

Abstract
CD8+ T-cells can cause hepatocellular injury by two distinct mechanisms. In addition to their direct cytotoxic effect, there is also collateral liver injury, which occurs when cells are killed in an antigen independent manner. While immune effector cytokines Interferon-gamma (IFNγ) and Tumor Necrosis Factor-alpha (TNFα) have both been implicated in various forms of hepatitis, their respective roles in direct and/or collateral liver damage remains unclear. In order to investigate these elements of liver injury, we have developed a new experimental model of CD8+ T-cell mediated hepatitis based on an Adeno-Associated Virus-based gene therapy vector. This vector is used to deliver antigen to hepatocytes, and CD8+ T-cells specific for the vector-encoded transgene are adoptively transferred to produce liver immunopathology. In this experimental model, CD8+ T-cell IFNγ acts on Kupffer cells, inducing TNFα secretion and liver injury. Both IFNγ and TNFα are important in this injury process, but TNFα acts as an autocrine amplifier of Kupffer cell function, rather than as a direct effector of hepatocellular damage. Conclusion: TNFα indirectly promotes liver damage, and is not a direct hepatotoxic agent. IFNγ also indirectly contributes to liver injury via Kupffer cell activation while, in parallel, directly promoting hepatitis via induction of hepatocyte MHC class I. In principle, it may be possible to ameliorate this immunopathologic indirect mechanism by developing therapies that target Kupffer cells, without impairing CD8+ T-cell mediated antiviral immunity. This would have great therapeutic potential in chronic viral hepatitis.
Keywords: Kupffer, macrophage, hepatitis, immunopathology, interferon-gamma
Ongoing infection of the liver often results in significant alteration in liver architecture and impaired liver function. Because liver infections such as Hepatitis B and C virus are non-cytolytic, the damage to the liver is mediated primarily by the immune response. Immune mediated liver injury occurs via two distinct mechanisms: direct antigen-specific Cytotoxic T Lymphocyte (CTL) mediated apoptosis and an indirect, or collateral damage, mechanism. Collateral damage accompanies T-cell activation but does not depend on direct recognition of the target cell by the CTL (1, 2).
Liver injury depends on CD8+ T-cells, as was documented in a study of Hepatitis B Virus infected chimpanzees, where CD8+ T-cell depletion, but not CD4+ T-cell depletion, delayed virus clearance and liver injury (3). While Hepatitis B infections place antigen in the hepatocytes, making direct and collateral mechanisms difficult to dissect, hepatitis can also be induced by extra-hepatic infection. In both humans and mice, influenza infection is accompanied by low-level liver injury, marked by the elevation of serum aminotransaminases (4). Because influenza does not replicate in the liver, this phenomenon reveals a strictly collateral mechanism of liver injury. Inflammatory foci were dependent on the presence of Kupffer cells and correlated with the frequency of virus specific CD8+ T-cells (4). These data suggest a link between activated antigen specific CD8+ T-cells and Kupffer cells in driving collateral liver injury.
CD8+ T-cell and Kupffer cell derived cytokines, Interferon-gamma (IFNγ) and Tumor Necrosis Factor-alpha (TNFα) respectively, are required to induce liver pathology in a number of T-cell driven liver injury models (2, 5–8). The requirement for these cytokines is independent of whether the antigen is expressed by bone marrow-derived cells or the liver parenchyma. While it is clear that IFNγ, TNFα and Kupffer cells are associated with liver pathology, their relative contributions to direct versus collateral injury are unclear.
To investigate how CD8+ T-cell recognition of antigen in the liver generates cytokine dependent liver injury, we developed an experimental model using a replication-defective recombinant Adeno-Associated Virus serotype 2 (rAAV2) vector. The rAAV2 vector encodes the green fluorescent protein (GFP) linked to a target antigen, the SIINFEKL peptide derived from ovalbumin, and is injected directly into the liver followed by adoptive transfer of antigen specific CD8+ T-cells from the OT-1 transgenic mouse. By using a replication deficient vector we are able to specifically target hepatocytes (9). Using mice deficient in the IFNγ Receptor (IFNγR) or the TNFα receptor-1 (TNFR1), the present study investigates the roles of IFNγ and TNFα in promoting liver injury. Our findings suggest that while IFNγ promotes injury via both direct and indirect mechanisms, TNFα only contributes to liver injury indirectly by acting as an amplifier of Kupffer cell activation, rather than by a direct cytotoxic effect on hepatocytes.
Materials and Methods
Mice
C57BL/6, B6.SJL-Ptprca Pepcb/BoyJ (CD45.1 transgenic), B6.129-Tnfrsf1atm1Mak/J (TNFR1 deficient), and B6.129S7-Ifngr1tm1Agt/J (IFNγR deficient) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). T-cell receptor transgenic OT-1 mice, specific for the SIINFEKL peptide derived from ovalbumin, were maintained in-house. All mice were sex and age matched and on a C57BL/6 background. All experiments were approved by the Institutional Animal Care and Use Committee.
AAV Vectors
Serotype 2 AAV vectors containing eGFP or an eGFP-SIINFEKL fusion sequence under the control of an EF1α promoter were obtained from the Columbus Children’s Research Institute, Viral Vector Core Facility (Columbus, OH). Briefly, plasmids encoding the EF1α promoter, together with eGFP or an eGFP-SIINFEKL fusion, the rep and cap genes from AAV, and neo/tk selection elements were transfected into HeLa-derived producer cells, which were subsequently infected with wild-type adenovirus type 5. Recombinant AAV2 (rAAV2) was purified from the lysed cells, and purified using heparin chromatography as described previously (10). Purified rAAV2 tested negative for adenovirus (≤1 Ad 5 PFU / 109 rAAV2 DNAse-resistant particles).
Intra-hepatic injection
Mice at 8 – 9 weeks of age were anesthetized with Avertin, and the right lobe of the liver was exposed via a 2-cm ventral midline incision. Using a 29-gauge insulin syringe, 60µl of vector suspension (5.88 × 1010 DNAse-resistant particles) was injected into the liver. The peritoneal cavity was sutured with Vicryl (Ethicon) and the skin closed with wound clips.
Bone Marrow Chimeras
Mice at 8 – 9 weeks of age were irradiated with 10.0 Gy using an RS2000 x-ray irradiator (Rad Source Technologies, Coral Springs, FL). T-cells were depleted from femoral and tibial bone marrow cells of donor mice using anti-CD4 (RL172.4) and anti-CD8 (3.155) antibodies (Abs), and guinea pig complement (Gibco-BRL/Invitrogen). Irradiated mice were injected with 9×106 T-depleted bone marrow cells via the tail vein. Mice received 0.16 mg/ml sulfamethoxazole and 0.32 mg/ml trimethoprim in their drinking water for 4 weeks, then were given 1mg of clodronate liposomes (Encapsula Nano Sciences, Nashville, TN) i.v. to remove radioresistant, as well as radiosensitive macrophages in the liver as previously described (11). Blood analysis showed 85 – 95% lymphocyte and ≥99% macrophages reconstitution by donor bone marrow.
OT-1 purification and adoptive transfer
Purified CD8+ T-cells were obtained from spleen and lymph node cells by depletion of B-cells, CD4+ T-cells, NK cells, and dendritic cells with an Ab mixture (212.A1; anti-MHC class II, 2.4-G2; anti-FcR, GK1.5; anti-CD4, and HB191; anti-NK1.1). Cells coated with primary Abs were removed with magnetic beads coated with the secondary Abs. Naïve B-cells were removed using beads coated with anti-IgM (Qiagen). Mice received 5×106 OT-1 cells (of >85% purity) via the tail vein.
Real-time PCR
RNA was extracted from liver tissue using TRIzol (Invitrogen), according to manufacturer’s instructions. Total RNA was converted to cDNA and used for real-time quantitative RT-PCR. TNFα, IFNγ, Caspase-12, CHOP, TRAIL, GAPDH, and H2-Kb assays were obtained from Applied Biosystems. The resulting relative expression values obtained by qRT-PCR were treated as non-parametric and analyzed using the Mann-Whitney test.
