

Abstract
We analyse the trade-off between the speed with which a gene can propagate information, the noise of its output and its metabolic cost. Our main finding is that for any given level of metabolic cost there is an optimal trade-off between noise and processing speed. Any system with a non-vanishing leak expression rate is suboptimal, i.e. it will exhibit higher noise and/or slower speed than leak-free systems with the same metabolic cost. We also show that there is an optimal Hill coefficient h which minimizes noise and metabolic cost at fixed speeds, and an optimal threshold K which minimizes noise.
Keywords: noise, speed, metabolic cost, trade-off
1. Introduction
In this article we will probe some of the fundamental limitations on the computational capabilities of gene networks. There is a variety of different mechanisms by which living organisms perform computations, and they do so at many different levels. Examples include the nervous system in higher organisms, signal transduction networks (Han et al. 2007) or even individual proteins (Bray 1995). Understanding what constrains the efficiency and the speed of these computations is not only of practical relevance (for example in the context of engineering purpose built novel life forms, i.e. synthetic biology), but will most of all also provide new insights into the design principles of living systems.
In the context of this article we will contribute to this larger field by considering the computational properties of a single gene. By ‘computation’ we mean that the gene maintains a functional relationship between the concentration of one or several types of input molecules and the concentration of the product of the gene. Instantaneous changes of the input conditions will normally be processed with a delay, namely the transient time required for the output concentration of the gene to reach the steady state corresponding to the new input (in reality the actual switching time will be slower, because inputs do not change instantaneously). From a computational point of view, this switching time is relevant because it imposes an upper limit on the frequency with which gene regulatory networks can detect and process changes in the environment. In a very direct sense, this limiting frequency can be seen as the computational speed of the gene.
Connected to the question of speed is the question of accuracy. Biochemical systems are usually affected by noise (Spudich & Koshland 1976; Arkin et al. 1998; Elowitz et al. 2002; Becskei et al. 2005; Rosenfeld et al. 2005; Dunlop et al. 2008). For a comprehensive review of noise in genetic systems see Kaern et al. (2005), Kaufmann & van Oudenaarden (2007) and Raj & van Oudenaarden (2008). If we assume that the concentration of a gene product acts as a signal further downstream, then noise will make it harder to detect the signal. There are at least two solutions to the noise problem: (i) to reduce the noise that affects the output of the gene, or (ii) to accept higher levels of noise and to average out random fluctuations by ‘measuring’ the signal over longer times. The first solution comes at a higher metabolic cost, whereas the second solution implies that the rate at which signals can be built up by the gene is reduced. This points to a trade-off between the processing speed of a gene, the noise it produces and the metabolic cost of the processing.
The main objective of this contribution is to describe this trade-off mathematically. To do this we will use a (well established) method based on van Kampen's linear noise approximation to calculate the noise in gene expression (Elf & Ehrenberg 2003; Paulsson 2005; van Kampen 2007) to a high degree of accuracy. We show that for a fixed metabolic cost the noise of the output of the computation can only be improved by reducing the speed of the computation, and vice versa. A notable result of our contribution is also that there is a theoretical performance limit for the computational efficiency of the gene when the leak expression rate is vanishing. Finally, we will also show that there are parameters for the gene regulation function that optimize the computational properties of a gene.
Gene activation functions are often approximated by Hill functions (Ackers et al. 1982; Bintu et al. 2005; Chu et al. 2009). A Hill function is characterized by two parameters, namely K and h (table 1). The latter determines how close the activation function is to a step function, whereas K is the ratio of the binding and the unbinding rate of the regulators to the operator site.
Table 1.
Nomenclature.
| H | high output of the gene |
| L | low output of the gene |
| xH | input to the gene such that the output of the gene is H |
| xL | input to the gene such that the output of the gene is L |
| f(x) | gene activation function |
| fH, fL | f(xH), f(xL) |
| μ | decay rate of gene product |
| τ | average life time (1/μ) |
| α | basal synthesis rate |
| β | regulation synthesis rate |
| Δβ | β (fH − fL)/μ |
| ϕ, ϕ̄ | the Hill activation/repression function |
| K | the regulation threshold |
| h | the Hill coefficient |
| Tgene | the time for gene output to reach θ % of H − L. |
| θ | see Tgene |
| σ2 | the (absolute) variance |
| 𝒩 | noise, i.e. variance normalized by signal strength (σy2/(H − L)2) |
| 𝒩in, 𝒩ex | the intrinsic/extrinsic noise |
| ζ | the metabolic cost of species y |
In what follows, we will limit our attention primarily to binary genes, i.e. regulated genes which have only two states of activation. An active gene is characterized by a high steady-state concentration (or particle number) H of its output product; the low state is characterized by a low concentration/particle number of L. In this setting, any downstream elements (such as for example other genes) then need to distinguish between the high and the low output of the gene. The problem is that this output is afflicted by noise which makes it harder to distinguish between the two states.
In biological cells, the metabolic cost of any process can be measured by the number of adenosine triphosphate molecules it requires (Akashi & Gojobori 2002). Here, we will not be interested in a quantitatively exact measure of the energy expense, but rather in how the cost scales as various parameters are changed. We will therefore assume that the metabolic cost of a particular gene is well measured by the maximum expression rate of the gene in question. Note that the real cost will depend on the average proportion of time over which this gene is activated, plus a number of additional factors, such as the length of the protein to be produced and so on. These complications, however, are irrelevant for our current purpose, where we consider a single gene only.
