals were kept under anesthesia maintained at a constant level with pentobarbital (15 mg · kg−1/h−1, i.v.). They were paralyzed with pancuronium (0.3 mg/h, i.v.) and artificially ventilated (3–3.5 ml; 55–65 strokes/min). End-tidal CO2 levels (kept at 4.0 ± 0.2%), heart rate, and electrocardiogram pattern were continuously monitored. Core body temperature was maintained at 37°C by means of a homeothermic blanket system. These measures ensured a constant internal state of body functions. Animals were mounted in a stereotaxic frame (David Kopf Instruments), and a small unilateral craniotomy was performed at the sutura frontoparietalis level.
Extracellular single-unit recordings.
Long-term extracellular recordings were made from single neurons in the BLA and medial (prelimbic) PFC with glass-insulated carbon filament electrodes (3–5 MΩ), using the following stereotaxic coordinates (in mm): posterior BLA, 2.5–3.0 caudal to bregma, 4.5–5.2 lateral to midline, 7.5–9 depth; prelimbic PFC, 3.0–3.2 rostral to bregma, 0.5–1.0 lateral to midline, 3–5 depth. Recordings were made in the right hemisphere to allow comparison with our previous studies that showed pain-related changes in the right, but not left, central nucleus of the amygdala (Ji and Neugebauer, 2009). Because connections between BLA and mPFC are predominantly ipsilateral (Bacon et al., 1996; Gabbott et al., 2006), recordings of BLA and mPFC neurons were made in the same (right) hemisphere. Recorded signals are amplified, displayed on storage oscilloscopes, fed into a window discriminator, digitized (1401plus interface; Cambridge Electronic Design), and recorded on a Pentium personal computer. Spike2 software (version 3; Cambridge Electronic Design) was used for online and offline analysis of single-unit activity (see next section).
Identification and classification of BLA and mPFC neurons.
An individual neuron was identified by the configuration, shape, and height of the recorded action potentials (spikes) that occurred spontaneously (background activity) or in response to mechanical (tissue compression) search stimuli (evoked responses). Spike detection and recoding was based on the waveform signal that crossed a trigger level and matched a preset shape or template that was created for the individual neuron at the beginning of the recording period.
BLA projection neurons were identified by antidromic activation using square-wave current pulses (50–500 μA, 150 μs) delivered with a monopolar stimulation electrode inserted into the mPFC, using the following coordinates (in mm): 3.2 rostral to bregma, 0.8 lateral to midline, 3–5 depth of tip. Criteria for antidromic activation were as follows (Neugebauer et al., 1999): constant latency of the evoked spike; ability to follow high-frequency (333 Hz) stimulation; and collision of orthodromic spikes with electrically evoked antidromic spikes. mPFC pyramidal cells were distinguished from inhibitory interneurons based on well established criteria (Constantinidis and Goldman-Rakic, 2002; Laviolette and Grace, 2006), including broader action potential waveform (spike width >500 μs) and lower baseline discharge rate (<10 Hz) .
In each animal, background activity and evoked responses of one neuron were recorded. Brief (15 s) mechanical test stimuli of innocuous (500 g/30 mm2) and noxious (2000 g/30 mm2) intensities were applied to the knee joint by means of a calibrated forceps equipped with a force transducer, whose output signal was amplified, displayed in grams on a liquid crystal display screen, digitized by the Cambridge Electronic Design interface, and recorded on the Pentium personal computer for online and offline analysis (Ji and Neugebauer, 2007). For the analysis of net evoked activity, background activity preceding the stimulus was subtracted from the total activity during stimulation. To determine changes in the arthritis pain model, one and the same neuron was recorded continuously before and after arthritis induction, allowing paired analysis.
Drug application by microdialysis.
Drugs were administered into the BLA (or CeA as a control) of the right hemisphere by microdialysis. A microdialysis probe [CMA11; (CMA/Microdialysis); membrane diameter, 250 μm; membrane length, 1 mm; 8 kDa cutoff, sufficient for the application of small molecules such as NBI27914 (5-chloro-4-[N-(cyclopropyl)methyl-N-propylamino]-2-methyl-6-(2,4,6-trichlorophenyl)amino-pyridine)] was positioned stereotaxically in the brain, using the following coordinates (in mm): BLA, 3.3 caudal to bregma, 5.0 lateral to midline, 9.0 depth; CeA, 2.3 caudal to bregma, 4.0 lateral to midline, 8.5 depth. The probe was connected to a Harvard Apparatus infusion pump and perfused with artificial CSF (ACSF) containing the following (in mm): 125.0 NaCl, 2.6 KCl, 2.5 NaH2PO4, 1.3 CaCl2, 0.9 MgCl2, 21.0 NaHCO3, and 3.5 glucose, oxygenated and equilibrated to pH 7.4 (at 5 μl/min. Throughout the experiment, ACSF was pumped through the fiber to establish equilibrium in the tissue. Drugs were administered for 20 min. They were dissolved in ACSF on the day of the experiment at a concentration 100-fold that predicted to be needed based on data from our previous microdialysis and in vitro studies and data in the literature (Ji and Neugebauer, 2007; Fu and Neugebauer, 2008). Drug concentration in the tissue is at least 100 times lower than in the microdialysis probe as a result of the concentration gradient across the dialysis membrane and diffusion in the tissue (Ji and Neugebauer, 2007). Numbers in the text refer to drug concentrations in the microdialysis fiber.
Electrophysiology in brain slices
Slice preparation.
Brain slices containing the amygdala or mPFC were obtained from normal (control) and arthritic rats (150–250 g) as described previously (Neugebauer et al., 2003; Orozco-Cabal et al., 2006; Fu and Neugebauer, 2008). Coronal brain slices (300–500 μm) containing the BLA were cut at 2.5–3.5 mm caudal to bregma using a vibrating microtome. Brain slices containing the PFC were cut at 3.0–3.2 mm rostral to bregma. At this level, slices contain both the prelimbic cortex (recording site) and infralimbic cortex (stimulation of BLA afferents passing through). A single brain slice was transferred to the recording chamber and submerged in ACSF (31 ± 1°C), which superfused the slice at ∼2 ml/min. ACSF contained the following (in mm): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose. The ACSF was oxygenated and equilibrated to pH 7.4 with a mixture of 95% O2/5% CO2. Only one or two brain slices per animal were used, one neuron was recorded in each slice, and a fresh slice was used for each new experimental protocol. Numbers in the text refer to the number of neurons tested for each parameter.
Patch-clamp recording.
Whole-cell patch-clamp recordings were obtained from BLA neurons using the blind-patch technique and from visualized PFC neurons using infrared differential interference contrast (DIC) video microscopy. Recordings were made in the right hemisphere. The boundaries of the different amygdalar nuclei are easily discerned under light microscopy (Fu and Neugebauer, 2008). Pyramidal cells in the mPFC were visualized in layer V (∼700 μm lateral to the interhemispheric fissure). Pyramidal cells in the BLA were identified by their accommodation properties to a sustained depolarizing intracellular current injection (Sah et al., 2003). Recording pipettes (3–5 MΩ tip resistance) made from borosilicate glass were filled with intracellular solution containing the following (in mm): 122 K-gluconate, 5 NaCl, 0.3 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, and 0.4 Na3-GTP, pH adjusted to 7.2–7.3 with KOH (osmolarity adjusted to 280 mOsm/kg with sucrose). Data acquisition and analysis of voltage and current signals was done using a dual four-pole Bessel filter (Warner Instruments), a low-noise Digidata 1322 interface (Molecular Devices), an Axoclamp-2B amplifier (Molecular Devices), a Pentium personal computer, and pClamp9 software (Molecular Devices). Head-stage voltage was monitored continuously on an oscilloscope to ensure precise performance of the amplifier. Neurons were voltage clamped at −70 or 0 mV (reversal potential of EPSCs) for the study of excitatory and inhibitory transmission, respectively. High (gigaohm) seal and low series (<20 MΩ) resistances were checked throughout the experiment (using pClamp9 membrane test function) to ensure high-quality recordings.
Synaptic transmission.
Using concentric bipolar stimulating electrodes (David Kopf Instruments), EPSCs were evoked in BLA neurons by focal electrical stimulation (Grass Instruments S88 stimulator) of inputs from the lateral amygdala. EPSCs and IPSCs were evoked in PFC neurons by focal electrical stimulation (150 μs square-wave pulses) of presumed BLA afferents in the infralimbic cortex as described by the Gallagher group (Orozco-Cabal et al., 2006). The stimulating electrode was placed in layer V (500 μm from the medial surface of the slice) of the infralimbic cortex in which BLA afferents were identified by the fluorescent signal originating from anterogradely labeled fibers after stereotaxic injections of a fluorescent tracer [1,1-dioctadecyl-3,3,3,3-tetramethylindocarbocyanine perchlorate (DiI) (Invitrogen); dissolved in N,N-dimethylformamide dimethyl acetal (Sigma-Aldrich); 2 μl at 40 nl/min) into the BLA (same coordinates as for microdialysis probes) 10–12 d before brain tissue was taken. The distance between stimulation and recording sites was 600–800 μm.
Spontaneous and miniature (in 1 μm TTX) EPSCs and IPSCs were recorded at −70 and 0 mV, respectively, as described previously (Fu and Neugebauer, 2008). A fixed length of traces (5 min) was analyzed for frequency and amplitude distributions using MiniAnalysis program 5.3 (Synaptosoft). The root mean square (RMS) of the background noise was computed for each set of data. The detection threshold for an event was set to three to four times the RMS value. Peaks were detected automatically, but each detected event was then visually inspected to prevent the inclusion of false data.
Drug application.
Drugs were applied by gravity-driven superfusion of the brain slice in the ACSF (∼2 ml/min). Solution flow into the recording chamber (1 ml volume) was controlled with a three-way stopcock.
Behavioral tests
Spinal reflexes.
Thresholds of hindlimb withdrawal reflexes evoked by mechanical stimulation of the knee joint were measured as described previously (Neugebauer et al., 2007). Mechanical stimuli of continuously increasing intensity were applied to the knee joint using a calibrated forceps (see above, Electrophysiology in vivo). Withdrawal threshold was defined as the minimum stimulus intensity that evoked a withdrawal reflex.
Vocalizations.
Audible and ultrasonic vocalizations were recorded and analyzed as described previously (Neugebauer et al., 2007; Fu and Neugebauer, 2008). The experimental setup (United States Patent 7,213,538) included a custom-designed recording chamber, a condenser microphone (audible range, 20 Hz to 16 kHz) connected to a preamplifier, an ultrasound detector (25 ± 4 kHz), filter and amplifier (UltraVox four-channel system; Noldus Information Technology), and data acquisition software (UltraVox 2.0; Noldus Information Technology), which automatically monitored the occurrence of vocalizations within user-defined frequencies and recorded number and duration of digitized events (vocalizations). The computerized recording system was set to suppress nonrelevant audible sounds (background noise) and to ignore ultrasounds outside the defined frequency range. Animals were placed in the recording chamber for acclimation 1 h before the vocalization measurements. Brief (15 s) innocuous (500 g/30 mm2) and noxious (2000 g/30 mm2) mechanical stimuli were applied to the knee, using a calibrated forceps (see above, Electrophysiology in vivo). The total duration of vocalizations (arithmetic sum of the duration of individual events) was recorded for 1 min, starting with the onset of the mechanical stimulus. Audible and ultrasonic vocalizations reflect supraspinally organized nocifensive and affective responses to aversive stimuli (Neugebauer et al., 2007).
Plus-maze test.
