

Abstract
PCR-based clinical and forensic tests often have low sensitivity or even false-negative results caused by potent PCR inhibitors found in blood and soil. It is widely accepted that purification of target DNA before PCR is necessary for successful amplification. In an attempt to overcome PCR inhibition, enhance PCR amplification, and simplify the PCR protocol, we demonstrate improved PCR-enhancing cocktails containing nonionic detergent, l-carnitine, d-(+)-trehalose, and heparin. These cocktails, in combination with two inhibitor-resistant Taq mutants, OmniTaq and Omni Klentaq, enabled efficient amplification of exogenous, endogenous, and high-GC content DNA targets directly from crude samples containing human plasma, serum, and whole blood without DNA purification. In the presence of these enhancer cocktails, the mutant enzymes were able to tolerate at least 25% plasma, serum, or whole blood and as high as 80% GC content templates in PCR reactions. These enhancer cocktails also improved the performance of the novel Taq mutants in real-time PCR amplification using crude samples, both in SYBR Green fluorescence detection and TaqMan assays. The novel enhancer mixes also facilitated DNA amplification from crude samples with various commercial Taq DNA polymerases.
A major problem with PCR-based diagnostic tests of blood samples is the false-negative or low-sensitivity reactions caused by PCR inhibitors. Blood samples are extensively used for PCR-based diagnosis of microbial infection, genetic disease, forensic analysis, and blood banking.1,2,3,4 Prenatal genetic diagnosis using maternal plasma or serum has been developed recently.5,6 However, when these techniques are applied to blood samples, it must be considered that PCR inhibitors in the specimens may lead to possible false-negative or reduced sensitivity despite advanced DNA purification methods. For example, even after DNA purification steps are used before PCR, a 14% false-negative rate has been observed for hepatitis B virus detection, most probably due to incomplete removal of PCR inhibitors.7 PCR inhibition is a common concern in detection of various pathogens, such as herpes, varicella, Epstein-Barr, polyoma, and cytomegalovirus viruses, bacteria, and fungi.2 Inhibition of RT-PCR in plasma samples has been reported to occur at a frequency of 0.34 to 2.4% of the tests in patients infected with HIV and hepatitis C virus, respectively.8,9 Among the most potent PCR inhibitors reported are hemoglobin/heme,10 leukocyte DNA,11 and an IgG fraction,12 in addition to anticoagulants such as EDTA, sodium citrate, and heparin, which also inhibit PCR.13 Other known inhibitors are bilirubin, bile salts, and lactoferrin.14 AmpliTaq Gold can be completely inhibited in the presence of less than 0.004% whole blood or traces of heme.14 Serum and plasma contain fewer inhibitors (heme/hemoglobin) than whole blood, but the detection rate of some pathogens in such specimens can be lower than that from whole blood as some pathogen fraction retains in the peripheral cells.15,16 Some PCR inhibitors such as hemoglobin can copurify with DNA and cause loss of detection of the targets. The inhibitory effect of blood on PCR is associated primarily with inactivation of the DNA polymerase and/or capturing or degradation of the target DNA and primers.10,14
Various methods have been developed to remove PCR inhibitors from blood samples.7,17,18 However, these various purification procedures are not always efficient and can lead to loss of the target DNA. Furthermore, these procedures are time-consuming and labor-intensive and increase cost. In addition, the multiple sample manipulations involved increase the risk of cross-contaminations.
Researchers have been trying to find additives to PCR that can relieve the inhibition and enhance amplification.19,20,21,22,23,24,25,26 The known PCR enhancers, however, usually cover only one aspect of the problem by working on GC-rich targets of purified DNA template or relieving the inhibition. As an alternative to the various time- and labor-consuming pre-PCR procedures needed with blood, we recently reported inhibition-resistant mutants of Taq DNA polymerase, OmniTaq (Taq-22) and Omni Klentaq (Klentaq-10), which can tolerate the major PCR inhibitors found in blood and soil.27
Here we report a novel PCR enhancer cocktail (PEC), which improves the performance of the Taq mutants, allowing direct amplification of targets from whole blood, serum, or plasma present in the final PCR volume at least at 25%, without DNA purification. This could especially benefit PCR applications in which the target concentration is low and a larger input amount of the crude sample is necessary for successful detection. With this enzyme-enhancer combination we were also able to amplify endogenous and exogenous, non-GC-rich and GC-rich targets directly from crude samples without DNA extraction. PEC was also efficient with most commercial Taq enzymes, although it performed optimally with the OmniTaq and Omni Klentaq enzymes, especially with crude blood. Therefore, the novel PCR enhancer combined with these mutants or other commercial Taqs should simplify, speed-up, and lower the cost of important clinical, forensic, and environmental PCR-based tests.
Materials and Methods
Primer Design
PCR primers for the amplification of targets were designed using the PrimerQuest online service or were from the literature.25 The primer sequences, the overall GC content, and the size of the expected amplicons are summarized in Table 1.
Table 1.
Primers Applied in This Project
| Gene symbol | GC % of amplicon | Amplicon length (bp) | Forward primer | Reverse primer |
|---|---|---|---|---|
| CCR5 | 47 | 630 | 5′-GCAGCGGCGGACCAGCCCCAAGATGCTATCT-3′ | 5′-TGGAACAAGATGGATTATCAAGTGTCAAGTCCA-3′ |
| HRES1 | 81 | 488 | 5′-TCCGGCCGCGCCACCGCCACCCTCA-3′ | 5′-ACCGCCAGAGCGCCCAGCCCGCGCA-3′ |
| β-Actin | 61 | 300 | 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ | 5′-TCACCCACACTGTGCCCATCTACG A-3′ |
| HV2S | 54 | 137 | 5′-GCTCGAGTGCGAAAAAACGTTC-3′ | 5′-TGCGGTTGATAAACGCGCAGT-3′ |
| DHBVP | 44 | 690 | 5′-ATGCCCCAACAATTGAAGCAATC-3′ | 5′-ACGTCCATTGATTTTGCTGGATG-3′ |
| SIM2 | 75 | 1202 | 5′-CCGTTTTCACGTGTGTGTGT-3′ | 5′-TTCCTTCTCCCTCCTGGTCT-3′ |
| DIP2A | 71 | 1185 | 5′-AGGGGAAGGAAGCAGGACT-3′ | 5′-CAGCTCAGCCAGGCTCTC-3′ |
| SLC19A* | 74 | 980 | 5′-CCTTCTGTTCTGTGCAGTGG-3′ | 5′-CGGACTCCGGGACTACAG-3′ |
| EV107 | 40 | 189 | 5′-AGAAGATATCAGACGATCCACAATC-3′ | 5′-GTAGAACG CGCCAGAATAAGAATA-3′ |
HV2S, herpes complex virus type 2.
GenBank identification no: NT011515.