Implicitly underlying the entire argument put forth in this article is the assumption that the cell is essentially a perfectly mixed reactor. This assumption is necessary to keep the mathematics tractable, and is commonly made. We expect that the conclusions about the trade-off between noise and speed are broadly valid in spatially extended systems as well (Newman et al. 2006). However, this is not addressed here. We also assume that the binding/unbinding dynamics of input molecules to the operator site of the gene in question is very fast compared with the transcription process; this assumption justifies the use of Hill-type gene activation functions.
Finally, we will model gene expression as a one-step process. This means that we essentially ignore post-transcriptional sources of noise, especially translation. Our results are therefore prima facie more about noise in mRNA signals, rather than noise in proteins. Yet, there is experimental evidence that mRNA production is indeed the dominant source of noise in the cell (Bar-Even et al. 2006; Newman et al. 2006). Moreover, it seems that typically translation merely scales the transcriptional noise (Bar-Even et al. 2006), and thus does not directly alter the computational properties of gene regulation.
2. Results
2.1. Deterministic time
We first consider the computational speed in a noiseless system. Our model system is a gene Gy with output y and regulated by a single input x. The system evolves according to the dynamical law given by the differential equation
| 2.1 |
Here α is the leak expression rate, (α + β) is the maximal expression rate of the gene, f(x) is the regulation function, x is the concentration of the regulator and μ is the degradation rate of the product of the gene. Clearly, this system is completely determined by the input to Gy, which we assume to be either xH (leading to the output of y = H) or xL (leading to y = L); we also assume H > L. We consider two families of regulation functions, namely f ≡ ϕ and ϕ̄:
Here, ϕ and ϕ̄ are the Hill and the repressor Hill function, respectively. Hill functions are customarily used to represent gene activation. They are sigmoid functions for h > 1 and approach a step function as h → ∞. If the transcription factors repressing/activating the gene are monomers, then the Hill coefficient h is bound from above by the number of binding sites.
At any time the actual metabolic expense attributable to a gene will be given by the time-dependent production rate of the system, α + βf(x(t)). Averaged over all environmental conditions, a gene will spend a certain fraction of time in the state H; the maximum production rate is therefore an indicator for the metabolic cost ζ associated with the gene:
| 2.2 |
Here, we use the shorthand fH≐f(xH) to simplify notation. In the case of α = 0, the metabolic cost becomes ζ = βfH.
Note that our notion of cost is an idealization relative to the real case. We only take into account production costs, whereas in reality there are a number of other costs related to the maintenance of protein signals. For example, if proteins are actively broken down (rather than just diluted) then this comes at an additional cost. This additional cost, however, is a constant factor and can be added to the production cost because ultimately every molecule that is produced will have to be broken down again. For some proteins the decrease in particle concentrations is mostly due to dilution, rather than active breakdown. When this is the case, then particle ‘decay’ is linked to growth and hence to metabolic cost. Yet, this link is not a necessary one. It is thus not relevant for the understanding of fundamental limitations constraining the noise, speed and metabolic cost of signals. Also, in addition to the cost of gene expression itself, the living cell has significant overhead metabolic costs associated with maintaining the necessary machinery of gene expression. These costs are equally not taken into account here because, again, they are not relevant to the fundamental limitation that is the main interest of this contribution. Instead, as far as costs are concerned the relevant measure is the marginal cost of protein production, rather than the total metabolic cost associated with protein levels. The production rate reflects this well and is thus a relevant indicator of cost when probing the fundamental limitations associated with the computational properties of genes.
Assuming the ideal case of an instantaneous change of the input signal from xL to xH so as to induce Gy at t = 0, the concentration of the output y will then evolve from its original steady state y0 = L at t = 0 according to
| 2.3 |
Here y* = H is the steady-state solution for equation (2.1):
We define the deterministic switching time, Tgene of the gene as the time required to reach the steady state to within a fraction θ of H − L (see figure 1). We can obtain from equation (2.3) the time required to reach a value of y0 + (y* − y0)θ given that we start from y0, by setting the left-hand-side equal to y0 + (y* − y0)θ and solve for t:
Figure 1.

Switching time. We set the cell volume to V = 8 × 10−16 l. The following set of parameters have been used: K = 0.5 µM and μ = 1 min−1. The two steady states are L = 0.2 µM and H = 0.8 µM. We consider both a switch (a) from low state to high state (L → H) and (b) from high state to low state (H → L). We chose θ = 0.9. Stochastic fluctuations do not influence significantly the time required to reach a fraction θ of the steady state concentration (solid line, deterministic; dashed line, stochastic).
Calling this solution Tgene we obtain the switching time of a gene:
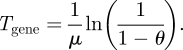 |
2.4 |
The only parameter which influences this switching time is the degradation rate of the product. Higher degradation rates generate faster responses. This suggests an immediate way for the deterministic gene to reduce its switching time, namely to increase the degradation rate μ as much as possible. Note that doing this also decreases the signal strength, H − L = β fH/μ.