Anxiety-like behavior was measured in the elevated plus-maze test as a decrease of open-arm choices (ratio of open-arm entries to the total number of entries expressed as percentage), using a computerized recording and analysis system (Multi-Varimex version 1.00; Columbus Instruments) as described previously (Ji et al., 2007). Entries were measured every 5 min for a total observation period of 45 min. Open-arm ratio during the first 5 min period was used for the assessment of anxiety-like behavior (Walf and Frye, 2007).
Gambling task.
Decision-making was measured in a behavioral task (Pais-Vieira et al., 2007, 2009; Neugebauer et al., 2009) using a custom-designed computerized behavioral system (Coulbourn Instruments). The system included a behavioral arena (octagon) with two levers on each side for automated delivery of chocolate-coated food pellets (Research Diets). The levers were separated by a Plexiglas divider so that the animal needed to make a choice between the two sides/levers. A runway connected to the octagon by an automated guillotine door housed the animal briefly (10 s) between each trial. The rat was given 20 s to choose between two levers to recover a food reward in 90 consecutive trials: one lever released three food pellets only in 3 of 10 trials (high risk); the other lever gave one food pellet in 9 of 10 trials (low risk). The side of the high-risk lever remained the same throughout the experiment but changed randomly between different animals to avoid any lateralization bias. Main outcome measure was the preference index defined as the proportion of choices between the levers during the gambling task. Preference index was calculated for each group of 10 consecutive trials using the formula: (low-risk lever choices − high-risk lever choices)/number of completed trials. A negative preference index reflects high-risk decision-making.
Drug application by microdialysis in awake behaving animals.
As described in detail previously (Ji et al., 2007; Neugebauer et al., 2007; Fu and Neugebauer, 2008), a guide cannula was implanted stereotaxically the day before behavioral measurements, using a stereotaxic apparatus (David Kopf Instruments). The animal was anesthetized with pentobarbital sodium (Nembutal, 50 mg/kg, i.p.), and a small unilateral craniotomy was performed at the sutura frontoparietalis level. The guide cannula was implanted on the dorsal margin of the BLA or CeA in the right hemisphere, using the following coordinates (in mm): BLA, 3.5 caudal to bregma, 5.0 lateral to midline, 7.0 depth; CeA, 2.0 caudal to bregma, 4.0 lateral to midline, 7.5 depth. The cannula was fixed to the skull with dental acrylic (Plastics One). Antibiotic ointment was applied to the exposed tissue to prevent infection. On the day of the experiment, a microdialysis probe (CMA/11; see above, Electrophysiology in vivo) was inserted through the guide cannula so that the probe protruded by 1 mm. The probe was connected to a Harvard Apparatus infusion pump and perfused with ACSF (oxygenated and equilibrated to pH 7.4). Before each drug application, ACSF was pumped through the fiber for at least 1 h to establish equilibrium in the tissue. Drugs were dissolved in ACSF on the day of the experiment at a concentration 100-fold that predicted to be needed because of the concentration gradient across the dialysis membrane and diffusion in the tissue (see above, Electrophysiology in vivo). Numbers in the text refer to drug concentrations in the microdialysis fiber.
Drugs
The following selective compounds purchased from Tocris Bioscience were used: NBI27914 [selective non-peptide corticotropin-releasing factor 1 (CRF1) receptor antagonist (Ji et al., 2007; Fu and Neugebauer, 2008)]; dl-2-amino-5-phosphonopentanoic acid (AP-5) (NMDA receptor antagonist); 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7- sulfonamide disodium salt (NBQX) (non-NMDA receptor antagonist); 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX) (non-NMDA receptor antagonist); and bicuculline ([R-(R*,S*)]-6-(5,6,7,8-tetrahydro-6-methyl-1,3-dioxolo[4,5-g]isoquinolin-5-yl)furo [3,4-e]-1,3-benzodioxol-8(6H)-one) (GABAA receptor antagonist). Drugs were dissolved in ACSF on the day of the experiment. ACSF served as vehicle control in all experiments.
Adrenocorticotropic hormone (ACTH) (Synacthen Depot, Novartis Portugal) was used for the assessment of hypothalamic–pituitary–adrenal (HPA) axis influences on anxiety-like and decision-making behaviors (see Fig. 4).
Figure 4.
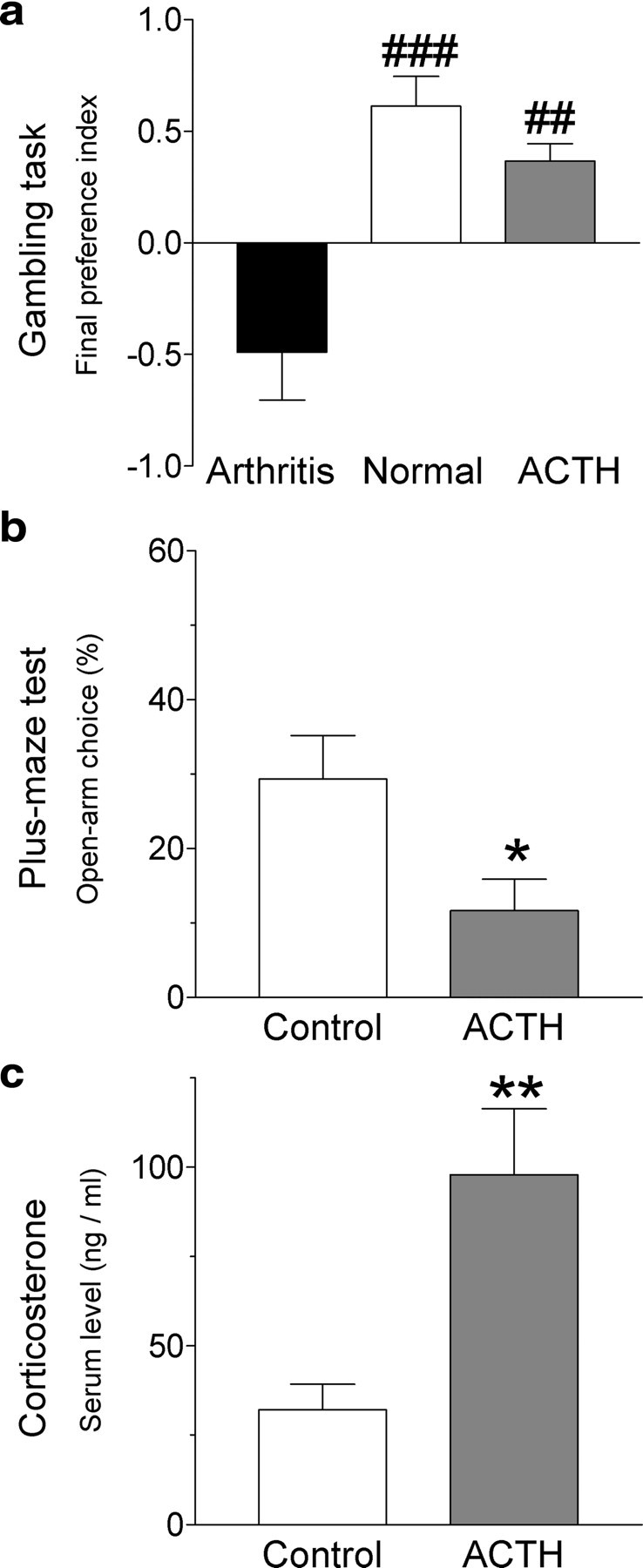
ACTH increases anxiety-like behavior and corticosterone blood levels but does not impair performance on the gambling task. ACTH (0.2 mg/kg in sterile physiological saline) or sterile saline (vehicle) was injected intraperitoneally every day for 4 d. Behavioral tests were performed 1 h after the final injection. a, Final preference index of ACTH-treated rats (n = 6) in the rodent gambling task (see Materials and Methods) was positive like that of normal control rats (n = 5), reflecting preference for the low-risk choice. Preference index of ACTH-treated rats and control rats was significantly different from the negative preference index of arthritic rats (n = 5). Final preference index was calculated for the last 30 consecutive trials using the following formula: (low-risk lever choices) − (high-risk lever choices)/number of completed trials. A negative preference index reflects high-risk decision-making (see Materials and Methods). b, In the elevated plus-maze test, open-arm preference (expressed as percentage; see Materials and Methods) of ACTH-treated rats (n = 6) was significantly decreased compared with vehicle-treated control rats (n = 6). c, Corticosterone serum levels (measured with radioimmunoassay kit; Siemens) (Ferreira et al., 2004) were significantly higher in ACTH-treated rats (n = 6) than in vehicle-treated control rats (n = 6). Bar histograms show mean ± SE. *,**p < 0.05, 0.01 (compared with control; t test); ##,###p < 0.01, 0.001 (compared with arthritis; Tukey's multiple comparison test).
Histology
At the end of an electrophysiological in vivo experiment, direct current (250 μA, 3 min) was passed through the recording electrode to create a small lesion to mark the recording site. The brain was removed and submerged in 10% Formalin and potassium ferrocyanide. At the end of a behavioral experiment, the animal was killed, and the brain was removed and submerged in 10% Formalin. Tissues were stored in 20% sucrose before they were frozen sectioned at 50 μm. Sections were stained with Neutral Red, mounted on gel-coated slides, and coverslipped. Positions of the microdialysis fibers and lesion/recording sites were identified under the microscope and plotted on standard diagrams (Ji et al., 2007; Fu and Neugebauer, 2008).
Statistical analysis
All averaged values are given as the mean ± SE. Statistical significance was accepted at the level p < 0.05. Prism 3.0 software (GraphPad Software) was used for all statistical analyses. For multiple comparisons, one-way ANOVA or two-way ANOVA was used with appropriate post hoc tests as indicated in the text and figure legends (Dunnett's test to compare all sets of data to a control value, Tukey's post hoc test to compare all pairs of data; and Bonferroni's test to compare selected pairs of data). Student's t test (paired or unpaired when appropriate) was used to compare two sets of data that have Gaussian distribution and similar variances (Prism 3.0 software; GraphPad Software).
Results
Pain-related plasticity in the BLA
As a first step in the analysis of amygdalo-cortical interactions in pain, we determined pain-related functional changes in the BLA. Extracellular single-unit recordings in anesthetized rats were made from individual BLA neurons (see location in Fig. 1e,f) before and after the induction of a localized arthritis in one knee joint by intra-articular injection of kaolin and carrageenan (see Materials and Methods) (Neugebauer et al., 2007). BLA neurons were activated by antidromic stimulation in the mPFC (Fig. 1b,g). Only one neuron was studied in each animal. Focus on a homogenous population of BLA projection neurons and use of a paired experimental paradigm in which each neuron serves as its own control form a highly sensitive assay that allows the detection of significant changes with relatively small numbers of animals and data points (Neugebauer and Li, 2003; Ji and Neugebauer, 2007).
Figure 1.
