

Abstract
One can manually isolate the giant oocyte nucleus or germinal vesicle (GV) of Xenopus from a living oocyte with nothing more complicated than jewelers’ forceps and a dissecting microscope. Similarly, one can remove the nuclear envelope by hand and allow the lampbrush chromosomes and other nuclear organelles to spread on a microscope slide. After centrifugation, the nuclear contents adhere tightly to the slide, where they can be subjected to immunostaining or fluorescent in situ hybridization for visualization by conventional or confocal microscopy. Preparations of isolated GV contents reveal details of nuclear structure that are almost impossible to attain by more conventional techniques.
Keywords: Cajal body (CB), germinal vesicle (GV), lampbrush chromosome (LBC), nucleolus, oocyte, Xenopus
Introduction
In his classic description of oogenesis in the chicken, the Czech scientist Jan Evangelista Purkinje was the first to describe the giant nucleus of an immature oocyte [1]. Writing in Latin he gave the name vesicula germinativa to this nucleus, because it was a clear, fluid filled vesicle inside the “germ” or future embryo. Translated into English vesicula germinativa became germinal vesicle or GV. It was not until three years later that the English botanist Robert Brown noticed a much smaller vesicle inside the cells of many plants, especially the Monocots [2]. He called this vesicle an areola or nucleus, alluding to the fact that it was more or less circular in outline and often in the center of the cell. Thus “GV” has roughly three years priority over “nucleus” as the name of the oocyte nucleus! Later in the 19th century the pioneering German cell biologist Walther Flemming studied sections of axolotl oocytes and realized that the giant GV contained equally giant chromosomes [3]. Because of the large size of these chromosomes, their detailed morphology was nearly impossible to study in sections. This inherent structural problem was recognized by another German, J. Rückert, who isolated GVs free-hand from shark oocytes and then fixed and stained them for observation as whole mounts [4]. In this way he could examine the entire set of chromosomes without resorting to sections. It was Rückert who first remarked on the similarity between an oocyte chromosome and a lampbrush (Lampencylinderputzer in German). Although lampbrushes for cleaning soot from the chimneys of oil lamps have long since disappeared, the name has persisted for fuzzy oocyte chromosomes with their characteristic lateral loops.
Some decades later the American biologist William Duryee revived Rückert’s method of isolating GVs from living oocytes, but with an important technical advance [5-7]. Once the GV was isolated, he removed the nuclear envelope by hand without fixation to allow direct access to the chromosomes and other nuclear organelles in a more or less living state (Figures 1 and 2). Duryee emphasized the importance of Ca++-free solutions for maintaining lifelike chromosome structure, and all subsequent protocols have been based on his crucial observation. Unfortunately, Duryee’s studies were made just before the introduction of phase contrast microscopy, and so he was not able to observe the finer details of the chromosomes and other nuclear organelles.
Figure 1.

“Living” oocytes and GV of X. laevis. A. A small portion of an ovary from an adult female, in OR2 medium. Oocytes in various stages of development are present. In the largest oocytes the animal hemisphere is black or brown due to pigment that covers the yolk, whereas the white or yellowish-white yolk is visible in the vegetal hemisphere. Bar = I mm. B. A single oocyte (left) and its nearly transparent GV (right), which was squeezed out through a small hole in the animal pole. Some yolk extrudes through the hole. This GV was isolated in oil, but its appearance is similar to a GV isolated in a saline solution. Reproduced with permission from Paine et al. [27]. Bar = 500 μm.
Figure 2.

Giant LBCs from salamanders, isolated in a saline solution. A. The earliest image of a LBC isolated in a saline solution, from the newt Cynops (Triturus) pyrrhogaster. The homologous chromosomes are being stretched between microneedles. Reproduced from Duryee [6] with permission from the University of Pennsylvania Press. B. A pair of homologous LBCs from the newt Notophthalmus viridescens, isolated in a saline solution but not centrifuged. This image was taken by flash photography to stop the vigorous Brownian motion of the chromatin loops that extend laterally from the chromosome axis. Bars = 100 μm.