DNA Template
Human genomic DNA was purchased from Novagen (Gibbstown, NJ). The pathogen DNA was extracted from whole blood, plasma, serum, or supernatant of cell cultures using a QIAamp DNA Micro Kit or QIAamp MinElute Virus Spin Kit (QIAGEN, Valencia, CA).
Virus Stock
Duck serum infected with duck hepatitis B virus (DHBV), herpes simplex type 2 (HVS2), and mousepox virus (Ectromelia virus) were kindly provided by Dr. John Tavis, Dr. Lynda Morrison, and Dr. Mark Buller, respectively, at St. Louis University. The mousepox virus concentration was quantitated by electron microscopy counting and an OD value of λ260 absorption. These virus titer values along with the dilution factor were used to determine the sensitivity of virus detection.
Blood Samples
Whole human blood and plasma, treated with 4.6 mmol/l EDTA, 21.3 U/ml of heparin, or 0.38% sodium citrate, and serum collected from the same donor were purchased from Innovative Research (Southfield, MI) and stored in aliquots at −70°C. Before PCR testing, the thawed blood samples were homogenized well by gentle mixing.
PCR Additives
l-Carnitine inner salt (C0158), d-(+)-trehalose dehydrate (T9531), betaine (B2629), heparin (H3393), and Nonidet P-40 (NP-40) were purchased from Sigma-Aldrich (St. Louis, MO). Three commercial PCR enhancers, GC Solution (used with FastStart Taq), Q-Solution (used with HotStarTaq Plus), and Hi-Spec Additive were obtained from Roche Applied Science (Indianapolis, IN), QIAGEN (Germantown, MD), and Bioline (Taunton, MA), respectively.
Taq DNA Polymerases
OmniTaq (OT) and Omni Klentaq (OKT) were invented and manufactured by DNA Polymerase Technology, Inc. (St. Louis, MO). Other commercial Taq enzymes used were FastStart Taq (Roche Applied Science), HotStarTaq Plus (QIAGEN), AmpliTaq Gold (ABI, Bedford, MA), JumpStart Taq (Sigma-Aldrich), and Taq DNA polymerase (New England Biolabs, Ipswich, MA).
Conventional PCR
PCR using OmniTaq or Omni Klentaq was performed in a 50-μl volume in 0.5-ml or 0.2-ml tubes. The reaction buffer contained 50 mmol/L Tris-HCl, pH 8.3 (OT) or 9.2 (OKT), 16 mmol/L ammonium sulfate, 0.1% (v/v) Tween 20, and 2.5 mmol/L (OT) or 3.5 mmol/L (OKT) magnesium chloride. The reactions contained 2 U of OT or OKT enzyme, 0.2 μmol of each primer, and 200 μmol of dNTP mix. The PCR conditions for other commercial Taqs were set according to the manufacturers' instructions. The reactions were performed in a RoboCycler (Stratagene, La Jolla, CA) or PTC-200 Peltier Thermocycler (MJ Research, Watertown, MA) with an initial denaturation for 10 minutes at 95°C followed by the thermal cycles as follows: denaturation step at 95°C for 30 to 40 seconds, an annealing step at 52 to 58°C for 40 to 60 seconds, and an elongation step at 70°C for 1 to 5 minutes, depending on the targets, for 30 to 40 cycles.
For some targets a “touch down” procedure was used with an initial PCR denaturation for 5 minutes at 95°C, 40 seconds at 72°C, 2 minutes at 72°C for 1 cycle followed by two steps of the thermal cycles as follows: denaturation at 95°C for 15 seconds, annealing at 64 to 54°C for 40 seconds with a progressive decrease of −0.5°C per cycle for 20 cycles and elongation at 72°C for 2 minutes; and then 20 to 30 cycles with denaturation at 95°C for 15 seconds and 54°C for 40 seconds and an elongation step at 72°C for 2 minutes with final elongation at 72°C for 5 minutes. The products were analyzed with 1.5 to 2% agarose gel electrophoresis and stained with ethidium bromide.
Real-Time PCR
The quantitative PCR (qPCR) was performed in an Opticon-2 cycler (Bio-Rad Laboratories, Hercules, CA). In SYBR Green fluorescent detection qPCR, a master mixture containing 5% whole blood (final concentration in PCR) was made, and aliquots of it were distributed to PCR tubes. The supernatant of HVS2 cell culture was fivefold serially diluted with water, from which dilutions of 1 μl were added to each reaction. A 137-bp HVS2 virus gene target was amplified from these samples. The reaction was performed in the presence or absence of PEC with an initial denaturation for 10 minutes at 95°C followed by a denaturation step at 95°C for 40 seconds, an annealing step at 54°C for 45 seconds, and an elongation step at 70°C for 2 minutes for 40 cycles, including 2 units of Taq mutants and 10× SYBR Green dye (10 ng/μl) (Invitrogen, Carlsbad, CA). Fluorescence was detected at each annealing step. For sensitivity comparison of purified DNA and crude blood samples, the virus particles were 10-fold serially diluted in 100 μl of whole heparinized blood samples, which finally contain 7.2 × 108 to 7.2 × 102 virus particles/ml. These virus-spiked blood samples were split in half, and virus DNA was purified from one-half (50 μl) of each sample with a QIAamp DNA Micro kit (QIAGEN). DNA from these samples was finally eluted in 50 μl of water, thus restoring the original blood volume. The other half of the crude blood samples was directly used in PCR, and 1.25 μl of each purified DNA and blood sample series (5% of the total volume of the PCR reaction) was used in parallel to amplify a 189-bp mousepox virus target (Ectromelia virus [Mousepox virus] 107 gene [EV107 gene]), using OmniTaq and SYBR Green detection in the presence of PEC.
In TaqMan assays, a qPCR Master Mix containing 10% human plasma (10% of total PCR volume) was made and aliquoted to the PCR tubes. A mousepox virus cell culture supernatant was 10-fold serially diluted with water, and the same target was amplified in the presence of PEC in 45 cycles. The target was amplified from an equivalent amount of purified DNA as well. AmpliTaq Gold was also applied in this assay as comparison. A CAL Fluor Gold 540/BHQ-labeled TaqMan probe (5′-GCCGGTGTAACATTCTCCACCAAACA-3′, Biosearch Technologies, Novata, CA) was included at 200 nmol/L concentration.