To determine the abundance of OT-1 T-cells in tissue, DNA was liberated from liver tissue using proteinase K (Qiagen), and treated with RNAse before qPCR analysis. Real-time PCR reactions were run using a MyiQ system (BioRad). Each reaction used sense and anti-sense primers for the genomic β-actin sequence (5’ CCCATCTACGAGGGCTATGC; 5’ CTCTCAGCTGTGGTGGTGAA), and genomic primers for the OT-1 transgene (5’ GATTCTCAGTCCAACAGTTTGATGA; 5’ CTGTTCATAATTGGCCCGA). Each reaction used probe sequences and cycle parameters, as previously described (12). Data were analyzed using BioRad MyiQ 2.0 software.
Measurement of serum aminotransaminases
Blood was collected via cardiac puncture. The samples coagulated at room temperature for 2–3 hours, and were then centrifuged to collect serum. Serum ALT was measured by the Strong Health Clinical Laboratories, Rochester, NY. The resulting data was analyzed for significance using the student’s t-test.
Histology
Formalin-fixed paraffin-embedded liver sections were sectioned (5µm) and stained with H&E. Sections were subjected to heat-induced epitope retrieval using Dako TRS for 4 minutes at 123 °C at 18 psi. Primary Abs were polyclonal rabbit anti-CD3 (1:200, Dako A0452), rat anti-F4/80 (1:400, eBioscience E016242, clone BM8), monoclonal rabbit anti-active caspase 3 (1:250, Cell Signaling 9664L). Secondary Abs included biotinylated donkey anti-rat (Jackson) at 1:200 dilution to detect F4/80, biotinylated goat anti-rabbit (Vector) at 1:200 dilution to detect the anti-active caspase 3, followed by ABC-Elite (Vector). To detect the anti-CD3, an anti-rabbit polymer (Dako, Envision Plus) was used. To detect GFP, antigen retrieval with 5 minutes of protease K digestion was followed by 1:1000 dilution of Abcam polyclonal rabbit Ab (AB6556-25). DAB+ (Dako) was used as the chromogenic substrate in all of the immunohistochemical assays. Hepatitis was evaluated by an experienced liver pathologist, who scored “blind” the H&E-stained sections using a 4-point semi-quantitative scale (0–3+) based on the prevalence and size of the inflammatory foci.
Liver lymphocyte isolation
Intrahepatic lymphocytes were isolated using a variant of a previously described method (9). Livers were perfused with 5 ml of PBS via the portal vein, mechanically homogenized, then digested in serum free RPMI 1640 containing 0.05% collagenase IV (Sigma) and 0.002% DNAse I (Sigma) at 37°C for 40 minutes with gentle agitation. The cells were washed and resuspended in 2.75 ml of 40% OptiPrep (Accurate Chemical) and 2.25 ml of serum free RPMI. The OptiPrep suspension was then layered under 2.0 ml of serum free RPMI 1640 and centrifuged at 1100 × g for 25 minutes at 4°C. The cells at the interface were collected for further analysis.
In vitro restimulation and intracellular cytokine staining
Liver leukocytes were resuspended in RPMI 1640 containing 10% FBS, 1µl/ml GolgiPlug (BD), 70 U/ml IL-2 (Endogen), and Penicillin-Streptomycin solution (Gibco/Invitrogen) with or without 10µg/ml SIINFEKL peptide. After restimulation for 4–5 hours at 37°C, 5% CO2, the cells were stained with LIVE/DEAD® stain (Invitrogen) according to manufacturer’s instructions plus 1µl/ml GolgiPlug. The cells were then stained for surface markers in HBSS containing 5% FBS plus 1µl/ml GolgiPlug. Cells were fixed in 2% formalin, permeabilized in PBS plus 1% saponin and 1% FBS, blocked with isotype Ab, and stained for intracellular IFNγ and TNFα for 30 minutes at 4°C. Liver leukocytes were washed twice with HBSS containing 5% FBS and analyzed by flow cytometry.
Flow cytometry
For surface staining, cells were stained in a HBSS solution containing 5% FBS. Intracellular staining was done in PBS with 1% saponin and 1% FBS. The following Abs were used: TCRβ (H57-597), NK1.1 (PK136), CD8 (53-6.7), IFNγ (XMG1.2), TNFα (MP6-XT22), and F4/80 (BM8). Abs were obtained from BD PharMingen or eBiosciences. Streptavidin-PE Texas Red (Caltag) and Aqua LIVE/DEAD stain (Invitrogen) were also used. Flow cytometric analysis was performed on a LSR II (BD Biosciences). Data were analyzed using FlowJo software (Treestar).
Results
Induction of liver immunopathology in AAV-transduced mice
To study the effects of CD8+ T-cell activation in the liver, we exposed the livers of C57BL/6 mice via laparotomy, and injected rAAV2 vectors expressing a GFP-SIINFEKL fusion protein, or GFP alone, under the control of the EF1α promoter. To document the extent of hepatocyte transduction, such livers were sectioned and subjected to immunohistochemical staining for GFP. The vector had dispersed throughout the hepatic parenchyma, transducing a subset of hepatocytes in multiple lobes, not only in the lobe injected. Representative data are shown in on-line supplementary Figure S1. After 3 weeks, 5 × 106 OT-1 CD8+ T-cells were given i.v. Histological evaluation of hematoxylin and eosin (H&E) stained liver sections revealed that in the presence of GFP-SIINFEKL, the introduction of OT-1 T-cells resulted in an active hepatitis, consisting of numerous inflammatory foci (Fig 1A). Liver H&E stained sections collected at 24-hour intervals post OT-1 transfer were also coded, scored “blind” to determine the severity of hepatitis, then decoded and plotted (Fig 1B). Control mice that received the rAAV2 GFP vector showed normal histology, or minimal hepatitis after OT-1 transfer. However, mice that received the rAAV2 GFP-SIINFEKL vector and OT-1 T-cells showed moderate hepatitis, maximal on day 3. These hepatitic foci consisted mostly of a mononuclear cell infiltrate, predominantly composed of CD3+ T-cells (Fig 2A) and F4/80+ Kupffer cells (Fig 2B). Immunohistochemical detection of cleaved/activated caspase 3 demonstrated numerous apoptotic cells within the inflammatory foci (Fig 2C). Although the apoptotic cells may have been of several types, it is clear that hepatocytes represented a significant proportion of the dying cells, based both on the large size of the active caspase 3-stained cells and on the presence of morphologically distinct Councilman bodies.
Figure 1. Histologic assessment of AAV-transduced livers.