With the advent of phase microscopy, the major features of lampbrush chromosomes (LBCs) were quickly worked out, leading to a generally accepted model of their structure that has been refined but not substantially altered in recent years [8-11]. The amplified nucleoli, Cajal bodies, and speckles also became the focus of numerous studies, based in part on observations of material from hand-isolated GVs. So long as observations were made on unfixed GV contents, or contents that had been lightly fixed with formaldehyde vapor, it was not necessary to have the material firmly attached to a microscope slide. Because the delicate nature of unfixed material precluded the addition of a coverslip, most early observations were made with special well slides that could be observed from below with an inverted microscope.
However, attachment of the chromosomes and other nuclear organelles to a glass slide is essential for conventional or immunofluorescence staining and particularly for in situ hybridization. Thus, all recent protocols include a centrifugation step to assure attachment of the GV contents to the slide. Although simple in principle, centrifugation of microscope slides requires special equipment, which may account in part for the fact that relatively few laboratories have carried out extensive studies on isolated GV contents.
Xenopus GVs
Most of the pioneering studies on isolated LBCs utilized salamanders as the experimental material, particularly Triturus and Pleurodeles in Europe and Notophthalmus in the United States (Figures 3 and 4) [12-15]. Tailed amphibians were preferred over frogs for the simple reason that the DNA contents of their nuclei are among the highest known, and LBC size correlates directly with the amount of DNA in the genome (the C-value). As Xenopus laevis became more and more popular as a model organism, attempts were made to study its lampbrush chromosomes and other nuclear organelles. In 1991 we published a detailed protocol for making spread preparations of Xenopus GV contents (Figures 5 and 6) [16]. At that time we noted three problems that usually frustrated the most determined attempts to make satisfactory preparations. First, Xenopus lampbrush chromosomes “shut down” transcription to a greater or lesser degree within minutes or hours after oocytes were removed from the animal, resulting in retraction of the loops and overall shortening of the chromosomes. Second, in the usual isolation media Xenopus GV contents formed a stiff gel that dispersed only partially or not at all. And finally, even when the GV contents dispersed well, they did not necessarily stick to the underlying substrate after centrifugation. Fortunately, we now know how to overcome these problems on a routine basis, making it possible to produce cytological preparations in which details of the chromosomes and other nuclear organelles are beautifully preserved and displayed. We will discuss each of these issues in detail, since they are relevant to studies that may be undertaken with other species.
Figure 3.
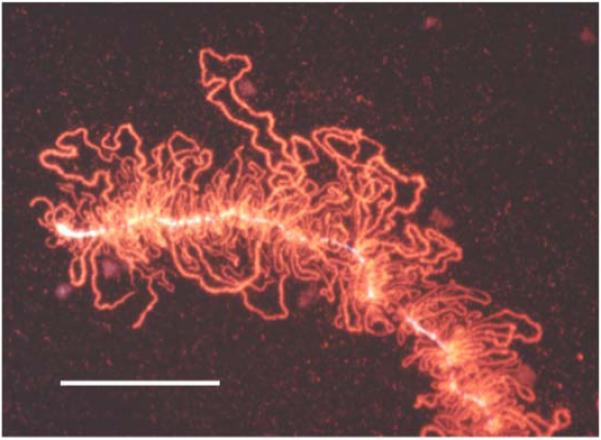
Effect of centrifugation on a LBC. A small segment of a LBC of the newt N. viridescens, from a preparation that had been centrifuged to attach the chromosomes to a glass microscope slide. Note that all the lateral loops of the chromosome are in essentially the same plane. This image was taken after immunofluorescent staining with an antibody against an abundant chromosomal protein. Bar = 50 μm.
Figure 4.
