

Abstract
Domestication has been practiced for centuries yet directed toward relatively few terrestrial crops and animals. While phenotypic and quantitative genetic changes associated with domestication have been amply documented, little is known about the molecular changes underlying the phenotypic evolution during the process. Here, we have investigated the brook charr (Salvelinus fontinalis) responses to artificial selection by means of transcriptional analysis of ∼32,000 cDNA features performed in both selected and control populations reared under identical environmental conditions during four generations. Our results indicate that selective breeding led to significant changes in the transcription of genes at the juvenile stage, where we observed 4.16% (156/3750) of differentially expressed genes between the two lines. No significant genes were revealed at the earlier life stage. Moreover, when comparing our results to those of previous studies on Atlantic salmon that compared lines that were selected for five to seven generations for similar traits (e.g., growth), genes with similar biological functions were found to be under selection in both studies. These observations indicate that (1) four generations of selection caused substantial changes in regulation of gene transcription between selected and control populations and (2) selective breeding for improving the same phenotypic traits (e.g., rapid growth) in brook charr and Atlantic salmon tended to select for the same changes in transcription profiles as the expression of a small and similar set of genes was affected by selection.
DOMESTICATION has been practiced for centuries but applied to relatively few terrestrial crops and animals (Giuffra et al. 2000; Diamond 2002; Burt 2005; Pozzi and Salamini 2007; Kovach et al. 2007). One of the main consequences of the domestication is the rapid phenotypic changes that wild species undergo through the process of artificial selection (Duarte et al. 2007). These substantial phenotypic changes are commonly referred to as the “domestication syndrome” (Hammer 1984). Phenotypic changes caused by domestication have been abundantly documented in many species and can be interpreted as fast human-induced evolutionary changes.
The quantitative genetic basis (e.g., heritability) underlying the variance of traits of interest has also been largely investigated and used in artificial selection programs. However, little is known about the molecular changes underlying the phenotypic evolution induced by domestication (Yamasaki et al. 2007). Such knowledge should reveal far more about the genes contributing to agronomic traits than has been learned to date (Ross-Ibarra et al. 2007). In wheat for example, this revealed that the domestication syndrome originates in “sudden” genetic events, controlled by few major pleiotropic genes (for review see Pozzi and Salamini 2007). Thus, the Q gene, conferring the free-threshing character, has been identified as largely responsible for the widespread cultivation of this crop (Simons et al. 2006). Knowledge about the genomic basis of domestication could be directly implemented in marker assisted selection (MAS) and also help to better predict the impact of the interaction of domesticated lines with their wild counterparts.
In animals, the number of newly domesticated species has, on average, grown by 3% annually since the early 1950s (Duarte et al. 2007). Yet, relatively speaking, domestication of freshwater and marine fishes is still in its infancy. The oldest directed selective breeding programs were initiated in 1971 on Atlantic salmon, Salmo salar, and rainbow trout, Oncorhynchus mykiss (Gjedrem 2000). However, production and economic importance of those species have increased very quickly. Worldwide Atlantic salmon production was estimated to 1.3 × 106 tons in 2006, for a value of $6.5 billion. Improvement programs of fish species have generally concentrated on growth rate and pathogen resistance as traits of highest economic importance. Here, the main objective is to reach higher yields by producing strains that grow faster under intensive rearing conditions and show better resistance to pathogens (Rougeot et al. 2007; Silverstein et al. 2009), as exemplified in the case of Atlantic salmon culture (Gjedrem 2000).
Selection of breeders in such domestication programs has led to rapid evolutionary changes at the phenotypic level (Allard and Wiley 1999; Fleming et al. 2000; Fleming et al. 2003; Glover et al. 2009). For instance Roberge et al. (2006) compared changes in gene transcription profiles between farmed Atlantic salmon (S. salar) fish and their wild counterparts. Results demonstrated that five to seven generations of artificial selection led to heritable changes in gene transcription. Moreover, evolutionary changes in domesticated lines lead to gene misregulation following the introgression of domestic genetic material in wild populations (Roberge et al. 2008; Normandeau et al. 2009). More recently, Glover et al. (2009) have compared several production and flesh quality traits between wild, farmed, and hybrid Atlantic salmon reared under farming conditions. They concluded that the differences observed were of genetic origin and that these were either the direct or indirect result of artificial selection. Finally, Devlin et al. (2008) showed in coho salmon (Oncorhynchus kisutch) that domestication and growth hormone transgenesis cause similar changes in expression for genes involved in energy metabolism of carbohydrates and lipids, protein synthesis, stress and immune functions, and cellular structure.