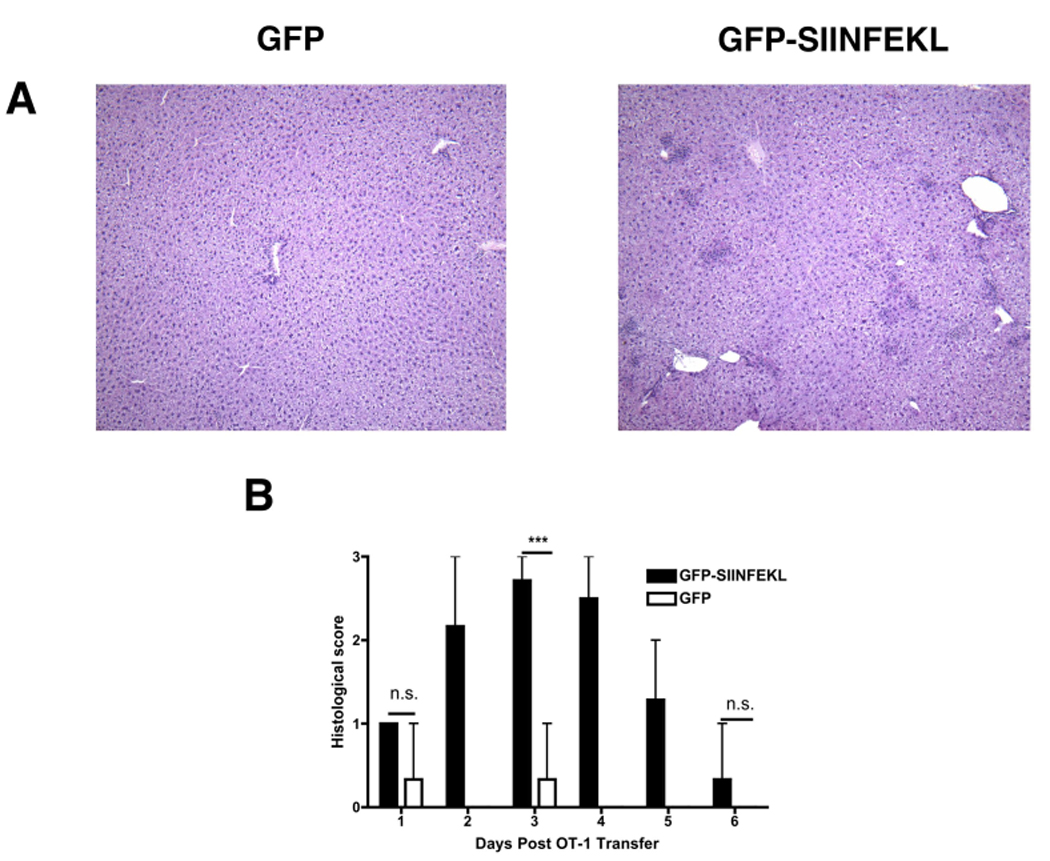
C57BL6 mice were transduced with either rAAV2 GFP or rAAV2 GFP-SIINFEKL vectors by direct intrahepatic injection. All mice received 5×106 OT-1 cells i.v. three weeks later. H&E-staining of the livers demonstrated the presence of numerous, discrete inflammatory foci in mice expressing the target antigen (A). The severity of liver injury was scored and plotted for 24-hr intervals post OT-1 transfer (B). Data points represent means ± range for n ≥ 3 animals per time point. ***, P < 0.0001; N.S, not significant. Data are the sum or representative of two independent experiments.
Figure 2. Histological staining for CD3, F4/80 and Caspase 3.
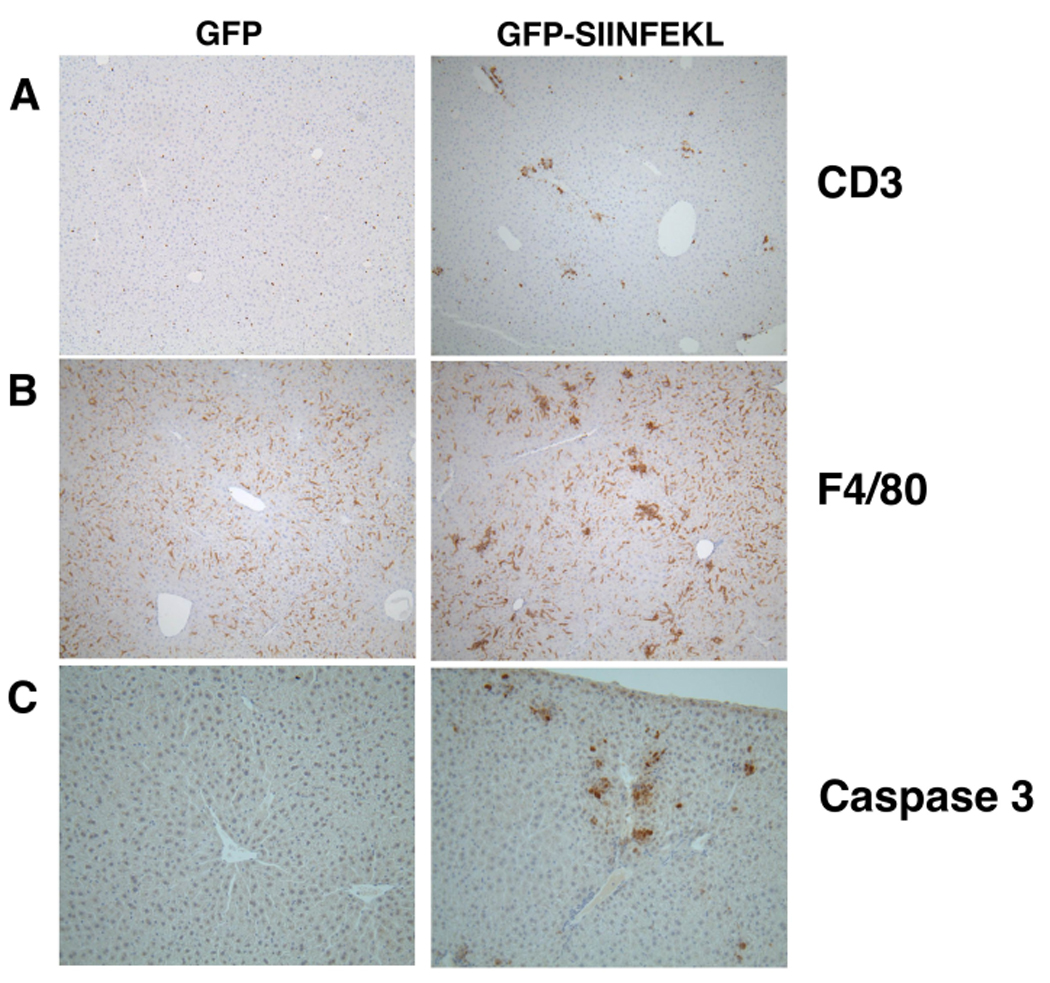
Liver sections from WT mice that had received rAAV2 GFP-SIINFEKL, or rAAV2 GFP, and OT-1 cells were stained for CD3 (A), F4/80 (B), or activated/cleaved Caspase 3 (C).
In parallel, we tested the serum levels of alanine aminotransaminase (ALT) at 24-hr intervals post OT-1 cell transfer. The serum ALT levels were greatest at 72 hours after OT-1 transfer (Fig 3A) and then resolved, returning almost to background levels by day 6. To quantify OT-1 T-cells accumulation in the liver, we performed real-time quantitative PCR on liver DNA, using primer / probe combinations specific for the transgenic OT-1 T-Cell Receptor-beta (TCRβ) chain (Fig 3B). The hepatitis severity scores shown in Figure 1B and the pattern of expansion and contraction of OT-1 cells correlated well with the serum ALT levels. To investigate the presence of IFNγ and TNFα in our system, we analyzed mRNA from liver homogenates each day after OT-1 transfer using quantitative real-time RT-PCR. We found that both IFNγ and TNFα transcripts were up-regulated during the first 48–72 hours, followed by a drop in expression (Fig 3C, D). These results are consistent with the involvement of IFNγ and TNFα in promoting hepatitis during the CD8+ T-cells response to AAV-encoded antigen.
Figure 3. Hepatitis in AAV-transduced Livers.