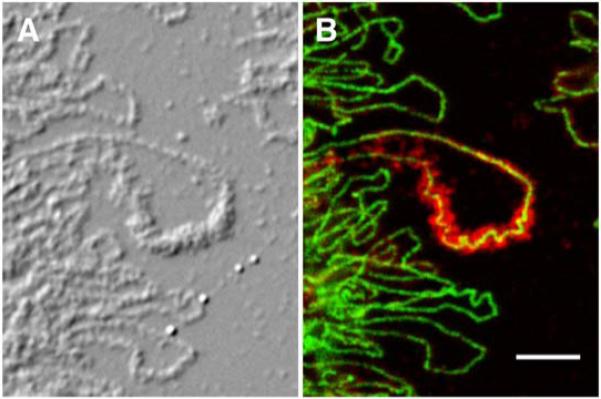
LBC preparations reveal details of chromosome structure at the molecular level. A. Differential interference contrast image of LBCs from the newt N. viridescens. B. The same region showing immunostaining with an antibody against RNA polymerase II (green) and cleavage stimulation factor 77 (red). Note that the green polymerase stain is limited to a diffraction-limited line in each lateral loop, especially well shown in the large loop in the center of the image. This line delineates the actively transcribing RNA polymerase molecules associated with the DNA axis of the loop. The red stain shows the nascent transcripts that coat the DNA axis. Modified from [32] with permission from Molecular Biology of the Cell. Bar = 5 μm.
Figure 5.
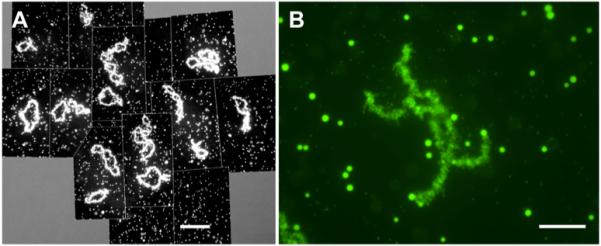
GV contents of X. laevis. A. Overall image of the contents of a single GV, showing the 18 lampbrush bivalents and several thousand nuclear bodies - primarily amplified nucleoli, Cajal bodies, and interchromatin granule clusters (speckles). Stained with mAb Y12, which reacts with small nuclear ribonucleoproteins. Bar = 100 μm. B. A single lampbrush bivalent with many nuclear speckles in the same field. Immunostained with mAb H5, which detects polymerase II in the chromosome loops and an unidentified epitope in the speckles. Bar = 10 μm.
Figure 6.

High magnification image of amplified nucleoli, Cajal bodies, and speckles. A. Differential interference contrast of nuclear organelles centrifuged onto a microscope slide after isolation in a saline solution. Each GV contains about 1000 nucleoli (N), 50-100 Cajal bodies (CBs) and several thousand interchromatin granule clusters or speckles (S). The contrast is high because the bodies are viewed in a medium of low refractive index. B. Phase contrast image of unfixed nuclear organelles in an oil-isolated GV. The organelles appear of low optical contrast because they are still in the nucleoplasm, which has a relatively high refractive index. As shown by fluorescence recovery after photobleaching (FRAP) studies, these organelles still exhibit active exchange of macromolecules with the surrounding medium [33, 34]. Panel B reproduced from [29] with permission from Molecular Biology of the Cell. Bars = 10 μm.
a) Transcription
The solution to the problem of chromosome shortening and loop retraction became apparent to us during experiments in which we injected oligodeoxynucleotides into freshly isolated oocytes and examined the chromosomes at various times thereafter [17]. Although the lampbrush chromosomes initially underwent contraction in both injected and control oocytes, they slowly “recovered” in the sense that they regained their overall length and re-extended transcriptionally active loops during the next 24 h. Earlier studies had shown a similar recovery of chromosome morphology after the more extreme contraction caused by actinomycin D [18, 19]. The essential conclusion is that oocytes removed from a female and held at 18°-22° C in a physiological saline solution until the next day usually display full-length lampbrush chromosomes with excellent loops.