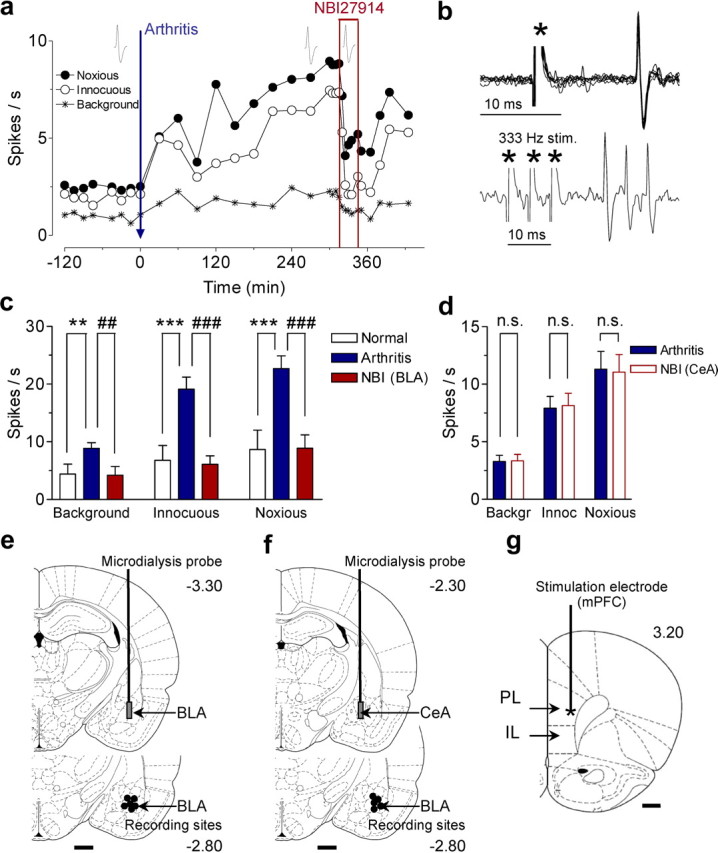
Pain-related sensitization of BLA neurons involves CRF1 receptors. a, Time course of activity changes in an individual BLA neuron recorded continuously before and after induction of an arthritis pain state (extracellular single-unit recordings in an anesthetized rat). Each symbol shows background activity or responses to innocuous and noxious stimuli applied to the knee joint. Net evoked activity was calculated as the difference between total activity during stimulation and preceding background activity. Administration of a CRF1 receptor antagonist (NBI27914, 10 μm, concentration in microdialysis fiber) into the BLA inhibited the increased activity in the pain state. b, Antidromic activation of BLA neuron by electrical stimulation in the prelimbic cortex (see Materials and Methods). Top, Six superimposed traces show constant latency of evoked spike; bottom, spikes follow high-frequency stimulation. * denotes stimulus artifact. c, Significant effects of arthritis (n = 6) and NBI27914 (n = 6) in the sample of neurons (Tukey's multiple comparison test). For each neuron, two to three values were averaged before arthritis induction, 5–6 h after induction, and during NBI27914 (>15 min). d, Control administrations of NBI27914 into the CeA had no effect on BLA neurons (n = 5; paired t test). Bar histograms in c and d show mean ± SE; ns, not significant; **,##p < 0.01, ***,###p < 0.001, compared with normal and predrug, respectively. e, f, Recording sites in the BLA and positions of microdialysis probes in the BLA (e) and CeA (f). Numbers indicate distance posterior to bregma. g, Stimulation site in the mPFC. Number indicates distance anterior to bregma. Scale bars, 1 mm.
Like CeA neurons (Neugebauer et al., 2004), BLA neurons with somatosensory receptive fields responded more strongly to noxious than innocuous stimuli (mechanical compression of peripheral tissue; n = 6 neurons). These neurons represent so-called “multireceptive” neurons based on the classification of nociceptive amygdala neurons (Neugebauer et al., 2004). After arthritis induction, background activity and responses to innocuous (500 g/30 mm2) and noxious (2000 g/30 mm2) stimulation of the knee increased significantly and reached a maximum plateau after 5 h (Fig. 1, see time course in a and summary data in c) (n = 6 neurons; q(10) = 5.739, background; q(10) = 8.792, innocuous; q(10) = 8.812, noxious; p < 0.01–0.001, Tukey's multiple comparison test).
Next we examined whether increased responsiveness was attributable to enhanced synaptic transmission and excitability changes in BLA neurons. Whole-cell patch-clamp recordings were made in brain slices from normal rats and from rats with arthritis (5–6 h after induction of kaolin/carrageenan arthritis). The 5–6 h time point was selected as in our previous studies (Neugebauer et al., 2003, 2004; Fu and Neugebauer, 2008) because continuous recordings in vivo (Neugebauer and Li, 2003; Neugebauer et al., 2004; Ji and Neugebauer, 2007) show that neuronal changes reach a maximum plateau at this stage (Fig. 1a).
BLA neurons in slices from normal and from arthritic rats selected for this study showed marked spike frequency accommodation in response to intracellular injection of a depolarizing current (200 pA, 500 ms) (Fig. 2a), suggesting that the same cell type was compared. Spike accommodation is a defining property of pyramidal neurons that constitute the predominant cell type in the BLA and function as projection neurons in this region of the amygdala (Washburn and Moises, 1992; Sah et al., 2003). BLA neurons from arthritic rats (n = 7 neurons) had a higher action potential firing rate and lower spike threshold than control neurons (n = 10) from normal animals (Fig. 2a) (p < 0.001, F(1,105) = 18.18, two-way ANOVA), which is consistent with increased neuronal excitability.
Figure 2.
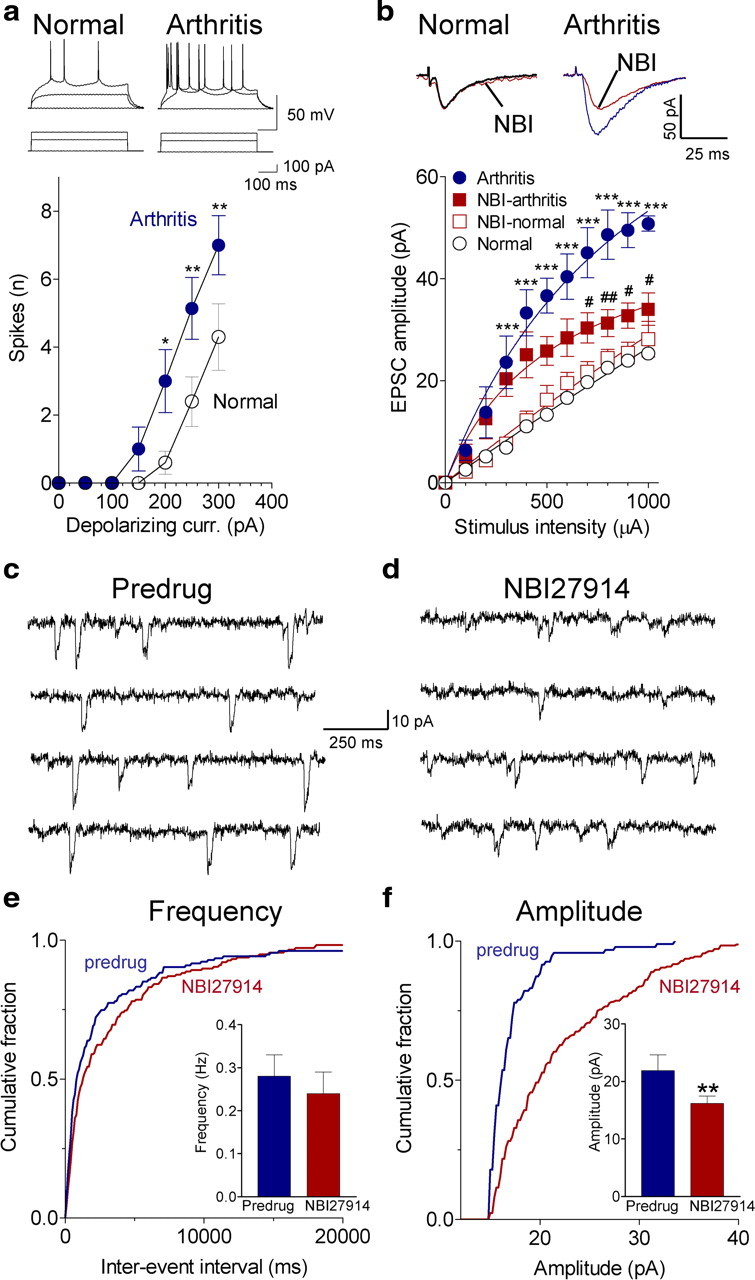
Pain-related changes in BLA neurons involves CRF1 receptors. a, Whole-cell current-clamp recordings of action potentials generated by intracellular current injections (500 ms) of increasing magnitude show significantly increased spike firing rate of BLA neurons in slices from arthritic rats (obtained 5–6 h after induction; n = 7 neurons) compared with normal controls (n = 10; Bonferroni's post hoc tests after two-way ANOVA; see Results). Symbols show mean ± SE number of spikes per 500 ms averaged for each sample of neurons. b, Whole-cell voltage-clamp recordings of monosynaptic EPSCs evoked at the LA–BLA synapse show that input–output function increased significantly (Bonferroni's post hoc tests after two-way ANOVA; see Results) in slices from arthritic rats (obtained 5–6 h after induction; n = 5 neurons) compared with control slices from normal rats (n = 5 neurons). NBI27914 (500 nm; 10–15 min) decreased EPSC amplitude in arthritis significantly (n = 5 neurons; Bonferroni's post hoc tests) but had no effect on normal transmission (n = 5 neurons). For each neuron, peak amplitudes of three to five EPSCs per stimulus intensity were averaged. Traces show averages of 8–10 evoked EPSCs. c, d, Original traces of miniature EPSCs recorded in TTX (1 μm) before and during application of NBI27914 (500 nm; 15 min) in a slice from an arthritic rat (5 h after induction). e, NBI27914 had no significant effect on cumulative interevent interval distribution and mean frequency of miniature EPSCs (n = 8 neurons; p > 0.05, paired t test; bar histograms). f, NBI27914 shifted cumulative amplitude distribution toward larger values, increasing mean amplitude significantly (n = 8 neurons; p < 0.01; bar histograms). e, f, Bar histograms shown mean ± SE before and during NBI27914. *,#p < 0.05, **,##p < 0.01, ***p < 0.001 (compared with normal and predrug, respectively).
Increased synaptic transmission was also measured in slices from arthritic rats compared with controls (Fig. 2b). Monosynaptic EPSCs were evoked by electrical stimulation in the LA. The LA–BLA synapse is part of the amygdala circuitry involved in aversive behavior (Sah et al., 2003; Maren and Quirk, 2004; Phelps and LeDoux, 2005; Ehrlich et al., 2009). EPSCs were mediated by non-NMDA receptors because they were completely blocked with CNQX (20 μm) (data not shown). Only one neuron was recorded in each slice, and only one slice was used from each animal. Input–output (I/O) function of excitatory transmission at the LA–BLA synapse was significantly increased in slices from arthritic rats (n = 5 neurons) compared with normal controls (n = 5 neurons; p < 0.001, F(1,88) = 233.75, two-way ANOVA) (Fig. 2b). I/O function is a well accepted measure of the sensitivity of a neuron to synaptic inputs (Carvalho and Buonomano, 2009) and has been used successfully in our previous studies to determine synaptic plasticity in ex vivo slice preparations in different disease models (Neugebauer et al., 1997, 2000, 2003; Bird et al., 2005; Fu and Neugebauer, 2008).
Neural changes are not observed in slices from sham animals with intra-articular saline injections (Neugebauer et al., 2003), suggesting that synaptic and excitability changes in slices from arthritic rats are the result of the arthritic lesion in the knee in this pain model. The results demonstrate pain-related synaptic plasticity in the BLA, as we showed before in the CeA (Neugebauer et al., 2004, 2009), because synaptic facilitation is preserved in the brain slice preparation independently of continued input from the site of injury (arthritic knee).
Blockade of CRF1 receptors restores normal activity in the BLA
To determine the correlation of neuronal activity in BLA with cortical function and behavior, we examined whether a selective antagonist for corticotropin-releasing factor 1 (CRF1) receptor (NBI27914) could be used to deactivate the BLA. CRF1 receptor was targeted because of its critical contribution to pain-related plasticity, but not normal processing, in the CeA as shown in our previous studies (Ji and Neugebauer, 2007; Fu and Neugebauer, 2008). CRF1 receptor activation is also important for BLA plasticity related to anxiety (Rainnie et al., 2004). However, pain-related CRF1 receptor function in the BLA has not been shown. Here we tested the hypothesis that CRF1 receptor is critical for pain-related plasticity in the BLA.
