

Abstract

An enantioselective synthesis of α-methylene-β-hydroxy carboxylic acid derivatives via a highly diastereoselective, one-pot syn-aldol and β-elimination sequence utilizing the chiral β-(phenylselenyl)propionyl imide 15 is described. This new method, which constitutes an alternative to the Baylis-Hillman reaction, has been applied to the synthesis of the C(15)-C(21) fragment of tedanolide C.
Tedanolide C (1) is the newest member of a family of marine natural products which include tedanolide (2), 13-deoxytedanolide (3) and the candidaspongiolides (4, Figure 1).1–4 Tedanolide C was isolated from a Papua New Guinea marine sponge of the Ircinia species, and its structure and relative stereochemistry were assigned by NMR methods in conjunction with molecular modeling and DFT calculations. Tedanolide C displays an IC50 value of 0.057 µg/mL (95 nM) against HCT-116 cells (colorectal cancer cell line), and also arrests growth of HCT-116 cells in the S-phase after 24 h exposure at 0.2 µg/mL. It has been suggested that tedanolide C, like 13-deoxytedanolide (3), may be a protein synthesis inhibitor.1
Figure 1.

Family of tedanolide natural products
In planning a synthesis of tedanolide C (1), we elected to target the enantiomeric structure 5 (Scheme 1). The C(10)–C(23) fragment has been identified as the key pharmacophoric unit of 13-deoxytedanolide,5 but the stereochemistry of the corresponding fragment has been given the enantiomeric configuration (except for the epoxide) in the proposed structure of tedanolide C (1);1 the absolute configuration of tedanolide C has not been assigned. Therefore, we selected the enantiomeric stucture 5 as the synthetic target in anticipation that this will lead to the biologically active, naturally occuring enantiomer.
Scheme 1.

Retrosynthetic anaylsis of the C(15)–C(23) fragment of tedanolide C (synthetic target 5).
Our retrosynthetic analysis (Scheme 1) of target 5 focuses on the formation of the tertiary hydroxy group at C(16) by a dihydroxylation reaction of alkene 7. In order to synthesize 7, we considered using the Baylis-Hillman reaction between epoxyaldehyde 8 and methyl acrylate (9).6,7 However, this reaction failed (Scheme 1), presumably because the valuable, synthetically advanced aldehyde intermediate 8 could not be used in large excess—as is generally required for bimolecular Baylis-Hillman reactions.6,7
We therefore considered the possibility of using an aldol reaction to prepare Baylis-Hillman type products in an enantioselective fashion.8 Specifically, we anticipated that an acyl oxazolidinone like 10, equiped with a leaving group at C(3) of the propionyl fragment, would be a suitable substrate for a boron-mediated Evans syn-aldol reaction (Scheme 2).9 This approach would have the advantage that both the masked Michael acceptor 10 and the aldehyde could be used in stoichiometric amounts.
Scheme 2.