b) Nuclear gel
The very stiff gel characteristic of Xenopus GVs presented a severe technical problem in earlier studies. The basic procedure for making a chromosome spread, long successful with newts, was to remove a GV from an oocyte in an isolation medium in which the GV did not disperse, dissect off the nuclear envelope at a leisurely pace, and transfer the still intact nuclear gel to a low ionic strength dispersal medium. Over a period of 10-30 min the nuclear gel dispersed in this medium, allowing the chromosomes and other nuclear structures to settle onto the underlying glass slide. Unfortunately, Xenopus GVs usually failed to disperse under these conditions. Numerous modifications of the dispersal medium were tried with no consistent success. Eventually it was found that Xenopus GVs will disperse satisfactorily, but only if they remain in the isolation medium for less than about 30 sec. During the first minute after isolation, the nucleoplasm of a Xenopus GV transforms from a near liquid to a firm gel. We have yet to find a medium that will disperse this gel, once it has formed, without damaging the morphology of the GV contents. Subsequent experiments showed that newt GVs undergo a similar gelling, but much more slowly. The practical conclusion is that one must work rapidly with Xenopus GVs, transferring them from the isolation medium to the dispersal medium within about 30 sec.
The stiffness of isolated nuclei is almost certainly related to their actin content. Actin is one of the two or three most abundant proteins in the GV [20-25]. Clark and Rosenbaum showed that about 63% of the actin in isolated Xenopus GVs is in the fibrous (F) rather than in the globular (G) form [22]. They further demonstrated that the ratio of F to G actin remained constant during the first 15 min after isolation. Because their first measurements were made within 60-90 sec, they concluded that polymerization did not take place after isolation of the GV. In fact, the GV is quite fluid while still in the oocyte but gels rapidly during the first minute after isolation. The most direct evidence for this fact comes from the behavior of GVs isolated under oil [26, 27]. GVs in oil are fluid-filled sacs that can be deformed with needles into long cylinders that return to a spherical shape when tension is removed. They retain this property for hours. If at any time after isolation in oil, the GV is transferred to an aqueous saline solution, it gels within seconds. The rapid change in consistency when a GV comes in contact with an aqueous medium suggests that gelling may be induced by some component of the medium or, conversely, by rapid loss of an endogenous inhibitor of gelling.
Of considerable interest is the fact that paraformaldehyde does not cause gelling of GV contents. Quite the contrary, it prevents gelling. This can be demonstrated by placing an intact oocyte in a solution of 4% paraformaldehyde in OR2 medium for 10 minutes. After this time the cytoplasm will be somewhat gelled, but the GV will be fluid and cannot be isolated in the usual way. Whether a paraformaldehyde-fixed GV is more fluid than an unfixed GV needs to be studied in more detail. Our impression is that they are similar. The only reason one can isolate the GV from an unfixed oocyte is that the isolation medium itself causes partial gelling during the time that it takes to find the GV and roll it away from the cytoplasm. To spread the GV contents one must transfer this partially gelled GV quickly into the spreading solution, which contains a little paraformaldehyde to prevent further gelling.
c. Adherence to glass substrate
An ongoing problem for many years has been to get the GV contents, especially the lampbrush chromosomes, to adhere to the glass slide during centrifugation. We tried various pretreatments of the slides with little improvement. Subbing the slides seemed to help, but increasing the concentration of subbing solution made little difference. For a while we thought that the time and speed of centrifugation were the critical factors. Since we cannot exceed 5,000 rpm with our standard rotor, we tried increasing the time of centrifugation with little or no improvement. The solution to the problem was found in the summer of 2008 - rigid control over the pH of the spreading solution. We have always kept the spreading solution at about pH 7, but usually did not control it carefully. We now find that LBCs of X. laevis adhere dramatically better at pH 6.6 - 6.8 than at pH 7.0 or higher. Preparations made at the lower pH show little Brownian motion of the loops and the LBCs are phase “dark.” At pH 7.4, there is obvious Brownian motion and the LBCs are pale by phase contrast. X. tropicalis GVs do not spread well at pH 6.6, but they do so at pH 7.0 and the LBCs stick tightly to the slide after centrifugation (Figure 7).