Administration of a CRF1 antagonist (NBI27914, 10 μm, concentration in microdialysis fiber; 20 min) into the BLA by microdialysis 5–6 h after arthritis induction inhibited background and evoked activity significantly (Fig. 1, see individual neuron in a and summary data in c,e) (n = 6 neurons; q(10) = 5.991, background; q(10) = 9.293, innocuous; q(10) = 8.664, noxious; p < 0.01–0.001, Tukey's multiple comparison test). Importantly, administration of NBI27914 into the CeA as a control for drug diffusion had no effect on BLA neurons 5–6 h after arthritis induction (Fig. 1d,f) (n = 5 neurons; t(4) = 0.3900, background; t(4) = 0.6038, innocuous; t(4) = 1.568; p > 0.05, paired t test) but inhibited pain-related sensitization of CeA neurons in our previous study (Ji and Neugebauer, 2007). The data suggest that inhibition of BLA activity resulted from CRF1 receptor blockade in the BLA.
To determine the synaptic site of CRF1 receptor action in the BLA, we tested the effect of NBI27914 on evoked and miniature EPSCs (Fig. 2). NBI27914 (500 nm; 15 min) inhibited synaptic plasticity in BLA neurons in slices from arthritic rats (Fig. 2b) (n = 5 neurons; p < 0.001, F(1,88) = 37.97, two-way ANOVA) but had no significant effect under normal conditions (n = 5 neurons; p > 0.05, F(1,88) = 4.89, two-way ANOVA). NBI27914 (500 nm; 15 min) decreased amplitude (Fig. 2f) but not frequency (Fig. 2e) of miniature EPSCs (in 1 μm TTX) (original traces in Fig. 2c,d) significantly (cumulative amplitude distribution, p < 0.05, Kolmogorov–Smirnov test; mean amplitude, p < 0.01, paired t test; n = 8 neurons). Miniature EPSC analysis was done only in slices from arthritic rats (obtained 5–6 h after induction) because NBI27914 had no effect on baseline synaptic transmission in slices from normal animals (Fig. 2b). The data are consistent with a postsynaptic action on BLA neurons as shown previously in the CeA (Fu and Neugebauer, 2008).
The pharmacological data show that postsynaptic CRF1 receptors contribute to synaptic facilitation and are useful targets to restore normal activity in the BLA (present study) or in the CeA (Ji and Neugebauer, 2007; Fu and Neugebauer, 2008) in the arthritis pain model.
Pharmacologic deactivation of BLA inhibits pain responses
Using the CRF1 receptor antagonist to restore normal function of the BLA in our pain model, we determined the contribution of BLA hyperactivity to nocifensive and affective pain behaviors (Fig. 3). Hindlimb withdrawal thresholds and audible vocalizations served as outcome measures of spinally and supraspinally, respectively, organized nocifensive responses. Ultrasonic vocalizations reflect affective responses. The elevated plus-maze test measured anxiety-like behavior. These tests have been used successfully in our previous studies to show the involvement of the CeA in nocifensive and affective components of arthritic pain (Ji et al., 2007; Neugebauer et al., 2007; Fu and Neugebauer, 2008). Baseline values under normal conditions in the absence of arthritis showed little variability in the sample of animals in this study (Fig. 3a–d).
Figure 3.
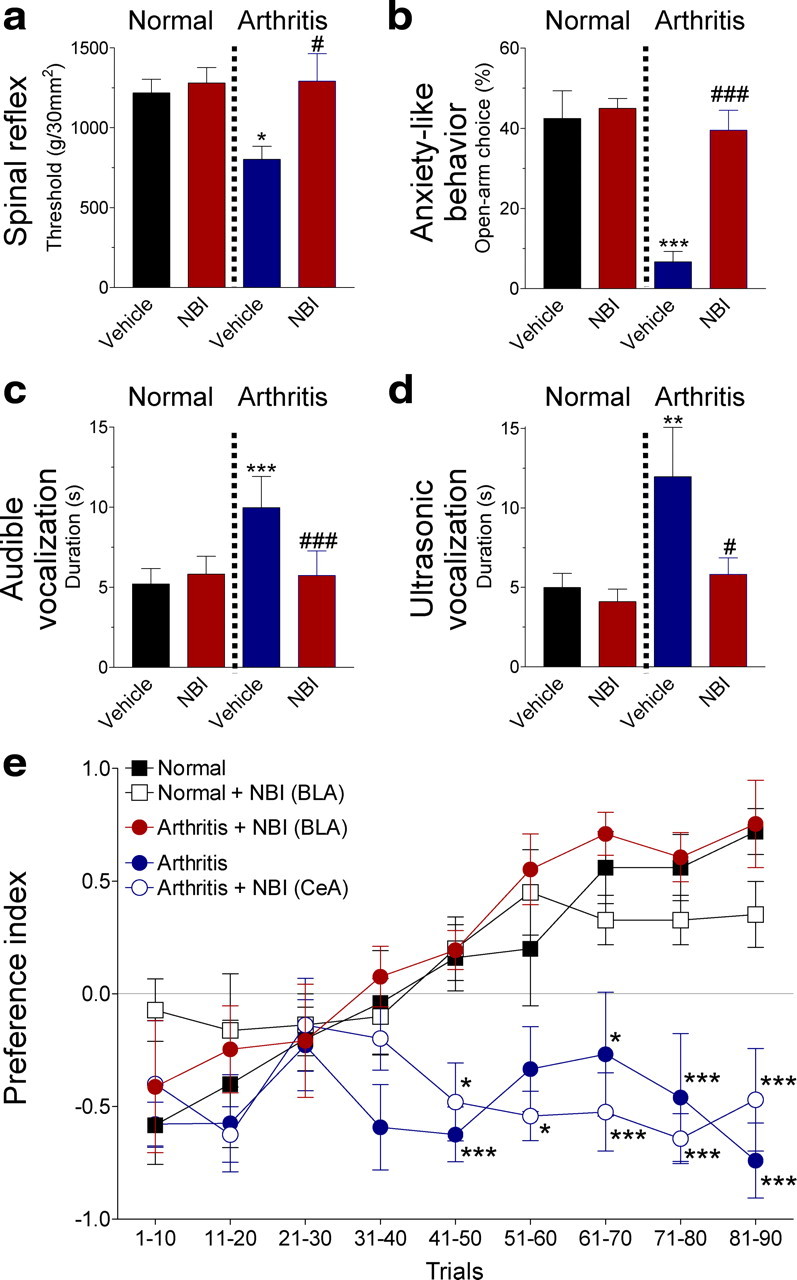
BLA deactivation inhibits nocifensive and affective pain behaviors and restores impaired decision-making. a, Administration of a CRF1 receptor antagonist (NBI27914, 10 μm; 15–20 min) into the BLA decreased hindlimb withdrawal thresholds in the arthritis pain state but not under normal conditions. The same animals (n = 5) were tested before and 5–6 h after arthritis induction. For each animal, three measurements were averaged. b, Anxiety-like behavior was measured in the elevated plus maze as decreased open-arm choice (ratio of open-arm entries to the total number of entries expressed as percentage; see Materials and Methods). Normal animals (n = 15) were tested with vehicle (ACSF) administration into the BLA and then assigned to one of three treatment groups: NB27914 (10 μm; 15–20 min) administration into BLA under normal conditions (n = 5), vehicle administration into the BLA 5–6 h after arthritis induction (n = 5), and NBI27914 administration into the BLA 5–6 h after arthritis induction (n = 5). c, d, Vocalizations in the audible and ultrasonic range were measured in the same animals before and 5–6 h after arthritis induction (n = 6). NBI27914 (10 μm; 15–20 min) inhibited vocalizations in arthritis but not under normal conditions. Bar histograms in a–d show means ± SE; two to three measurements were averaged for each animal. e, Deactivation of the BLA, but not CeA, with NBI27914 (10 μm, 20 min) restored normal reward-based decision-making in arthritic rats (5–6 h after induction). Symbols (mean ± SE) show the preference index [(low-risk − high-risk choices)/number of trials] averaged for 10 consecutive trials of animals in five experimental groups (n = 5 each). Animals chose between low-risk (1 food pellet in 9 of 10 visits) and high-risk (3 pellets in 3 of 10 visits) levers. Preference index of normal animals and arthritic animals with deactivated BLA was not significantly different. Preference index of arthritic rats with or without deactivated CeA was significantly different from normal animals. a–d, *,#p < 0.05, **p < 0.01, ***,###p < 0.001 (compared with normal and predrug, respectively; Tukey's multiple comparison test). e, *,***p < 0.05, 0.001 (compared with normal; Dunnett's multiple comparison test).
In the arthritis pain state (5–6 h after induction), withdrawal thresholds decreased, indicating increased spinal nocifensive reflexes (Fig. 3a) (n = 5; p < 0.05, Tukey's multiple comparison test). Duration of audible (Fig. 3c) and ultrasonic (Fig. 3d) vocalizations increased (n = 6; p < 0.001 and p < 0.01, respectively, Tukey's multiple comparison test). Animals spent less time exploring the open arm of the plus maze, which is consistent with anxiety-like behavior (Fig. 3b) (n = 5; p < 0.001, Tukey's multiple comparison test). Deactivation of the BLA with NBI27914 (10 μm, 15–20 min) reversed pain-related changes of spinal reflexes (Fig. 3a) (n = 5; p < 0.05) and audible (Fig. 3c) and ultrasonic (Fig. 3d) vocalizations (n = 6; p < 0.001 and p < 0.05, respectively, Tukey's multiple comparison test). NBI27914 also had anxiolytic-like effects in arthritic animals (Fig. 3b) (n = 5; p < 0.001, Tukey's multiple comparison test). NBI27914 did not affect behavioral outcome measures under normal conditions (Fig. 3a–d).
The behavioral effects of NBI27914 in the BLA were similar to those in the CeA reported in our previous studies (Ji et al., 2007; Fu and Neugebauer, 2008). The results show for the first time that pain-related plasticity in the BLA (this study) as well as in the CeA (our previous studies) contributes to nocifensive and affective components of pain behavior, which is consistent with a serial model of amygdala function involving BLA to CeA projections to execute behavioral responses (Sah et al., 2003; Phelps and LeDoux, 2005).
Pharmacologic deactivation of BLA, but not CeA, reverses pain-related decision-making deficits
Next, we tested the hypothesis that pain-related hyperactivity in the BLA leads to cognitive deficits associated with pain, because the BLA is a major source of input to the mPFC, which plays a key role in cognitive functions such as decision-making and avoidance of emotion-based risky choices (see Introduction). Pain-related cognitive impairment was determined in a computerized behavioral task in which rats decided between high-risk and low-risk strategies (Fig. 3e). This rodent “gambling task” is based on serial choices between food rewards of different value and probability (Pais-Vieira et al., 2007, 2009). In 90 consecutive trials, a rat enters the test arena for 20 s to press one of two levers associated with a food reward before it is returned to the entry chamber. After the non-gambling training phase in which each of two levers provided one food pellet in 9 of 10 visits, animals were tested on decision-making in a single session of 90 consecutive trials in which one lever was altered to return three pellets in 3 of 10 trials (high-risk choice), whereas the other lever continued to deliver one pellet in 9 of 10 trials (low-risk choice).
Normal rats (n = 5) developed, after ∼40 trials, a preference for the low-risk lever that provided smaller but more reliable rewards (Fig. 3e). In contrast, rats with arthritis (5–6 h after induction; n = 5) failed to change strategies and perseverated in preferring the high-risk lever associated with larger but infrequent rewards, which is reflected in a negative preference index (Fig. 3e). Preference index of arthritic rats was significantly different from that of normal rats after 40 trials (p < 0.05–0.001, Dunnett's multiple comparison test for each group of 10 trials). A similar decision-making deficit has been observed in rats with PFC lesions (Pais-Vieira et al., 2007) or chronic pain (Pais-Vieira et al., 2009). Impaired decision-making was not attributable to pain-related motor or attention deficits, because reaching each lever involved the same movements and choices were not cue-based in this task.