Brook charr, Salvelinus fontinalis, is one of the most economically important species for freshwater aquaculture in Canada, where it is farmed both for food and population stocking purposes (Page and Burr 1991). In Quebec in particular, domestication of two wild populations of brook charr from the Rupert and Laval rivers has been initiated with the aim of increasing growth rates and delaying sexual maturation of these strains (Martin et al. 1997; Audet and Bernatchez 2004). For the Laval river population, the domestication program also included an experimental selection regime whereby “control” families have been maintained up to four generations under the same environmental conditions as the selected ones. Mating random individuals, while still avoiding the cross of closely related individuals, generated these control individuals. This system thus represents a rare opportunity to rigorously assess the early steps of evolutionary changes induced by domestication.
The aim of this study was to document the effects of domestication in brook charr by measuring changes in gene expression to identify biological processes targeted by the selection regime and to elucidate their patterns of transcription. Our main goals were (1) to study the impact of artificial selection during domestication in the species at two-life stages to determine whether selective pressures act mostly in the early or later stage; (2) to identify potential targets of this artificial selection; and (3) to analyze the expression pattern of candidate genes involved in multifactorial traits and regulatory pathways between the selected and the control lines. We also compared our results to previous studies of gene expression that documented rapid evolutionary changes in the Atlantic salmon (Roberge et al. 2008; Normandeau et al. 2009). Here the goal was to identify candidate genes associated with domestication in salmonids and to test if the same candidate genes can be detected in distinct species under domestication.
MATERIAL AND METHODS
Fish crosses and sampling:
A selective breeding program was initiated using wild brook charr from the Laval River (Québec, 48°44′ N, 68°05′ W; Martin et al. 1997; Savaria 1998). In 1994, wild fish (F0) were used to perform 12 crosses between 6 females and 8 males, which produced 12 families at the F1 population. On the basis of selection protocol explained below, F1 individuals were retained to produce the F2 generation of the selected population. Thus, 11 pairs of selected F1 individuals were mated to produce 11 full-sib families at the F2 generation. On the basis of the same selection protocol, 10 pairs of selected individuals were mated to produce 12 full-sib families at the F3. At all generations, fish from different families were distinctively tagged and reared in a common environment.
We applied a combined between- and within-family selection protocol (Falconer and Mackay 1996) on the basis of the absence of precocious sexual maturation at 22 months and growth performance. The number of fish selected by family (NSi) was determined according to
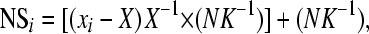 |
where xi is the mean of weight for family i, X is the general mean for the population, N is the number of breeders considered necessary, and K is the number of families. The proportion of fish selected in this way for each family varied between 4.1 and 14.2%.
A control group was created and maintained over the same period at the F2 and F3 selection generations. First, 10 fish from each family in the F1 population were randomly chosen before selection to create the F1 control population. Mixed breedings were performed using 11 pairs of individuals from different families selected randomly within each family to produce 11 full-sib F2 families. This same procedure was repeated to produce 11 full-sibs control families at the F3 generation. Observations after three generations clearly illustrate the pronounced phenotypic effect of the selective breeding program: the average weight gain of fish in the selected population relative to the control one was 34.7% while a substantial reduction in sexually mature fish at 1 year old has also been observed (61.4%; Bastien 2010).
For this study, we used progeny of F4 generation. From both control and selected F3 populations, we produced five sets of half-sib families: a total of 10 dams and 5 sires per population; 5 paired of families issued from different dams but sharing the same sire. For each F4 progeny, sire and dam were issued from different F3 families. Fish were sampled at two life stages: (1) sexually undifferentiated fry were sampled (n = 2 individuals per family) at the yolk-sac resorption stage between March 14 and 19, 2007, before exogenous feeding and (2) at the juvenile stage (n = 2 individuals per family), 10 weeks later. Experiments were conducted on whole individual yolk-sac resorption stage fry and on entire liver and pyloric cecum of individual fish at the juvenile stage. These two tissues are the seats of most metabolic pathways and therefore represent a target of choice in the “genomewide” study of genes expression. Fish of both life stages were killed prior to sampling and tissues were individually stored at −80°.
RNA extraction and cDNA hybridization:
All RNA extractions were performed on ice (4°) and following the same protocol to reduce technical biases in genes expression measures. Entire “control” and “selected” fry were individually homogenized in a mixture composed of 4 ml Trizol reagent (Sigma) and 20 μl isopropanol (Sigma), using a Qiagen homogenizer, whereas approximately 150 mg of liver and pyloric caeca of individual juveniles were treated in the same way. Total RNA was extracted using the Purelink Micro to midi total RNA purification system kit (Invitrogen), according to the manufacturer's recommendations. Subsequently, RNA integrity and quantity were controlled using a Nanodrop instrument (ND-1000, Nanodrop). Messenger RNA from each sample was reverse transcribed and labeled using the Genisphere 3DNA Array 50 kit provided by Invitrogen, using the Superscript II retrotranscriptase enzyme and Cy3/Cy5 dyes (Genisphere), according to the manufacturer's recommendations (Invitrogen protocol: http://web.uvic.ca/grasp/microarray/array.html). Then, cDNA was purified using Microcon YM30 (Millipore, Bedford, MA).