Figure 7.

A single LBC from a mature GV of X. tropicalis. An advantage of X. tropicalis over X. laevis for the study of LBCs is the smaller chromosome number (n = 10 vs n = 18) and the fact that the genome has been sequenced (http://www.xenbase.org). Working maps of the LBCs have also been published recently [35]. A. The longest chromosome of the set, stained with the DNA-specific dye DAPI. B. Immunofluorescent staining (red) with an antibody that recognizes RNA polymerase III. A few interstitial sites of pol III transcription are evident. The nature of the stained spherical objects on the chromosome ends is not known. Counterstained with DAPI (blue). C. The same chromosome showing staining for pol II (green) and pol III (red). The majority of loops are transcribed by pol II. Bar = 20 μm.
Techniques
A. Isolation of the GV and its contents in an aqueous medium
The contents of a Xenopus GV can be spread for microscopic examination according to the following protocol, which is similar to one published earlier [28], but includes some critical new insights.
1. Remove an ovary sample from a mature Xenopus female [16] and place it in OR2 medium in a Petri dish (60 X 15 mm). Hold the oocytes at 18°-22° C (not 4° C) for at least 18 h before use.
The transcriptional state of the chromosomes is highly variable in oocytes examined during the first few hours after removal of the ovary. When held at 18°-22° C for 18 h, oocytes will almost invariably contain large, transcriptionally active lampbrush chromosomes. If the medium is replaced daily, oocytes will remain usable for up to 3-4 days. It is not necessary to treat the female with gonadotrophin, as previously recommended.
2. Select an oocyte with a diameter of 0.9-1.2 mm and transfer it to a small Petri dish (35 × 10 mm) containing “5:1” isolation medium.
3. With two pairs of No. 5 jeweler’s forceps or one pair of forceps and a sharpened tungsten needle, make a large tear in the animal hemisphere of the oocyte. Find the GV within the protruding cytoplasm and roll it away. Suck the GV in and out of a pipette once or twice to remove yolk if necessary, but don’t worry if some yolk remains attached to the envelope.
4. Within 20-30 sec transfer the GV to a small Petri dish of dispersal medium. Speed is absolutely critical. If the GV remains for more than 40-60 sec in the isolation medium, it will form a stiff gel that will not disperse during the following steps.
5. Working rapidly, remove the nuclear envelope with two pairs of forceps or with one pair of forceps and a fine needle. At this point the nuclear contents are a gelatinous ball, but they will become quite soft within 2-3 min.
An alternative procedure is to remove the nuclear envelope in the dispersal chamber (described below). In this case, transfer the GV from the isolation medium to dispersal medium in a Petri dish, but then transfer it immediately to the dispersal chamber, where the envelope can be removed. The intermediate wash ensures complete replacement of the isolation medium. Removing the envelope in the dispersal chamber usually results in more yolk granules contaminating the final preparation.
6. Pick up the gel with a pipette and transfer to a dispersal chamber previously filled with dispersal medium. The liquid in the well should have a slightly convex surface. An uncovered preparation can be observed with a low magnification lens (10 X), but an 18-mm2 coverslip should be added for higher magnification observations and for subsequent centrifugation.
Seal the coverslip with petroleum jelly (Vaseline). To do this, remove excess liquid from the chamber by placing a piece of filter paper on top of the coverslip and pressing gently. Then apply spots of petroleum jelly to the four edges of the coverslip and melt with a heated metal rod. Don’t heat the contents of the well.
The dispersal chamber is made by boring a 5 mm round hole in the middle of a 25-mm square of Plexiglas (1-mm thickness). Attach the plastic square to the middle of a standard glass microscope slide with a 1:1 mixture of petroleum jelly/paraffin wax. To do this, first place two or three small drops of melted petroleum jelly/paraffin wax on a slide. The drops will immediately solidify. Press the square onto the solidified wax drops and then place the slide on a hot plate. When the wax melts, it should spread evenly to the edges of the plastic square, but should not penetrate into the central hole. With a little practice, one can judge the correct amount of wax to start with. We routinely use subbed slides to enhance sticking of the nuclear contents (see details below).