Pharmacologic deactivation of the BLA in arthritic rats (5–6 h after induction; n = 5) with NBI27914 (10 μm, concentration in microdialysis fiber; 20 min) restored normal decision-making (Fig. 3e). Preference index of arthritic rats with deactivated BLA was not significantly different from that of normal rats (p > 0.05, Dunnett's multiple comparison test). Administration of NBI27914 into the BLA of normal rats (n = 5) had no effect on their performance in the decision-making task, which is consistent with the lack of involvement of CRF1 receptor in normal processing in the BLA (Fig. 2b).
Importantly, deactivation of the CeA in arthritic rats (5–6 h after induction; n = 5) with NBI27914 did not restore normal decision-making, although administration of NBI27914 into the CeA restores normal function of CeA neurons and inhibits nocifensive and affective behaviors in the arthritis pain model (Ji and Neugebauer, 2007; Fu and Neugebauer, 2008). Preference index of arthritic rats with deactivated CeA was significantly different from that of normal rats (p < 0.05–0.001, Dunnett's multiple comparison test). The data suggest that hyperactivity in the BLA, but not CeA, contributes to impaired decision-making in the arthritis pain model.
Because the CeA can facilitate the activation of the HPA axis in response to stress (Phelps and LeDoux, 2005; Myers and Greenwood-Van Meerveld, 2007), we considered the possibility that CRF1 receptor antagonist effects were attributable to alterations of HPA axis function and stress response. Electrophysiological data in this (Figs. 1, 2) and our previous study (Fu and Neugebauer, 2008) show a direct action on amygdala neurons. Differential effects of site-specific antagonist administration into BLA and CeA further argue against a generalized action on the HPA axis. To better understand the contribution of HPA axis-mediated stress responses to cognitive deficits observed in the arthritis pain model, we measured the effect of ACTH treatment on the performance in the gambling task and elevated plus maze (Fig. 4a,b). ACTH challenge is commonly used to induce rapid increases of corticosterone levels (Murakami and Takahashi, 1983; Ferreira et al., 2004). Rats treated with systemic injections of ACTH (0.2 mg/kg, i.p.) showed significantly higher corticosterone blood levels (Fig. 4c) and increased anxiety-like behavior (Fig. 4b) as expected (Myers and Greenwood-Van Meerveld, 2007) but were not impaired in the decision-making task (Fig. 4a). Preference index of ACTH-treated rats, like that of normal rats, was significantly different from that of arthritic rats (p < 0.01, Dunnett's multiple comparison test). The data strongly suggest that HPA axis activation and anxiety are not sufficient to explain cognitive impairment in pain.
Pain-related deactivation of mPFC neurons is reversed by deactivation of BLA but not CeA
The medial regions of the PFC are important for emotional value-based decision-making, which is impaired in pain patients (Apkarian et al., 2004b). Pain-related morphological changes have been observed in the mPFC of animals (Metz et al., 2009) and humans (Apkarian et al., 2004a). We tested the hypothesis that mPFC function is impaired in pain through a mechanism that involves amygdala hyperactivity (see Introduction).
Extracellular single-unit recordings in anesthetized rats were made from individual neurons in the prelimbic part of the mPFC (n = 26 neurons) (location shown in Fig. 5d). Action potential duration (540 ± 20 μs peak-to-valley) and firing rate (2.9 ± 0.3 spikes/s) were consistent with presumed pyramidal cells rather than fast-spiking interneurons, the latter having a higher baseline firing rate (>10 Hz) and narrower spike waveform (<300 μs) (Constantinidis and Goldman-Rakic, 2002; Laviolette and Grace, 2006). Background activity and responses to innocuous (500 g/30 mm2) and noxious (2000 g/30 mm2) stimuli were measured repeatedly in the same mPFC neurons (n = 26) before and for several hours after induction of an arthritis pain state (see individual neurons in Figs. 4b, 5a). Background and evoked activity decreased in the sample of neurons (n = 26) significantly (p < 0.05–0.01, paired t test, comparing 5–6 h post-arthritis with pre-arthritis values) (Fig. 5c).
Figure 5.
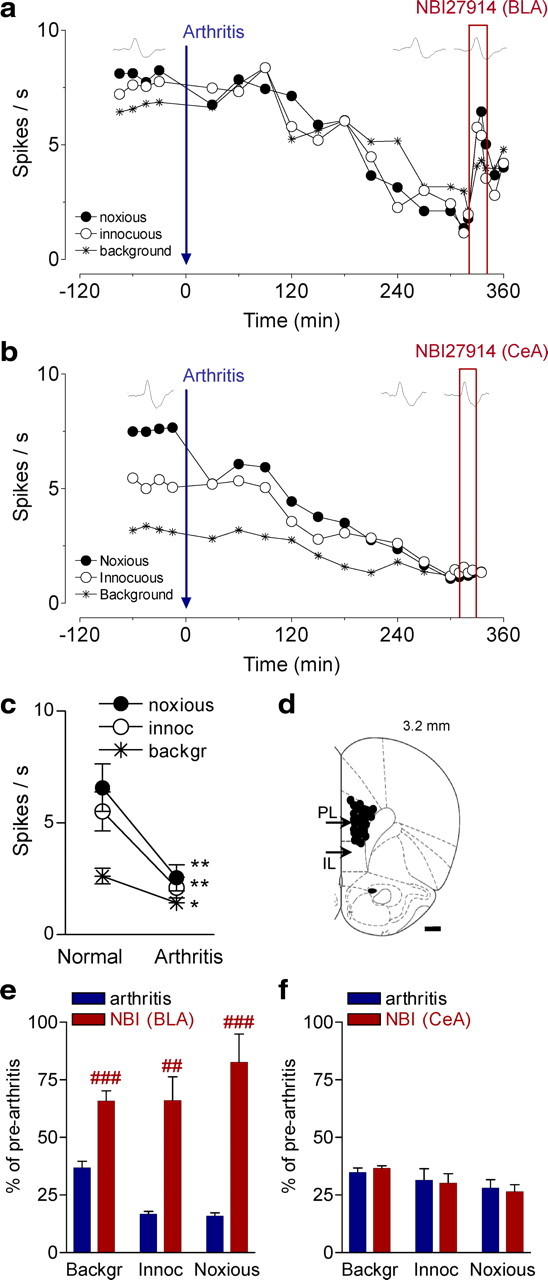
Deactivation of BLA restores normal activity in PFC neurons. a, Time course of activity changes in an individual prelimbic mPFC neuron recorded continuously before and after induction of an arthritis pain state (extracellular single-unit recordings in an anesthetized rat). Each symbol shows background activity or responses to innocuous and noxious stimuli applied to the knee joint (see Fig. 1). Administration of a CRF1 receptor antagonist (NBI27914, 10 μm, concentration in microdialysis fiber; 20 min) into the BLA increased the activity of the mPFC neuron. b, Time course of activity changes in another prelimbic mPFC neuron recorded before and after arthritis induction. Administration of NBI27914 (10 μm) into the CeA had no effect. c, Significant decrease of background and evoked activity in the mPFC neurons 5–6 h after arthritis induction (n = 26 neurons). *p < 0.05, **p < 0.01 (compared with normal; paired t test). d, Recording sites in the prelimbic cortex. PL, Prelimbic part of the mPFC; IL, infralimbic part of the mPFC. e, Deactivation of the BLA with NBI27914 (10 μm) increased the background activity of mPFC neurons and their responses to innocuous and noxious stimuli significantly (n = 5 neurons; p < 0.01–0.001, paired t test). f, Administration of NBI27914 into the CeA had no effect on mPFC neurons (n = 5). e, f, Bar histograms show mean ± SE normalized to control values before arthritis (set to 100%). For each neuron, two to three data points were averaged before arthritis, 5–6 h after induction of arthritis, and during drug application (>15 min). ##,###p < 0.01, 0.001 (compared with predrug; paired t test).
Pharmacologic deactivation of the BLA with NBI27914 (10 μm) increased the background and evoked activity of prelimbic mPFC neurons in the arthritis pain state (5–6 h after induction) significantly (n = 5; p < 0.01–0.001, paired t test). An individual example is shown in Figure 5a, and normalized data for the sample of neurons are summarized in Figure 5e. In contrast, administration of NBI27914 into the CeA had no effect on the pain-related decrease of mPFC activity 5–6 h after induction of arthritis (n = 5 neurons) (Fig. 5, see individual neuron in b and summary data in f).
These results provide the first direct evidence that the amygdala (BLA) contributes to mPFC deactivation and dysfunction in pain, because blocking pain-related hyperactivity of BLA neurons restored normal activity in the prelimbic cortex.
Pain-related increase of glutamate-driven synaptic inhibition of mPFC neurons
Using whole-cell patch recordings in a brain slice preparation containing the mPFC (Orozco-Cabal et al., 2006), we studied synaptic mechanisms of BLA-induced PFC inhibition. Specifically, we tested the hypothesis that activation of inhibitory synaptic transmission to mPFC pyramidal cells is increased in the arthritis pain state and involves a glutamatergic mechanism. Our hypothesis was based on anatomical evidence for direct, primarily ipsilateral projections from BLA to mPFC, which form glutamatergic contacts with layer V pyramidal cells as well as with local circuit inhibitory neurons in the mPFC (Kita and Kitai, 1990; Gabbott et al., 2006).
EPSCs and IPSCs were evoked in visually identified prelimbic layer V pyramidal cells (Fig. 6a) by stimulation of presumed afferents from the BLA that traverse the infralimbic cortex as described previously (Orozco-Cabal et al., 2006). The stimulation electrode was positioned on the track of anterogradely labeled fibers from the BLA (Fig. 6b–d). Low stimulus intensities of twice the threshold were used to avoid current spread and activation of nearby fibers. Monosynaptic EPSCs were mediated by non-NMDA receptors because they were blocked significantly (p < 0.05–0.001, compared with predrug control, paired t test) by NBQX (2 μm, n = 6; 10 μm, n = 6 neurons) or CNQX (20 μm, n = 5 neurons) but not by an NMDA receptor antagonist (AP-5, n = 4 neurons) (Fig. 6e,f). GABAergic IPSCs were blocked by a GABAA receptor antagonist (bicuculline, 10 μm, n = 6 neurons) (Fig. 6e). IPSCs recorded at 0 mV holding potential were also reduced by non-NMDA receptor antagonists (NBQX, 2 μm, n = 7 neurons; 10 μm, n = 12; CNQX, 20 μm, n = 4; p < 0.05–0.01, compared with predrug, paired t test) (Fig. 6e,g), indicating that they were polysynaptic. NMDA receptor blockade with AP-5 had no effect (n = 4 neurons) (Fig. 6e,g).
Figure 6.