Results
Formulation of PCR Enhancer Cocktail
In an attempt to find an efficient additive to PCR that would facilitate the enzyme performance, especially with crude samples containing potent PCR inhibitors, we selected and focused on d-(+)-trehalose, l-carnitine, and a detergent, NP-40. d-(+)-Trehalose enables thermostable enzymes to maintain their normal activity (thermostabilization) or even to increase their activity at high temperatures (thermoactivation).28 Some detergents such as NP-40 and Tween 20 have been reported to improve DNA sequencing by reducing the frequency of nonspecific bands.29,30 No application of l-carnitine in PCR has been reported. This compound, however, is known as an effective osmoprotectant, like betaine and proline.31 Therefore, we assumed that it might enhance the PCR reaction in a fashion similar to that of betaine and proline. In initial experiments we tested various combinations of d-(+)-trehalose, l-carnitine, and NP-40 to develop a reliable PEC. In some of the tests we applied the PEC in the amplification of a 630-bp target of human HIV coreceptor gene (CCR5) (GC content 47%) and a 488-bp fragment of human T-lymphotropic virus-1 related sequence (HRES1) (GC content 81%). The two targets were directly amplified from whole blood present at a 10% final PCR concentration using the OT and OKT mutant enzymes in the presence of different combinations of PEC. Results indicated that when the concentrations of NP-40 and l-carnitine were constant, the optimal concentrations of d-(+)-trehalose were 0.5 to 0.8 mol/L for the non-GC-rich target CCR5 and 0.4 to 0.8 mol/L for the GC-rich HRES1 target. When the concentrations of NP-40 and d-(+)-trehalose were constant, the optimal concentrations of l-carnitine were 0.12 to 0.72 and 0.48 to 1.2 mol/L for these two targets, respectively (data not shown).
Furthermore, the 488-bp HRES1 target was amplified from different concentrations of blood with d-(+)-trehalose, l-carnitine, or NP-40 individually, as well as combined in the cocktail. The results showed that the Taq mutants were able to amplify this GC-rich target with 1.25 to 20% blood content in the PCR reaction in the presence of the cocktail, whereas nonspecific products or lower yields were observed with d-(+)-trehalose, l-carnitine, or NP-40 alone (data not shown). Based on these results, we formulated the PCR cocktail enhancer as a 2× solution, containing 0.6 mol/L d-(+)-trehalose, 0.48 mol/L l-carnitine, and 0.8% NP-40, which was used in the future experiments.
Effect of PEC on PCR Specificity and Sensitivity
The impact of PEC on PCR specificity (Figure 1A) was assessed by amplification of the GC-rich HRES1 target from crude samples containing serial dilutions of whole blood, using OT and OKT in the absence or presence of PEC. A comparison of 1.3 mol/L of betaine was also included. The results showed that in the presence of PEC, OT and OKT enzymes were able to specifically amplify this target from 4 to 0.0013% blood, whereas some nonspecific products were observed with betaine, especially in lower concentrations of template (Figure 1A). We also addressed the question of whether or not sensitivity is compromised in blood samples because of PCR inhibitors. The results with the same GC-rich target showed that the sensitivity of detection from purified DNA and blood samples containing equivalent amounts of DNA was very similar (Figure 1B).
Figure 1.
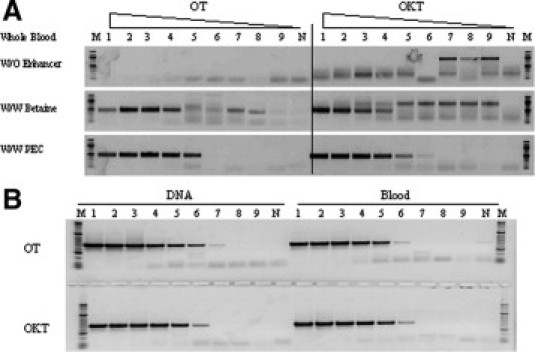
Effect of PEC on PCR specificity and sensitivity. A: The impact of PEC on PCR specificity was assessed by directly amplification of the HRES1 target from blood samples in the absence (W/O) or presence (W/W) of the enhancer; 1.3 mol/L of betaine was included for comparison. The blood sample was serially fivefold diluted from 4 to 0.00001% (final concentration in PCR, lanes 1 to 9), and the negative control contained no DNA or blood (N). A 488-bp HRES1 target (GC content is 81%) was amplified from different concentrations of blood sample. OT, OmniTaq; OKT, Omni Klentaq. B: PCR sensitivity in the blood sample was also evaluated by amplification of the same target from both purified DNA and blood. The blood sample was serially fivefold diluted from 4 to 0.00001% (final concentration in PCR, lanes 1 to 9), and a 488-bp HRES1 target was amplified both from purified DNA and blood containing the same amount of DNA in presence of PEC. The negative control contained no DNA or blood (N). PCR products were resolved in a 1.5% agarose gel. Lanes M, DNA standards ladder.
Impact of Anticoagulants on PCR Reaction and Blood Tolerance of OmniTaq and Omni Klentaq
Citrate, EDTA, and heparin are the most commonly used anticoagulants in clinical blood samples. To evaluate whether PEC can overcome the known inhibitory effect of these anticoagulants on PCR, the HRES1 target and a 300-bp β-actin target were amplified from various concentrations of blood (1.25 to 25%) treated with each anticoagulant. As shown in Figure 2A, both OT and OKT required PEC to efficiently amplify the two human genomic targets. With the GC-rich target, there was a higher tolerance of heparinized blood compared with the EDTA or citrate-treated blood (25 versus 20% blood, respectively) (Figure 2B). In the absence of PEC, the enzymes were functional in relatively lower blood concentrations, 5 to 10%, predominantly with no GC-rich target. The results indicate that the anticoagulants tested do not significantly affect the amplification from whole blood samples with our combination of Taq mutants plus PEC and also demonstrate that PCR could be performed in at least 25% whole blood in the presence of PEC + OT or PEC + OKT. To determine whether the effect of PEC in amplification was subject to target dependence, we also tested other endogenous (human genes) and exogenous (pathogen) targets and obtained similar results.
Figure 2.
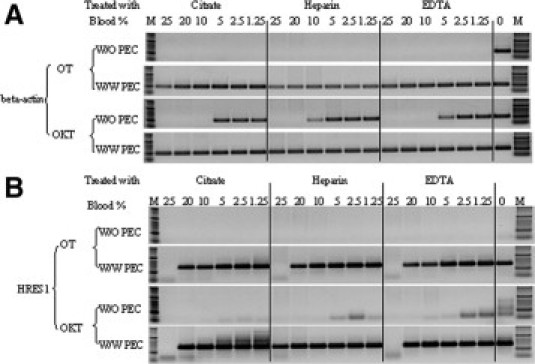
Tolerance of OmniTaq (OT) and Omni Klentaq (OKT) to blood treated with different anticoagulants. Two endogenous targets, 300 bp of the β-actin gene (A) and 488 bp of the HRES1 gene (B), were amplified from 1.25 to 25% (of total PCR volume) from a whole blood sample treated with citrate, heparin, or EDTA, separately, using OT or OKT in the absence (W/O) or presence (W/W) of PEC. The positive control contained 4 ng of purified DNA (0). The products were analyzed in 1.5% agarose gel electrophoresis. Lanes M, DNA standards ladder.
