

Abstract
The involvement of myeloperoxidase (MPO) in various inflammatory conditions has been the scope of many recent studies. Besides its well studied catalytic activity, the role of its overall structure and glycosylation pattern in biological function is barely known. Here, the N-glycan composition of native dimeric human MPO purified from neutrophils and of monomeric MPO recombinantly expressed in Chinese hamster ovary cells has been investigated. Analyses showed the presence of five N-glycans at positions 323, 355, 391, 483, 729 in both proteins. Site by site analysis demonstrated a well conserved micro- and macro-heterogeneity and more complex-type N-glycans for the recombinant form. Comparison of biological functionality of glycosylated and deglycosylated recombinant MPO suggests that glycosylation is required for optimal enzymatic activity. Data are discussed with regard to biosynthesis and the three-dimensional structure of MPO.
Keywords: Glycosylation, Inflammation, Lipoprotein, Mass Spectrometry (MS), Reactive Oxygen Species (ROS), Myeloperoxidase
Introduction
Myeloperoxidase (MPO,5 EC 1.11.1.7) is a heme-containing glycosylated oxidoreductase that catalyzes the production of hypochlorous acid (HOCl) in the presence of hydrogen peroxide (H2O2) and chloride anions (1). The macromolecule is a homodimer with each monomer consisting of a light and a heavy chain. The dimer is stabilized by a disulfide bridge and by several interactions between the carbohydrate moieties located at the dimer interface (2). MPO is a lysosomal enzyme present in the azurophilic granules of human neutrophils and monocytes and is synthesized in the promyelocytic state during maturation of the polymorphonuclear (PMN) leukocytes (3).
During phagocytosis of exogenous structures, neutrophils release their granule content (e.g. MPO) into the phagosome. Respiratory burst is initiated by activation of NADPH-oxidase that reduces molecular oxygen and releases superoxide radical anions. Upon superoxide dismutation, hydrogen peroxide is produced that mediates MPO-driven oxidation of chloride to antimicrobial HOCl.
More recent data have indicated that these reactions are not restricted to the phago-lysosome. Indeed, under oxidative stress (i.e. excessive and uncontrolled production of various reactive oxygen-derived species, ROS), MPO can escape from neutrophils and circulate in extracellular fluids. Thereby it might cause oxidative modification to host tissues and biomolecules. The potential deleterious effect of this “circulating” MPO has been demonstrated in various pathological conditions involving chronic inflammation, such as atherosclerosis, demyelinating disorders of the central nervous system or end-stage renal disease (4–9).
In many of these experiments, recombinant human MPO (r-MPO) expressed in a Chinese hamster ovary (CHO) cell line has been used (10) because recombinant production delivers higher amounts of protein as compared with the classical purification from neutrophils (11). However, although having the same catalytic activity as the dimeric leukocyte enzyme (h-MPO) (12–14), biosynthesis of r-MPO in CHO cell lines lacks some proteolytic steps. As a consequence, r-MPO is monomeric and differs in glycan structure.
Recently Lau et al. (15) reported the implication of leukocyte MPO in the activation of neutrophils mediated by CD11b/CD18 integrins. Moreover, r-MPO was shown to activate endothelial cells in a proatherogenic process of lipoprotein oxidation (16). Evidence was provided that the observed phenomena might be correlated with surface structures of both forms of MPO, especially with their glycosylation pattern. This motivated us to comprehensively investigate the glycan structures of h-MPO and r-MPO and their micro- and macro-heterogeneity. Another aim was to clarify controversial opinions about the number of glycosylation sites in h-MPO and r-MPO. Whereas Johnson et al. (17) predicted 4 sites (Asn323, Asn355, Asn391, and Asn729, excluding Asn483), others like Hanson et al. (18) proposed Asn323, Asn355, Asn391, and Asn483 (excluding 729). Similarly, crystallographic data suggested three (355, 391, 483) (19) or four N-glycosylation sites (323, 355, 391, 483) (20). Finally, the impact of glycosylation on MPO functionality (chlorination activity, LDL oxidation, ROS release from endothelial cells) was investigated.
EXPERIMENTAL PROCEDURES
Materials
h-MPO was kindly provided by Zentech (product B-AC-001; Angleur, Belgium) and obtained according to a previously described procedure (11). Recombinant human MPO (r-MPO) was expressed in a CHO cell line and prepared as previously described (10). The corresponding protocols for both proteins were detailed in the supporting material section. Each batch was characterized according to its protein concentration (mg/ml), its activity (units/ml), and its specific activity (unit/mg). The chlorination activity was determined according to Hewson et al. (21).
Digestions
N-terminal amino acid sequence determination of MPO was obtained after protein denaturation and SDS/PAGE was performed by the method of Laemmli (22). The mini-slab gel (0.75 × 50 mm) consisted of 7.5% or 10% separating gel with a 3.5% stacking gel. The heaviest bands (94 and 84 kDa) were picked up and analyzed by automated Edman's degradation by Eurogentec (Seraing, Belgium).
Glycans release from glycoprotein was obtained with PNGase-F digestion of glycoprotein and performed on 100 μg of r-MPO and 50 μg of h-MPO according to the manufacturing protocol (Roche Molecular Biochemicals). The released glycans were desalted on column of non-porous graphitized carbon (Alltech, Deerfield, IL), as previously described (23).