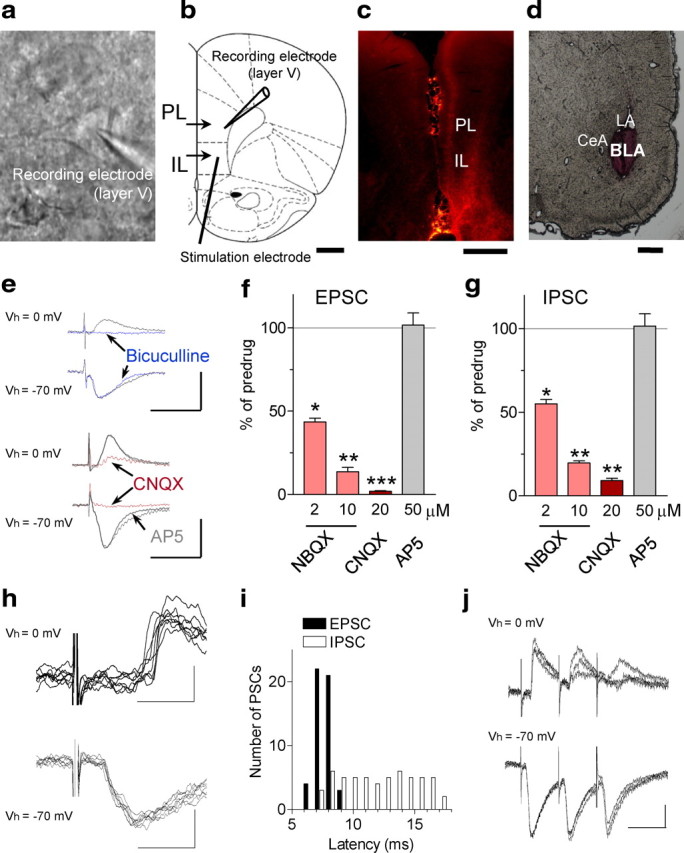
Glutamate-driven inhibitory synaptic transmission in the mPFC. Whole-cell voltage-clamp recordings of layer V mPFC pyramidal neurons. a, Individual pyramidal cell visually identified with infrared DIC video microscopy. b, Positions of recording electrode in prelimbic cortex (PL) and stimulation electrode on presumed BLA afferents in infralimbic cortex (IL). Level is 3.2 mm anterior to bregma. c, Identification of BLA afferents by anterograde labeling using tracer injections (DiI). Confocal microscopy analysis of coronal brain slices obtained 10–12 d after BLA injection shows fluorescent labeling of fiber bundles that divide to reach layers II and V of the infralimbic and prelimbic cortices, as described previously (Gabbott et al., 2006; Orozco-Cabal et al., 2006). Labeling of fibers is only ipsilateral to the injection site, and no cell bodies are stained in layer V (arguing against retrograde labeling). d, Tracer injection site in the BLA (2.3 posterior to bregma). Scale bars (in b–d), 1 mm. e–g, EPSCs and IPSCs evoked by stimulating presumed BLA afferents (see b–f). e, Individual traces (average of 8–10) evoked at −70 mV (downward deflections, EPSCs) or at 0 mV (upward deflections, IPSCs; blocked by bicuculline, 10 μm). Calibration: 50 pA, 20 ms. f, EPSCs were inhibited by NBQX (2 μm, n = 6; 10 μm, n = 6) and CNQX (20 μm, n = 5) but not by AP-5 (50 μm, n = 4). g, IPSCs were also inhibited by NBQX (2 μm, n = 7; 10 μm, n = 12) and CNQX (20 μm, n = 4) but not by AP-5 (50 μm, n = 4). f, g, Bar histograms show means ± SE. *,**,***p < 0.05, 0.01, 0.001 (compared with predrug; paired t test). h, Individual IPSCs and EPSCs evoked with twice-threshold stimulation. Latencies of IPSCs were longer and more variable. Calibration: 50 pA, 10 ms. i, Distribution of IPSC and EPSC latencies measured from onset of stimulus artifact to onset of synaptic current in one neuron (n = 50 events). j, Monosynaptic EPSCs, but not polysynaptic IPSCs, follow high-frequency stimulation (20 Hz; 3 individual traces each).
Average latency (from onset of stimulus artifact to onset of synaptic current) of monosynaptic EPSCs was significantly (p < 0.001, t test; n = 23 neurons) shorter (6.2 ± 0.3 ms) than that of polysynaptic IPSCs (8.3 ± 0.4 ms) (Fig. 6h,i). Synaptic jitter (SD of variation in latency) of IPSCs was significantly higher (874 ± 96 μs) than that of EPSCs (331 ± 29 μs; p < 0.001, t test; n = 23 neurons). Average failure rate of IPSCs (27 ± 3%) was significantly higher than that of EPSCs (8 ± 1.6%) with low stimulus intensities of twice the threshold (n = 23 neurons; p < 0.001, t test). EPSCs, but not IPSCs, followed high-frequency (10 Hz) synaptic stimulation.
The pharmacological profile (blockade by NBQX/CNQX and bicuculline) and synaptic characteristics (high jitter, longer latency, higher failure rate, inability to follow high-frequency stimulation) suggest that the IPSCs were polysynaptic, involving a glutamatergic synapse. In this serial two-step pathway, NBQX blocked an initial unobserved EPSC that was responsible for evoking the bicuculline-sensitive GABAergic IPSC (for discussion, see Doyle and Andresen, 2001). The results are consistent with morphological evidence for glutamatergic projections from BLA to GABAergic interneurons in the mPFC (Gabbott et al., 2006), suggesting glutamate-driven inhibition of prelimbic mPFC neurons by input from presumed BLA afferents.
Next we determined whether synaptic inhibition of PFC neurons increased in the arthritis pain model as a mechanism of pain-related PFC deactivation and dysfunction. I/O function of IPSCs (Fig. 7a,c), but not EPSCs (Fig. 7b,d), increased significantly (p < 0.0001, two-way ANOVA, F(1,242) = 55.16) in slices from arthritic rats (6 h after induction; IPSCs, n = 7 neurons; EPSCs, n = 9) compared with normal controls (IPSCs, n = 17 neurons; EPSCs, n = 21). The data suggest that the strength of inhibitory rather than excitatory transmission to PFC pyramidal cells increased in the arthritis pain state.
Figure 7.
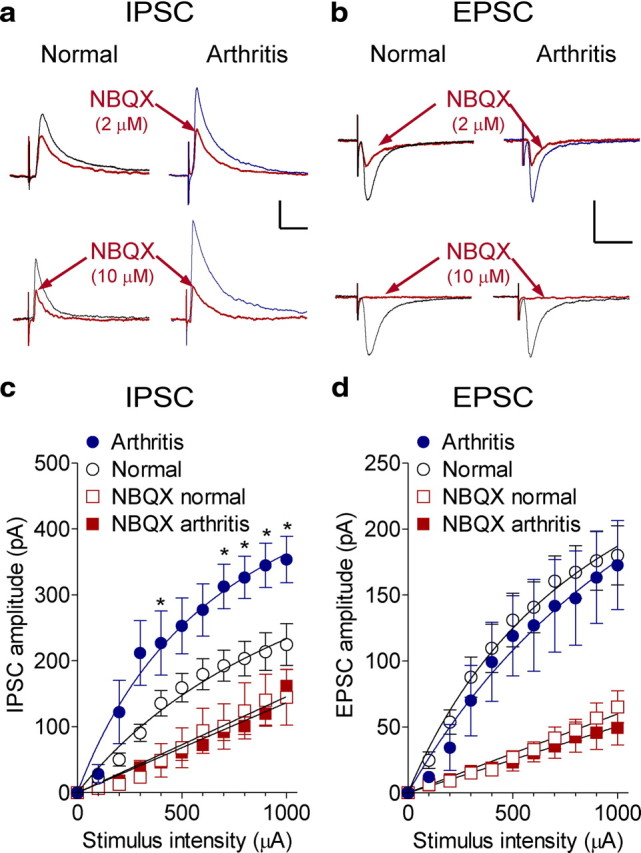
Increased synaptic inhibition of PFC neurons in arthritis pain model. Whole-cell voltage-clamp recordings of visually identified prelimbic layer V pyramidal cells as in Figure 6. a, Evoked IPSCs (recorded at 0 mV) increased in arthritis and were decreased by NBQX (2 and 10 μm). b, Evoked EPSCs (recorded at −70 mV) were inhibited by NBQX (2 and 10 μm) in control slices and in slices from arthritic rats (6 h after induction). Calibration: 100 pA, 50 ms. Individual traces are averages of 8–10 IPSCs. c, Input–output functions of IPSCs increased in slices from arthritic rats (5–6 h after induction; n = 7 neurons) compared with normal controls (n = 17 neurons) significantly (two-way ANOVA, F(1,242) = 55.16, p < 0.0001). NBQX (2 μm) significantly decreased IPSCs in control slices (n = 7 neurons; two-way ANOVA, F(1,242) = 32.98, p < 0.0001) and in slices from arthritic rats (n = 5 neurons; two-way ANOVA, F(1,110) = 129.91, p < 0.0001). d, Input–output functions of evoked EPSCs in slices from arthritic rats (n = 9 neurons) and from normal controls (n = 21 neurons) were not significantly different (two-way ANOVA, F(1,308) = 1.84, p > 0.05). NBQX (2 μm) had significant inhibitory effects under normal conditions (n = 6 neurons; two-way ANOVA, F(1,275) = 70.69, p < 0.0001) and in arthritis (n = 6 neurons; two-way ANOVA, F(1,143) = 46.25, p < 0.0001). *p < 0.05 (compared with normal; Bonferroni's post hoc test).
Increased synaptic inhibition of PFC pyramidal cells was driven by a glutamatergic mechanism involving non-NMDA receptors. Blockade of non-NMDA receptors with NBQX (2 μm) decreased evoked IPSCs (Fig. 7a,c) significantly in control slices (n = 7 neurons; p < 0.0001, two-way ANOVA, F(1,242) = 32.98) and in slices from arthritic rats (n = 5 neurons; p < 0.0001, two-way ANOVA, F(1,110) = 129.91). In the presence of NBQX, I/O functions of inhibitory transmission under normal conditions and in arthritis were not significantly different (p > 0.05, two-way ANOVA, F(1,110) = 0.10), suggesting that the pain-related increase of synaptic inhibition required non-NMDA receptor activation. Accordingly, inhibitory effects of NBQX (2 μm) on IPSCs were significantly greater in slices from arthritic rats (29 ± 2.3% of predrug; n = 5 neurons) compared with normal controls (55 ± 7.3% of predrug; n = 7; p < 0.05, t test). A general reduction of excitability by the blockade of postsynaptic AMPA receptors on pyramidal cells cannot account for the results. Inhibitory effects of NBQX on inhibitory synaptic transmission were recorded at the reversal potential for AMPA receptors (0 mV), and AMPA receptor-mediated excitatory transmission was not increased in the slices from arthritic animals.
These results are consistent with increased non-NMDA receptor function in the modulation of polysynaptic inhibitory transmission. The glutamate-driven increase of inhibitory, but not excitatory, transmission to mPFC neurons results in a net shift toward inhibition and is consistent with mPFC deactivation shown in our in vivo studies.
Discussion
Pain as a multidimensional experience offers valuable insight into neurobiological mechanisms of sensory, motor, emotional-affective, and cognitive aspects of behavior. Whereas sensorimotor aspects of pain have been studied in great detail and progress has been made in the better understanding of emotional-affective pain processes, mechanistic analysis of cognition-related brain functions and dysfunctions in pain is lagging behind (Neugebauer et al., 2009). This study tested the hypothesis that pain-related hyperactivity of the BLA, like the CeA, contributes to emotional-affective aspects of pain; however, unlike the CeA, BLA hyperactivity deactivates the mPFC through glutamate-driven synaptic inhibition, resulting in decision-making deficits. The rationale was based on the accumulating evidence for cognitive impairment in pain in humans (Apkarian et al., 2004b) and animals (Pais-Vieira et al., 2009), role of the PFC (Bechara et al., 1999; Vertes, 2006; Pais-Vieira et al., 2007; Stalnaker et al., 2007a; Kouneiher et al., 2009) and amygdala (Bechara et al., 1999, 2003; Stalnaker et al., 2007b; Seymour and Dolan, 2008) in emotion-based decision-making, implication of the PFC in pain-related cognitive deficits (Apkarian et al., 2004a; Pais-Vieira et al., 2009), and anatomical connections (Kita and Kitai, 1990; McDonald, 1991; Bacon et al., 1996; Gabbott et al., 2006) and functional interactions (Holland and Gallagher, 2004; McGaugh, 2004; Laviolette and Grace, 2006; Stalnaker et al., 2007a; Herry et al., 2008; Roozendaal et al., 2009) between amygdala and mPFC as a neurobiological basis for emotional learning and behavior.