In a ‘deterministic world’ very small signals can be discerned without any problems and do not limit the computational usefulness of the gene. We can therefore conclude from equation (2.4) that the time required to build up a signal can be decreased (and hence the computing speed increased) arbitrarily, as long as the gene output is noise-free and varies continuously. In deterministic systems, there is no need to increase the production rate (and hence the cost ζ) to offset any increase in μ. It will become clear below, however, that in real systems that are discrete and afflicted by noise the signal strength sets a fundamental computational performance limit for the genes.
2.2. Stochastic case
The products of real genes come in discrete units and their copy number is subject to random fluctuations. The quantized nature of molecules means that there is a logical minimum signal strength of H − L = 1 molecule. More importantly, for low signal strengths (even when they are well above the logical minimum) the presence of stochastic fluctuations may make it difficult to distinguish between the input and the output states. It is thus no longer possible to arbitrarily increase the computational speed of a gene by increasing the degradation rate. In order to maintain a signal that is large compared to stochastic fluctuations around the steady-state concentrations, the production rate must be increased when the degradation rate is increased. Unfortunately, this comes at the cost of an increased metabolic rate.
We will develop a quantitative understanding of the trade-off between speed, noise and the metabolic cost of a ‘computation’. Following Paulsson (2004, 2005) who in turn used van Kampen's linear noise approximation (Elf & Ehrenberg 2003; van Kampen 2007), we can derive the variance σ2 at steady state of a stochastic version of the system described by equation (2.1) by applying the fluctuation dissipation theorem (FDT; see §4). In first-order approximation this predicts noise to be a Gaussian distribution with mean 〈y〉 and variance σ2. In reality, the distribution will be a correction to the Gaussian. Yet, as long as the mean is sufficiently high, simulations and theoretical considerations (van Kampen 2007, ch. 10) show that the Gaussian approximation is very good; see figure 2. The linear noise approximation is only valid when the mean of the stochastic system corresponds to the solution of the deterministic system, which is the case here. The transition from the deterministic system to the stochastic system also requires that we consider particle numbers, rather than concentrations. For simplicity, we will not reflect this distinction in our notation; however, it is implicitly understood that stochastic systems/simulations always refer to particle numbers, rather than concentrations.
Figure 2.

Comparison between analytical solution and simulation data. We considered a cell volume of V = 8 × 10−16 l. The following set of parameters have been used: h = 3, K = 0.5 µM, L = 0.2 µM, H = 0.8 µM and μ = 1 min−1. At steady state, the two species have the following concentrations: x = 0.2 µM and y = 0.8 µM. The probability distribution of species x and y can be approximated by normal distributions.
The variance, which is a measure of the absolute fluctuations in particle numbers, will be much higher in the H state than in the L state. The distinguishability between the two possible output states is therefore poorest in the case of H; we will henceforth only use this pessimistic estimate. In the context of this contribution, we are interested in how noise affects our ability to distinguish between the two known output states, H and L. To get a meaningful measure of this, we will subtly change the definition of noise with respect to convention. Normally, noise is defined as the variance normalized by the square of the mean value of the varying quantity. Since we are interested in how noise affects our ability to distinguish between two known values, we will adjust this and use as the relevant measure of noise the variance normalized by the square of the signal strength, 𝒩≐σH2/(H − L)2, rather than by the square of the mean (which is often used as a definition of noise):
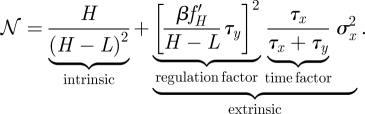 |
2.5 |
Here σx2 is the variance of the input signal, H and L the steady-state particle number of the output species for inputs xH and xL, respectively, and we denote by τx = 1/μx and τy = 1/μy, the average life time of the two species; finally, f′ denotes the derivative of f with respect to its argument. The first term on the right-hand-side of the normalized variance of 𝒩 in equation (2.5) represents the intrinsic noise (𝒩in), while the second term represents the extrinsic noise (𝒩ex). The extrinsic noise has two components, the first of which is the time factor, which can be thought of as the time over which the gene averages its input. The other term is the regulation factor (see Paulsson 2005; Pedraza & van Oudenaarden 2005; Shibata & Fujimoto 2005; Shibata & Ueda 2008).
We checked the accuracy of the linear noise approximation by performing extensive simulations using Gillespie's algorithms (see the electronic supplementary material and figure 2 for details). A comparison with analytic results shows excellent agreement between the noise obtained from simulation and calculated values of the noise.
2.3. Noise, time and cost
We will first make the idealizing assumption that there is no leak expression, i.e. α = 0, and we will further assume that L = 0. The (mean) high state H can be calculated from the deterministic equation to H = β fH/μ. Following equation (2.5) the noise can be written as
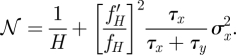 |
2.6 |
We first consider the case of scaling the production rate by a factor of γ, i.e.
The average particle number at the high state H scales by the same factor γ, i.e. Hβ′ = γHβ. Here the subscript β indicates that the relevant mean particle number is generated by a system with production rate β.
According to equation (2.6), scaling β leaves the extrinsic noise, 𝒩ex, unaffected. However, the intrinsic noise, 𝒩in, scales with γ−1:
| 2.7 |
As expected, increasing the production rate of a gene product increases the cost, but reduces the noise correspondingly. This suggests a noise–metabolic cost trade-off.