7. After about 10 min the nuclear contents should spread evenly over the floor of the dispersal chamber. The chromosomes and other nuclear organelles can be observed with a low power phase contrast objective, but the thickness of the plastic square prevents one from using a standard high power objective at this point. Wait until the chromosomes are flat against the slide before centrifuging.
8. Centrifuge the slides at 5,000 rpm (4,800 g) for 20-40 min at 5°-10° C. We use specially constructed holders for the Sorvall HS-4 rotor, as described earlier [16]. However, several types of large centrifuge buckets can be adapted to hold microscope slides, such as those in the Sorvall T-6000 bench top refrigerated centrifuge or the Beckman-Coulter Allegra X-15R.
8. Place the slides vertically in a staining dish containing PBS + 2% paraformaldehyde. Remove the coverslips by pushing them laterally with blunt forceps. Leave the preparations undisturbed for 10-15 min.
9. Transfer the slides to a second staining dish containing PBS only. Remove the spreading chambers with a razor blade.
10. Further treatment of the nuclear contents depends on the nature of the experiment. After a brief rinse in PBS to remove the paraformaldehyde, the slide can be stained for immunofluorescence. Alternatively it can be dehydrated through an ethanol series and stored in 70% ethanol for in situ hybridization.
We have found that most antibodies work best after a brief fixation in paraformaldehyde. Longer fixation (1 hr - overnight) gives poor staining with some antibodies, although others are unaffected. Very short fixation is required in some cases. For example, several antibodies against nucleolar proteins, such as mAb 72B9 against fibrillarin, are very weak after I h fixation, but give excellent results with 10-15 min fixation. A few antibodies work best if paraformaldehyde fixation is omitted entirely, the slides being placed directly into 70% ethanol after centrifugation. Fixation in either paraformaldehyde or ethanol after centrifugation helps to prevent the material from coming off the slide during immunostaining.
More detailed discussion of materials and procedures are given in Callan’s monograph [8] and in Gall et al. [16].
B. Isolation and observation of GVs in oil
A GV can be isolated in oil according to a procedure published by Paine and his collaborators (Figure 1B) [26, 27]. The method is very simple and yields GVs that retain physiological activity for several hours. Because the chromosomes and nuclear bodies are viewed while suspended in the nucleoplasm, which has a relatively high refractive index, all structures appear of low contrast by DIC or phase contrast microscopy (Figure 6B) [29].
1. Remove one or two oocytes from the OR2 solution and transfer to the edge of a 1-inch square of nylon mesh. Remove as much of the aqueous medium as possible by touching the underside of the mesh with a piece of Whatman #1 filter paper. Roll the oocyte(s) into mineral oil in a small Petri dish (35 × 10 mm). We use Sigma Mineral Oil M-5904, but other brands of paraffin oil or mineral oil may be satisfactory.
2. Make a small hole in the animal hemisphere with a needle or the tip of a #5 jeweler’s forceps. Gently squeeze the oocyte until you can just see the transparent GV begin to protrude through the hole. Continue squeezing and at the same time roll the GV away from the oocyte. This is a somewhat tricky maneuver, because the GV is more liquid than solid and is easily deformable. Almost invariably some yolk remains attached to the GV.
3. Pick up the GV in a Pipetteman along with exactly 5 μl of oil. Transfer to the center of a standard 3” X 1” microscope slide. Gently lower a 22-mm2 coverslip onto the GV and allow the oil to spread to the edges of the coverslip (the preparation is now nominally 10 μm thick). If the preparation is viewed by phase contrast or DIC, nucleoli are easy to find, but Cajal bodies and chromosomes are very faint (Figure 6B). Because yolk granules on the surface of the GV are highly refractile, they interfere with viewing. Oil-isolated GVs are ideal for observing fluorescein- or Alexa-labeled RNAs or GFP-proteins by epifluorescence microscopy.