The key result of the present study is that pain-related hyperactivity of BLA neurons contributes not only to emotional-affective aspects of pain but also to pain-related decision-making deficits through amygdala–cortical interactions that involve increased glutamate-driven GABAergic synaptic inhibition of PFC neurons. Specifically, our data show that BLA neurons, like CeA neurons (Neugebauer et al., 2009), develop central sensitization and synaptic plasticity in an animal model of arthritis pain. Pain-related hyperactivity of BLA neurons, like CeA neurons (Ji and Neugebauer, 2007; Fu and Neugebauer, 2008), involves CRF1 receptors. Deactivation of the BLA with a CRF1 receptor antagonist, like CeA deactivation (Ji et al., 2007; Fu and Neugebauer, 2008), inhibits nocifensive and affective pain responses and pain-related anxiety-like behavior. This first set of data is consistent with a serial model that involves BLA to CeA projections to execute appropriate emotional responses (Sah et al., 2003; Maren and Quirk, 2004; Phelps and LeDoux, 2005; Seymour and Dolan, 2008; Ehrlich et al., 2009). The result is novel and important because pain-related functional changes in the BLA were not known and previous evidence suggested potentially different roles of BLA and CeA in pain modulation (Manning and Mayer, 1995; Tanimoto et al., 2003; Carrasquillo and Gereau, 2007; Ortiz et al., 2007) and in certain aspects of emotion-related behaviors (Seymour and Dolan, 2008; Roozendaal et al., 2009).
Our second set of data shows that hyperactivity in the BLA, but not CeA, is critically involved in pain-related decision-making deficits and deactivation of the medial PFC. Pharmacologic deactivation of BLA, but not CeA, restores normal behavior and cortical activity. Differential effects of CRF1 receptor antagonist administration into BLA versus CeA (Fig. 3) and lack of decision-making deficits in animals with ACTH-induced anxiety (Fig. 4) argue against more generalized antagonist actions on the HPA axis and suggest that ACTH-induced stress response and anxiety are not sufficient to explain the cognitive impairment observed in the arthritis pain model. The conclusion about differential roles of BLA and CeA in amygdalo-cortical pain mechanisms rests on the selective pharmacological deactivation of BLA versus CeA in electrophysiological in vivo and behavioral studies. The differential effects of CRF1 receptor antagonist administration into the BLA versus CeA on mPFC activity and on decision-making behavior argue against the spread of drug from the injection site to the other nucleus. As another important control for drug diffusion, administration of the antagonist into the CeA did not affect activity of BLA neurons. Conversely, drug administration into the BLA (this study) or CeA (previous studies) had similar inhibitory effects on neuronal activity in the respective nucleus and on nocifensive and affective behaviors. In the slice preparation, the CRF1 antagonist acted postsynaptically directly on BLA neurons in this study and on CeA neurons in the previous study. Thus, both nuclei have functional CRF1 receptors that can be targeted to deactivate the neurons, but the consequences are nucleus specific.
The underlying mechanism of pain-related BLA-driven cortical deactivation and cognitive deficits is increased synaptic inhibition of mPFC pyramidal cells. Although details of the cortical circuitry remain to be determined, pharmacological and electrophysiological data of our study are consistent with a polysynaptic pathway, in which glutamate activates GABAergic transmission onto pyramidal cells through non-NMDA receptors. Disynaptic “feedforward inhibition” is found in various brain areas and involves activation of inhibitory interneurons and their target cells by the same excitatory input (Doyle and Andresen, 2001; Silberberg and Markram, 2007; Ferrante et al., 2009). In the rat somatosensory cortex, GABAergic inhibitory transmission to layer V pyramidal cells is driven via glutamatergic afferents arriving at non-NMDA receptors on interneurons (Ling and Benardo, 1995). In contrast, axo-axonic ionotropic glutamate receptor-mediated excitation of the nerve terminals of inhibitory interneurons and subsequent synaptic inhibition of nearby pyramidal cells that was reported in the mouse visual cortex (Ren et al., 2007) has been questioned recently (Hull et al., 2009).
Our model of glutamate-driven synaptic inhibition is supported by anatomical data showing that pyramidal cells in the BLA, but not CeA, form monosynaptic glutamatergic connections not only with layer V pyramidal cells but also nearby GABAergic interneurons in the mPFC (Kita and Kitai, 1990; Bacon et al., 1996; Gabbott et al., 2006). Additional evidence for the glutamatergic drive originating in the BLA is based on our in vivo studies showing that deactivation of BLA projection neurons increased mPFC activity. The time course of BLA hyperactivity and of mPFC deactivation in the arthritis model was comparable. In the slice preparation, the stimulation site for evoking synaptic responses was selected based on fluorescent labeling of fibers after anterograde tracer (DiI) injections into the BLA. The course of the fibers through the infralimbic cortex has been shown in previous studies of BLA–PFC projections (Gabbott et al., 2006; Orozco-Cabal et al., 2006). Although activation of nearby fibers attributable to current spread cannot be ruled out, the use of low stimulus intensities (Fig. 6) and evaluation of the full range of input–output relationships (Fig. 7) makes a confounding effect of multiple pathway activation unlikely.
Although the proposed model of glutamatergic BLA projection to GABAergic mPFC interneurons can explain BLA-driven cortical deactivation, it remains to be determined why excitatory transmission to mPFC pyramidal cells did not change in the arthritis pain model. One possible explanation is that separate lines of input account for excitatory transmission to pyramidal cells and to inhibitory interneurons. Another possibility is that glutamate-driven GABAergic transmission controls the direct excitatory input to pyramidal cells, and pain-related increase of the GABAergic tone prevents increased glutamatergic drive onto pyramidal cells. In support of this hypothesis, preliminary data suggest that blockade of GABA receptors in the mPFC increases monosynaptic excitatory transmission to pyramidal cells (H. Sun and V. Neugebauer, unpublished observation). GABAergic control of excitatory inputs to pyramidal cells may involve presynaptic GABA receptors on the axon terminal (Miller, 1998) and/or postsynaptic GABA receptors on the soma that would occlude or shunt an (increased) effect of excitatory drive. Greater sensitivity of cortical interneurons than pyramidal cells to excitatory inputs attributable to cell-type-specific differences in the relative kinetics of excitatory and inhibitory synaptic currents (Cruikshank et al., 2007) would further explain why inhibitory interneurons can control excitatory transmission onto pyramidal cells. Expression of faster gating AMPA receptors with higher Ca2+ permeability in interneurons than pyramidal cells (Markram et al., 2004) contributes to the faster synaptic kinetics of inhibitory interneurons (Cruikshank et al., 2007).
The novelty of these results is the functional changes of amygdalo-cortical interactions in pain and the contribution of these upstream projections to decision-making deficits. Previous studies showed that a majority of orbitofrontal neurons respond to noxious cutaneous (Snow et al., 1992) and/or visceral (Yang and Follett, 1998) stimuli. The present study not only identified neurons in the mPFC that respond to noxious stimuli but also showed for the first time that activity of these neurons is inhibited in a model of persistent pain. Reduced mPFC activity was also observed in conditioned fear, but the neural mechanism remains to be determined (Garcia et al., 1999). Inhibitory, but not excitatory, synaptic transmission to layer V pyramidal cells increased in our arthritis pain model. In a model of neuropathic pain, the NMDA, but not non-NMDA, receptor component of excitatory synaptic transmission increased in layer II/III pyramidal cells, inhibitory transmission was not measured, and underlying mechanisms and behavioral consequences remain to be determined (Metz et al., 2009). These results may suggest differential changes in functionally distinct subsets of mPFC neurons in different pain models.
Pain-related mPFC inhibition is the result of BLA hyperactivity. In this relationship, the amygdala modulates cortical activity through upstream projections, whereas the traditional view has been top-down cortical (PFC) regulation of the amygdala (Urry et al., 2006; Delgado et al., 2008; Herry et al., 2008). However, facilitatory upstream interactions between amygdala and cortex are important for emotional learning and memory (Roozendaal et al., 2009), including BLA-dependent plasticity of mPFC neurons in olfactory fear conditioning (Laviolette and Grace, 2006). The amygdala also influences sensory cortical plasticity and facilitates attention and perception to emotionally significant stimuli (Phelps and LeDoux, 2005). Conversely, an inverse correlation between amygdala and PFC has been found in the regulation of negative affect (Urry et al., 2006), conditioned fear (Garcia et al., 1999), and fear extinction (Milad et al., 2007) but also in cue-evoked reward-seeking behavior (Ishikawa et al., 2008).
In our study, the inverse relationship between BLA and mPFC produced cortical deactivation and decision-making deficits in pain. The correlation of decreased PFC activity with impaired cognitive function is supported by neuroimaging (Breiter et al., 2001; Daw et al., 2006; Hampton et al., 2006; Taylor et al., 2006) and electrophysiological (Oya et al., 2005) data in humans. Cocaine-induced decision-making deficits (impaired cognitive flexibility on reversal learning and gambling tasks) involve a loss of outcome-expectant encoding in the orbitofrontal cortex and subsequently inflexible encoding of cue significance in the BLA (Stalnaker et al., 2007b). Impaired cognitive flexibility induced by orbitofrontal cortical lesions is improved by BLA ablation (Stalnaker et al., 2007a). Cocaine-induced decision-making deficits (cognitive inflexibility) are reminiscent of those observed in the present study (failure to switch strategies), and both involve abnormal amygdalar activity. However, BLA miscoding was the consequence of cocaine-induced orbitofrontal dysfunction (Stalnaker et al., 2007b), whereas our data show that abnormal BLA activity drives mPFC dysfunction in pain. The inverse BLA–mPFC interaction in pain is consistent with BLA-mediated depression of mPFC activity in conditioned fear (Garcia et al., 1999) and decreased amygdala activation but increased ventromedial PFC activation during fear extinction (Milad et al., 2007).
This study is the first demonstration of a neural mechanism by which the amygdala impairs cortical and cognitive function in pain. Our results suggest a parallel model of BLA–mPFC interaction for cognitive aspects of pain and BLA–CeA circuits involved in emotional-affective pain responses. BLA encodes emotions in their context and signals stimulus-specific information (Amaral et al., 2003; Balleine and Killcross, 2006; Herry et al., 2008). Connections with CeA execute appropriate emotional behavior (Sah et al., 2003; Phelps and LeDoux, 2005), whereas connections with PFC produce goal-directed actions (Seymour and Dolan, 2008) and are important for reappraisal of emotional stimuli, extinction learning, and cognitive emotion-regulation strategies (Phelps and LeDoux, 2005; Delgado et al., 2008; Herry et al., 2008), which may be impaired in pain and other disorders.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS-38261 and NS-11255. We thank Drs. Richard E. Coggeshall and William D. Willis Jr. (University of Texas Medical Branch, Galveston, TX) and Dr. Brad Keele (Baylor University, Waco, TX) for critical reading of and valuable comments on this manuscript. We also thank Margarida Dourado for excellent technical experimental assistance.