We next scale the degradation rate, which we take to be a free parameter:
Figure 3 illustrates this equation for various costs (see also the electronic supplementary material). From the steady-state equation it is clear that μ and y scale in opposite directions. Scaling the degradation rate leads to an overall change of the noise of Gy even at constant cost ζ as follows:
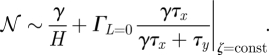 |
2.8 |
Figure 3.

Relation between noise (per molecule based on the concentrations given below and the typical volume of E. coli), deterministic time, and metabolic cost. The metabolic cost of each point is indicated by a shade of grey. The equal cost points fall onto a line. Different costs were achieved by varying the maximum rate of production of β. The following set of parameters have been used: x = 0.2 µM, α = 0, K = 0.5 µM, h = 3. The degradation rate was varied in interval μ ∈ [0.36,1] (min−1) and the synthesis rate in interval β ∈ [0.34,0.85] (µM min−1). These are results obtained from the analytic solution (equation (2.9)); we found close agreement with results from stochastic simulations (see the electronic supplementary material). The time was computed for θ = 0.9. We considered a cell volume of V = 8 × 10−16 l.
Here Γ summarizes terms in equation (2.5) that remain constant under the scaling
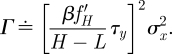 |
(Note that τy appears in Γ but scales into opposite direction to H − L and hence Γ as a whole is constant under the scaling.) Equation (2.8) establishes a degradation rate–noise trade-off. Formally equivalent, but of more intuitive appeal is to consider scaling the inverse of the degradation rate, namely
Remembering the inverse relationship between τy and μ, the scaling can be obtained from
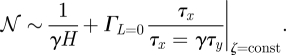 |
2.9 |
The inverse degradation rate is—up to a factor—equal to the deterministic switching time (see equation (2.4)) which in turn is an indicator of the computational speed of the gene. Hence, this establishes the noise–speed trade-off at constant cost. We will show below that equation (2.9) describes a theoretical computational performance limit of the gene at fixed cost, in the sense that the noise and speed characteristics cannot be simultaneously improved, without also increasing the metabolic cost.
2.3.1. Noise in the case of non-vanishing leak expression.
So far we have assumed that α = 0. We will now relax this assumption and assume a non-vanishing α. A consequence of this is that L > 0. For any fixed value of α there exists a (non-optimal) set of noise–speed trade-offs at fixed cost. This can be obtained by modifying equation (2.9):
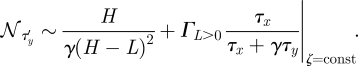 |
2.10 |
The main difference between equations (2.9) and (2.10) is the first term. In the case of L = 0—that is in equation (2.9)—this term reduces to 1/γH. For a given cost ζ, the set of possible noise–speed trade-offs with α > 0 is ‘worse’ than the optimal noise–speed trade-off set in the sense that for a fixed noise, the speed is always lower, and vice versa; this is illustrated for some parameters in figure 4.
Figure 4.

Comparison of various α at fixed metabolic cost. We plot the speed and noise (per molecule based on the concentrations given below and the typical volume of E. coli) trade-off for different leak rates under fixed metabolic cost. The metabolic cost of the three curves is constant, ζ = 0.8 µM min−1. We consider three cases: αy = 0 µM min−1 (solid line), αy = 0.05 µM min−1 (dashed line) and αy = 0.10 µM min−1 (dashed dotted line). The following set of parameters have been used: x = 0.2 µM, K = 0.5 µM, h = 3. The degradation rate was varied in interval μ ∈ [1,0.36] (min−1). We considered a cell volume of V = 8 × 10−16 l.
In general the suboptimality of α > 0 can be directly seen from the expression for the noise (equation (2.5)) as follows: the extrinsic noise is unaffected by α, so we only need to consider the intrinsic noise given by
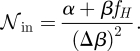 |
2.11 |
Here Δβ is a shorthand for β(fH − fL)/μ. From equation (2.11) it is clear that the 𝒩in and hence the noise increases with α. At the same time, increasing α also increases the cost; this follows from the definition of the metabolic cost in equation (2.2). Altogether, this shows that any binary gene with non-vanishing α is sub-optimal with respect to its noise and cost characteristics.
We now consider how the noise scales with β when α > 0. If β is scaled by γ and α = 0, then the corresponding change of the total cost will be exactly offset by an inverse scaling in the noise. When there is a leak expression, that is α > 0, then this is no longer the case. Instead, if β′ = γβ, then
where δ is given by
From this we can obtain the scaling property of the intrinsic noise (see appendix B)
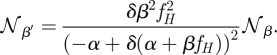 |
For α = 0 this scaling factor reduces to 1/δ, which in turn reduces to 1/γ, thus recovering the above scaling from equation (2.7). This scaling relation implies (see appendix B for details) that systems with α > 0 have a more favourable scaling behaviour than leak-free systems, in the sense that for a given increase of the cost, leak systems show a more pronounced decrease of noise than leak-free systems. A corollary of this is that for increasing expression rates the systems with and without leak become more similar.