Rationale of applying aldol chemisty for the preparation of Baylis-Hillman type products.
As additional design criteria, the leaving group “X” in 10 was required to be stable under the aldol reaction conditions but to undergo β-elimination during reaction work-up. We anticipated that the phenylselenyl group, as in 15, would satisfy these criteria.8 Accordingly, acyl oxazolidinone 15 was synthesized in high yield by 1,4-addition of PhSeH to known acryloyl imide 14 and subsequent re-crystallization.10
Oxazolidinone 15 was readily converted into the corresponding boron enolate upon treatment with Bu2BOTf and NEt3 under standard conditions (c = 0.2 M in CH2Cl2, −78 °C).9a Addition of isobutyraldehyde to the enolate solution at −78 °C to promote the aldol reaction (6 h at up to 0 °C), followed by oxidative workup with H2O2 and pyridine at 0 °C resulted in the oxidation of the phenylselenide to the selenoxide which underwent a subsequent β-elimination to give the allylic alcohol 13a in 81% yield.
A wide variety of aldehydes are compatible with this methodology (Table 1). Simple aliphatic aldehydes such as isobutyraldehyde and propionaldehyde gave α-methylene-β-hydroxy imides 13a and 13b in 81–93% yield (entries 1, 2). Use of the stereochemically demanding pivaldehyde as substrate gave product 13c in 56% yield after conversion to the TBS ether (entry 3).
Table 1.
Synthesis of α-methylene-β-hydroxy acyl oxazolidinones 13a–i via an aldol-elimination sequencea
| entry | aldehyde | product b,c | yield d | |
|---|---|---|---|---|
| 1 | 13a | 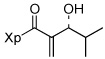 |
81% | |
| 2 | 13b | 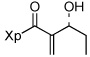 |
93% | |
| 3 | 13c | 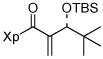 |
56%e | |
| 4 | 13d | 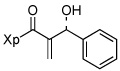 |
86% | |
| 5 | 13e | 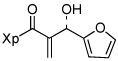 |
86% | |
| 6 | 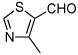 |
13f | 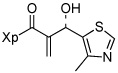 |
84% |
| 7 | 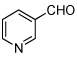 |
13g | 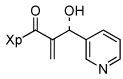 |
88% |
| 8 | 13h | 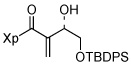 |
88% | |
| 9 | 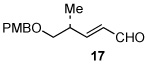 |
13i | 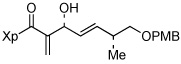 |
76% |
General procedure for the aldol-elimination sequence: Formation of the boron enolate from imide 15 (1.0 equiv), Bu2BOTf (1.2 equiv) and NEt3(1.8 equiv) in CH2Cl2(c = 0.2 M) at −78 °C; addition of aldehyde (1.1 equiv) at −78 °C; oxidation of the aldol product in CH2Cl2 and pyridine (2.0 equiv) at 0 °C with H2O2 (approx. 3–5 equiv); all products were isolated by column chromatography;
Xp = (4S)-(4-benzyloxazolidinone)-5-yl;
dr >20:1;
Isolated yields of the indicated products;
13a was isolated after protection as the TBS ether.
Various aromatic aldehydes were excellent substrates: aldol-elimination reactions of benzaldehyde, 2-furaldehyde, 4-methylthiazole-5-carboxaldehyde and 3-pyridinecarboxaldehyde gave products 13d (86%), 13e (86%), 13f (84%), and 13g (88%), respectively (entries 4 – 7). Due to the mild oxidative work-up conditions, pyridine-N-oxide formation was not observed in the latter case.
Finally, the silyl-protected α-hydroxyaldehyde 1611 gave 13h in 88% yield (entry 8). In addition, α,β-unsaturated aldehydes such as 1712 could also be used as substrate (76% yield; entry 9); unsaturated aldehydes are typically avoided in Baylis-Hillman reactions.6 The only substrate that worked poorly in our hands was acrolein, which gave a multitude of side products that were difficult to separate (data not shown). In all cases, the isolated allylic alcohols 13a–13i were obtained as single diasteromers.
We expected from the outset that acyl oxazolidinone 15 would undergo aldol reaction via the corresponding Z(O)-boron-enolate 11 (X = SePh) and provide syn-aldol products prior to oxidative workup. This was confirmed in the case of aldol 18, which was isolated from an aldol reaction of 15 and isobutryraldehyde following mild basic (but non-oxidative) work-up. Treatment of 18 with LiBH4 and protection of the resulting 1,3-diol gave p-methoxyphenyl (PMP) acetal 19. The Ha–Hb coupling constant of 19 (3J = 1.9 Hz) as well as 1H NOE data provide the basis for this assignment (Scheme 3).
Scheme 3.

Stereochemical assignments
The absolute configuration of aldol 18 was determined independently via the advanced Mosher ester method,13 as well as by reduction of the derived allylic alcohol 13a to the known diol 20 upon treatment with NaBH4 in the presence of CeCl3 .7H2O.14,15
Application of this new methodology to the synthesis of the C(15)–C(21) fragment 27 of tedanolide C is presented in Scheme 4. Aldehyde 8 was synthesized starting from the known aldehyde 21. Thus, standard Wittig reaction of 21 with Ph3P=CHCO2Et followed by DIBAL reduction of the ester provided allylic alcohol 22 (80%). Subjection of 22 to the Sharpless asymmetric epoxidation conditions16 followed by a Parikh-Doering oxidation of epoxyalcohol 23 provided the epoxyaldehyde 8 in 79% yield from 22. Use of epoxyaldehyde 8 as the substrate for aldol reaction with 15, followed by oxidative workup, provided allylic alcohol 24 in 79% yield with > 20:1 distereoselectivity. Treatment of 24 with NaBH4 and CeCl3.7H2O14b and subsequent protection of the bis-allylic alcohol 25 with 2 equiv of TESCl gave the bis-silyl ether 26 in 91% yield for the two steps. Finally, asymmetric dihydroxylation of 26 by using Sharpless’ AD-mix β17 gave diol 27 (75%, 95% b.r.s.m.) as the major component of a 4.5:1 mixture of diastereomers.18,19
Scheme 4.