Permethylation of N-glycans released by PNGase digestion was carried out with the sodium hydroxide procedure was performed according to Ciucanu and Kerek (24). After derivatization, the reaction products were dissolved in 200 μl of methanol and further purified on a C18 Sep-Pak (Waters, Milford, MA).
Linkage analysis of N-glycans was endorsed by GC-MS linkage analysis as previously described (25). Partially methylated alditol acetates, were prepared from permethylated samples. GC-MS analyses were recorded using an Automass II 30 quadrupole mass spectrometer interfaced with a Carlo Erba 8000 Top gas chromatograph (Finnigan, Argenteuil, France).
Reduction, alkylation, and trypsin hydrolysis of the proteins were carried out on 500 μg of h-MPO or 1 mg of r-MPO according to standard protocols. Proteins were denatured by guanidine, and reduced by dithiothreitol (Bio-Rad). Alkylation was obtained with iodoacetamide (Bio-Rad) before extensive dialysis against ammonium bicarbonate and lyophilization. Proteolysis was performed by adding Tos-Phe-CH2Cl-treated trypsin (Sigma Chemicals) at an enzyme-substrate ratio of 1:50. The tryptic fragments obtained were then purified by C18 Sep-Pak cartridge, eluted with 80% acetonitrile in water containing 0.1% trifluoroacetic acid and finally lyophilized. The detailed protocol is available in supporting material section. The purified peptides were ready to be separated either by affinity chromatography using lectin-coupled Sepharose or by reverse-phase (RP) chromatography.
Deglycosylation of recombinant-MPO (∼2 mg/ml, deglyMPO) was obtained according to the standard protocol of a glycoprotein deglycosylation kit (Calbiochem). The protocol was detailed in the supplemental materials. Another sample from the same batch was treated similarly but in the absence of any glycosidase (MPO37°). The protein concentration of each sample was measured by the Lowry assay (26).
To assess the deglycosylation, the three samples were alkylated, reduced, digested, and glycopeptides enriched according to the protocols described above. The peptides were analyzed by LC/ESI/(MS)MS with the QTOF6520 (Agilent, Palo Alto, CA).
Analytical Procedures
Enrichment of glycopeptides was obtained by affinity chromatography on immobilized lectin. The peptide digest was loaded onto a concanavalin A (Con A)-Sepharose column (Amersham Biosciences), and the glycopeptides were enriched by elution with α-methyl-d-glucoside solutions at different concentrations. The protocol was detailed in supplemental materials.
The tryptic digest was separated by RP-HPLC applied to a C18 3.5 μm XTerra column (2.1 × 150 mm) (Waters, Milford, MA) equipped with a guard column Optiguard 1 mm C18 (Interchim, Montluçon, France) and equilibrated with 0.1% trifluoroacetic acid in water. The elution was obtained by a gradient mode with a 0.1% trifluoroacetic acid solution applied for 15 min, followed by a linear gradient of acetonitrile from 0 to 40% over 90 min, then from 40 to 70% over 30 min at a flow rate of 0.150 ml/min. Eluates were monitored at 220 nm. Glycopeptide-containing fractions were characterized by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS).
PNGase-F digestion of tryptic glycopeptides was performed in 50 mm ammonium bicarbonate at 37 °C for 24 h. The products were purified on a C18 Sep-Pak equilibrated in 5% acetic acid. The PNGase-F-released N-glycans, and peptides were lyophilized. The glycans were further permethylated as described above.
SDS-PAGE of MPO was carried out on an aliquot that was denatured in Laemmli buffer (22) at 100 °C for 5 min and run on 10% acrylamide gel (acrylamide/bis-acrylamide, 29:1 ratio). Electrophoresis was conducted using Mini-Protean III (Bio-Rad) under constant current (30 mA) for 2 h. The gel was stained with Coomassie Blue R250 and destained with a mixture of methanol-acetic acid water (20:7:73; v/v/v).
In-gel PNGase-F and tryptic digestion was performed by excising gel pieces that were first in-gel N-deglycosylated using PNGase-F. Trypsin was then used for in-gel proteolysis after extraction of released N-glycans as previously described (27). In an independent experiment, gel pieces were incubated only with trypsin. Peptides were dissolved in 0.1% trifluoroacetic acid, and an aliquot was desalted using RP C18 Zip-Tip (Millipore) and eluted with 60% acetonitrile in water containing 0.1% trifluoroacetic acid and 10 mg/ml α-cinnamic acid, i.e. the matrix for MALDI-MS analysis.
Matrix-assisted laser desorption/ionisation mass spectrometry (MALDI-MS) experiments were carried out on a Voyager Elite DE-STR Pro instrument (Perseptive Biosystems, Framingham, MA) in reflectron mode with delayed extraction. Sample preparation is detailed in supplemental materials.