2.3.2. Noise and the Hill parameter K.
In the case of the repressed gene (i.e. f ≡ ϕ̄), it may be the case that L > 0 due to incomplete repression (even when α = 0). It is clear from the shape of the repression function ϕ̄ that complete repression can only be achieved in the limiting cases of either an infinite number of repressor molecules or an infinite Hill coefficient h. Neither is realizable. If we assume the input signal (i.e. xH and xL) to be fixed then the leak rate will depend on the Hill parameter K. Physically, this parameter is in essence the fraction of the association and dissociation rate constants of the regulatory protein to/from the specific binding site in the operator. Clearly, the lower K the fewer molecules are required to achieve a certain level of repression. The value of K which maximizes signal strength H − L is given by 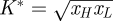 . This
. This  is not identical to the value of K that optimizes noise, which is given by the solution to
is not identical to the value of K that optimizes noise, which is given by the solution to
| 2.12 |
The corresponding formula is too complicated to be useful, but can be calculated numerically; it will typically be similar, but not equal, to the value that optimizes signal strength. Moreover, a numerical analysis suggests that around the optimal value of K the noise depends only very weakly on K, particularly when the signal strength of the input, |xL − xH| is large (see figure 5).
Figure 5.

The total noise 𝒩in + 𝒩ex as a function of K in the repressor case. We used the following set of parameters: h = 3, xH = 0.2 µM, τx = 1 min, τy = 1 min, σx2 = 96. We compute the noise per molecule based on the concentrations given above and a volume of V = 8 × 10−16 l (solid line, xL = 3 µM; long-dashed line, xL = 4 µM; short-dashed line, xL = 5 µM; dotted line, xL = 6 µM).
2.3.3. Dependence of noise on h.
To understand the dependence of noise on h it is necessary to consider f ≡ ϕ and ϕ̄ separately. Since we always consider the noise at y = H, the scaling relation of the repressor needs to be evaluated at very low particle numbers of x, whereas the noise of the activator needs to be evaluated at high x. In the cases of f ≡ ϕ̄ and ϕ the noise changes with h like
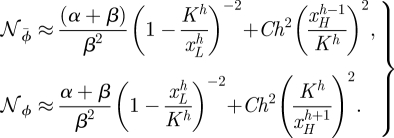 |
2.13 |
In the repression case we assumed xH≪K≪xL, and in the case of activation we assumed xH≫K≫xL. The details of the approximation can be found in appendix C.
Figure 6 illustrates how the noise depends on h. For a specific example the graph suggests that improvements in the noise for increased h diminish fast as h increases. In general, this diminishing effect of h can be seen directly from equation (2.13). In the second term on the right-hand-side in both equations the factors next to h2 are very small in both cases and decrease with h faster than h2; C summarizes terms in the noise equation that do not change with h. Hence, altogether the extrinsic noise tends to zero as h increases.
Figure 6.

The noise (per molecule based on the concentrations given below and the typical volume of E. coli) as a function of h. We used the following set of parameters: K = 0.5 µM, xH = 0.2 µM, τx = 1 min, τy = 1 min, σx2 = 96, xL = 4 µM, α = 0.05 µM min−1, β = 0.8 µM min−1. We also use a cell volume of 8 × 10−16 l (solid line, intrinsic noise; long dashed line, extrinsic noise; short dashed line, total noise).
This begs the question of the metabolic cost of increasing h. An analysis of this needs to consider, for a given K, how many molecules of activator are necessary to guarantee occupation of the binding site within a fixed time. An analysis shows that the cost strictly increases with h (see appendix A). The increase in the metabolic cost combined with the diminishing efficiency in noise reduction suggests that there is an optimal Hill coefficient beyond which a further increase is not cost-effective. We conclude that there exists an optimum value of h.
3. Discussion
While in an ideal, deterministic system, binary genes could be driven at an arbitrary speed with minimal cost, noise and discrete output states impose strict limits on the computational efficiency of real genes. We found that there is a three-way trade-off between the output noise of a gene, its switching time and the metabolic cost necessary to maintain it.
For a given metabolic cost, there is a noise–computational time trade-off. The set of possible noise–speed trade-offs, as defined by equations (2.9) and (2.10), represents the possible noise–time pairs a gene can take at a given cost. The trade-off set is optimal if α = 0, i.e. for a given metabolic cost, there are no solutions that have both better noise characteristics and higher computational speed. Figure 3 illustrates ideal trade-off sets, for α = 0 for various costs (indicated by the shades of grey of the points). Figure 4, on the other hand, illustrates that the trade-off set for α > 0 has worse noise–time characteristics than the leak-free system. The suboptimality of genes with α > 0 is also directly demonstrated by equation (2.11), which shows that increasing α leads to an increase in both the cost and the intrinsic noise; the extrinsic noise is unaffected by α.
A non-vanishing α is suboptimal; however, it can be improved upon cheaper than optimal sets, by increasing the production rate β. Hence, for a fixed leak rate, the optimal noise–speed trade-off can be approached by increasing the total energy expenditure (see equation (B 1) in appendix B).
In repressed genes, there will be a base-level of gene expression even at the maximum concentration of the repressor, which is due to the nature of the Hill repression function. The amount of the residual gene expression depends on the input signal (xL) and is not necessarily under the control of the cell. In any case, if we assume the strength of the input signal to be fixed, then there is an optimal value of K that minimizes output noise (see equation (2.12)). Similarly, according to equations (2.13) there is an optimal value of the Hill coefficient, that minimizes noise and metabolic expense. In a system of binary genes, there are therefore optimal values for the parameters α, K and h, whereas there is a trade-off for β and μ.
In this contribution we have considered only binary genes, with a single species of input. Real genes are typically controlled by more than only one species of regulatory molecules, often both activators and repressors. The expression level of the gene will depend on the combination of the inputs. Our analysis would then still be valid for each regulator species, given that the concentration of all other species is constant.
