

Abstract
The constraint-based reconstruction and analysis approach has recently been extended to describe Escherichia coli's transcriptional and translational machinery. Here, we introduce the concept of reaction coupling to represent the dependency between protein synthesis and utilization. These coupling constraints lead to a significant contraction of the feasible set of steady-state fluxes. The subset of alternate optimal solutions (AOS) consistent with maximal ribosome production was calculated. The majority of transcriptional and translational reactions were active for all of these AOS, showing that the network has a low degree of redundancy. Furthermore, all calculated AOS contained the qualitative expression of at least 92% of the known essential genes. Principal component analysis of AOS demonstrated that energy currencies (ATP, GTP, and phosphate) dominate the network's capability to produce ribosomes. Additionally, we identified regulatory control points of the network, which include the transcription reactions of σ70 (RpoD) as well as that of a degradosome component (Rne) and of tRNA charging (ValS). These reactions contribute significant variance among AOS. These results show that constraint-based modeling can be applied to gain insight into the systemic properties of E. coli's transcriptional and translational machinery.
Introduction
Kinetic models of transcription (1,2), translation (1,3), and the cell cycle (4) have been formulated with systems of ordinary differential equations. These models describe the temporal changes in concentration accompanying production, degradation, transport, or modification of the molecules in the network. Although this modeling approach has been shown to be very useful and mechanistically insightful for small-scale Escherichia coli networks, such as those of the Trp operon (5) and the Lac operon (6), it cannot be readily applied for large-scale, sequence-dependent networks due to the paucity of experimentally measured kinetic parameters.
Constraint-based reconstruction and analysis (COBRA) can be used to model biological systems without the use of kinetic parameters. In this approach, the network is formulated as a set of linear equations describing the biochemical transformations taking place within a cell. The networks are constructed in a bottom-up fashion based on available genomic, biochemical, and bibliomic data (BiGG) (7–10). Information about reaction rates can be incorporated into the COBRA approach as constraints (bounds) on network reactions (9,11). This approach is well established for metabolic networks (12). More recently, the COBRA approach has been extended to the study of other cellular functions such as signaling (13,14), transcriptional regulation (15), and protein synthesis (16).
Flux balance analysis (FBA) is a constraint-based optimization approach, in which the flux through a particular network reaction is optimized while ensuring that the applied biological and physico-chemical constraints are obeyed (11). FBA relies on linear programming to find the optimal solution of a given objective function that maximizes or minimizes a particular flux. Depending on the properties of the model, however, the identified solution may not be unique—meaning that there may be an infinite number of different flux vectors giving an identical optimal objective value (Fig. 1).
Figure 1.
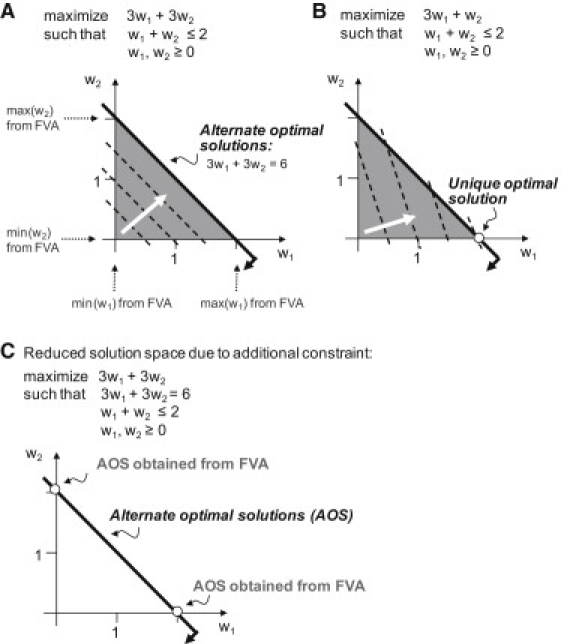
(A–C) Schematic illustration of alternate optimal solutions (AOS), unique solutions, and results of flux variability analysis (FVA) on a linear toy problem is shown.
In the context of metabolic models, these flux vectors are called alternate optimal solutions (AOS) or equivalent phenotypic states (17–19). The presence of AOS in constraint-based models was realized in the early 90s when FBA was applied to biologically realistic networks (20). Consider the example shown in Fig. 1 A. An infinite number of AOS lies on the line with optimal value for the objective function 3w1 + 3w2, in which the vector for each AOS is different. Therefore, not all AOS can be determined, but a representative subset of AOS can be calculated. Different mathematical methods have been used to determine subsets of AOS, e.g., vertex enumeration (19) or flux variability analysis (FVA (21)). Challenges associated with computing AOS in genome-scale metabolic networks are due to redundant, alternate pathways (18). Reed and Palsson (19) calculated subsets of AOS for E. coli's metabolic network that differ in at least one active reaction at different growth environments, and they determined correlated reaction sets. This computation is very time-consuming. In this study, we use FVA to determine AOS that correspond to a subset of extreme points of the steady-state solution space. In Fig. 1 C, such extreme points are highlighted.