Electrospray Tandem Mass Spectrometry (nano-ESI-MS/MS)
Glycopeptide identifications were performed using Q-Star Pulsar hybrid quadrupole/time-of-flight (Q/TOF) mass spectrometer (Applied Biosystems/MDS Sciex, Toronto, Ontario, Canada) fitted with a nano-electrospray probe as the ion source (nano-ESI) (Protana, Odense, Denmark). For the recording of conventional mass spectra, TOF-MS data were acquired by accumulation of 50 multiple channel acquisition (MCA). For the recording of CID-MS/MS spectra, TOF-MS data were acquired by accumulation of 60 MCA.
Liquid Chromatography Coupled to Electrospray (Tandem) Mass Spectrometry (LC/ESI/MSMS)
Analyses of MPO glycosylation and deglycosylation were also done by a liquid chromatographic system (LC12000, Agilent) equipped with a rapid resolution column (Zorbax Eclipse plus 2.1 × 5 mm, 1.8 μm) and a quadrupole/time-of-flight (QTOF 6520) mass spectrometer (Agilent). The analyses were performed in MS and MSMS according to the protocols detailed in supplemental materials.
Measurements of the MPO Activity
Measurement of MPO activity has been firstly carried out according to a standard protocol that follows chlorination of monochlorodimedone (MCD) (20) and detailed in supplemental materials. The activity was calculated by linear fitting of the initial (4–90 s) absorbance decrease at 290 nm. The specific activity was expressed as units (i.e. ΔA290/min) per mg of protein.
MPO-mediated oxidation of LDL was carried out as previously described (8). LDL oxidation by the MPO/H2O2/Cl− system was followed by an ELISA. The latter was based on a mouse monoclonal antibody (Mab AG9) that specifically recognizes oxidized APO B-100 on LDL and on staining with an anti-mouse immunoglobulin G coupled with alkaline phosphatase (7). For details, see supplemental materials.
The generation of ROS mediated by MPO in endothelial cells (Eahy926 cells) was measured by a fluorescent probe. The fluorescent dyes H2DCF-DA and Bodipy® were purchased from Invitrogen (Carlsbad, CA). Eahy926 cells were incubated with 20 μmol/liter dichloro-dihydro-fluorescein diacetate (H2DCF-DA) for 30 min at 37 °C in the dark before stimulation with r-MPO (10 ng/ml). After 30 min, cells were rinsed and the intracellular oxidation of H2DCF by ROS was quantified by measuring the DCF fluorescence at 520 nm (λexc., 485 nm) with a Fluoroskan Ascent fluorimeter (Thermo Scientific, Waltham, MA). The protein concentration was measured by the Bradford method after lysis in 1 m NaOH, and the fluorescence was expressed as relative fluorescence in arbitrary units per mg protein.
RESULTS
Structure of N-Glycans Released by PNGase-F
The general strategy employed for glycosylation analysis is outlined in supplemental materials S1. The glycan structures were determined after tryptic digestion of the reduced carboxamido-methylated proteins. Glycopeptides were enriched by Con A-immobilized affinity chromatography, and fractionated and analyzed by RP-C18 HPLC. Glycopeptides as well as their N-deglycosylated forms were then identified by MALDI-MS. Released glycans were separated from the peptide moiety using a C18 Sep-Pak cartridge, permethylated, and profiled by MALDI-MS. The amino acid sequences of N-deglycosylated peptides were finally obtained by Q/TOF tandem mass spectrometry (MS/MS).
MALDI-MS Analysis of N-Glycans Released by PNGase-F
The first part of this study has focused on the general glycosylation pattern of both MPO forms. Data presented in Table 1 and Fig. 1 clearly indicate that both h-MPO and r-MPO are characterized by the presence of high-mannose structures (Hex3–7HexNAc2) accompanied by variable proportions of biantennary and triantennary structures, with the latter less abundant. These results are in agreement with the currently accepted model of eukaryotic N-glycan biosynthesis. The major difference between the two MPO forms concerns the occurrence of higher amount of complex-type glycans in r-MPO (Fig. 1B), e.g. the presence of the di- and trisialylated triantennary structures in r-MPO. The predominant glycan in both MPO forms was Hex6HexNAc2 (Man6, m/z 1784) (100%), followed by Hex5HexNAc2 (Man 5, m/z 1580) and Hex7HexNAc2 (Man7, m/z 1988), with the latter two having the relative abundance of 81 and 19% for h-MPO, and 72 and 13% for r-MPO, respectively. The complex-type glycans predominantly found in r-MPO were more branched but less sialylated than in h-MPO. Indeed, a very heterogeneous mixture of complex-type glycans was observed in the glycoprofile of r-MPO, dominated by a strong signal at m/z 2606 (NeuAc1dHex1Hex5HexNAc5), corresponding to a monosialylated fucosylated biantennary bigalactosylated structure. This abundant glycoform was accompanied by minor incompletely sialylated fucosylated triantennary structures, as observed at m/z 2694 (dHex1Hex5HexNAc5), m/z 3055 (NeuAc1dHex1Hex5HexNAc5), and m/z 3416 (NeuAc2dHex1Hex5HexNAc5), corresponding to asialyl, monosialyl, and disialyl glycoforms, the latter two being not observed in the MALDI mass glycoprofile of h-MPO.
TABLE 1.