Microarrays (32k cDNA array) were provided by the cGRASP (Consortium for Genomics Research on All Salmonids Project; Rise et al. 2004). These 32,000 cDNAs resulted from the assembly of more than 700,000 expressed sequences tags (EST) obtained from a variety of cDNA libraries and should comprise the majority of all cDNA expressed, at least in Atlantic salmon. Gene annotation is provided in the cGRASP database for a proportion of these transcripts (http://web.uvic.ca/grasp/). Moreover, the cGRASP microarray has been applied successfully in a previous study on brook charr (Mavarez et al. 2009). Microarray were hybridized using Cy3/Cy5 dyes (Genisphere) as detailed in St-Cyr et al. (2008).
To analyze gene expression at both the post-yolk-sac resorption stage and juvenile stage, two individuals were randomly chosen from each of the 10 control and the 10 selected families for a total of 40 individuals analyzed at each life stage. Experiments were conducted on whole (unpooled) individuals at the yolk-sac resorption stage and on entire liver and pyloric cecum of individual (unpooled) fish at the juvenile stage. For a given life stage, one individual of a control family was hybridized on a same array with one individual of a selected family and compared directly. This was repeated for all 20 control and 20 selected individuals at each life stage for a total of 40 paired, direct comparisons of microarray hybridizations. Dyes were swapped between the 2 individuals of a same family. Such direct comparison of gene expression between two groups is one of the most powerful experimental designs in microarray studies (Churchill 2002).
Gene expression measurement and data analysis:
Transcription profiles from selected and control lines were measured using the Scanarray laser instrument (Packard Bioscience). Pictures were analyzed and gene expression levels quantified using the Quantarray software with the histogram quantification method. Data from the two life stages were exported in text format and were separately analyzed using a mixed model ANOVA and the R/MAANOVA package (Kerr and Churchill 2000; Wu et al. 2002). On each array, genes showing a mean intensity lower than control empty features plus twice their standard deviations in both channels were removed from the analysis. Moreover, over the 20 arrays of each life stage, expression data of a feature were discarded if two or more measures did not shown reliable or intense enough data.
Missing data were imputed using the K nearest neighbor algorithm with 10 neighbors and then transformed by a base 2 logarithm. A regional LOWESS correction was used to correct for intensity distortions as well as spatial variation in signal level across the array. Given that the same amounts of mRNA were used in every sample, the total quantity of expressed transcripts should be the same in all of them. The terms “dye” and “line” were included in the ANOVA model as fixed whereas the “array” and “sample” terms were included as random factors to assess the presence of significant differences in expression level between the two lines. Permutation-based F-tests with 1000 sample-based permutations were performed to solve the mixed-model equations for each of the analyzed genes. For each gene differentially expressed between the two lines, the average normalized intensity data of each line were back transformed (unlogged) to compute the fold change ratio.
A false discovery rate (FDR) procedure was used to correct for multiple testing before obtaining the list of candidate transcripts differentially regulated between the control and domesticated lines at the two stages. The Q-value R package (Storey et al. 2004) was used to obtain the list of transcripts containing an estimated 10% of false positives (FDR = 0.10).
Gene ontology:
Among the differentially expressed genes between the two lines for each life stage, those possessing a Unigene number were used to evaluate the overrepresentation of biological processes. Unigene numbers were converted to Entrez GeneID numbers using the online David Gene ID Conversion tool (http://david.abcc.ncifcrf.gov/conversion.jsp). Then, the online Panther Classification System gene list comparison tool (http://www.pantherdb.org/tools/compareToRefListForm.jsp) was used to identify biological processes that were overrepresented in the two comparisons. The proportional representation of all biological processes in expressed transcripts was used as an expected proportion under a random sampling hypothesis. Biological processes represented by only one transcript were discarded from the analysis to minimize the occurrence of false positives. The biological processes presented in the results are those for which the proportion of genes with differences in expression between the lines was significantly different than expected by chance, within at least one of the two life stages being compared (Fisher's exact proportion test, α = 0.05).
Cluster analysis:
Using the averaged unlogged LOWESS-corrected data, a cluster tree was built by using the average linking method (Eisen et al. 1998) with pairwise distances estimated from the Pearson correlation coefficient (Qu and Xu 2006) using the Genesight software (Biodiscovery). To reduce the bias introduced by differential array fluorescence that systematically tends to group individuals paired on the same array, we applied the conservative approach detailed in St-Cyr et al. (2008). Briefly, data were normalized by dividing expression values from one individual by expression values on its paired individual. In this way, gene expression values represent relative gene expression levels and not absolute levels.