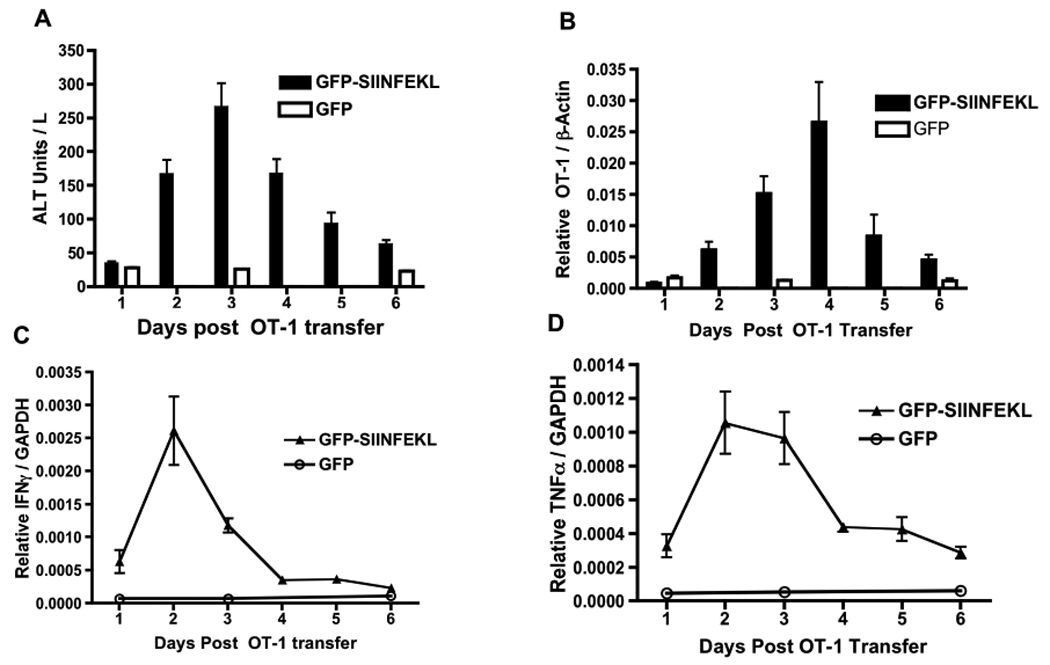
C57BL6 mice were injected with either rAAV2 GFP or rAAV2 GFP-SIINFEKL vectors followed by OT-1 T-cells as described in Figure 1. Serum was harvested and analyzed for ALT (A) at 24-hour intervals post OT-1 transfer. Genomic DNA from the liver was isolated and the OT-1 TCRβ chain measured by qPCR. (B). Liver mRNA for IFNγ (C) or TNFα (D) was measured by qRT-PCR. Data points represent means ± SEM for n ≥ 3 animals per time point. Data are the sum or representative of two independent experiments.
Significance of IFNγ and TNFα
To test the significance of IFNγ and TNFα during T cell-driven liver immunopathology, we injected the rAAV2 vectors into mice deficient in either the IFNγR or the TNFR1, and compared them to wild-type (WT) controls. Because the peaks for serum ALT and TNFα expression were all on day 3, we used this time-point to analyze the difference between WT and deficient mouse strains. In these experiments, the injection of OT-1 T-cells into rAAV2 GFP-SIINFEKL vector transduced IFNγR-deficient (Fig 4A) or TNFR1-deficient mice (Fig 4B) resulted in less liver injury as compared to WT controls. We next measured the expression of TNFα mRNA in both IFNγR and TNFR1 deficient animals, using GAPDH as an internal standard. OT-1 T-cell transfer into WT antigen-expressing mice resulted in a 20-fold increase in TNFα message. When we analyzed IFNγR and TNFR1 deficient mice given the rAAV2 GFP-SIINFEKL vector plus OT-1 T-cells, there was reduced upregulation of TNFα expression in both gene-targeted strains compared to WT controls (Fig 4). These data suggest that IFNγ and TNFα signaling are both required for optimal liver injury and TNFα expression.
Figure 4. Significance of IFNγR and TNFR1 in TNFα expression and liver injury.
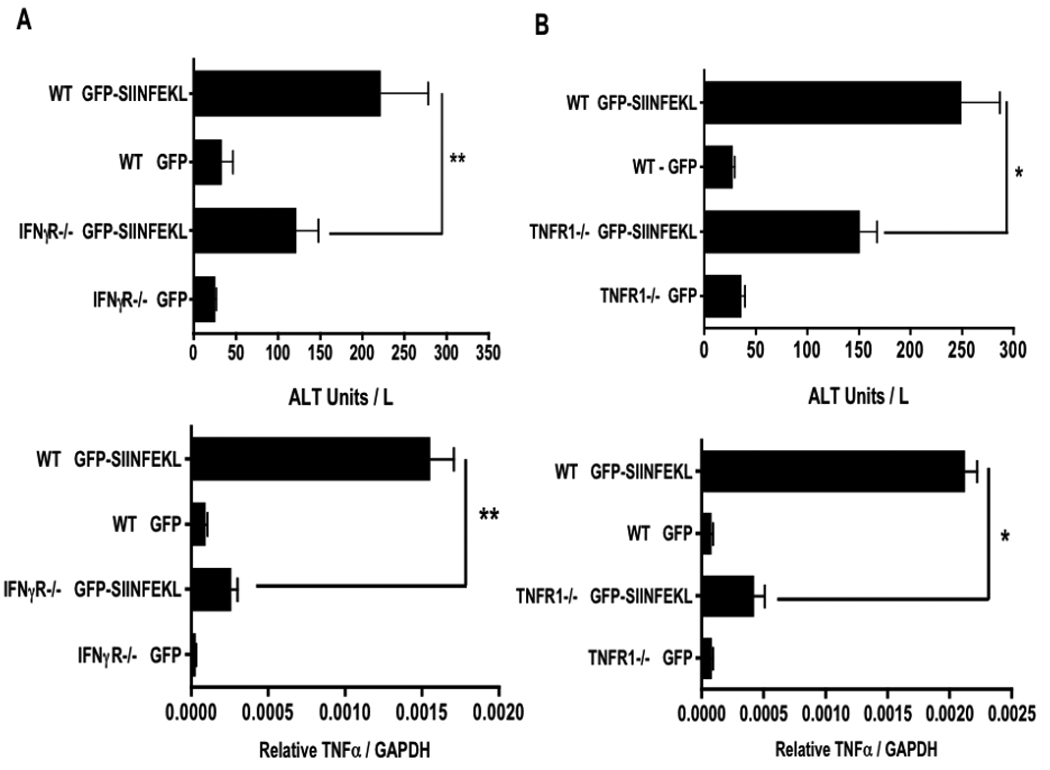
Genetically deficient and control mice were treated with rAAV2 GFP or GFP-SIINFEKL vectors and given OT-1 cells as described in Figure 1. Serum ALT and liver TNFα message expression were measured on day 3 post OT-1 transfer in WT versus IFNγR −/− mice, (A) and WT versus TNFR1 −/− mice (B). Data points represent means ± SEM for n ≥ 2 in rAAV2 GFP controls and n ≥ 3 in rAAV2 GFP-SIINFEKL groups. *, P < 0.05; **, P < 0.01. n ≥ 3 Data are the sum or representative of two independent experiments.
OT-1 T-cells and liver macrophages produce IFNγ and TNFα after restimulation
To formulate an accurate model of how IFNγ and TNFα contribute to liver injury, it was important to establish which cell types may be producing these cytokines. Based on other T-cell dependent models of hepatitis, it is possible that T-cells, NK cells, NKT cells and/or Kupffer cells could contribute to the local concentration of these molecules (5, 7, 13–15). To evaluate the cellular sources of the cytokines, we performed in vitro restimulation in the presence of Brefeldin A, IL-2 and the SIINFEKL peptide. Isolated liver leukocytes from rAAV2 vector transduced WT mice 3 days post OT-1 transfer, were restimulated for 4–5 hours at 37°C and then stained for cell surface identification markers and for the presence of intracellular IFNγ and TNFα. When gating on live liver lymphocytes, CD8+ cells were the primary population showing positive IFNγ staining (Fig 5B). This staining was significantly reduced when the SIINFEKL peptide was absent from the restimulation culture. Gating on NK1.1+ TCRβ− cells or NK1.1+ TCRβ+ cells showed less than 2% of the populations expressing either TNFα or IFNγ (Fig 5C, D). Analysis of TNFα production showed that the majority of cytokine positive cells were positive for F4/80 (Fig 5A). These data therefore support a model of injury that is dependent on IFNγ production by activated CD8+ T-cells and on TNFα production by activated Kupffer cells.
Figure 5. Cytoplasmic staining for IFNγ and TNFα.