References
- Amaral DG, Bauman MD, Schumann CM. The amygdala and autism: implications from non-human primate studies. Genes Brain Behav. 2003;2:295–302. doi: 10.1034/j.1601-183x.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci. 2004a;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task. Pain. 2004b;108:129–136. doi: 10.1016/j.pain.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Bacon SJ, Headlam AJ, Gabbott PL, Smith AD. Amygdala input to medial prefrontal cortex (mPFC) in the rat: a light and electron microscope study. Brain Res. 1996;720:211–219. doi: 10.1016/0006-8993(96)00155-2. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Killcross S. Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 2006;29:272–279. doi: 10.1016/j.tins.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR, Lee GP. Different contributions of the human amygdala and ventromedial prefrontal cortex to decision-making. J Neurosci. 1999;19:5473–5481. doi: 10.1523/JNEUROSCI.19-13-05473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR. Role of the amygdala in decision-making. Ann N Y Acad Sci. 2003;985:356–369. doi: 10.1111/j.1749-6632.2003.tb07094.x. [DOI] [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol. 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiter HC, Aharon I, Kahneman D, Dale A, Shizgal P. Functional imaging of neural responses to expectancy and experience of monetary gains and losses. Neuron. 2001;30:619–639. doi: 10.1016/s0896-6273(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho TP, Buonomano DV. Differential effects of excitatory and inhibitory plasticity on synaptically driven neuronal input-output functions. Neuron. 2009;61:774–785. doi: 10.1016/j.neuron.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinidis C, Goldman-Rakic PS. Correlated discharges among putative pyramidal neurons and interneurons in the primate prefrontal cortex. J Neurophysiol. 2002;88:3487–3497. doi: 10.1152/jn.00188.2002. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Daw ND, O'Doherty JP, Dayan P, Seymour B, Dolan RJ. Cortical substrates for exploratory decisions in humans. Nature. 2006;441:876–879. doi: 10.1038/nature04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MR, Nearing KI, Ledoux JE, Phelps EA. Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron. 2008;59:829–838. doi: 10.1016/j.neuron.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Ferrante M, Migliore M, Ascoli GA. Feed-forward inhibition as a buffer of the neuronal input-output relation. Proc Natl Acad Sci U S A. 2009;106:18004–18009. doi: 10.1073/pnas.0904784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JG, Cruz C, Vinson GP, Pignatelli D. ACTH modulates ERK phosphorylation in the adrenal gland in a time-dependent manner. Endocr Res. 2004;30:661–666. doi: 10.1081/erc-200043952. [DOI] [PubMed] [Google Scholar]
- Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci. 2008;28:3861–3876. doi: 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Busby SJ. Amygdala input monosynaptically innervates parvalbumin immunoreactive local circuit neurons in rat medial prefrontal cortex. Neuroscience. 2006;139:1039–1048. doi: 10.1016/j.neuroscience.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF. The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature. 1999;402:294–296. doi: 10.1038/46286. [DOI] [PubMed] [Google Scholar]
- Hampton AN, Bossaerts P, O'Doherty JP. The role of the ventromedial prefrontal cortex in abstract state-based inference during decision making in humans. J Neurosci. 2006;26:8360–8367. doi: 10.1523/JNEUROSCI.1010-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Holland PC, Gallagher M. Amygdala-frontal interactions and reward expectancy. Curr Opin Neurobiol. 2004;14:148–155. doi: 10.1016/j.conb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Hull C, Adesnik H, Scanziani M. Neocortical disynaptic inhibition requires somatodendritic integration in interneurons. J Neurosci. 2009;29:8991–8995. doi: 10.1523/JNEUROSCI.5717-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda R, Takahashi Y, Inoue K, Kato F. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain. 2007;127:161–172. doi: 10.1016/j.pain.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Ambroggi F, Nicola SM, Fields HL. Contributions of the amygdala and medial prefrontal cortex to incentive cue responding. Neuroscience. 2008;155:573–584. doi: 10.1016/j.neuroscience.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Rabkin SD, Granato A, Boudah A, Ohara PT. Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature. 2003;424:316–320. doi: 10.1038/nature01808. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol. 2007;97:3893–3904. doi: 10.1152/jn.00135.2007. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol. 2009;102:2253–2264. doi: 10.1152/jn.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Fu Y, Ruppert KA, Neugebauer V. Pain-related anxiety-like behavior requires CRF1 receptors in the amygdala. Mol Pain. 2007;3:13–17. doi: 10.1186/1744-8069-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Kitai ST. Amygdaloid projections to the frontal cortex and the striatum in the rat. J Comp Neurol. 1990;298:40–49. doi: 10.1002/cne.902980104. [DOI] [PubMed] [Google Scholar]
- Kouneiher F, Charron S, Koechlin E. Motivation and cognitive control in the human prefrontal cortex. Nat Neurosci. 2009;12:939–945. doi: 10.1038/nn.2321. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA. Cannabinoids potentiate emotional learning plasticity in neurons of the medial prefrontal cortex through basolateral amygdala inputs. J Neurosci. 2006;26:6458–6468. doi: 10.1523/JNEUROSCI.0707-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS. Recruitment of GABAA inhibition in rat neocortex is limited and not NMDA dependent. J Neurophysiol. 1995;74:2329–2335. doi: 10.1152/jn.1995.74.6.2329. [DOI] [PubMed] [Google Scholar]
- Manning BH, Mayer DJ. The central nucleus of the amygdala contributes to the production of morphine antinociception in the rat tail-flick test. J Neurosci. 1995;15:8199–8213. doi: 10.1523/JNEUROSCI.15-12-08199.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience. 1991;44:1–14. doi: 10.1016/0306-4522(91)90247-l. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Heinricher MM. Microinjection of morphine into various amygdaloid nuclei differentially affects nociceptive responsiveness and RVM neuronal activity. Pain. 2002;96:153–162. doi: 10.1016/s0304-3959(01)00440-7. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci U S A. 2009;106:2423–2428. doi: 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annu Rev Pharmacol Toxicol. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Murakami N, Takahashi K. Enhancement of corticosterone release by repeated injections of ACTH in the dexamethasone pre-treated rat. Acta Endocrinol (Copenh) 1983;102:583–588. doi: 10.1530/acta.0.1020583. [DOI] [PubMed] [Google Scholar]
- Myers B, Greenwood-Van Meerveld B. Corticosteroid receptor-mediated mechanisms in the amygdala regulate anxiety and colonic sensitivity. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1622–G1629. doi: 10.1152/ajpgi.00080.2007. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W. Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol. 2003;89:716–727. doi: 10.1152/jn.00799.2002. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Keele NB, Shinnick-Gallagher P. Epileptogenesis in vivo enhances the sensitivity of inhibitory presynaptic metabotropic glutamate receptors in basolateral amygdala neurons in vitro. J Neurosci. 1997;17:983–995. doi: 10.1523/JNEUROSCI.17-03-00983.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Chen PS, Willis WD. Role of metabotropic glutamate receptor subtype mGluR1 in brief nociception and central sensitization of primate STT cells. J Neurophysiol. 1999;82:272–282. doi: 10.1152/jn.1999.82.1.272. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Zinebi F, Russell R, Gallagher JP, Shinnick-Gallagher P. Cocaine and kindling alter the sensitivity of group II and III metabotropic glutamate receptors in the central amygdala. J Neurophysiol. 2000;84:759–770. doi: 10.1152/jn.2000.84.2.759. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RW., 4th Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Han JS, Adwanikar H, Fu Y, Ji G. Techniques for assessing knee joint pain in arthritis. Mol Pain. 2007;3:8–20. doi: 10.1186/1744-8069-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Galhardo V, Maione S, Mackey SC. Forebrain pain mechanisms. Brain Res Rev. 2009;60:226–242. doi: 10.1016/j.brainresrev.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco-Cabal L, Pollandt S, Liu J, Vergara L, Shinnick-Gallagher P, Gallagher JP. A novel rat medial prefrontal cortical slice preparation to investigate synaptic transmission from amygdala to layer V prelimbic pyramidal neurons. J Neurosci Methods. 2006;151:148–158. doi: 10.1016/j.jneumeth.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Ortiz JP, Heinricher MM, Selden NR. Noradrenergic agonist administration into the central nucleus of the amygdala increases the tail-flick latency in lightly anesthetized rats. Neuroscience. 2007;148:737–743. doi: 10.1016/j.neuroscience.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oya H, Adolphs R, Kawasaki H, Bechara A, Damasio A, Howard MA., 3rd Electrophysiological correlates of reward prediction error recorded in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2005;102:8351–8356. doi: 10.1073/pnas.0500899102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais-Vieira M, Lima D, Galhardo V. Orbitofrontal cortex lesions disrupt risk assessment in a novel serial decision-making task for rats. Neuroscience. 2007;145:225–231. doi: 10.1016/j.neuroscience.2006.11.058. [DOI] [PubMed] [Google Scholar]
- Pais-Vieira M, Mendes-Pinto MM, Lima D, Galhardo V. Cognitive impairment of prefrontal-dependent decision-making in rats after the onset of chronic pain. Neuroscience. 2009;161:671–679. doi: 10.1016/j.neuroscience.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Paulus MP. Decision-making dysfunctions in psychiatry–altered homeostatic processing? Science. 2007;318:602–606. doi: 10.1126/science.1142997. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Yoshimura Y, Takada N, Horibe S, Komatsu Y. Specialized inhibitory synaptic actions between nearby neocortical pyramidal neurons. Science. 2007;316:758–761. doi: 10.1126/science.1135468. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Seymour B, Dolan R. Emotion, decision making, and the amygdala. Neuron. 2008;58:662–671. doi: 10.1016/j.neuron.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Snow PJ, Lumb BM, Cervero F. The representation of prolonged and intense, noxious somatic and visceral stimuli in the ventrolateral orbital cortex of the cat. Pain. 1992;48:89–99. doi: 10.1016/0304-3959(92)90135-X. [DOI] [PubMed] [Google Scholar]
- Stalnaker TA, Franz TM, Singh T, Schoenbaum G. Basolateral amygdala lesions abolish orbitofrontal-dependent reversal impairments. Neuron. 2007a;54:51–58. doi: 10.1016/j.neuron.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Stalnaker TA, Roesch MR, Franz TM, Calu DJ, Singh T, Schoenbaum G. Cocaine-induced decision-making deficits are mediated by miscoding in basolateral amygdala. Nat Neurosci. 2007b;10:949–951. doi: 10.1038/nn1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto S, Nakagawa T, Yamauchi Y, Minami M, Satoh M. Differential contributions of the basolateral and central nuclei of the amygdala in the negative affective component of chemical somatic and visceral pains in rats. Eur J Neurosci. 2003;18:2343–2350. doi: 10.1046/j.1460-9568.2003.02952.x. [DOI] [PubMed] [Google Scholar]
- Taylor SF, Martis B, Fitzgerald KD, Welsh RC, Abelson JL, Liberzon I, Himle JA, Gehring WJ. Medial frontal cortex activity and loss-related responses to errors. J Neurosci. 2006;26:4063–4070. doi: 10.1523/JNEUROSCI.4709-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urry HL, van Reekum CM, Johnstone T, Kalin NH, Thurow ME, Schaefer HS, Jackson CA, Frye CJ, Greischar LL, Alexander AL, Davidson RJ. Amygdala and ventromedial prefrontal cortex are inversely coupled during regulation of negative affect and predict the diurnal pattern of cortisol secretion among older adults. J Neurosci. 2006;26:4415–4425. doi: 10.1523/JNEUROSCI.3215-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142:1–20. doi: 10.1016/j.neuroscience.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2:322–328. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Follett KA. The effect of morphine on responses of ventrolateral orbital cortex (VLO) neurons to colorectal distension in the rat. Brain Res. 1998;808:101–105. doi: 10.1016/s0006-8993(98)00804-x. [DOI] [PubMed] [Google Scholar]