RESULTS
Analysis of variance:
A total of 3481 cDNA features were used for the analysis of the post-yolk-sac resorption life stage, whereas 3750 features were retained in the juvenile stage. The ANOVA revealed that 248 (7.12%) and 523 (13.9%) genes were differentially expressed in the two populations respectively. The P-values were imported in the Q-value R package to obtain the list of transcripts containing an estimated 10% of false positives (FDR = 0.10). Following this procedure, 0 and 156 (4.16%) genes were differentially regulated between controlled and selected population at the post-yolk-sac resumption and the juvenile stages, respectively. As a result, further investigations were conducted only on the data obtained from the juvenile life stage. Here, the maximum P-value associated to a FDR correction of 0.10 was P = 0.0049. The list of differentially regulated transcripts was then statistically compared to a reference list to look for under- and overrepresented functional categories, which consisted of the set of analyzed transcripts in the ANOVA.
Gene ontology and genes representation:
Unigene numbers were obtained for 21.7% (816) of all the analyzed genes. Of these 816 genes, 156 (19%) were differentially expressed between control and selected populations at the juvenile stage. The gene ontology analysis using Panther classified 52 genes into five biological processes shown to be significantly under- or overrepresented between the control and selected groups (P < 0.05) compared to chance expectation (Table 1). Further analyses only consider those 52 as it was too hazardous to interpret the results obtained for the other 104 differentially expressed genes, given the total absence of annotation information. Overrepresented biological processes included: (1) G-protein mediated signaling (GPCRs), also known as seven-transmembrane domain receptors (n = 4); (2) other coenzyme and prosthetic group metabolism (n = 1); and (3) protein metabolism and modification (n = 35). Underrepresented biological processes corresponded to (1) immunity and defense (n = 3) and (2) nucleoside, nucleotide, and nucleic acid metabolism (n = 9). Detailed gene functions and associated P-values are reported in Table 1. The minimal and maximal fold change ratios in expression values for these 52 genes were 0.688 and 1.595, respectively (supporting information, Table S1). The lowest fold change was observed for the 40S ribosomal protein S5 clone whereas the highest ratio was found for the activator of 90-kDa heat shock protein ATPase homolog 1 clone.
TABLE 1.
Biological processes that are overrepresented relative to chance expectation and showing differentially expressed genes at the juvenile stage between control and selected line of Laval River brook charr
| Biological process | Reference list (n = 295) | List of differentially expressed genes (n = 52) | Expected frequency in the set of differentially expressed genes | Genes representation (over +/under −) | Associated P value |
|---|---|---|---|---|---|
| Immunity and defense | 59 | 3 | 11.11 | — | 3.20 × 10−03 |
| Nucleoside, nucleotide, and nucleic acid metabolism | 80 | 9 | 15.06 | — | 2.70 × 10−02 |
| G-protein-mediated signaling | 7 | 4 | 1.32 | + | 4.38 × 10−02 |
| Other coenzyme and prosthetic group metabolism | 2 | 1 | 0.38 | + | 4.51 × 10−02 |
| Protein metabolism and modification | 130 | 35 | 24.48 | + | 4.59 × 10−02 |
The reference list was built using the list of the analyzed transcript (n = 3750) that belonged to the five significantly over- and underrepresented biological processes.
Cluster analysis:
Cluster analysis performed on the subset of 52 genes associated with one of the five under- or overrepresented biological processes is presented in Figure 1. The heat map obtained almost completely separated the fish into two different groups corresponding to the two populations (control/selected), except for 8 of 40 individuals, which showed transcription profiles similar to the opposite population. Second, the genes also grouped into two distinct clusters. The 28 genes composing cluster 1 showed lower level of expression in the selected fish relative to the control fish, whereas the 24 genes composing cluster 2 showed higher level of expression in the selected fish relative to the control fish. Genes from the five biological processes were present in both gene clusters, except for “other coenzyme and prosthetic group metabolism,” for which the two related clones belong to cluster 2. However, genes that are involved in growth pathways were generally expressed at higher levels in the selected population, whereas genes associated with other biological functions were generally expressed at lower levels in the selected population relative to the control populations. For example, in cluster 1, genes related to growth (e.g., precursors of Meprin A subunits α, capable of cleaving growth factors) were grouped together and highly expressed in control population, showed opposite transcriptional patterns to other genes with antagonistic role and related to the same trait, in cluster 2 (e.g., transforming growth factor β early inducible response). Moreover, genes associated to the mechanisms of translation (eukaryotic translation initiation factor 3 subunit D; 28S, 40S, and 60S ribosomal subunits; elongation factor 1 β, EF1b) were grouped together in cluster 2 (Figure 1). Also, three genes associated to protection against oxidative stress (precursor of hemopexin, heme-binding protein 2, precursor of fibrinogen γ chain, and precursor of the inter-α trypsin inhibitor heavy chain H2), were upregulated in selected fish and closely clustered together.