PCR Performance with GC-Rich Targets of OT and OKT Versus Commercial Taq Enzymes
We also challenged OT, OKT, and several commercial Taq polymerase enzymes with three GC-rich targets. The gene targets SIM2 (human homolog of single-minded 2), DIP2A (human disco-interacting protein 2A), and SLC19A (human chromosome 21 genome region) were amplified from purified human genomic DNA and 5% whole blood with different DNA polymerases in the absence or presence of PCR enhancer. FastStart Taq was applied with GC Solution, HotStarTaq Plus with Q-Solution, plain Taq with Hi-Spec Additive, and OT and OKT with PEC. As shown in Figure 3, A and B, with purified DNA none of the enzymes could yield specific products in the absence of enhancer, and some commercial enzymes failed to do so even in the presence of it. All commercial enzymes failed regardless of the addition of the corresponding enhancer when whole blood was used instead of purified DNA. In contrast, in the presence of PEC, OT and OKT produced specific amplified products, both from purified DNA and blood. These results demonstrate two aspects of PEC, acting as both a general PCR enhancer and a facilitator of PCR in the presence of inhibitors.
Figure 3.
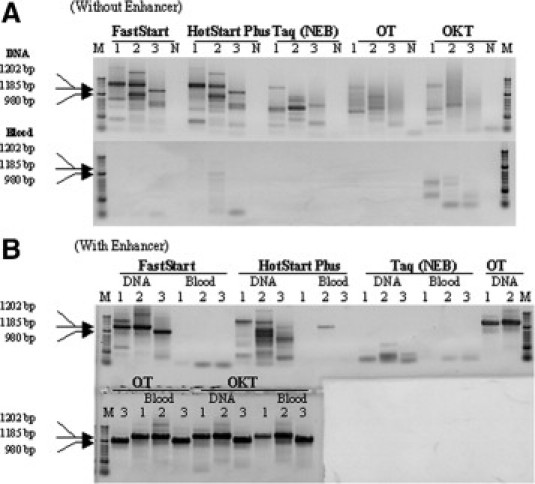
Performance comparison of OT and OKT versus commercial enzymes and their PCR enhancers in amplification of GC-rich targets. The gene targets of SIM2 (1202 bp, GC content 75%, lane 1), DIP2A (1185 bp, GC content 71%, lane 2), and SLC19A (980 bp, GC content 74%, lane 3) were amplified from purified DNA or 5% whole blood (final concentration in PCR) in the absence (A) or in the presence (B) of PCR enhancers. FastStart Taq was combined with GC Solution, HotStarTaq Plus with Q-Solution, plain Taq with Hi Spec Additive, and OT and OKT with PEC. A negative control contained no DNA and blood (N). The position of the specific products 1 to 3 is shown by arrows.
Beneficial Effect of Heparin on DNA Amplification from Plasma and Serum
We observed that OT and OKT could amplify targets from whole blood treated with citrate, heparin, and EDTA, but with some targets they were, surprisingly, functional only in heparinized plasma but not in plasma treated with EDTA or citrate and serum. To test whether heparin could have an inhibition relieving effect, we added it to the enhancer cocktail. A 189-bp mousepox virus target was amplified from purified DNA or crude samples, containing 10% (final concentration of total PCR volume) whole blood, serum, heparinized plasma, and plasma treated with citrate or EDTA, using an OT/OKT mix (1:1). The reaction was performed in the absence of an enhancer and in the presence of PEC, PEC plus heparin (PEC-Plus), heparin alone, and a combination of heparin and d-(+)-trehalose. As shown in Figure 4, heparin alone was able to reverse the inhibitory effect of plasma treated with citrate and EDTA but did not interfere with the inhibition by serum and whole blood. Furthermore, the combination of heparin and d-(+)-trehalose was effective in relieving the inhibitory effect of whole blood, plasma, and serum; however, it inhibited the amplification of purified DNA. In the presence of PEC alone, the target was amplified from purified DNA, whole blood, and heparin-treated plasma, but not from citrate- and EDTA-treated plasma and serum. Finally, only with the combination of PEC plus heparin (at concentration 5 to 20 U/ml plasma or serum) the target was amplified from DNA and all crude samples tested.
Figure 4.
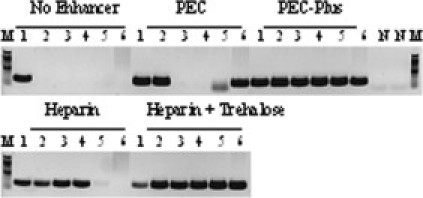
Application of PEC-Plus in PCR in plasma and serum. A 189-bp target of mousepox virus (EV107 gene) was amplified by the OT/OKT mix (1:1) from 0.1 ng of viral DNA and crude samples containing virus particles, including 10% whole blood, serum, or plasma (final concentration in PCR) treated with heparin, citrate, and EDTA. The reactions were performed in the absence of enhancer or in the presence of heparin alone, a combination of heparin and trehalose, PEC, and PEC-Plus (PEC supplemented with heparin). Negative controls contained no virus DNA (N). Lane 1, DNA; lane 2, heparin-treated plasma; lane 3, citrate-treated plasma; lane 4, EDTA-treated plasma; lane 5, serum; and lane 6, heparin-treated blood. Lanes M, DNA standards ladder.
Application of PEC and PEC-Plus in Real-Time PCR
It was important to determine whether the formulated enhancer cocktail could be applied in real-time PCR analysis of crude samples. In preliminary experiments we tested various concentrations of SYBR Green and found that 1× dye was optimal for 1 to 10% plasma and serum, and 10× or 20× SYBR Green dye was optimal for 5 and 10% whole human blood, respectively. Also, 200 and 500 nmol/L TaqMan probe (CAL Fluor Gold 540 BHQ-labeled) was optimal for 1 to 10% plasma and serum and 5% whole blood, respectively (data not shown). These concentrations were used in real-time PCR of crude blood samples. A 137-bp HVS2 target was amplified from 5% blood spiked with serially diluted virus particles by OT (Figure 5A and 5B) or OKT (Figure 5C and 5D) using SYBR Green detection in the absence or in the presence of PEC. Although in clinical samples HSV is typically detected in tissue or smear specimens rather than in blood, we added whole blood for the purpose of challenging the reaction with more PCR inhibitors. The results showed that the two enzymes benefited from the addition of PEC, and they were able to amplify this target from 5% whole blood containing different template concentrations. PEC was necessary for OT to work, whereas with OTK, it improved the enzyme sensitivity, as judged by shifting the threshold cycle (Ct) to lower values.
Figure 5.