Assignment of pseudomolecular ion [M+Na]+ observed in MALDI-MS spectrum of the PNGase-F-released N-glycans from both MPO forms
Symbols: blue square, N-acetylglucosamine (HexNAc); green circle, mannose (Hex); yellow circle, galactose (Hex); red triangle, fucose (dHex); and purple diamond, N-acetyl neuraminic acid (NeuAc).

FIGURE 1.

MALDI-TOF mass spectrum of permethylated N-glycans derived from human mature myeloperoxidase (h-MPO) (A) and recombinant myeloperoxidase (r-MPO) (B).
Linkage Analysis of the Glycans Released by PNGase-F
For further analysis of obtained glycosylation patterns a GC-MS analysis of the partially methylated alditol acetate derivatives was carried out. This analysis made possible the identification of the position of interglycosidic bonds and the confirmation of the N-glycan structures previously observed on both MPO forms. As shown in Table 2, h-MPO is characterized by a high abundance of 2-linked mannose (relative abundance is 1.00), which is related to the presence of high-mannose structures and biantennary structures. This abundance is additionally confirmed by the high proportion of t-mannose (0.56) and of 3,6-linked mannose (0.22). These data are congruent with those obtained by the MALDI mass glycoprofiling. Moreover, the very low level of 2,4- and 2,6-linked mannose (respectively, 0.03 and 0.11) confirms the low abundance of tri- and/or tetraantennary structures. The presence of t-fucose (0.17) concomitantly with 4,6-linked GlcNAc (0.01) confirms the presence of (α1–6)-core-fucosylated glycans. Concerning bi- and triantennary complex glycans, 3- and 6-linked galactose (0.15 and 0.14) are well represented as compared with terminal galactose (0.08), underlining the predominance of sialylated toward non-sialylated complex structures. The same analysis performed on r-MPO is summarized in Table 2. As a matter of fact, 2-linked mannose, t-mannose and 3,6-linked mannose are also abundant (1.00, 1.32, and 1.06, respectively), confirming the presence of major high-mannose and biantennary structures. The low amount of 2,4- and 2,6-linked mannose (0.035 and n.d., respectively) is congruent with a low level of tri- and/or tetraantennary structures. However, the proportion of terminal galactose (0.56) and of 3-linked galactose (0.17) is in favor of terminal galactose. This is related to the predominance of nonsialylated galactose illustrated by the abundance of dHex1Hex5HexNAc4 (m/z 2245). Moreover, 6-linked galactose is absent, which is in accordance with the absence of (α2–6) sialyltransferase in the CHO cell line (28).
TABLE 2.
Linkage analysis of partially methylated alditol acetate derivatives of the PNGase-F-released N-glycans from both MPO forms
| Assignments | Characteristic fragment ions | Elution time | Relative abundance |
|
|---|---|---|---|---|
| h-MPO | r-MPO | |||
| min | ||||
| Terminal fucose | 115,118,131,162,175 | 14:00 | 0.169 | 0.005 |
| Terminal mannose | 102,118,129,145,161,162,205 | 18:33 | 0.557 | 1.324 |
| Terminal galactose | 102,118,129,145,161,162,206 | 19:25 | 0.079 | 0.558 |
| 2-Linked mannose | 129,130,161,190,234 | 22:31 | 1.000 | 1.000 |
| 3-Linked galactose | 101,118,129,161,234 | 23:22 | 0.154 | 0.171 |
| 6-Linked galactose | 118,162,189,233 | 25:19 | 0.135 | - |
| 2,4-Linked mannose | 130,190,233 | 26:35 | 0.028 | 0.035 |
| 2,6-Linked mannose | 129,130,189,190 | 28:09 | 0.113 | - |
| 3,6-Linked mannose | 118,129,189,234 | 28:32 | 0.223 | 1.059 |
| Terminal GlcNAc | 117,159,203,205 | 31:06 | 0.119 | 0.121 |
| 4-Linked GlcNAc | 117,159,233 | 33:31 | 0.509 | 1.246 |
| 4,6-Linked GlcNAc | 117,159,261 | 37:00 | 0.007 | 0.043 |
Site-specific N-Glycosylation
After investigation of the general glycosylation profile of both MPO forms, site by site analysis of glycosylation was performed. For this reason, glycopeptides from r-MPO have been isolated from the tryptic digest by Con A-affinity chromatography that allows separation of glycopeptides from peptides. Elution with 10 and 300 mm of α-methyl-d-glucose allowed differentiation between low and high affinity fractions (see supplemental materials S2). Both fractions were separated by RP-C18 HPLC giving at least seven defined peaks in the chromatogram at 45.5, 55.2, 65.8, 81.0, 86.4, 115.4, and 119.6 min (supplemental materials S2) that have been analyzed by MALDI-TOF MS. Glycopeptides were found only in the first five fractions (45.5, 55.2, 65.8, 81.0, and 86.4 min). After the release of the N-glycans by incubation with PNGase-F, the peptidic aglycones were purified on a C18 ZipTip and sequenced by nano-ESI-MS/MS. As shown in Fig. 2, the MS/MS analyses revealed five peptidic sequences corresponding to Asn483 (m/z 1053), Asn323 (m/z 1434), Asn355 (m/z 1575, oxidized methionine), Asn391 (m/z 2125), and Asn729 (m/z 1964) (see supplemental materials S3 and S4). This allowed correlation between glycopeptides and fractions of the