Figure 1.—

Cluster tree for 52 genes showing the patterns of regulation between control (C1–C20) and selected animals (S1–S20). Genes names are reported on the right of the figure. The dendogram on top (Experimental Condition Clusters) groups individuals based on similar patterns of transcription. Each individual is named according to its group: C for control individual and S for selected. On the left side, the dendrogram (Gene Cluster) represents the clustering of genes showing similar patterns of regulation. Red represents higher expression values whereas values shown in green are relatively lower in the experimental pairwise comparison. Hierarchical clustering was performed using the average linking method (Eisen et al. 1998), while pairwise distances were calculated using the Pearson correlation coefficients (Qu and Xu 2006).
DISCUSSION
Our results show that three generations of selective breeding resulted in significant changes in regulation of gene transcription between control and selected brook charr. Of the genes that significantly differed between both lines, those belonging to biological functions that were overrepresented relative to chance expectations are the most likely to have diverged under the effect of selective breeding, as opposed to random genetic drift effect. However, significant changes were observed at the juvenile stage only. This corroborates the results obtained in a recent transcriptomic study of the limnetic and benthic forms of another salmonid, the lake whitefish, Coregonus clupeaformis (Nolte et al. 2009). These authors reported very few genes (n = 5) showing difference in expression at the embryonic stage relative to juvenile (16 weeks) for which over 500 genes were differentially expressed. To some extent, this system is analogous to the one used in the present study, given that: (1) the two forms of whitefish have evolved under selective (albeit natural rather than artificial) pressures that mainly affected genes related to energetic metabolism and growth differences between forms and (2) the fish were sampled at similar life stages in both systems.
A total of 52 differentially regulated genes that were annotated were classified into five biological processes. Arguably, these differences are specific to the two organs analyzed and it is likely that selected and control fish could also differ in other functions being expressed in other tissues. The observed pattern of expression of genes belonging to these biological processes appears congruent with the expected effect of directional selection for faster growth. Namely, there were more genes related to the metabolism of coenzymes, protein signaling, and protein metabolism and modifications that were differentially expressed relative to the number that was expected by chance. Examples of such genes are three RNA subunits (28S, 40S, and 60S), EF1b, and the translation initiation factor 3 (eIF3). The EF1b is a highly conserved protein that catalyzes the exchange of bound GDP for GTP, a required step to ensure continued protein synthesis and cell growth (Carr-Schmid et al. 1999). The three ribosomal subunits and eIF3 are also closely linked in their molecular functions. The eIF3 complex is thought to be composed of essential core subunits required for global protein synthesis and nonessential subunits that may modulate mRNA specificity. This complex of several polypeptides plays at least two important roles in protein synthesis: first, eIF3 binds to the 40S ribosomal subunit and facilitates loading of the Met–tRNA/eIF2 and also apparently assists eIF4 in recruiting mRNAs to the 43S complex, involved in the synthesis of protein related to cell growth (Zhou et al. 2005). Moreover, the C-terminal extension of eIF3 appears to play a role in the dissociation of Met–tRNA bound to 28S subunits in the absence of mRNA (Haque et al. 2008).
On the other hand, we also identified biological processes for which there were a lower number of genes showing differences in expression between control and selected fish than were expected by chance, as observed for the immune system and defense function. This could be a consequence of the common controlled environmental conditions where fish of both lines were presumably exposed to weak selective constraints compared to natural conditions, resulting in relaxation on expression regulation of the genes related to immunity. Alternatively, this could also reflect a drawback of the selective breeding process. For instance, it has previously been suggested that fish selected for faster growth may suffer from a deficiency in the immune response and reduced adaptive potential to pathogens exposition (Glover et al. 2006a,b).
The heatmap based on the analysis of the 52 annotated genes that were differentially expressed revealed two distinct gene clusters, one composed of 28 genes generally showing a higher level of transcription in control individuals and a second one composed of 24 genes that were generally overexpressed in selected fish and belonging to different functional categories. Thus, four genes were involved in oxidative stress protection (hemopexin precursor, heme binding protein, inter-α trypsin inhibitor, and fibrinogen γ chain precursor; Stanford et al. 2003), four others were related to essential proteins in the ribosomal subunits or related to transcription (e.g., eukaryotic translation initiation factor), while other genes were associated to the proliferation and development of cells and include genes such as the transforming early growth factor β and the T complex protein 1. Overexpression of these genes is consistent with a higher growth rate observed in the selected line compared to control fish (Bastien 2010).