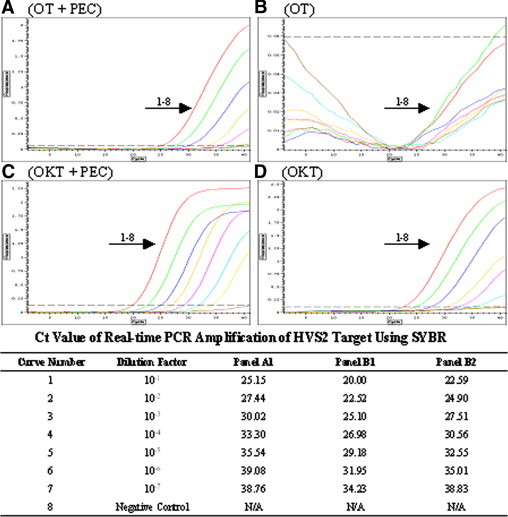
Application of PEC in SYBR Green real-time PCR. A master mixture containing 5% whole blood (in total PCR volume) was made, and aliquots of it were distributed to PCR tubes. The supernatant of the HVS2 cell culture was fivefold serially diluted with water, from which dilutions of 1 μl were added to each reaction. A 137-bp HVS2 target gene was amplified from these samples using OT (A and B) or OKT (C and D) and SYBR green detection in the presence (A and C) or absence of PEC (B and D). PCR was performed for 40 cycles in an Option 2 real-time cycler in the presence of 10× SYBR Green. A negative control contained no virus. The Ct values for the panels where the enzymes were functional (A, C, and D) are provided at the bottom.
In another set of experiments a 189-bp target of mousepox virus was amplified from 10% heparin-treated plasma by an OT/OKT (1:1) enzyme mixture and detected in a TaqMan assay in the presence of PEC. The sensitivity of detection was similar to that with purified viral DNA (Figure 6A and 6B). In a parallel test AmpliTaq Gold completely failed to amplify the target in plasma, and it was functional only with purified DNA (Figure 6C and 6D).
Figure 6.
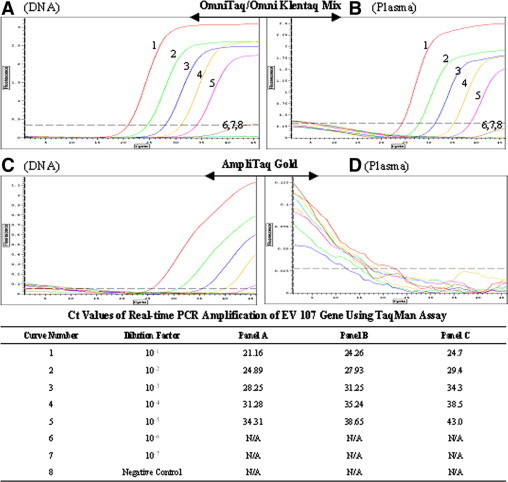
Application of PEC in a real-time PCR TaqMan assay. A qPCR Master Mix containing 10% human plasma (10% of total PCR volume) was made and aliquoted to the PCR tubes. A mousepox virus cell culture supernatant was 10-fold serially diluted with water and a 189-bp mousepox virus gene (EV107 gene) was amplified from these crude samples. A parallel sample series contained equivalent amounts of purified virus DNA dilutions. An OT/OKT enzyme mixture (1:1) was used and the reactions contained PEC (A and B). The same virus target was also amplified with AmpliTaq Gold as a comparison (C and D). PCR was performed for 45 cycles in an Option 2 real-time cycler, and the amplification was detected with 200 nmol/L TaqMan probe. A negative control contained no virus or DNA. The Ct values are shown at the bottom.
An important issue for clinical practice in testing directly crude samples with PCR is whether the sensitivity of detection of the pathogen is compromised after skipping the DNA extraction steps. We addressed this issue by performing qPCR with SYBR Green detection of mousepox virus (the same target used in Figure 6) in whole blood. Two parallel serial dilutions of the virus in human blood were prepared, and one of them went through to a conventional procedure where each sample was treated with a blood DNA extraction kit, whereas the other dilution series was used directly in qPCR. As shown in Figure 7, the sensitivity of PCR with or without DNA extraction was quite comparable, detecting in both cases six dilutions of the virus down to 7 copies/μl. With the lowest concentration of the purified DNA we observed a nonspecific product (Figure 7A, curve 7), as confirmed by the melting curve analysis and agarose gel electrophoresis (not shown). However, a specific amplification was still observed in the blood sample at this dilution even though the signal was very low (Figure 7B).
Figure 7.
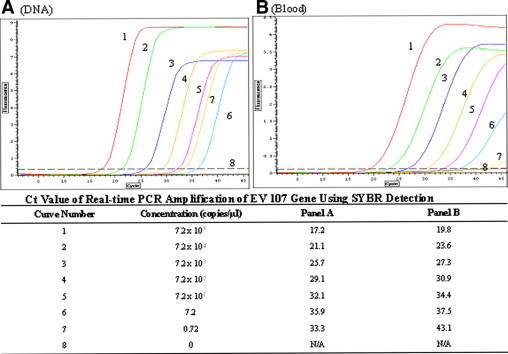
qPCR assessment of sensitivity of pathogen detection in crude samples containing whole blood. Ten-fold serial dilutions of mousepox virus were made in 100 μl of heparinized blood (final virus concentration ranged from 7.2 × 105 to 0.72 copies/μl). These virus-spiked blood samples were split in half and virus DNA was purified from 50 μl of each sample with a QIAGEN DNA extraction kit (A). DNA purified from these samples was finally eluted in 50 μl of water, thus restoring the original blood volume. The other half of the crude blood samples was directly used in PCR (B), and 1.25 μl of each purified DNA and blood sample series (5% final volume in PCR) were used in parallel to amplify a 189-bp mousepox virus target (EV107 gene), using Omni Taq and SYBR Green detection in the presence of PEC. A negative control contained no virus or DNA. The concentrations of the virus particles and the corresponding Ct values are shown at the bottom. With both purified DNA and blood seven virus copies/μl were detected (A and B and curves 6).
In these real-time protocols, using SYBR Green or TaqMan probe, we also observed a slight shift in the Ct values when crude sample (whole blood or plasma) was used, an effect, however, that did not compromise the sensitivity of the assay, as the same dilutions of the virus target were detected with purified DNA and with the crude sample. These data demonstrate that the components of PEC do not negatively interfere with the fluorescence of SYBR Green or the TaqMan probe, which extends the application of the novel enhancer cocktail in real-time PCR applications with crude clinical samples, omitting nucleic acid extraction.
Discussion
In this study, three (PEC) or four chemicals (PEC-Plus) were selected to formulate PCR enhancer cocktails. These cocktails, when combined with two novel Taq mutants resistant to PCR inhibitors, OmniTaq and Omni Klentaq, enabled amplification of exogenous and endogenous targets, including GC-rich targets from purified DNA or from crude samples such as blood and derivates without DNA purification before conventional or real-time PCR. In the presence of PEC, OT and OKT could tolerate at least 25% of whole blood that was inhibitory to wild-type TaqDNA polymerase. PEC also improved the performance of various commercially available Taq enzymes.