chromatograms. Consequently, the total tryptic digests of both MPO forms were injected in the same chromatographic system and fractions around the selected time of glycopeptides (45.5, 55.2, 65.8, 81.0, 86.4 min.) were analyzed by MALDI-TOF MS. Fractions containing glycopeptides were digested using PNGase-F to characterize, on one hand, the carbohydrate composition of each glycosylation site and to identify, on the other hand, the peptide sequence. The site by site N-glycan composition of h-MPO and r-MPO is presented in Fig. 3 and detailed in supplemental materials S5. The site-specific N-glycosylation appears to be relatively conserved in r-MPO. Starting from the N-terminal residue, the first site is Asn323 being glycosylated with a variety of complex-type hyposialylated structures in r-MPO. However, Hex4HexNAc2 (Man4, m/z 1374) was the major glycan in both enzymes (Fig. 3 and supplemental materials S5A). The second site (Asn355) is characterized by the presence of high-mannose structures (Hex4–7HexNAc2; Man4 to Man7, m/z 1374–1988) with Hex6HexNAc2 occurring in h-MPO nearly twice the number of r-MPO, which was mainly glycosylated by Hex4HexNAc2 (Man4, m/z 1384; Fig. 3 and supplemental materials S5B). The third N-glycosylation site (Asn391) is well conserved in r-MPO comprising mainly Hex5HexNAc2 (Man5, m/z 1580), while Hex6HexNAc2 (Man6, m/z 1784) was most abundant in h-MPO (Fig. 3 and supplemental materials S5C). Asparagine 483, the third site, holds a wide variety of biantennary complex-type structures, a fucosylated pentasaccharidic (dHex1Hex3HexNAc2; a fucosylated Man3, m/z 1345) core and a fucosylated asialobiantennary (dHex1Hex5HexNAc4, m/z 2245) structure forming the major glycoform in this site in both h-MPO and r-MPO (Fig. 3 and supplemental materials S5D). Finally, discrepancies were observed at Asn729. Recombinant MPO has been shown to contain biantennary complex-type structures that were absent in h-MPO. By contrast, the leukocyte protein has only high-mannose type glycans. Moreover, Hex5HexNAc4 (Man5, m/z 1580) was the most abundant structure in r-MPO while Hex4HexNAc4 (Man4, m/z 1374) was the major glycan found in h-MPO at this site (Fig. 3 and supplemental materials S5E).
FIGURE 2.

CID MS/MS mass spectrum of the parent ions of the human mature myeloperoxidase peptide moiety of the glycopeptide from the RP-C18 HPLC fractions respectively eluted at: A, 45.5 min (m/z 1053 MH+); B, 55.2 min (m/z 1433 MH+); C, 65.8 min (m/z 1575 MH+); D, 81.0 min (m/z 2125 MH+); E, 86.4 min (m/z 1964 MH+). Glycopeptides from the different fractions were deglycosylated using PNGase-F. The peptidic aglycone was purified on to a C18 Zip-tip and then analyzed by nano-ESI-MS/MS under 40 eV as collision energy. The Asn residues were converted to aspartic residues after PNGase-F treatment (For detailed information and raw data, see supplemental materials S2 and S3).
FIGURE 3.

Comparison of the microheterogeneity of N-glycans of mature dimeric myeloperoxidase (h-MPO) and recombinant myeloperoxidase (r-MPO). Colors and form codes are as in Table 1.
During the first experiments, the propeptide of r-MPO could not be isolated. However, the N-terminal amino acid sequencing of recombinant MPO demonstrated the (partial) presence of the pro-peptide S(A49)-P(A50)-P51-A52-V53, as previously described (Moguilevsky et al., Ref. 10). Analysis of the putative glycosylation at Asn139 by direct LC/ESI/MSMS analyses revealed the presence of glycan structures in the propeptide.
In-gel Analysis of Peptidic Sequence and Relative Site Occupancy
The in-gel analysis of MPO allowed the identification of band sequences as well as the estimation of the relative occupancy of glycosylation sites. As illustrated in supplemental materials S6, the electrophorograms of both MPO forms showed three bands after denaturation (57, 37, and 15 kDa) with the two upper bands corresponding to the heavy and the 15-kDa band to the light chain.
To estimate the relative occupancy of each site, the peptide mass fingerprints of the heavy chain of both MPO forms were determined (upper band, 57 kDa). The signal intensity of the non-glycosylated peptides containing each glycosylation site was measured before and after PNGase-F treatment and normalized to that of the abundant signals at m/z 1508 and 1973, which are known to lack a glycosylation site. The relative occupancy (%) was estimated using Equation 1,
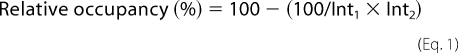 |
with Int1 and Int2 representing the relative signal intensities before and after incubation with PNGase-F. As summarized in Table 3, h-MPO is characterized by about 100% occupancy of all N-glycosylation sites except for Asn729 where no occupancy could be calculated. Concerning r-MPO, a relative occupancy of 100% was calculated for Asn323 and Asn483 whereas Asn355, Asn391, and Asn729 had a relative occupancy of 58, 96, and 75%, respectively.