We also observed several genes of potentially antagonistic functions that were expressed differentially between selected and control lines, which would be more consistent with the expected effect of selection than mere genetic drift. Of particular interest were three clones related to the meprin A subunit precursor gene that were more highly expressed in control relative to selected fish. Meprins are multidomain extracellular metalloproteases and capable of cleaving growth factors and would therefore be expected to have an antagonistic effect on genes related to growth that were more highly expressed in selected fish (e.g., transforming growth factor β early inducible response).
Taken together, our results demonstrate that selection over three generations, in a controlled environment, has led to significant and heritable differences in genes transcription in the brook charr. This also corroborates results observed in a recent study that investigated transcriptional changes induced by domestication in Atlantic salmon (Roberge et al. 2006). Significantly differentially expressed genes in Atlantic salmon were classified into several biological functions including transcription regulation, protein synthesis, signal transduction, cellular growth, and division (Roberge et al. 2006). In our study, differentially expressed genes primarily represented very similar biological functions, including protein metabolism and modification, nucleic acids metabolism or protein signaling. Yet, specific genes involved in these biological functions were not the same for both brook charr and Atlantic salmon. This could be mainly attributable to (1) statistical issues, as analytical methods have changed over time to handle microarray data (e.g., introduction of FDR correction; Storey and Tibshirani 2003), (2) the different microarray version used in both experiments, and (3) the results between the Atlantic salmon and brook charr can simply reflect that different genes are under selection in those two species.
Our results and those on Atlantic salmon clearly show that a small number of generations of selective breeding for improved growth can lead to significant evolutionary change in gene transcription between selected and control fish. The fact that artificially selected brook charr are intensively used for stocking wild populations raises the issue of potentially negative consequences associated with reduction in genomic integrity and adaptative capacity of wild populations (Leary et al. 1983; Gharrett et al. 1999; Gilk et al. 2004; Roberge et al. 2006). For example, it has been shown in Atlantic salmon that genetic introgressive hybridization between farmed and wild fish substantially alters the genetic control of gene transcription within natural populations. In brook charr, it has also been shown that crosses between sympatric anadromous and resident populations that diverged <12,000 years ago cause major disruption of gene expression in the F1 hybrid progeny concordant with the Dobzhansky–Muller model of genetic incompatibilities (Mavarez et al. 2009).
In summary, this study provided a list of candidate genes for which the control of expression has been altered by artificial selection and deserving further investigations. Namely, allelic variation in genetic markers linked to the variation of traits of economic interest provides information about the inheritance of the considered traits that can then be used in a marker-assisted selection program. Consequently, one of the next logical steps in the study of brook charr selection is the identification and mapping of molecular markers (e.g., SNP) linked to the candidate genes identified in this study. These markers could then be linked to phenotypic traits of interest through quantitative trait loci. This study is currently underway. Finally, they could be implemented in a marker-assisted selection program to enhance a sustainable production capability for the brook charr aquaculture.
Acknowledgments
The authors are very grateful to staff of the Station aquicole de l'Institut des sciences de la mer de Rimouski for their invaluable help in rearing the fish used in this experiment. We thank Guillaume Côté for his help with the fish sampling process. We also thank D. Grunwald and two anonymous reviewers for their most useful and constructive comments.This work was financially supported by a Partnership grant (Strategic program) of the Conseil de Recherches en Sciences Naturelles et en Génie (CRSNG) to L.B., N.D., and C.A. This is a contribution to the research program of Réseau Aquaculture Québec (RAQ).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.115071/DC1.
References
- Allard, R. W., 1999. Principles of Plant Breeding, Ed. 2. Wiley, New York.