An immediate extension of the ideas presented here is to consider networks of genes. We would expect that such networks have a number of additional constraints as far as their computational properties are concerned. Both from a computational, but also a synthetic biology point of view, understanding the computational properties of such networks is of fundamental interest. In this context Shannon's information theory could provide an interesting alternative perspective, in that it could provide a theoretical framework to investigate signal transmission in cells. A gene could then be considered a communication channel and one could measure the channel's capacity, i.e. the maximum information in bits per second that a communications channel can handle.
4. Material and Methods
4.1. Simulations
In all figures we used the repressed gene f ≡ ϕ̄ for illustration. In figures 3 and 4 the noise was computed analytically, where we assumed fL = 0, L = α/μy (µM) and the parameters, h = 3, x = 0.2 µM, and K = 0.5 µM.
The error bars in figure 4, and in the electronic supplementary material were obtained by stochastic simulations. We used the Dizzy (Ramsey et al. 2005) implementation of the Gillespie algorithm (Gillespie 1977; Gibson & Bruck 2000). For all simulations we used the following set of chemical reactions:
| 4.1 |
For figure 2 the simulations were run for 106 min. The error bars from figure 4 represent the range of a sample of 10 simulations each run for 105 min. In the figure caption we measured the abundance of species x and y in concentrations instead of particle numbers. Using a cell volume of V = 8 × 10−16 l, which is the average volume of E. coli (Santillan & Mackey 2004), we converted from concentrations into number of molecules using x = x̃VNA, where x̃ is the concentration of x and NA is the Avogadro number (NA = 6.02214179 × 1023 mol−1).
4.2. Fluctuation dissipation theorem
We indicate here briefly how to derive the expression for the variance of the output of a gene using the FDT. At steady state we have (Paulsson 2004, 2005)
| 4.2 |
where A is the drift matrix, B the diffusion matrix and C the covariance matrix. The matrix A is the following:
| 4.3 |
Here Ji− is the elimination flux of species i, and Ji+ the production flux of species i. For chemical reaction system (4.1), A becomes
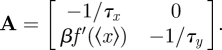 |
In the case when each chemical reaction adds or removes only one molecule, B becomes diagonal, with Bii = 〈Ji+〉 + 〈Ji−〉. For system 1, B yields
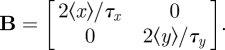 |
Having determined A and B, one can solve equation (4.2) and completely determine the covariance matrix C. The variance of species y, σy2, yields
| 4.4 |
Using stochastic simulations we show that for most parameters this equation estimates the noise very accurately for the systems we consider (see the electronic supplementary material). We also found that when the Hill parameter K is close to xH or xL then the accuracy of the method suffers. This is expected, since in this case, the mean behaviour of the stochastic system may deviate from the behaviour of the deterministic system.
Appendix A. Dependence of cost on Hill coefficient
The gene activation function f(x) is commonly modelled as a Hill function. As long as the transcription factor (TF) regulating the gene is a monomer, the Hill coefficient is bounded from above by the number of regulatory binding sites. Hence, if the operator site has two binding sites, then h ≤ 2. The case of h = 2 will only be reached in the limiting case of infinite cooperativity between the two sites. In essence, this means that the average binding time of a single TF is zero unless both sites are occupied. Hence, infinite cooperativity requires that all binding sites are occupied simultaneously. For large, but finite cooperativity, this condition can be relaxed and even if not all binding sites are occupied individual TFs will remain bound for a finite (albeit short) period of time.
Here, we assume that regulatory proteins are bound for a small amount of time T only, unless all binding sites are occupied; furthermore, the number of binding sites is h. Hence, in order to activate/repress the regulated gene it is necessary that, within the time period T of the first protein binding, the remaining (h − 1) sites will be occupied as well. The basic idea is as follows: the more binding sites there are, the more unlikely it is that all binding sites are occupied within T. To put it differently, in order to keep the waiting time for occupation of all h sites constant, a higher number of regulatory sites needs to be compensated by a higher concentration of regulatory proteins, which comes at a metabolic cost. Since the binding and un-binding dynamics of TFs to/from the operator site happens at a faster time scale than gene expression, the particular time to achieve full occupation does not matter as long as it is above a certain threshold that guarantees the separation of time scales between gene expression and the operator dynamics.
To make the argument more formal, assume now, that at some time t = 0 a first TF binds to one of the binding sites and that this first TF remains bound, on average, for a time period of T. Any subsequent TF also remains bound for a time T unless full occupation is reached; in this case then all TFs remain bound for a time period ≫T. We denote the average number of binding events per unit time for TFs to any of the free binding sites as r. Strictly speaking, this r will decrease as more and more binding sites are occupied, but we will ignore this complication for now (that is we assume that the copy number of the TF is very large compared to h).
Note that r is an indicator for metabolic cost in the following sense: it reflects the rate with which individual molecules collide with the binding site and for a fixed geometry of the cell it depends solely on the TF concentration. Hence the cell can increase r by increasing the number of regulatory proteins, which comes at a cost. However, note that r is not identical to the cost ζ above, but instead related to the cost of maintaining a sufficiently high concentration of TFs in the cell, so as to achieve that the probability per time unit that all binding sites are occupied is at least equal to a certain threshold value η that guarantees the above-mentioned separation of time scales, i.e. Pall ≥ η.