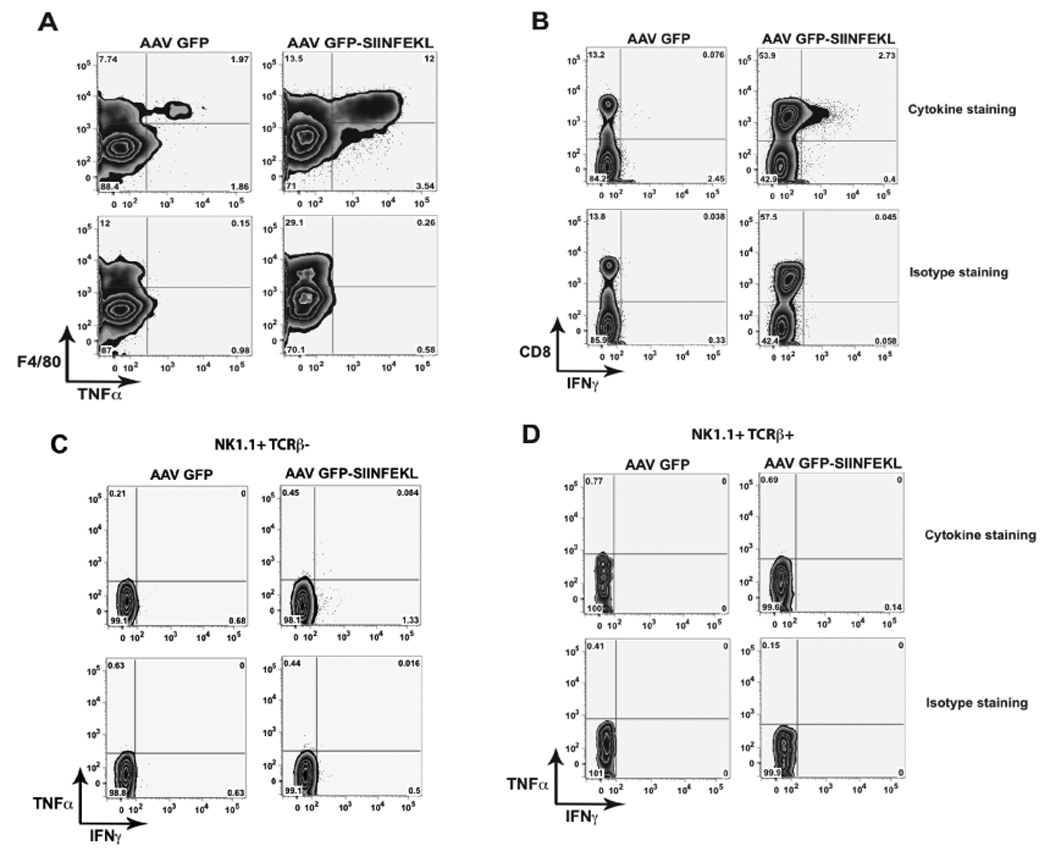
Liver lymphocytes isolated on day 3 from mice treated with rAAV2 vectors and OT-1 T-cells as described in Figure 1, were restimulated in media with 70 U/ml IL-2, 10 µg/ml SIINFEKL peptide, and 1 µl/ml GolgiPlug-BD for 4–5 hours. Lymphocytes were stained for cell identification markers and TNFα (A) and IFNγ (B). Cytokine expression in NK1.1+ TCRβ− (C) or NK1.1+ TCRβ+ cells (D). Data are representative of 3 separate experiments.
Cellular targets of IFNγ signaling
IFNγ contributes to liver injury in various experimental systems (2, 5, 8, 16). However, it is not clear whether its contribution is mediated by effects on the parenchyma, the liver leukocytes, or both. To test this we generated bone marrow chimeras where the bone marrow-derived cells were replaced with IFNγR deficient bone marrow. To ensure complete replacement of Kupffer cells, radiation chimeras were treated with clodronate-loaded liposomes 4 weeks after bone marrow transfer. Subsequent, secondary repopulation of the liver resulted in an animal with >99% of Kupffer cells derived from donor bone marrow as previously described (11). Day 3 analysis of such chimeric mice given the rAAV2 GFP-SIINFEKL vector, followed by OT-1 T-cells, showed significant reduction in serum ALT in mice that received IFNγR deficient bone marrow, compared to control mice that received WT bone marrow (Fig 6A). Quantitation of liver homogenate mRNA also showed a 10-fold reduction of TNFα gene expression in IFNγR−/−→WT mice. These data support the hypothesis that CD8+ T-cell IFNγ drives Kupffer cell TNFα production, and that this signaling event contributes to liver injury.
Figure 6. Effect of IFNγ on bone marrow-derived cells.
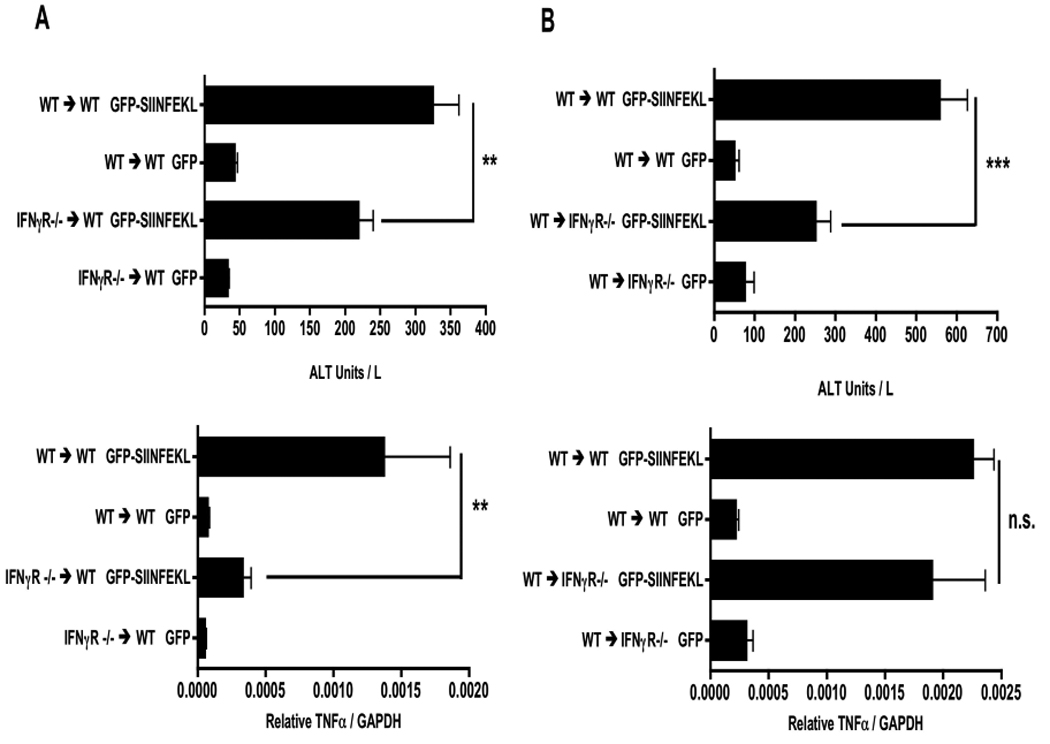
IFNγR−/− chimeric mice and their WT controls were treated with rAAV2 vectors as described in Figure 1. Day 3 post OT-1 transfer, serum and liver mRNA were harvested and measured for ALT and TNFα expression in IFNγR −/− → WT chimera (A) and WT → IFNγR −/− chimera (B) experiments. Data points represent means ± SEM for n ≥ 5 in each group. **, P < 0.01; ***, P < 0.001; N.S, not significant. Data are the sum of two independent experiments.
In the reverse chimera experiment, in which WT bone marrow was transferred into IFNγR deficient mice, we also observed reduced liver injury (Fig 6B). However, unlike what was seen in Figure 6A, IFNγR deficiency in solid tissues had no effect on the level of TNFα message expression. Together these data demonstrate the presence of an indirect mechanism of injury that depends on Kupffer cell activation by IFNγ as well as a direct injury pathway that is promoted by IFNγ signaling on the liver parenchyma.