- Audet, C., and L. Bernatchez, 2004. La souche Laval, le pourquoi et le comment. L'Aquicole, Bulletin Assoc Aquaculteurs Québec 10 9–11. [Google Scholar]
- Bastien, A., 2010. Evaluation of a selection program and identification of physiological traits linked to anadromous in the brook trout (Salvelinus fontinalis). Ph.D. Thesis, Université du Québec à Rimouski, Canada
- Burt, D. W., 2005. Chicken genome: current status and future opportunities. Genome Res 15 1692–1698. [DOI] [PubMed] [Google Scholar]
- Carr-Schmid, A., L. Valente, V. I. Loik, T. Williams, L. M. Starita et al., 1999. Mutations in elongation factor 1beta, a guanine nucleotide exchange factor, enhance translational fidelity. Mol. Cell. Biol. 19 5257–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., 2002. Fundamentals of experimental design for cDNA microarrays. Nat. Genet. 32(Suppl.): 490–495. [DOI] [PubMed] [Google Scholar]
- Devlin, R., H. D. Sakhrani, W. Tymchuk, E., M. L. Rise and B. Goh, 2008. Domestication and growth hormone transgenesis cause similar changes in gene expression in coho salmon (Oncorhynchus kisutch). Proc. Natl. Acad. Sci. USA 106 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, J., 2002. Evolution, consequences and future of plant and animal domestication. Nature 418 700–707. [DOI] [PubMed] [Google Scholar]
- Duarte, C. M., N. Marba and M. Holmer, 2007. Rapid domestication of marine species. Science 316 382–383. [DOI] [PubMed] [Google Scholar]
- Eisen, M. B., P. T. Spellman, P. O. Brown and D. Botstein, 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman Group, Essex, UK.
- Fleming, I. A., S. Einum, B. Jonsson and N. Jonsson, 2003. Comment on “rapid evolution of egg size in captive Salmon.” Science 302 59. [DOI] [PubMed] [Google Scholar]
- Fleming, I. A., K. Hindar, I. B. Mjølnerød, B. Jonsson, T. Balstad et al., 2000. Lifetime success and interactions of farm salmon invading a native population. Proc. R. Soc. B. Biol. Sci. 267 1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharrett, A. J., W. W. Smoker, R. R. Reisenbichler and S. G. Taylor, 1999. Outbreeding depression in hybrids between odd- and even-broodyear pink salmon. Aquaculture 173 117–129. [Google Scholar]
- Gilk, S. E., I. A. Wang, C. L. Hoover, W. W. Smoker, S. G. Taylor et al., 2004. Outbreeding depression in hybrids between spatially separated pink salmon, Oncorhynchus gorbuscha, populations: marine survival, homing ability, and variability in family size. Environ. Biol. Fish 69 287–297. [Google Scholar]
- Giuffra, E., J. M. H. Kijas, V. Amarger, O. Carlborg, J.-T. Jeon et al., 2000. The Origin of the domestic pig: independent domestication and subsequent introgression. Genetics 154 1785–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjedrem, T., 2000. Genetic improvement of cold-water fish species. Aquaculture Res. 31 25–33. [Google Scholar]
- Glover, K. A., Ÿ. Bergh, H. Rudra and Ÿ. Skaala, 2006. a Juvenile growth and susceptibility to Aeromonas salmonicida subsp. salmonicida in Atlantic salmon (Salmo salar L.) of farmed, hybrid and wild parentage. Aquaculture 254 72–81. [Google Scholar]
- Glover, K. A., C. Skâr, K. E. Christie, J. Glette, H. Rudra et al., 2006. b Size-dependent susceptibility to infectious salmon anemia virus (ISAV) in Atlantic salmon (Salmo salar L.) of farm, hybrid and wild parentage. Aquaculture 254 82–91. [Google Scholar]
- Glover, K. A., H. Otterâ, R. E. Olsen, E. Slinde, G. L. Taranger et al., 2009. A comparison of farmed, wild and hybrid Atlantic salmon (Salmo salar L.) reared under farming conditions. Aquaculture 286 203–210. [Google Scholar]
- Hammer, K., 1984. Das Domestikationsyndrom. Kulturpflanze 32 11–34. [Google Scholar]
- Haque, M. E., D. Grasso and L. L. Spremulli, 2008. The interaction of mammalian mitochondrial translational initiation factor 3 with ribosomes: evolution of terminal extensions in IF3mt. Nucleic Acids Res. 36 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, K. M., and G. A. Churchill, 2000. Analysis of variance for gene expression microarray data. J. Comp. Biol. 7 819–837. [DOI] [PubMed] [Google Scholar]
- Kovach, M. J., M. T. Sweeney and S. R. McCouch, 2007. New insights into the history of rice domestication. Trends Genet. 23 578–587. [DOI] [PubMed] [Google Scholar]
- Leary, R. F., F. W. Allendorf and K. L. Knudsen, 1983. Developmental stability and enzyme heterozygosity in rainbow trout. Nature 301 71–72. [DOI] [PubMed] [Google Scholar]
- Martin, S., J.-Y. Savaria, C. Audet and L. Bernatchez, 1997. Microsatellites reveal no evidence for inbreeding effects but low-inter-stock genetic diversity among brook charr stocks used for production in Québec. Bull. Aquaculture Assoc. Canada 97 21–23. [Google Scholar]
- Mavarez, J., C. Audet and L. Bernatchez, 2009. Major disruption of gene expression in hybrids between young sympatric anadromous and resident populations of brook charr (Salvelinus fontinalis Mitchill). J. Evol. Biol. 22 1708–1720. [DOI] [PubMed] [Google Scholar]
- Nolte, A., S. Renaut and L. Bernatchez, 2009. Divergence in gene regulation at young life history stages of whitefish (Coregonus sp.) and the emergence of genomic isolation. BMC Evol. Biol. 9 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normandeau, E., J. A. Hutchings and D. J. Fraser, 2009. Population specific gene-expression responses to hybridization between farm and wild Atlantic salmon. Evol. Appl. 2 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, L. M., and B. M. Burr, 1991. A Field Guide to Freshwater Fishes of North America North of Mexico. Houghton Mifflin, Boston.