TABLE 3.
Relative occupancy for the five N-glycan sites obtained with or without in-gel PNGase-F treatment on the first band (57 kDa)
Int1 is the relative intensity of the signal peptide with PNGase-F treatment and Int2 without PNGase-F treatment.
| Peptide mass (m/z) | Glycosylation site | h-MPO |
r-MPO |
||||
|---|---|---|---|---|---|---|---|
| Int1 | Int2 | Occupancy | Int1 | Int2 | Occupancy | ||
| % | % | ||||||
| 1433 | Asn-323 | 3 | 0 | 100 | 3 | 0 | 100 |
| 1558/1575/1958 | Asn-355 | 3 | 0 | 100 | 33 | 14 | 58 |
| 2126 | Asn-391 | 12 | 0 | 100 | 48 | 2 | 96 |
| 1053 | Asn-483 | 6 | 0 | 100 | 3 | 0 | 100 |
| 1965 | Asn-729 | 0 | 0 | NDa | 93 | 23 | 75 |
a Not determined.
Deglycosylation of MPO and Functional Analysis
Considering the established glycosylation profile of MPO, the impact of the deglycosylation on the enzyme activity was further studied. Actually, the deglycosylation of MPO was followed by LC/ESI/MSMS (supplemental materials S7). According to the recovery of MPO sequences obtained in the enriched glycopeptide fraction, Asn355 and Asn391 were deglycosylated. Furthermore, the samples were also analyzed by LC/ESI/MS and the total ion chromatogram (TIC) extracted at the masses corresponding to the deglycosylated peptides. Obtained data suggest that only Asn355 was totally deglycosylated as the peak disappeared in the chromatogram corresponding to MPO treated by glycosidases (supplemental materials S8).
Concerning the functional analysis, the activity of MPO was measured in three different in vitro models. As shown in Fig. 4A, there was a significant decrease (37%) in chlorination activity of deglycosylated r-MPO (MPOdegly, 39 ± 3 units/mg, 63%) as compared with untreated r-MPO (63 ± 4 units/mg, 100%) and MPO treated in the absence of glycosidases (MPO37°, 55 ± 2 units/mg, 56%). Similarly, a comparable decrease was monitored in MPO-mediated LDL oxidation (72 ± 12% versus 100 ± 5%, p < 0.05; Fig. 4B). Finally, ROS production induced by MPO in endothelial cells was measured. A first experiment was carried out with an inactive form of recombinant MPO (Q257T) that showed a background level production similar to native r-MPO (supplemental materials S9). However deglycosylated r-MPO exhibited a 50% decrease in ROS generation as compared with untreated MPO (Eahy926, Fig. 4C).
FIGURE 4.

Measurement of MPO-mediated monochlorodimedone chlorination (A); measurement of MPO-mediated LDL oxidation by ELISA (B); and measurement of ROS generated in endothelial cells by MPO (C). MPO: untreated recombinant MPO; MPO37°: sample treated at 37 °C in the absence of glycosidases; MPOdegly: sample treated at 37 °C in the presence of glycosidases.
DISCUSSION
The carbohydrate moiety of glycoproteins plays important roles in biosynthesis (targeting and processing) and influences properties of the mature protein such as solubility, stability, immunogenicity, or molecular recognition (29–32). As far as MPO is concerned, the catalytic activity of the enzyme is located in the heme pocket deeply embedded in the inner protein core (33). So far, post-translational modifications (e.g. glycosylation) occurring at the protein surface were thought to be irrelevant for the enzymatic activity. However, there is nowadays a growing body of evidence that MPO might contribute to the inflammatory process by interacting with neutrophils and endothelial cells. Leukocyte MPO was shown to act as an autocrine stimulator of PMN, thereby promoting degranulation, integrin expression and NAPDH oxidase activation (15), and recombinant MPO was demonstrated to increase LDL oxidation at the surface of endothelial cells in the absence of any stimulating factor (16).
Comprehensive analysis and comparison of glycosylation of h-MPO and r-MPO revealed striking observations. N-Glycans are largely represented by high-mannose structures in h-MPO and minor biantennary complex glycans are largely sialylated. On the other hand, r-MPO is characterized by the abundance of 2-linked mannose and t-mannose also reflecting oligomannose and biantennary structures. These observations reflect some conservation of the N-glycans types in the recombinant form. However, the sialylation of r-MPO is markedly increased. There is a larger amount of complex structures in r-MPO that might be related to its processing. Indeed, h-MPO is rapidly synthesized in the early stages of the promyelocytic maturation and is stored in the primary granules of PMNs (3). Such a processing is not in favor of the synthesis of complex structures. In contrast, r-MPO is slowly processed in CHO cell lines in a mannose-supplemented medium that allows maturation of the protein through an efficient N-glycan synthesis and promotes synthesis of complex glycan structures (34).