We can estimate the probability that within a time period T all h binding sites are occupied. This is the probability that within T there are (h − 1) or more binding events. Binding events are Poisson distributed and thus the probability of all sites being occupied within T is given by
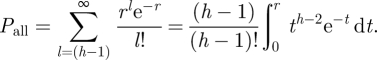 |
A 1 |
Here we chose units such that T = 1 to simplify the notation. This integral cannot be solved for arbitrary h. It is instructive to consider specific solutions. For example, for h = 3 one obtains
A closer inspection of Pall reveals that it defines a family of sigmoid functions parametrized by h; see figure 7.
Figure 7.

The probability of all binding sites occupied versus the cost. We consider this function for various h (solid line, h = 3; long-dashed line, h = 5; short-dashed line, h = 7; dotted line, h = 9; dashed-dotted line, h = 11).
The precise value of Pall is immaterial for gene expression as long as this process happens at a time scale that is fast compared with the typical time of gene expression. We therefore assume that Pall is kept at some (unspecified) threshold value η that guarantees efficient regulation of the operon in question.
In order to understand how Hill coefficients increase the cost, we need to find h as a function of r for fixed η. Unfortunately, it is not possible to derive this from equation (A 1) analytically. From figure 7, it is however possible to understand the dependence graphically. To do this, we choose an η, for example η = 0.5. Looking at the graph it is clear that for increasing h the fixed value of η is reached for increasingly high values of r and hence cost. A numerical solution of equation (A 1) indicates that this dependence is approximately linear for moderate values of h (data and calculation not shown).
This establishes the dependence of the metabolic cost on h.
Appendix B. Scaling of β in the case of non-zero α
Consider the scaling of β → β′ = γβ. This leads then to a corresponding increase of the total cost to ζ = α+ γβfH. With β scaling by γ the total cost scales by a factor of δ, defined by
Solving for γ, we obtain
Considering that the intrinsic noise is given by
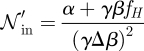 |
one can see that the intrinsic noise scales as
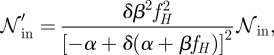 |
B 1 |
whereas the total cost scales as ζβ′ = δζβ. If we assume that δ > 1, i.e. we increase the cost, then the scaling term is smaller than 1/δ:
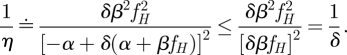 |
This implies that η ≥ δ, or that the actual decrease in noise is more than the inverse of the increase in metabolic cost.
Appendix C. Dependence of noise on h
C.1. Repression
We consider the case of f ≡ ϕ̄. In this case, the intrinsic noise is given by
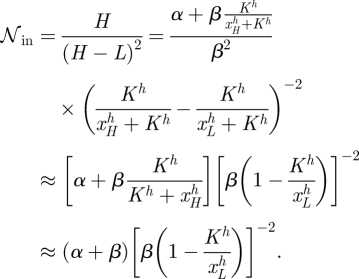 |
C 1 |
Here approximated using xH≪K, and xL≫K. Similarly, if C summarizes factors in the extrinsic noise that are not affected by h, then the extrinsic noise scales as
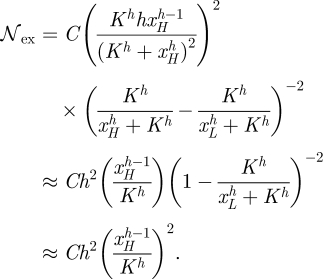 |
C 2 |
Both the intrinsic and the extrinsic noise are decreasing functions of h; however, it is clear from the equations that for higher values of h the slopes of both equation (C 1) and equation (C 2) approach 0 relatively rapidly. Hence, with increasing h, the dependence of the noise on h becomes increasingly weak. Altogether, we therefore obtain for the noise to change as a function of h like (see figure 6)
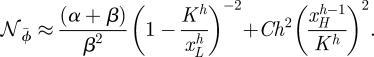 |
C 3 |
C.2. Activation
Using analogous approaches, but now assuming that xH≫K, and xL≪K and f ≡ ϕ, we obtain for the intrinsic noise the expression
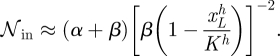 |
The extrinsic noise can be written as
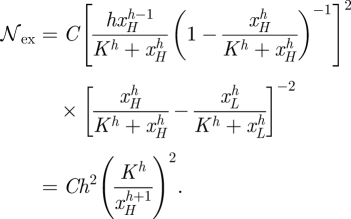 |
Hence, in the activation case, the noise depends on h as follows:
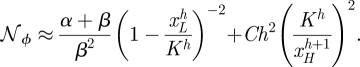 |
C 4 |
References
- Ackers G. K., Johnson A. D., Shea M. A. 1982. Quantitative model for gene regulation by lambda phage repressor. Proc. Natl Acad. Sci. USA 79, 1129–1133. ( 10.1073/pnas.79.4.1129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H., Gojobori T. 2002. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl Acad. Sci. USA 99, 3695–3700. ( 10.1073/pnas.062526999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin A., Ross J., McAdams H. H. 1998. Stochastic kinetic analysis of developmental pathway bifurcation in phage l-infected Escherichia coli cells. Genetics 149, 1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Even A., Paulsson J., Maheshri N., Carmi M., O'Shea E., Pilpel Y., Barka N. 2006. Noise in protein expression scales with natural protein abundance. Nat. Genet. 38, 636–643. ( 10.1038/ng1807) [DOI] [PubMed] [Google Scholar]