It has been previously suggested that IFNγ can be directly cytotoxic to the liver by inducing endoplasmic reticulum (ER) stress mediated apoptosis (17). Caspase-12 and the CCAAT/enhancer-binding protein homologous protein (CHOP) have both been associated with ER stress induced apoptosis in hepatocytes (18, 19). Thus, to test whether IFNγ was inducing ER stress within the liver, we measured Caspase-12 and CHOP mRNA from liver homogenates. Caspase-12 transcript was induced in control WT→WT chimeras, consistent with a direct effect of IFNγ on the hepatocytes (Figure 7A). The absence of the IFNγR from bone marrow-derived cells in IFNγR−/−→WT chimeras did not compromise caspase-12 induction, compared to the effect in WT→WT controls (Fig 7A). This also was consistent with a direct action of IFNγ on hepatocytes. In the WT→IFNγR−/− chimeric mice we observed a modest (around 20%), yet statistically significant reduction in caspase-12 mRNA (Fig 7B). Therefore, although liver injury was causing the expression of caspase-12 and an element of this was dependent on the IFNγR on solid tissues, the majority of caspase-12 induction could not be attributed to the direct action of IFNγ on hepatocytes.
Figure 7. IFNγ mediated effects on MHC class I H2-Kb induction and ER-stress.
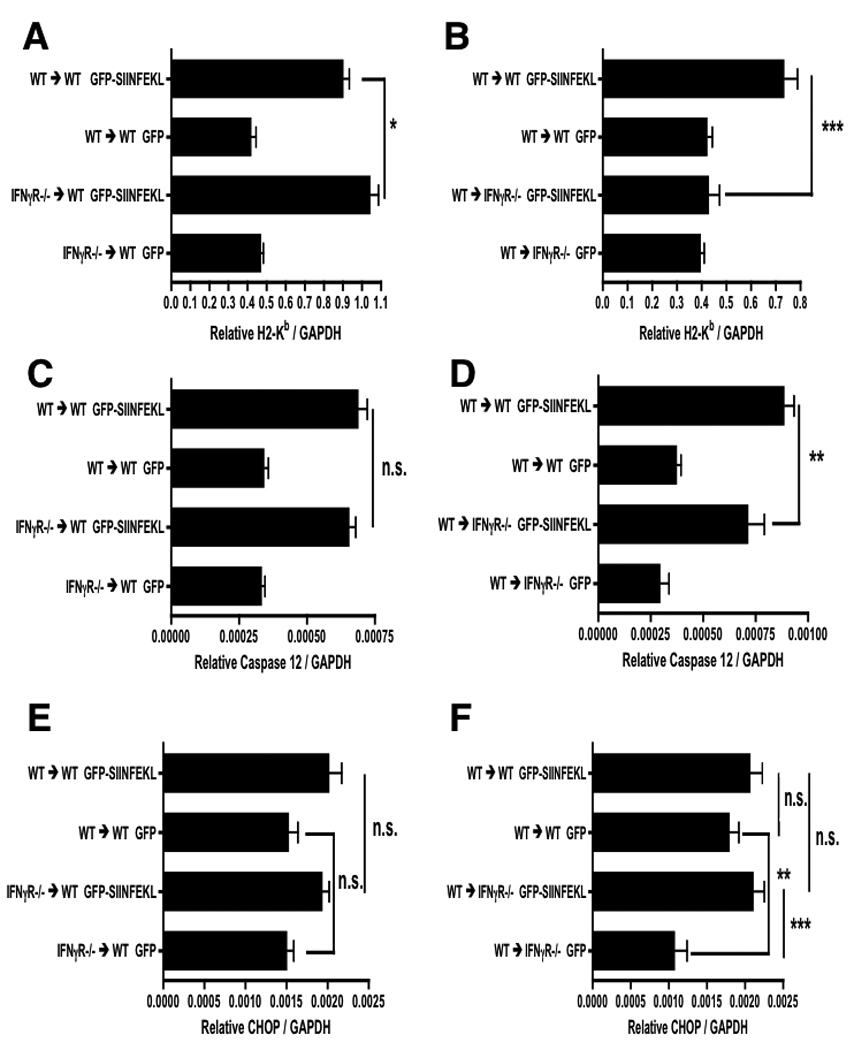
IFNγR−/− chimeric mice and their WT controls were treated with rAAV2 vectors as described in Figure 1. Day 3 post OT-1 transfer liver mRNA was harvested and measured for H2-Kb expression in WT → IFNγR −/− chimeras (A) and IFNγR −/− → WT chimeras (B). Markers of ER-stress, caspase-12 (C, D) and CHOP (E, F), were also measured in each chimera. Data points represent means ± SEM for n ≥ 2 in rAAV2 GFP controls and n ≥ 3 in rAAV2 GFP-SIINFEKL groups. *, P < 0.05; **, P < 0.01; ***, P < 0.001; N.S. not significant. Data are representative of two independent experiments.
Expression of CHOP was only weakly induced in this system, with an increase to approximately 20% above background, and the lack of IFNγR on either bone marrow derived cells (Fig 7C), or the liver parenchyma (Fig 7D), did not significantly change the level of CHOP transcript in antigen expressing mice. Taken together, the data suggest that if there is a direct effect of IFNγ in promoting ER stress in hepatocytes, this is a very small effect. Most of the induction of both caspase-12 and CHOP message is clearly independent of an action of IFNγ on hepatocytes.
One common effect of IFNγ is the up-regulation of MHC molecules. We therefore considered the hypothesis that the decreased liver injury seen in IFNγR deficient mice resulted from reduced antigen availability, due to the loss of IFNγ mediated induction of the MHC class I antigen presentation pathway. To test this hypothesis, we measured MHC class I H2-Kb transcript levels in the livers of mice from the previous IFNγR chimeras. As shown in Figure 7F, antigen expressing WT→IFNγR−/− mice did not induce H2-Kb. Conversely, in IFNγR−/−→WT mice the induction of H2-Kb was preserved (Fig 7E). These data suggest that H2-Kb induction is driven by IFNγ, acting via IFNγR expressed on the liver parenchyma.
How does TNFα contribute to liver injury?
The potential role of TNFα in liver injury is complex to dissect. The cytotoxic and anti-apoptotic functions of TNFα have been studied in various contexts (7, 20, 21). Data from experiments with TNFR1 deficient mice in Figure 4 showed that TNFα signaling was required for optimal TNFα expression and liver injury. Several reports show that autocrine TNFα signaling is required for complete IFNγ-dependent macrophage activation (22, 23). Therefore, TNFα signaling may be needed for Kupffer cell activation within the liver, and this activation could then contribute to liver injury. Conversely, TNFα signaling on the liver parenchyma might promote liver injury as demonstrated by experiments that couple TNFα with a transcription inhibitor (6, 24).
To test whether TNFα causes direct cytolytic damage to the liver parenchyma, we generated bone marrow chimeras using TNFR1 deficient mice as hosts, and gave them WT bone marrow followed by clodronate liposomes, rAAV2 vectors, and OT-1 T-cells as described for the experiments shown in Figure 6. We found no significant difference in either serum ALT or TNFα message expression between WT→TNFR1 deficient mice and the WT→WT control chimeras (Fig 8). These data suggest that TNFα does not directly act on hepatocytes, or on other radioresistant cells, to promote liver injury. While TRAIL expressing Kupffer cells have been shown to be cytotoxic (25) and TRAIL has been implicated in viral hepatitis (26), the expression of TRAIL was not altered by the lack of TNFR1 in either experiment (S.Fig 2), making it an unlikely agent of liver injury.
Figure 8. Does TNFα cause direct liver parenchyma cytotoxicity?