Synthesis of the C(15)–C(21) fragment 27 of tedanolide C
In summary, we developed an efficient and highly stereoselective procedure for synthesis of α-methylene-β-hydroxy acyl oxazolidinones - so-called Baylis-Hillman adducts - via an enantioselective syn-aldol/β-elimination sequence using β-(phenylselenyl)propionyl imide 15 as the key reagent. This method allows for the use of 15 and the aldehyde reaction partner in stoichiometric amounts—which is not possible in conventional Baylis-Hillman reactions. This new sequence has been applied to the synthesis of the C(15)–C(21) fragment 27 of tedanolide C (target structure 5). Further elaborations of 27 towards tedanolide C will be reported in due course.
Supplementary Material
Acknowledgment
Financial support provided by the National Institutes of Health (GM038436) is gratefully acknowledged. R. B. thanks the Austrian Science Fund (FWF) for a Schrödinger fellowship.
Footnotes
Supporting Information Available: Experimental procedures and copies of 1H NMR and 13C NMR spectra of new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Tedanolide C (1): Chevallier C, Bugni TS, Feng X, Harper MK, Orendt AM, Ireland CM. J. Org. Chem. 2006;71:2510. doi: 10.1021/jo052285+.
- 2.Tedanolide (2): Schmitz FJ, Gunasekera SP, Yalamanchili G, Bilayet Hossain M, Van Der Helm D. J. Am. Chem. Soc. 1984;106:7251.
- 3.13-Deoxytedanolide (3): Fusetani N, Sugawara T, Matsunaga S, Hirota H. J. Org. Chem. 1991;56:4971.
- 4.Candidaspongiolide (4): Meragelman TL, Willis RH, Woldemichael GM, Heaton A, Murphy PT, Snader KM, Newman DL, van Soest R, Boyd MR, Cardellina JH, II, McKee TC. J. Nat. Prod. 2007;70:1133. doi: 10.1021/np0700974.
- 5.Nishimura S, Matsunaga S, Yoshida S, Nakao Y, Hirota H, Fusetani N. Bioorg. Med. Chem. 2005;13:455. doi: 10.1016/j.bmc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 6. Baylis AB, Hillman MED. 2,155,113. Ger. Pat. 1972 For reviews of the Baylis-Hillman reacdtion, see: Ciganek E. Org. React. 1997;51:201. Basavaiah D, Rao AJ, Satyanarayana T. Chem. Rev. 2003;103:811. doi: 10.1021/cr010043d. Krishna PR, Sachwani R, Reddy PS. Synlett. 2008:2897. Ma G-N, Jiang J-J, Shi M, Wei Y. Chem. Commun. 2009:5496. doi: 10.1039/b909405a.
- 7.For reports of enantioselective Baylis-Hillman reactions using chiral acylate derivatives: Brzezinski LJ, Rafel S, Leahy JW. J. Am. Chem. Soc. 1997;119:4317. and references therein. ; Brzezinski LJ, Rafel S, Leahy JW. Tetrahedron. 1997;53:16423. [Google Scholar]
- 8.For previous syntheses of racemic Baylis-Hillman adducts from aldol reactions of β-selenophenyl enolboranes: Leonard WR, Livinghouse T. J. Org. Chem. 1985;50:730. ; Leonard WR, Livinghouse T. Tetrahedron Lett. 1985;26:6431. [Google Scholar]
- 9.(a) Evans DA, Bartroli J, Shih TL. J. Am. Chem. Soc. 1981;103:2127. [Google Scholar]; (b) Ager DJ, Prakash I, Schaad DR. Aldrichimica Acta. 1997;30:3. [Google Scholar]
- 10.Miyashita M, Yoshikoshi A. Synthesis. 1980:664. [Google Scholar]
- 11.Nicolaou KC, Liu J-J, Yang Z, Ueno H, Sorensen EJ, Claiborne CF, Guy RK, Hwang C-K, Nakada M, Nantermet PG. J. Am. Chem. Soc. 1995;117:634. [Google Scholar]
- 12.Ramachandran P, Burghardt TE, Reddy MVR. J. Org. Chem. 2005;70:2329. doi: 10.1021/jo048144+. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512. [Google Scholar]; (b) Hoye TR, Jeffrey CS, Shao F. Nature Protocols. 2007;2:2451. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 14.(a) The reductive cleavage of the auxiliary with LiBH4 gave a mixture of the 1,2-and the 1,4-reduced products in a 1:1 ratio; Luche J-L. J. Am. Chem. Soc. 1978;100:2226.
- 15.Enders D, Voith M. Synthesis. 2002:1571. [Google Scholar]
- 16.Katsuki T, Sharpless KB. J. Am. Chem. Soc. 1980;102:5974. [Google Scholar]
- 17.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483. [Google Scholar]
- 18.The stereochemistry of the C(16) hydroxy group was assigned based on the usual sense of asymmetric induction in Sharpless asymmetric dihydroxylation reactions, and was not rigorously determined at this stage.
- 19.The two diastereomers were inseparable by standard column chromatography. Purification of the major diastereomer was performed by HPLC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.