The present study also demonstrated for the first time occupancy of all five Asn glycosylation sites (323, 355, 391, 483, and 729) on the heavy polypeptide of both MPO forms. The occupancy of four of them was confirmed by crystallographic analysis (323, 355, 391, and 483) (20) and biochemical studies (18, 20). Thus, in accordance with crystallographic data (2, 33), dimeric h-MPO has at least 3 glycosylation sites that are able to interact with receptors or other membrane proteins (Asn355, Asn391, and Asn729). Asn323 and Asn483 contribute to dimer stabilization due to their location at the interface (2, 20, 33). The present site-by-site N-glycosylation analysis demonstrates that most of the N-glycosylation sites are well conserved. Concerning the occupancy, in-gel experiments indicated partial glycosylation at Asn355, Asn391, and Asn729 in r-MPO, whereas h-MPO showed a total (100%) occupancy of these sites.
Recombinant monomeric MPO (N-terminal Ala49) carries part of the propeptide (10) that includes glycosylated Asn139 (10). This was confirmed by N-terminal sequencing and LC/ESI/MSMS showing the presence of Asn139 in the glycopeptide fraction.
Surprisingly, the enzyme activity was affected by N-glycosylation. MPO is characterized by rigid heme architecture, as the heme is covalently linked to Asp260, Glu408, and Met409 and the proximal histidine (His502) is hydrogen-bonded with an asparagine (Asn587). The roof of the distal heme pocket is formed by an arginine (Arg405) and the distal histidine (His261) that is close to calcium-binding Asp262. Additional Ca2+-binding residues are found in the sequence Thr334Ser335Phe336Val337Asp338 Ala339Ser340 that is connected to Asn355 by a α-helix (20, 33). Thus, residues in heme to protein linkages and Ca2+ binding are positioned both in the N- and C-terminal region of the polypeptide (light and heavy subunit in h-MPO). Because glycosylation affects the enzymatic activity, it is reasonable to assume that modification of the overall protein structure is also reflected by (minor) changes in the heme cavity architecture and/or the access channel. Indeed, despite the long incubation time that also affects MPO activity to some extent, deglycosylation caused a significant decrease in activity. Such an effect could have important consequences on the in vivo activity of MPO. For example, it is generally accepted that MPO is adsorbed on LDL and specifically oxidizes the apolipoprotein B100 (7, 35). This interaction might be modulated by glycosylation since deglycosylated MPO is less active in LDL oxidation than native peroxidase. In this model, decreased activity could be caused by alterations of heme or substrate channel architecture or by modification of the interaction with the substrate. In case of MPO-mediated ROS production in endothelial cells, deglycosylation clearly reduced the enzymatic activity of r-MPO because the heme lacking glycosylated variant could not mediate ROS production.
In summary, the macro- and micro-heterogeneity of N-glycans is well conserved in recombinant monomeric MPO as compared with the mature dimeric leukocyte protein (h-MPO). All predicted N-asparagine glycosylation sites are occupied and oligomannose structures are dominant among the N-glycans. Recombinant MPO carries more complex structures like bi- and triantennary N-glycans. Deglycosylation of especially Asn355 lowers enzyme activity, suggesting the importance of intact glycosylation for enzyme activity.
Supplementary Material
Acknowledgments
We thank Professor Rolando and Ing. Page from the Mass Spectrometry Centre of the Université des Sciences et des Technologies of Lille 1 (France) for the facilities. Thanks are also expressed to Yves Leroy for helpful assistance during the GC-MS experimentations and Alain Bosseloir from Zentech S.A. for providing h-MPO. We have also appreciated the support of Prof. Jean Ducobu from the Laboratory of Experimental Medicine, and Dr. Paul Delree from the Institute of Pathology and Genetics (IRSPG, Loverval, Belgium).
This work was supported by Grants from the Belgian Fund for Scientific Research (FRS-FNRS), number 2006/V 3/5/244-IB/BT-13924 and number 34553.08, and a grant from the Dept. of International Relationship (ULB).

The on-line version of this article (available at http://www.jbc.org) contains supplemental materials S1–S9.
- MPO
- myeloperoxidase
- MALDI
- matrix-assisted laser desorption
- ESI
- electrospray ionization
- MS
- mass spectrometry
- LDL
- low-density lipoprotein
- ROS
- reactive oxygen species
- Q/TOF
- quadrupole/time-of-flight.