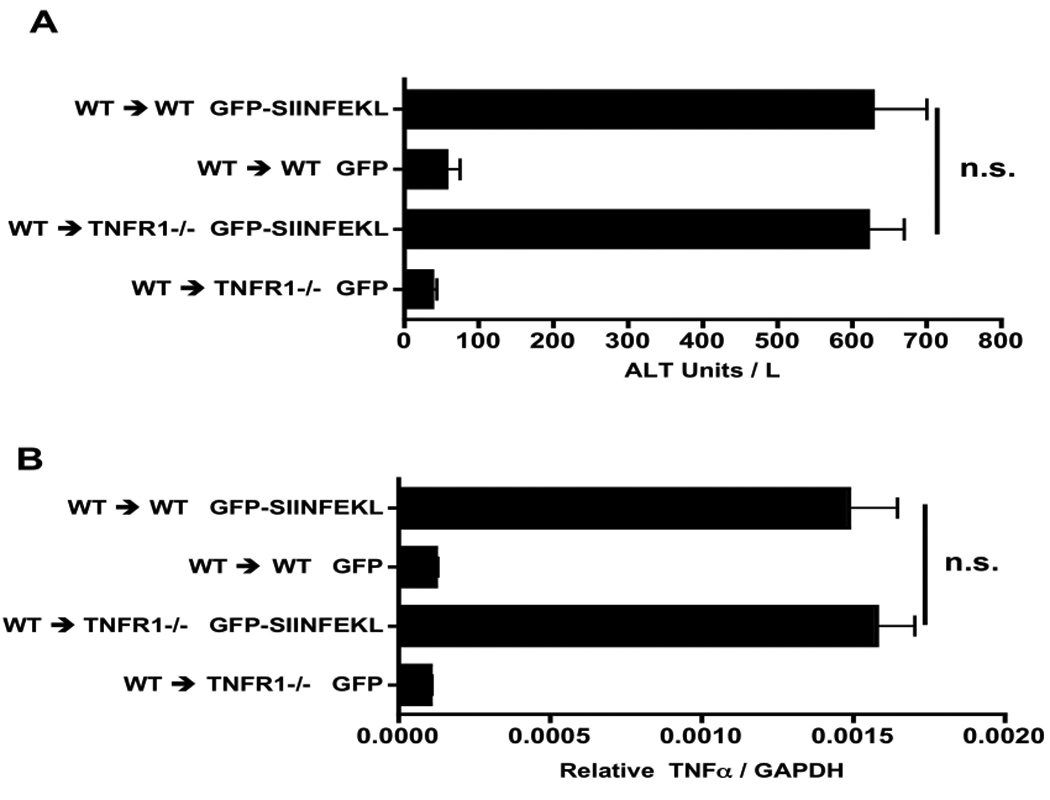
WT → TNFR1 −/− chimeras and their WT controls were treated as described in Figure 1. Day 3 post OT-1 cell transfer, ALT (A) and TNFα expression (B) were measured. Data points represent means ± SEM for n ≥ 5 per group. N.S, not significant. Data are the sum of two independent experiments.
Discussion
To investigate the roles of IFNγ and TNFα in the context of CD8+ T cell-dependent liver injury, we used hepatic injections of an antigen encoding rAAV2 vector which transduces hepatocytes (9). Experiments comparing IFNγR or TNFR1 deficient mice with WT confirmed the importance of these cytokines as suggested by other reports (7, 8, 16). Intracellular cytokine staining of liver leukocytes showed that the F4/80+ population was the primary TNFα producer, which is consistent with reports of Kupffer cell TNFα expression in other liver injury models (7, 13). Evaluation of intracellular IFNγ staining showed that OT-1 cells were the major IFNγ source while less than 2% of the other populations stained positive for IFNγ.
The reduction in serum ALT in radiation bone marrow chimeras, in which the IFNγR was absent from either hematopoietic tissues or parenchyma, revealed IFNγ signaling to promote both direct and indirect forms of liver injury in parallel. With respect to direct IFNγ signaling on hepatocytes, we evaluated the possibility that IFNγ had a direct cytotoxic effect on the hepatocytes by promoting ER stress mediated cell death (17). However, the data showed minimal induction of CHOP and caspase-12 mRNA during liver injury. More significantly, the presence of the IFNγR on bone marrow cells was irrelevant, and its expression on the liver parenchyma did not strongly impact the expression of either CHOP or caspase-12. Overall, these data would suggest that a direct action of IFNγ in inducing hepatocyte ER stress is, at most, a small contributor to liver injury.
Alternatively, IFNγ signaling on hepatocytes may promote apoptosis by increasing hepatocyte susceptibility to CTL mediated death mechanisms (5, 8, 27, 28). While the investigation of Fas or perforin-mediated injury is beyond the scope of this study, up-regulation of liver H2-Kb expression was dependent on IFNγR expression on the radio-resistant host cells, and not on bone marrow-derived cells. This would suggest that MHC class I up-regulation may contribute to direct IFNγ mediated liver injury, presumably by increasing the expression of target MHC-peptide complexes.
In contrast, removal of IFNγR from bone marrow-derived cells reduced serum ALT and Kupffer cell TNFα expression; which was also evident in TNFR1-deficient mice. The lack of any significant difference between serum ALT or TNFα expression in TNFR1 chimeras when compared to control chimeras supports the idea of a TNFα dependent autocrine loop which facilitates optimal TNFα expression. This feedback loop is likely to be acting on the Kupffer cells themselves, since macrophage activation may be driven by TNFR1 autocrine signaling (22, 23). Furthermore, these data demonstrate that TNFα does not have a direct cytotoxic effect on the hepatocytes. This stands in contrast to experiments conducted in an adenovirus model, in which the conclusion of a direct effect of TNFα was based on in vitro cytotoxicity data (27). While TRAIL expression was not affected by the loss of TNFR1 signaling, other Kupffer cell derived cytotoxic molecules, such as reactive oxygen species or nitric oxide (29, 30), may be responsible for an indirect form of liver damage.
The liver injury we had investigated depends entirely on the introduction of exogenous CD8+ T cells that recognize the AAV vector-encoded target antigen. In normal mice, AAV tranduction of hepatocytes normally results in long-lived vector expression, arguing that the endogenous T cell response is weak (31, 32). However, the immunopathology we investigate here takes place in the context of an endogenous innate immune response, characterized by transient, local cytokine expression (33). In the future, it will be important to determine whether this innate response is important in the induction of CD8+ T cell-mediated immunopathology.
In summary, we propose a model in which IFNγ promotes liver injury by two parallel mechanisms, while TNFα is exclusively involved in Kupffer cell dependent damage. This mechanism begins with CD8+ T-cell production of IFNγ, resulting in Kupffer cell TNFα expression and sensitization of hepatocytes to CTL by increasing antigen availability via the MHC class I pathway. While TNFα itself is not directly cytotoxic, we believe that its role lies in promoting Kupffer cell activation via an autocrine positive feedback loop that promotes optimal TNFα expression and subsequently results in liver damage. Further investigation is required to determine whether this TNFα dependent, Kupffer cell mediated injury acts via a collateral mechanism, or enhances direct liver injury driven by CTL recognition of transduced hepatocytes. In the context of a hepatic viral infection, interference with direct forms of CTL mediated liver injury may have the undesirable effect of inhibiting viral clearance. However, targeting collateral liver injury pathways may be beneficial to reduce liver damage while preserving, and perhaps enhancing, antiviral immunity. Further analysis of these mechanisms therefore holds therapeutic potential.
Supplementary Material
(A) C57BL6 mice were given an intrahepatic injection of the rAAV2 GFP vector and > 3 weeks later liver tissue was harvested and sections were stained for GFP. (B) C57BL6 mice were given the rAAV2 GFP-SIINFEKL vector plus 5×106 OT-1 cells approximately 3 weeks later. Day 3 post OT-1 transfer, harvested liver sections were stained for CD3.
Three days post OT-1 cell transfer TNFR1 −/− (A), WT → TNFR1 −/− chimeras (B) and their WT controls had liver mRNA measured for TRAIL expression. N.S. not significant. Data are representative, or the sum, of two independent experiments.