To see the chromosomes in oil-isolated GVs, it is necessary to make a thicker preparation, as described by Michel Bellini [30].
C. Solutions
OR2 medium [31]
82.5 mM NaCl
2.5 mM KCl
1.0 mM CaCl2
1.0 mM MgCl2
1.0 mM Na2HPO4
5.0 mM HEPES
Use a HEPES stock of 500 mM, pH 8.3. The final pH of the OR2 will then be about 7.8. Add penicillin and streptomycin (100 mg/ml) to retard bacterial growth.
“5:1” isolation medium. Adjust to pH 7.0
83.0 mM KCl
17.0 mM NaCl
6.5 mM Na2HPO4
3.5 mM KH2PO4
1.0 mM MgCl2
1.0 mM dithiothreitol (DTT)
Dispersal Medium
Adjust to pH 6.6-6.8 for X. laevis or pH 7.0 for X. tropicalis (critical!)
20.7 mM KCl
4.3 mM NaCl
1.6 mM Na2HPO4
0.9 mM KH2PO4
1.0 mM MgCl2
1.0 mM dithiothreitol (DTT)
0.1% paraformaldehyde
Subbed slides
Wash commercial 3” X 1” glass microscope slides in a dilute detergent solution, rubbing them to remove any surface contaminants. Rinse thoroughly to remove the detergent and place the still wet slides in a slide rack. Dip them for a few seconds in subbing solution in a staining dish. Remove the slide rack from the staining dish and allow the slides to dry at an angle. Bake the dry slides at 60° for a few hours or overnight. Subbing solution is made as follows:
Place 0.5 g gelatin in a flask, add 500 ml of boiling water, and swirl to dissolve. The gelatin dissolves quicker if it is allowed to swell for a few minutes in 20 ml cold water before adding the boiling water. Cool and add 50 mg of “ chrome alum” (chromium potassium sulfate, CrK(SO4)2.12 H2O) previously dissolved in a minimal amount of H20. Filter with suction. Subbing solution can be stored at 4° and reused several times.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Purkinje JE. Symbolae ad ovi avium historiam ante incubationem. Leopold Vossi; Leipzig: 1830. [Google Scholar]
- [2].Brown R. Trans. Linnean Soc. Lond. 1833;16:685–745. [Google Scholar]
- [3].Flemming W. Zellsubstanz, Kern und Zelltheilung. F. C. W. Vogel; Leipzig: 1882. [Google Scholar]
- [4].Rückert J. Anat. Anz. 1892;7:107–158. [Google Scholar]
- [5].Duryee WR. Arch. exp. Zellforsch. 1937;19:171–176. [Google Scholar]
- [6].Duryee WR. Cytology, Genetics, and Evolution. University of Pennsylvania Bicentennial Conference, University of Pennsylvania Press; 1941. pp. 129–141. [Google Scholar]
- [7].Duryee WR. Ann. N. Y. Acad. Sci. 1950;50:920–953. [Google Scholar]
- [8].Callan HG. Lampbrush Chromosomes. Springer-Verlag; Berlin: 1986. [Google Scholar]
- [9].Morgan GT. Chromosome Res. 2002;10:177–200. doi: 10.1023/a:1015227020652. [DOI] [PubMed] [Google Scholar]
- [10].Gall JG, Wu Z, Murphy C, Gao H. Exp. Cell Res. 2004;296:28–34. doi: 10.1016/j.yexcr.2004.03.017. [DOI] [PubMed] [Google Scholar]
- [11].Gaginskaya E, Kulikova T, Krasikova A. Cytogenet. Genome Res. 2009;124:251–267. doi: 10.1159/000218130. [DOI] [PubMed] [Google Scholar]
- [12].Gall JG. J. Morphol. 1954;94:283–352. [Google Scholar]
- [13].Callan HG, Lloyd L. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1960;243:135–219. [Google Scholar]