Innis et al32 and Bachmann et al29 reported that 0.5% NP-40 or 0.5% Tween 20, either individually or combined, can stimulate DNA polymerase activity at high temperature in PCR reactions and sequencing. In our study we added 0.8, 1.6, 2.4, and 3.2% of NP-40 in the enhancer cocktail to amplify the DHBVP, CCR5, and HRES1 gene targets. The results showed that all detergent concentrations performed similarly (data not shown), so we selected 0.8% NP-40 as one constant component of PEC. Spiess et al33 reported that 0.2 mol/L d-(+)- trehalose could enhance the yield of the amplified product in PCR up to 200-fold, most probably by lowering the template melting temperature and by elimination of secondary structures. In our experiments the optimal concentration of d-(+)-trehalose in PCR ranged from 0.5 to 0.8 mol/L when used either alone or combined with l-carnitine and NP-40, with a concentration range similar to that in the report of Carninci et al.28 In our tests d-(+)-trehalose alone increased the PCR yield, but it did not help much in improving specificity, especially with a GC-rich target (data not shown).
There have been no reports on an enhancing effect of l-carnitine in PCR. l-Carnitine is an effective osmoprotectant, like betaine and proline, which have been shown to stimulate PCR.31 We anticipated that l-carnitine could facilitate PCR as well. This proved true, and we found that the optimal concentration of l-carnitine in PCR was 0.12 to 0.72 mol/L for non-GC-rich targets and 0.48 to 1.2 mol/L for GC-rich targets.
The PCR additives reported previously usually cover one aspect, helping either the amplification from GC-rich targets or facilitating the reaction in the presence of PCR inhibitors. In contrast, the novel PEC described here provides a double benefit by combining these aspects. It consists of d-(+)-trehalose, l-carnitine, and NP-40. Trehalose stimulated amplification of relatively easy DNA targets, but for more difficult and GC-rich targets the addition of l-carnitine was crucial. The detergent NP-40 may help lyse the blood cells or viral capsids in crude clinical samples. None of the PEC components alone was sufficient to elicit the potent enhancing effect observed with the triple combination, especially with crude samples and GC-rich targets.
Our novel enzyme/enhancer PCR system presumes no DNA extraction steps before amplification. In such a protocol, it is important to determine whether the efficiency/sensitivity of target detection in the crude sample is compromised compared with purified DNA. Endogenous targets usually bind to proteins, and it is critical to free targets from proteins and to release them to the reaction mixture. We found that initial heating at 95°C for 10 minutes is more helpful than 3 minutes of heating for crude samples (data not shown). Similar sensitivity was obtained with the 488-bp HRES1 target amplified from purified DNA and blood sample containing an equivalent amount of human genomic DNA, meaning that the longer initial heating together with PEC was sufficient to release DNA from cells and efficiently overcome the inhibition.
It is necessary to use a relatively larger volume of crude clinical sample in PCR when the pathogen concentration is lower, in which case there is demand for a more efficient PCR enhancer to overcome the higher amounts of PCR inhibitors. We demonstrated that, in the presence of PEC, the reaction could tolerate 25% whole blood (Figure 2).
Citrate, EDTA, and heparin are the most common anticoagulants used for blood treatment in diagnostics. Citrate-treated blood requires an unusually high salt buffer for PCR and leukocytes in EDTA-treated blood are difficult to lyse.34 An inhibitory effect of heparin on Taq in PCR with purified DNA or RNA substrate has been documented.35 Our data demonstrated that PEC helps PCR with all anticoagulants.
Betaine is one of the most common enhancers in PCR that can lower melting temperature and eliminate second structures in the template.22,24,36 Our earlier experiments showed that the enhancing effect of betaine was target-dependent. We found that PEC was able to lower the melting temperature as well and outperformed betaine with GC-rich targets (Figure 1).
Our data showed that PEC could improve the performance of the blood inhibition-resistant mutants OT and OKT, allowing even higher tolerance of these enzymes to at least 25% whole blood, plasma, or serum present in PCR. The enhancer cocktail probably acted by eliminating second structures along the template and neutralizing the effect of PCR inhibitors. At present the mechanism of the inhibition neutralizing effect is unknown. It was suggested that enzyme degradation can take place in amplification of whole blood samples.19 One possibility could be that PEC components may inhibit certain proteases in blood. Additional experiments with isolated blood inhibitors, such as heme, hemin, lactoferrin, and IgG fraction may help in elucidating the role of PEC in overcoming PCR inhibition.
We recently found that blood PCR inhibitors can predominantly reduce the DNA extension speed of the wild-type Taq polymerase compared with the OT and OKT enzymes that seem to be capable of more efficiently traversing inhibitor-blocked DNA templates.27 It remains to be determined whether PEC can facilitate PCR of crude samples by affecting the enzyme speed and/or processivity.
In control experiments with β-actin gene amplification, we have not observed significant variations when using fresh or frozen and thawed blood in PCR in terms of PCR inhibition, most likely because the blood cells will eventually completely lyse anyway in the first heating step in PCR. However, we observed a higher fluorescent quenching rate in real-time PCR after five or more blood freezing/melting cycles. We speculate that with prelysed/frozen blood the heme released from erythrocytes, which is mainly responsible for that quenching effect, inactivates/blocks the fluorescent dye more efficiently early in the reaction. Accordingly, one can compensate for this by increasing the SYBR Green concentration in the real-time PCR protocol, a modification that we found to work very efficiently (present data).27
Furthermore, PEC was active with various commercial Taq enzymes, rendering them functional in crude blood samples, demonstrating the potential of its use as a universal additive to PCR. One exception for which PEC showed some inhibitory effect was with AmpliTaq Gold. This may have to do with the fact that this enzyme is chemically modified, and it could not possibly tolerate PEC.