Acknowledgments
Funding: This work was supported by grants from the NIH (R01AI064463 and R01DK075274). M. Giannandrea was supported by the NIH Training Program in Viral Disease, Vaccines / Biodefense (T32 AI 007 169).
Nonstandard Abbreviations
- Abs
Antibodies
- CTL
Cytotoxic T-Lymphocyte
- IFNγ
Interferon-gamma
- TNFα
Tumor Necrosis Factor-alpha
- rAAV2
recombinant Adeno-Associated Virus Serotype 2
- GFP
Green Fluorescent Protein
- IFNγR
Interferon-gamma Receptor
- TNFR1
Tumor Necrosis Factor Receptor 1
- ALT
Alanine Aminotransaminase
- TCRβ
T-Cell Receptor-beta
Contributor Information
Matthew Giannandrea, The David H Smith Center for Vaccine Biology and Immunology, Aab Institute for Biomedical Research, University of Rochester Medical Center, 601 Elmwood Avenue, Rochester NY 14642, USA Matthew_Giannandrea@urmc.rochester.edu.
Robert H. Pierce, Schering-Plough Biopharma, 901 California Ave, Palo Alto, CA 94304, USA. Robert.Pierce@spcorp.com
Ian Nicholas Crispe, The Seattle Biomedical Research Institute, 307 North Westlake Avenue, Seattle, WA 98019 nick.crispe@sbri.org.
References
- 1.Mehal WZ, Azzaroli F, Crispe IN. Antigen presentation by liver cells controls intrahepatic T cell trapping, whereas bone marrow-derived cells preferentially promote intrahepatic T cell apoptosis. J Immunol. 2001;167:667–673. doi: 10.4049/jimmunol.167.2.667. [DOI] [PubMed] [Google Scholar]
- 2.Bowen DG, Warren A, Davis T, Hoffmann MW, McCaughan GW, Fazekas de St Groth B, et al. Cytokine-dependent bystander hepatitis due to intrahepatic murine CD8 T-cell activation by bone marrow-derived cells. Gastroenterology. 2002;123:1252–1264. doi: 10.1053/gast.2002.36058. [DOI] [PubMed] [Google Scholar]
- 3.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, et al. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polakos NK, Cornejo JC, Murray DA, Wright KO, Treanor JJ, Crispe IN, et al. Kupffer cell-dependent hepatitis occurs during influenza infection. Am J Pathol. 2006;168:1169–1178. doi: 10.2353/ajpath.2006.050875. quiz 1404-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth E, Pircher H. IFN-gamma promotes Fas ligand- and perforin-mediated liver cell destruction by cytotoxic CD8 T cells. J Immunol. 2004;172:1588–1594. doi: 10.4049/jimmunol.172.3.1588. [DOI] [PubMed] [Google Scholar]
- 6.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 7.Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, et al. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity. 2005;22:93–104. doi: 10.1016/j.immuni.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 9.Wuensch SA, Pierce RH, Crispe IN. Local intrahepatic CD8+ T cell activation by a non-self-antigen results in full functional differentiation. J Immunol. 2006;177:1689–1697. doi: 10.4049/jimmunol.177.3.1689. [DOI] [PubMed] [Google Scholar]
- 10.Clark KR, Liu X, McGrath JP, Johnson PR. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- 11.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright KO, Murray DA, Crispe NI, Pierce RH. Quantitative PCR for detection of the OT-1 transgene. BMC Immunol. 2005;6:20. doi: 10.1186/1471-2172-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N, et al. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol. 2000;157:1671–1683. doi: 10.1016/S0002-9440(10)64804-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu ZX, Govindarajan S, Okamoto S, Dennert G. NK cells cause liver injury and facilitate the induction of T cell-mediated immunity to a viral liver infection. J Immunol. 2000;164:6480–6486. doi: 10.4049/jimmunol.164.12.6480. [DOI] [PubMed] [Google Scholar]
- 15.Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 2000;97:5498–5503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kusters S, Gantner F, Kunstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 1996;111:462–471. doi: 10.1053/gast.1996.v111.pm8690213. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe Y, Suzuki O, Haruyama T, Akaike T. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89:244–253. doi: 10.1002/jcb.10501. [DOI] [PubMed] [Google Scholar]
- 18.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 20.Sass G, Shembade ND, Haimerl F, Lamoureux N, Hashemolhosseini S, Tannapfel A, et al. TNF pretreatment interferes with mitochondrial apoptosis in the mouse liver by A20-mediated down-regulation of Bax. J Immunol. 2007;179:7042–7049. doi: 10.4049/jimmunol.179.10.7042. [DOI] [PubMed] [Google Scholar]
- 21.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 22.Calder CJ, Nicholson LB, Dick AD. A selective role for the TNF p55 receptor in autocrine signaling following IFN-gamma stimulation in experimental autoimmune uveoretinitis. J Immunol. 2005;175:6286–6293. doi: 10.4049/jimmunol.175.10.6286. [DOI] [PubMed] [Google Scholar]
- 23.Benveniste EN, Nguyen VT, Wesemann DR. Molecular regulation of CD40 gene expression in macrophages and microglia. Brain Behav Immun. 2004;18:7–12. doi: 10.1016/j.bbi.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann V, Freudenberg MA, Galanos C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer R, Cariers A, Reinehr R, Haussinger D. Caspase 9-dependent killing of hepatic stellate cells by activated Kupffer cells. Gastroenterology. 2002;123:845–861. doi: 10.1053/gast.2002.35384. [DOI] [PubMed] [Google Scholar]
- 26.Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204:667–680. doi: 10.1084/jem.20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kafrouni MI, Brown GR, Thiele DL. Virally infected hepatocytes are resistant to perforin-dependent CTL effector mechanisms. J Immunol. 2001;167:1566–1574. doi: 10.4049/jimmunol.167.3.1566. [DOI] [PubMed] [Google Scholar]
- 28.Liu ZX, Govindarajan S, Okamoto S, Dennert G. Fas- and tumor necrosis factor receptor 1-dependent but not perforin-dependent pathways cause injury in livers infected with an adenovirus construct in mice. Hepatology. 2000;31:665–673. doi: 10.1002/hep.510310317. [DOI] [PubMed] [Google Scholar]
- 29.Medan D, Wang L, Toledo D, Lu B, Stehlik C, Jiang BH, et al. Regulation of Fas (CD95)-induced apoptotic and necrotic cell death by reactive oxygen species in macrophages. J Cell Physiol. 2005;203:78–84. doi: 10.1002/jcp.20201. [DOI] [PubMed] [Google Scholar]
- 30.Griffon B, Cillard J, Chevanne M, Morel I, Cillard P, Sergent O. Macrophage-induced inhibition of nitric oxide production in primary rat hepatocyte cultures via prostaglandin E2 release. Hepatology. 1998;28:1300–1308. doi: 10.1002/hep.510280519. [DOI] [PubMed] [Google Scholar]
- 31.Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. 2008;16:280–289. doi: 10.1038/sj.mt.6300355. [DOI] [PubMed] [Google Scholar]
- 32.Ding Z, Georgiev P, Thony B. Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Ther. 2006;13:587–593. doi: 10.1038/sj.gt.3302684. [DOI] [PubMed] [Google Scholar]
- 33.Zaiss AK, Liu Q, Bowen GP, Wong NC, Bartlett JS, Muruve DA. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J Virol. 2002;76:4580–4590. doi: 10.1128/JVI.76.9.4580-4590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) C57BL6 mice were given an intrahepatic injection of the rAAV2 GFP vector and > 3 weeks later liver tissue was harvested and sections were stained for GFP. (B) C57BL6 mice were given the rAAV2 GFP-SIINFEKL vector plus 5×106 OT-1 cells approximately 3 weeks later. Day 3 post OT-1 transfer, harvested liver sections were stained for CD3.
Three days post OT-1 cell transfer TNFR1 −/− (A), WT → TNFR1 −/− chimeras (B) and their WT controls had liver mRNA measured for TRAIL expression. N.S. not significant. Data are representative, or the sum, of two independent experiments.