REFERENCES
- 1.Klebanoff S. J. (2005) J. Leukoc. Biol. 77, 598–625 [DOI] [PubMed] [Google Scholar]
- 2.Zeng J., Fenna R. E. (1992) J. Mol. Biol. 226, 185–207 [DOI] [PubMed] [Google Scholar]
- 3.Bainton D. F., Ullyot J. L., Farquhar M. G. (1971) J. Exp. Med. 134, 907–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi D. K., Pennathur S., Perier C., Tieu K., Teismann P., Wu D. C., Jackson-Lewis V., Vila M., Vonsattel J. P., Heinecke J. W., Przedborski S. (2005) J. Neurosci. 25, 6594–6600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malle E., Waeg G., Schreiber R., Gröne E. F., Sattler W., Gröne H. J. (2000) Eur. J. Biochem. 267, 4495–4503 [DOI] [PubMed] [Google Scholar]
- 6.Maruyama Y., Lindholm B., Stenvinkel P. (2004) J. Nephrol. 17, S72–S76 [PubMed] [Google Scholar]
- 7.Moguilevsky N., Zouaoui Boudjeltia K., Babar S., Delrée P., Legssyer I., Carpentier Y., Vanhaeverbeek M., Ducobu J. (2004) Biochem. Biophys. Res. Commun. 323, 1223–1228 [DOI] [PubMed] [Google Scholar]
- 8.Van Antwerpen P., Zouaoui Boudjeltia K. Z., Babar S., Legssyer I., Moreau P., Moguilevsky N., Vanhaeverbeek M., Ducobu J., Nève J. (2005) Biochem. Biophys. Res. Commun. 337, 82–88 [DOI] [PubMed] [Google Scholar]
- 9.Zouaoui Boudjeltia K., Roumeguere T., Delree P., Moguilevsky N., Ducobu J., Vanhaeverbeek M., Wespes E. (2007) Eur. Urology 51, 262–269 [DOI] [PubMed] [Google Scholar]
- 10.Moguilevsky N., Garcia-Quintana L., Jacquet A., Tournay C., Fabry L., Piérard L., Bollen A. (1991) Eur. J. Biochem. 197, 605–614 [DOI] [PubMed] [Google Scholar]
- 11.Bakkenist A. R., Wever R., Vulsma T., Plat H., van Gelder B. F. (1978) Biochim. Biophys. Acta 524, 45–54 [DOI] [PubMed] [Google Scholar]
- 12.Furtmüller P. G., Jantschko W., Regelsberger G., Jakopitsch C., Moguilevsky N., Obinger C. (2001) FEBS Lett. 503, 147–150 [DOI] [PubMed] [Google Scholar]
- 13.Kooter I. M., Pierik A. J., Merkx M., Averill B. A., Moguilevsky N., Bollen A., Wever R. (1997) J. Am. Chem. Soc. 119, 11542–11543 [Google Scholar]
- 14.Kooter I. M., Moguilevsky N., Bollen A., Sijtsema N. M., Otto C., Wever R. (1997) J. Biol. Inorg. Chem. 2, 191–197 [Google Scholar]
- 15.Lau D., Mollnau H., Eiserich J. P., Freeman B. A., Daiber A., Gehling U. M., Brümmer J., Rudolph V., Münzel T., Heitzer T., Meinertz T., Baldus S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boudjeltia K. Z., Legssyer I., Van Antwerpen P., Kisoka R. L., Babar S., Moguilevsky N., Delree P., Ducobu J., Remacle C., Vanhaeverbeek M., Brohee D. (2006) Biochem. Cell Biol. 84, 805–812 [DOI] [PubMed] [Google Scholar]
- 17.Johnson K. R., Nauseef W. M., Care A., Wheelock M. J., Shane S., Hudson S., Koeffler H. P., Selsted M., Miller C., Rovera G. (1987) Nucleic Acids Res. 15, 2013–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansson M., Olsson I., Nauseef W. M. (2006) Arch. Biochem. Biophys. 445, 214–224 [DOI] [PubMed] [Google Scholar]
- 19.Blair-Johnson M., Fiedler T., Fenna R. (2001) Biochemistry 40, 13990–13997 [DOI] [PubMed] [Google Scholar]
- 20.Carpena X., Vidossich P., Schroettner K., Calisto B. M., Banerjee S., Stampler J., Soudi M., Furtmüller P. G., Rovira C., Fita I., Obinger C. (2009) J. Biol. Chem. 284, 25929–25937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hewson W. D., Hager L. P. (1979) J. Biol. Chem. 254, 3175–3181 [PubMed] [Google Scholar]
- 22.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 23.Morelle W., Michalski J. C. (2005) Curr. Pharm. Des. 11, 2615–2645 [DOI] [PubMed] [Google Scholar]
- 24.Ciucanu I., Kerek F. (1984) Carbohydr. Res. 131, 209–217 [Google Scholar]
- 25.Geyer R., Geyer H. (1994) Methods Enzymol. 230, 86–108 [DOI] [PubMed] [Google Scholar]
- 26.Peterson G. L. (1977) Anal. Biochem. 83, 346–356 [DOI] [PubMed] [Google Scholar]
- 27.Slomianny M. C., Dupont A., Bouanou F., Beseme O., Guihot A. L., Amouyel P., Michalski J. C., Pinet F. (2006) Proteomics 6, 2365–2375 [DOI] [PubMed] [Google Scholar]
- 28.Bragonzi A., Distefano G., Buckberry L. D., Acerbis G., Foglieni C., Lamotte D., Campi G., Marc A., Soria M. R., Jenkins N., Monaco L. (2000) Biochim. Biophys. Acta 1474, 273–282 [DOI] [PubMed] [Google Scholar]
- 29.Helenius A., Aebi M. (2001) Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 30.Helenius A., Aebi M. (2004) Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 31.Trombetta E. S. (2003) Glycobiology 13, 77R–91R [DOI] [PubMed] [Google Scholar]
- 32.Varki A. (1993) Glycobiology 3, 97–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiedler T. J., Davey C. A., Fenna R. E. (2000) J. Biol. Chem. 275, 11964–11971 [DOI] [PubMed] [Google Scholar]
- 34.Butler M. (2006) Cytotechnology 50, 57–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carr A. C., Myzak M. C., Stocker R., McCall M. R., Frei B. (2000) FEBS Lett. 487, 176–180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.