- Becskei A., Kaufmann B. B., van Oudenaarden A. 2005. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nat. Genet. 37, 937–944. ( 10.1038/ng1616) [DOI] [PubMed] [Google Scholar]
- Bintu L., Buchler N. E., Garcia H. G., Gerland U., Hwa T., Kondev J., Phillips R. 2005. Transcriptional regulation by the numbers: models. Curr. Opin. Genet. Dev. 15, 116–124. ( 10.1016/j.gde.2005.02.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D. 1995. Protein molecules as computational elements in living cells. Nature 376, 307–313. ( 10.1038/376307a0) [DOI] [PubMed] [Google Scholar]
- Chu D., Zabet N. R., Mitavskiy B. 2009. Models of transcription factor binding: sensitivity of activation functions to model assumptions. J. Theor. Biol. 257, 419–429. ( 10.1016/j.jtbi.2008.11.026) [DOI] [PubMed] [Google Scholar]
- Dunlop M. J., Cox R. S., III, Levine J. H., Murray R. M., Elowitz M. B. 2008. Regulatory activity revealed by dynamic correlations in gene expression noise. Nat. Genet. 40, 1493–1498. ( 10.1038/ng.281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J., Ehrenberg M. 2003. Fast evaluation of fluctuations in biochemical networks with the linear noise approximation. Genome Res. 13, 2475–2484. ( 10.1101/gr.1196503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M. B., Levine A. J., Siggia E. D., Swain P. S. 2002. Stochastic gene expression in a single cell. Science 297, 1183–1186. ( 10.1126/science.1070919) [DOI] [PubMed] [Google Scholar]
- Gibson M. A., Bruck J. 2000. Efficient exact stochastic simulation of chemical systems with many species and many channels. J. Phys. Chem. A 104, 1876–1889. ( 10.1021/jp993732q) [DOI] [Google Scholar]
- Gillespie D. T. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361. ( 10.1021/j100540a008) [DOI] [Google Scholar]
- Han Z., Vondriska T. M., Yang L., MacLellan W. R., Weiss J. N., Qu Z. 2007. Signal transduction network motifs and biological memory. J. Theor. Biol. 246, 755–761. ( 10.1016/j.jtbi.2007.01.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaern M., Elston T. C., Blake W. J., Collins J. J. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464. ( 10.1038/nrg1615) [DOI] [PubMed] [Google Scholar]
- Kaufmann B. B., van Oudenaarden A. 2007. Stochastic gene expression: from single molecules to the proteome. Curr. Opin. Genet. Dev. 17, 107–112. ( 10.1016/j.gde.2007.02.007) [DOI] [PubMed] [Google Scholar]
- Newman J. R. S., Ghaemmaghami S., Ihmels J., Breslow D. K., Noble M., DeRisi J. L., Weissman J. S. 2006. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846. ( 10.1038/nature04785) [DOI] [PubMed] [Google Scholar]
- Paulsson J. 2004. Summing up the noise in gene networks. Nature 427, 415–418. ( 10.1038/nature02257) [DOI] [PubMed] [Google Scholar]
- Paulsson J. 2005. Models of stochastic gene expression. Phys. Life Rev. 2, 157–175. ( 10.1016/j.plrev.2005.03.003) [DOI] [Google Scholar]
- Pedraza J. M., van Oudenaarden A. 2005. Noise propagation in gene networks. Science 307, 1965–1969. ( 10.1126/science.1109090) [DOI] [PubMed] [Google Scholar]
- Raj A., van Oudenaarden A. 2008. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. ( 10.1016/j.cell.2008.09.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey S., Orrell D., Bolouri H. 2005. Dizzy: stochastic simulation of large-scale genetic regulatory networks. J. Bioinformatics Comput. Biol. 3, 415–436. ( 10.1142/S0219720005001132) [DOI] [PubMed] [Google Scholar]
- Rosenfeld N., Young J. W., Alon U., Swain P. S., Elowitz M. B. 2005. Gene regulation at the single-cell level. Science 307, 1962–1965. ( 10.1126/science.1106914) [DOI] [PubMed] [Google Scholar]
- Santillan M., Mackey M. C. 2004. Influence of catabolite repression and inducer exclusion on the bistable behavior of the lac operon. Biophys. J. 86, 1282–1292. ( 10.1016/S0006-3495(04)74202-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T., Fujimoto K. 2005. Noisy signal amplification in ultrasensitive signal transduction. Proc. Natl Acad. Sci. USA 102, 331–336. ( 10.1073/pnas.0403350102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T., Ueda M. 2008. Noise generation, amplification and propagation in chemotactic signaling systems of living cells. BioSystems 93, 126–132. ( 10.1016/j.biosystems.2008.04.003) [DOI] [PubMed] [Google Scholar]
- Spudich J. L., Koshland D. E. 1976. Non-genetic individuality: chance in the single cell. Nature 262, 467–471. ( 10.1038/262467a0) [DOI] [PubMed] [Google Scholar]
- van Kampen N. G. 2007. Stochastic processes in physics and chemistry, 3rd edn North-Holland Personal Library Amsterdam, The Netherlands: North-Holland. [Google Scholar]