Plasma and serum contain less heme/hemoglobin than whole blood and should have a less inhibitory effect on PCR performance than whole blood. Surprisingly, it was observed that OT and OKT could amplify targets from whole blood treated with three anticoagulants, whereas, with some targets, they were only active in heparin-treated plasma, not in the citrate- and EDTA-treated plasma. This gave us some clue that heparin may play a role in overcoming PCR inhibition. Indeed, heparin alone was able to relieve the inhibitory action at least in citrate- and EDTA-treated plasma samples here, and adding heparin to PEC (PEC-Plus) allowed efficient amplification from all crude specimens by OT and OKT. Another experiment showed that heparin alone could relieve the inhibition in serum samples (not shown). This finding was intriguing because heparin is known as an inhibitor of Taq polymerase and PCR.37 Heparin also inhibits restriction enzymes and polymerases because of its high-affinity binding to these enzymes and potentially competing for their interaction with DNA.34,35,37 The inhibitory effect of heparin in PCR, however, was only demonstrable with purified DNA templates not with crude blood samples. We assume that the highly negatively charged heparin may bind and inactivate some of the PCR inhibitors found in blood, plasma, and serum, consistent with the observation mentioned above that the heparin pretreated plasma is less inhibitory. We confirmed this assumption by a pulldown test, using heparin-agarose beads to deplete plasma for inhibitor(s) of PCR. Next we tested individually three known PCR inhibitors (human IgG fraction, lactoferrin, and hemin), in a PCR of purified DNA in the presence of heparin and found that heparin does indeed relieve the inhibitory effect of IgG and lactoferrin but not of hemin (data not shown). This finding partially explains the mechanism of heparin stimulation of PCR in the crude sample. The stimulation effect of heparin on DNA amplification in crude samples was dose-dependent, and we also saw inhibition at heparin concentrations exceeding 20 U/ml in plasma or serum. In addition, in this line of tests we found that the blood-resistant Taq mutants OT and OKT can better tolerate heparin compared with wild-type Taq (not shown), which may contribute to the optimal performance of PEC-Plus with these enzymes.
Real-time PCR detection from blood samples is more challenging than conventional PCR because blood components such as heme/hemoglobin quench the signal from fluorescence dyes. The SYBR Green I dye is inhibitory to Taq enzyme, which may impose certain limitations on the fluorescent signal and sensitivity of detection.38,39 We found earlier that the Taq mutants OT and OKT were able to tolerate extremely high concentrations of SYBR Green in crude samples, especially blood.27 This finding was very helpful, because using an excess of the dye could compensate for the quenching effect of the blood components. Our data with PEC showed that the enhancer performed efficiently in both SYBR Green detection real-time PCR and TaqMan assay, indicating that the PEC components do not compromise the fluorescence signal or generate a higher background.
In summary, we have demonstrated that the combination of d-(+)-trehalose, l-carnitine, and NP-40 (PEC), or these three compounds supplemented with heparin (PEC-Plus) can provide a potent and reliable enhancing effect on PCR amplification compared with that of other existing PCR additives, especially when crude clinical samples are applied in the PCR reaction. PEC reveals a universal effect in improving the performance of various commercial TaqDNA polymerases, especially in crude samples or GC-rich targets. When combined with the recently developed inhibition-resistant mutants of Taq, OmniTaq and Omni KlenTaq, PEC provides a unique protocol for simplified, fast, and cost-effective amplification of clinical and forensic samples from crude specimens without extracting template DNA.
Acknowledgements
We thank Lyubka Kirolova for her assistance in this project. We thank Dr. John E. Tavis, Dr. Lynda Morrison, and Dr. Mark Buller for kindly providing duck hepatitis B virus, herpes simplex, and mousepox virus, respectively, and helpful comments.
Footnotes
Supported by Small Business Innovation Research grant 2R44GM073401 from The National Institutes of Health.
Z.Z. and M.K. have applied for a patent for PCR enhancers (PEC, PEC-Plus, and their compounds) described in this article.
References
- 1.Cursons RT, Jeyerajah E, Sleigh JW. The use of polymerase chain reaction to detect septicemia in critically ill patients. Crit Care Med. 1999;27:937–940. doi: 10.1097/00003246-199905000-00029. [DOI] [PubMed] [Google Scholar]
- 2.Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR, Smith TF. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165–256. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rautenberg P, Lubbert C, Weers W, Boetel E, Schweichler J, Zhou L, Costard-Jackle A, Kraemer-Hansen H, Harder TC. Evaluation of the AmpliSensor PCR and the SHARP signal detection system for the early prediction of symptomatic CMV infection in solid transplant recipients. J Clin Virol. 1999;13:81–94. doi: 10.1016/s1386-6532(99)00013-x. [DOI] [PubMed] [Google Scholar]
- 4.Robertson JM, Walsh-Weller J. An introduction to PCR primer design and optimization of amplification reactions. Methods Mol Biol. 1998;98:121–154. doi: 10.1385/0-89603-443-7:121. [DOI] [PubMed] [Google Scholar]
- 5.Bussani C, Cioni R, Mattei A, Fambrini M, Marchionni M, Scarselli G. Prenatal diagnosis of common aneuploidies in transcervical samples using quantitative fluorescent-PCR analysis. Mol Diagn Ther. 2007;11:117–121. doi: 10.1007/BF03256231. [DOI] [PubMed] [Google Scholar]
- 6.Deng Z, Wu G, Li Q, Zhang X, Liang Y, Li D, Gao S, Lan Y. Noninvasive genotyping of 9 Y-chromosome specific STR loci using circulatory fetal DNA in maternal plasma by multiplex PCR. Prenat Diagn. 2006;26:362–368. doi: 10.1002/pd.1422. [DOI] [PubMed] [Google Scholar]
- 7.Kramvis A, Bukofzer S, Kew MC. Comparison of hepatitis B virus DNA extractions from serum by the QIAamp blood kit. GeneReleaser, and the phenol-chloroform method. J Clin Microbiol. 1996;34:2731–2733. doi: 10.1128/jcm.34.11.2731-2733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drosten C, Seifried E, Roth WK. TaqMan 5′-nuclease human immunodeficiency virus type 1 PCR assay with phage-packaged competitive internal control for high-throughput blood donor screening. J Clin Microbiol. 2001;39:4302–4308. doi: 10.1128/JCM.39.12.4302-4308.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolte FS, Fried MW, Shiffman ML, Ferreira-Gonzalez A, Garrett CT, Schiff ER, Polyak SJ, Gretech DR. Prospective multicenter clinical evaluation of AMPLICOR and COBAS AMPLICOR hepatitis C virus tests. J Clin Microbiol. 2001;39:4005–4012. doi: 10.1128/JCM.39.11.4005-4012.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akane A, Matsubara K, Nakamura H, Takahashi S, Kimura K. Identification of the heme compound copurified with deoxyribonucleic acid (DNA) from bloodstains, a major inhibitor of polymerase chain reaction (PCR) amplification. J Forensic Sci. 1994;39:362–372. [PubMed] [Google Scholar]