- Pozzi, C., and F. Salamini, 2007. Genomics of wheat domestication, pp. 453–481 in Genomics-Assisted Crop Improvement. Volume 2: Genomics Applications in Crops, edited by R. K. Varshney and R. Tuberosa. Springer-Verlag, New York.
- Qu, Y., and S. Xu, 2006. Quantitative trait associated microarray gene expression data analysis. Mol. Biol. Evol. 23 1558–1573. [DOI] [PubMed] [Google Scholar]
- Rise, M. L., K. R. von Schalburg, G. D. Brown, M. A. Mawer, R. H. Devlin et al., 2004. Development and application of a salmonid EST database and cDNA microarray: data mining and interspecific hybridization characteristics. Genome Res. 14 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberge, C., S. Einum, H. Guderley and L. Bernatchez, 2006. Rapid parallel evolutionary changes of gene transcription profiles in farmed Atlantic salmon. Mol. Ecol. 15 9–20. [DOI] [PubMed] [Google Scholar]
- Roberge, C., E. Normandeau, S. Einum, H. Guderley and L. Bernatchez, 2008. Genetic consequences of interbreeding between farmed and wild Atlantic salmon: insights from the transcriptome. Mol. Ecol. 17 314–324. [DOI] [PubMed] [Google Scholar]
- Ross-Ibarra, J., P. L. Morrell and B. S. Gaut, 2007. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc. Natl. Acad. Sci. USA 104(Suppl. 1): 8641–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeot, C., C. Bervillers, C. Prignon, D. Gustin, M. Del Giudice et al., 2007. Growth improvement of Eurasian perch (Perca fluviatilis) using domesticated strains under intensive rearing conditions. Aquaculture 272 S306. [Google Scholar]
- Savaria, J.-Y., 1998. Amorce d'un programme de sélection génétique chez deux souches d'ombles de fontaine en fonction des critères de croissance et de l'âge à la maturation sexuelle. M.Sc. Thesis, Université du Québec à Rimouski, Canada.
- Silverstein, J. T., R. L. Vallejo, Y. Palti, T. D. Leeds, C. E. Rexroad, III et al., 2009. Rainbow trout resistance to bacterial cold-water disease is moderately heritable and is not adversely correlated with growth. J. Anim. Sci. 87 860–867. [DOI] [PubMed] [Google Scholar]
- Simons, K. J., J. P. Fellers, H. N. Trick, Z. Zhang, Y.-S. Tai et al., 2006. Molecular characterization of the major wheat somestication Gene Q. Genet. 172 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Cyr, J., N. Derome and L. Bernatchez, 2008. The transcriptomics of life-history trade-offs in whitefish species pairs (Coregonus sp.). Mol. Ecol. 17 1850–1870. [DOI] [PubMed] [Google Scholar]
- Stanford, S. J., M. J. Walters and A. A. Hislop, 2003. Heme oxygenase is expressed in human pulmonary artery smooth muscle where carbon monoxide has an anti-proliferative role. Eur. J. Pharmacol. 473 135–141. [DOI] [PubMed] [Google Scholar]
- Storey, J. D., and R. Tibshirani, 2003. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey, J. D., J. E. Taylor and D. Siegmund, 2004. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J. R. Statist. Soc. Ser. B. Statist. Methodol. 66 187–205. [Google Scholar]
- Wu, H., K. M. Kerr and G. A. Churchill, 2002. MAANOVA: a software package for the analysis of spotted cDNA microarray experiments, pp. 313–316 in The Analysis of Gene Expression Data: Methods and Software, edited by H. Wu, K. Kerr, X. Cui and G. A. Churchill. Springer-Verlag, New York.
- Yamasaki, M., S. I. Wright and M. D. McMullen, 2007. Genomic screening for artificial selection during domestication and improvement in maize. Ann. Bot. 100 967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C., F. Arslan, S. Wee, S. Krishnan, A. Ivanov et al., 2005. PCI proteins eIF3e and eIF3m define distinct translation initiation factor 3 complexes. BMC Biol. 3 14. [DOI] [PMC free article] [PubMed] [Google Scholar]