- [14].Lacroix J-C, Loones MT. Chromosoma. 1971;36:112–118. doi: 10.1007/BF00326426. [DOI] [PubMed] [Google Scholar]
- [15].Scheer U, Spring H, Trendelenburg MF. Cell Nucleus. 1979;7:3–47. [Google Scholar]
- [16].Gall JG, Callan HG, Wu Z, Murphy C. In: Xenopus laevis: Practical Uses in Cell and Molecular Biology. Kay BK, Peng HB, editors. Vol. 36. Academic Press; San Diego, CA: 1991. pp. 149–166. [Google Scholar]
- [17].Tsvetkov A, Jantsch M, Wu Z, Murphy C, Gall JG. Mol. Biol. Cell. 1992;3:249–261. doi: 10.1091/mbc.3.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Snow MHL, Callan HG. J. Cell Sci. 1969;5:1–25. doi: 10.1242/jcs.5.1.1. [DOI] [PubMed] [Google Scholar]
- [19].Scheer U. Biol. Cell. 1987;59:33–42. [Google Scholar]
- [20].Clark TG, Merriam RW. Cell. 1977;12:883–891. doi: 10.1016/0092-8674(77)90152-0. [DOI] [PubMed] [Google Scholar]
- [21].De Robertis EM, Longthorne RF, Gurdon JB. Nature. 1978;272:254–256. doi: 10.1038/272254a0. [DOI] [PubMed] [Google Scholar]
- [22].Clark TG, Rosenbaum JL. Cell. 1979;18:1101–1108. doi: 10.1016/0092-8674(79)90223-x. [DOI] [PubMed] [Google Scholar]
- [23].Krohne G, Franke WW. Exp. Cell Res. 1980;129:167–189. doi: 10.1016/0014-4827(80)90341-9. [DOI] [PubMed] [Google Scholar]
- [24].Bohnsack MT, Stuven T, Kuhn C, Cordes VC, Görlich D. Nature Cell Biol. 2006;8:257–263. doi: 10.1038/ncb1357. [DOI] [PubMed] [Google Scholar]
- [25].Gall JG. Nature Cell Biol. 2006;8:205–207. doi: 10.1038/ncb0306-205. [DOI] [PubMed] [Google Scholar]
- [26].Lund E, Paine PL. Methods Enzymol. 1990;181:36–43. doi: 10.1016/0076-6879(90)81110-g. [DOI] [PubMed] [Google Scholar]
- [27].Paine PL, Johnson ME, Lau YT, Tluczek LJ, Miller DS. Biotechniques. 1992;13:238–246. [PubMed] [Google Scholar]
- [28].Gall JG. In: Cells: A Laboratory Manual. Spector DL, Goldman RD, Leinwand LA, editors. Vol. 1. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1998. pp. 52.51–52.54. [Google Scholar]
- [29].Handwerger KE, Cordero JA, Gall JG. Mol. Biol. Cell. 2005;16:202–211. doi: 10.1091/mbc.E04-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Patel S, Novikova N, Beenders B, Austin C, Bellini M. Chromosome Res. 2008;16:223–232. doi: 10.1007/s10577-007-1189-z. [DOI] [PubMed] [Google Scholar]
- [31].Wallace RA, Jared DW, Dumont JN, Sega MW. J. Exp. Zool. 1973;184:321–333. doi: 10.1002/jez.1401840305. [DOI] [PubMed] [Google Scholar]
- [32].Gall JG, Bellini M, Wu Z, Murphy C. Mol. Biol. Cell. 1999;10:4385–4402. doi: 10.1091/mbc.10.12.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Handwerger KE, Murphy C, Gall JG. J. Cell Biol. 2003;160:495–504. doi: 10.1083/jcb.200212024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deryusheva S, Gall JG. Proc. Natl. Acad. Sci USA. 2004;101:4810–4814. doi: 10.1073/pnas.0401106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Penrad-Mobayed M, El Jamil A, Kanhoush R, Perrin C. Devel. Dyn. 2009;238:1492–1501. doi: 10.1002/dvdy.21930. [DOI] [PubMed] [Google Scholar]