- 11.Morata P, Queipo-Ortuno MI, De Dios CJ. Strategy for optimizing DNA amplification in a peripheral blood PCR assay used for diagnosis of human brucellosis. J Clin Microbiol. 1998;36:2443–2446. doi: 10.1128/jcm.36.9.2443-2446.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Soud WA, Jonsson LJ, Radstrom P. Identification and characterization of immunoglobulin G in blood as a major inhibitor of diagnostic PCR. J Clin Microbiol. 2000;38:345–350. doi: 10.1128/jcm.38.1.345-350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satsangi J, Jewell DP, Welsh K, Bunce M, Bell JI. Effect of heparin on polymerase chain reaction. Lancet. 1994;343:1509–1510. doi: 10.1016/s0140-6736(94)92622-0. [DOI] [PubMed] [Google Scholar]
- 14.Al-Soud WA, Radstrom P. Purification and characterization of PCR-inhibitory components in blood cells. J Clin Microbiol. 2001;39:485–493. doi: 10.1128/JCM.39.2.485-493.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watkins-Riedel T, Ferenci P, Steindl-Munda P, Gschwantler M, Mueller C, Woegerbauer M. Early prediction of hepatitis C virus (HCV) infection relapse in nonresponders to primary interferon therapy by means of HCV RNA whole-blood analysis. Clin Infect Dis. 2004;39:1754–1760. doi: 10.1086/425614. [DOI] [PubMed] [Google Scholar]
- 16.Klungthong C, Gibbons RV, Thaisomboonsuk B, Nisalak A, Kalayanarooj S, Thirawuth V, Nutkumhang N, Manmen MP, Jr, Jarman RG. Dengue virus detection using whole blood for reverse transcriptase PCR and virus isolation. J Clin Microbiol. 2007;45:2480–2485. doi: 10.1128/JCM.00305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein A, Barsuk R, Dagan S, Nusbaum O, Shouval D, Galun E. Comparison of methods for extraction of nucleic acid from hemolytic serum for PCR amplification of hepatitis B virus DNA sequences. J Clin Microbiol. 1997;35:1897–1899. doi: 10.1128/jcm.35.7.1897-1899.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cattaneo C, Craig OE, James NT, Sokol RJ. Comparison of three DNA extraction methods on bone and blood stains up to 43 years old and amplification of three different gene sequences. J Forensic Sci. 1997;42:1126–1135. [PubMed] [Google Scholar]
- 19.Al-Soud WA, Rådström P. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J Clin Microbiol. 2000;38:4463–4470. doi: 10.1128/jcm.38.12.4463-4470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarti R, Schutt CE. The enhancement of PCR amplification by low molecular weight amides. Nucleic Acids Res. 2001;29:2377–2381. doi: 10.1093/nar/29.11.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakrabarti R, Schutt CE. Novel sulfoxides facilitate GC-rich template amplification. Biotechniques. 2002;32:866. doi: 10.2144/02324rr04. 868, 870, 872, 874. [DOI] [PubMed] [Google Scholar]
- 22.Henke W, Herdel K, Jung K, Schnorr D, Loening SA. Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res. 1997;25:3957–3958. doi: 10.1093/nar/25.19.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang J, Lee MS, Gorenstein DG. The enhancement of PCR amplification of a random sequence DNA library by DMSO and betaine: application to in vitro combinatorial selection of aptamers. J Biochem Biophys Methods. 2005;64:147–151. doi: 10.1016/j.jbbm.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Musso M, Bocciardi R, Parodi S, Ravazzolo R, Ceccherini I. Betaine, dimethyl sulfoxide, and 7-deaza-dGTP, a powerful mixture for amplification of GC-rich DNA sequences. J Mol Diagn. 2006;8:544–550. doi: 10.2353/jmoldx.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ralser M, Querfurth R, Warnatz HJ, Lehrach H, Yaspo ML, Krobitsch S. An efficient and economic enhancer mix for PCR. Biochem Biophys Res Commun. 2006;347:747–751. doi: 10.1016/j.bbrc.2006.06.151. [DOI] [PubMed] [Google Scholar]
- 26.Schnoor M, Voss P, Cullen P, Boking T, Galla HJ, Galinski EA, Lorkowski S. Characterization of the synthetic compatible solute homoectoine as a potent PCR enhancer. Biochem Biophys Res Commun. 2004;322:867–872. doi: 10.1016/j.bbrc.2004.07.200. [DOI] [PubMed] [Google Scholar]
- 27.Kermekchiev MB, Kirilova LI, Vail EE, Barnes WM. Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res. 2009;37:e40. doi: 10.1093/nar/gkn1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carninci P, Nishiyama Y, Westover A, Itoh M, Nagaoka S, Sasaki N, Okazaki Y, Muramatsu M, Hayashizaki Y. Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc Natl Acad Sci USA. 1998;95:520–524. doi: 10.1073/pnas.95.2.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bachmann B, Luke W, Hunsmann G. Improvement of PCR amplified DNA sequencing with the aid of detergents. Nucleic Acids Res. 1990;18:1309. doi: 10.1093/nar/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petry H, Bachmann B, Luke W, Hunsmann G. PCR sequencing with the aid of detergents. Methods Mol Biol. 1996;65:105–109. doi: 10.1385/0-89603-344-9:105. [DOI] [PubMed] [Google Scholar]
- 31.Beumer RR, Te Giffel MC, Cox LJ, Rombouts FM, Abee T. Effect of exogenous proline, betaine, and carnitine on growth of Listeria monocytogenes in a minimal medium. Appl Environ Microbiol. 1994;60:1359–1363. doi: 10.1128/aem.60.4.1359-1363.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Innis MS, Myambo KB, Gelfand DH, Brown MA. DNA sequencing with Thermus acquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA. 1988. Biotechnology. 1992;24:6–10. [PubMed] [Google Scholar]
- 33.Spiess AN, Mueller N, Ivell R. Trehalose is a potent PCR enhancer: lowering of DNA melting temperature and thermal stabilization of Taq polymerase by the disaccharide trehalose. Clin Chem. 2004;50:1256–1259. doi: 10.1373/clinchem.2004.031336. [DOI] [PubMed] [Google Scholar]
- 34.Burckhardt J. Amplification of DNA from whole blood. PCR Methods Appl. 1994;3:239–243. doi: 10.1101/gr.3.4.239. [DOI] [PubMed] [Google Scholar]
- 35.Chen JZ, Herzenberg LA, Herzenberg LA. Heparin inhibits EcoRI endonuclease cleavage of DNA at certain EcoRI sites. Nucleic Acids Res. 1990;18:3255–3260. doi: 10.1093/nar/18.11.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henke W, Loening SA. Recently, betaine has been introduced as an additive in different PCR strategies. Nucleic Acids Res. 1998;26:687. [PubMed] [Google Scholar]
- 37.Ghadessy FJ, Ong JL, Holliger P. Directed evolution of polymerase function by compartmentalized self-replication. Proc Natl Acad Sci USA. 2001;98:4552–4557. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nath K, Sarosy JW, Hahn J, Di Como CJ. Effects of ethidium bromide and SYBR Green I on different polymerase chain reaction systems. J Biochem Biophys Methods. 2000;42:15–29. doi: 10.1016/s0165-022x(99)00033-0. [DOI] [PubMed] [Google Scholar]
- 39.Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]