Recently, we reconstructed the first genome-scale network of the transcriptional and translational (tr/tr) machinery (16). This comprehensive reconstruction, named the expression or E-matrix, accounts for the sequence-specific synthesis reaction matrix of 423 functional gene products, including rRNAs, tRNAs, ribosomes, and RNA polymerases. It is well known that the growth rate of E. coli, as well as that of other organisms, directly correlates with the cellular abundance of its protein synthesis machinery (22). Although the E-matrix does not account for metabolism, it does contain exchange reactions that supply the network with precursors (i.e., amino acids (aa), nucleotide triphosphates (NTP)) and remove metabolic by-products from the network (i.e., nucleotide monophosphates (NMP) and orthophosphate (Pi)) (16). When defining systems boundaries around protein synthesis, one can use these exchange reactions to determine the dependency between tr/tr and metabolism, in silico, under various environmental conditions. In this study, we determine the AOS of the E-matrix, characterize their properties, and compare the in silico expressed genes with experimental gene essentiality data (23).
Material and Methods
Reconstruction
We used the recently published reconstruction of E. coli's transcriptional and translational machinery, termed E-matrix (16). Briefly, 13,694 reactions and 11,991 components (i.e., metabolites, proteins, RNA molecules, and intermediate complexes) describe the sequence-specific synthesis reactions and cellular functions of 423 known gene products involved in this protein synthesis machinery (Table S1). Gene products include 86 tRNAs, proteins such as ribosomes (with rRNA incorporated), RNA polymerase, transcription, and translation factors. I.e., each transcription and translation reaction is gene-sequence specific, accounting for all tr/tr necessary (e.g., RNA polymerase, ribosomes) and NTP/aa requirements. Note that transcriptional regulators were not accounted for in the E-matrix. A more detailed description of the network content can be found in Thiele et al. (16). For modeling purposes, proteins and mRNA species are represented in the E-matrix in two forms: Protein_active/Protein_inactive, and mRNA_1/mRNA_2. These two forms have no correspondence in nature, but do allow the modeling of a synthesized protein or transcript that can be used more than once before mRNA degradation, as found in cells. (16).
Constraint-based modeling
The E-matrix reconstruction can be converted into a mathematical format as stoichiometric matrix, S ∈ ℜm, n, where each row corresponds to a network component and each column corresponds to a network reaction. By definition, the stoichiometric coefficients for substrates are negative numbers, whereas products are positive coefficients. For the analysis of the network properties, we assume that the system is at steady state, therefore
| (1) |
where v is a flux vector (n × 1) and dx/dt is the rate of change in concentration of a component x over time, which is zero in steady state.
The E-matrix is underdetermined, as there are more variables (reactions) than equations (mass-balances). Therefore, a unique solution to this set of linear equations does not exist (Fig. 1). The addition of further inequalities (e.g., reaction rates) reduces the set of feasible solutions.
Network constraints
Other constraints may include the directionality of a reaction, vi, based on thermodynamic information (e.g., the ATP-dependent phosphorylation of glucose to glucose-6-phosphate is effectively irreversible) or environmental constraints for the availability of a nutrient in the medium (e.g., restricting glucose to be the sole carbon source by constraining all uptake fluxes for other carbon sources to be zero). By changing the set of inequality constraints applied to the model, different subsets of the steady-state feasible set are obtained and their properties can be studied using mathematical tools.
Network boundaries
The inputs to the E-matrix are biosynthetic precursors, such as amino acids and NTPs, which are provided to the network via exchange reactions. In the E-matrix, by-products of protein synthesis, such as NMP and Pi, are also removed from the system (16). For every protein and tRNA species, a demand reaction was included to mimic the requirement of that component for growth. The steady-state assumption does not allow for accumulation of intracellular components, but cell doubling does include a doubling of the proteome; therefore, these demand reactions represent the newly produced proteome of the in silico cell.
Objective function
The demand reaction of ribosomal 50S subunit production (DM_rib_50) was chosen as an objective function for the model, as the ribosome content of the cell is correlated to the growth rate (22). Using the synthesis reaction of the 50S as an objective function is equal to using the reaction of the 30S ribosomal subunit, since both subunits are present in cells in equal amounts. In contrast, the whole 70S ribosome leaves the mRNA after termination of translation and is dissociated through binding of IF1 and IF3 to the 30S subunit (16). By choosing 50S subunit (or 30S subunit), we can investigate the active ribosome subunit synthesis in the model, but do not require that all synthesized ribosomes are used for translation. This is in agreement with the duplication of the ribosome number in the dividing cell.
The optimization problem is formulated as
| (2) |
| (3) |
| (4) |
where cT is a vector (1 × n), indicating the objective reaction with a nonzero entry.
Simulation constraints
To model the E-matrix corresponding to different doubling times, we calculated the maximal possible stable RNA transcription initiation rates based on data given in Neidhardt (24) (Table S2).
The total transcription initiation rate for stable RNA gene i is given by
| (5) |
where irrn is the initiation rate per ribosomal RNA copy (initiation × min−1 × gene−1) (Table S2). To account for the gene-dosage effect, we multiplied irrn by gi (gene × cell−1), which is the gene copy number. The number of gene copies depends on the number of replication forks, which creates multiple copies of the chromosome within one cell. Therefore, the copy number of a gene depends on its genome position (m′i) and doubling time (t). The value gi is given by
| (6) |
where D is the time necessary to replicate the chromosome (D = 0.3314 × t + 32.564, t in minutes), C is lag time between chromosome replications (C = 0.0898 × t + 21.238, t in minutes), and t is the doubling time (in minutes) (24).
The total transcription initiation rate of stable RNA can be converted into an nmol × × h−1 rate by multiplying Eq. 5 by the scaling factor
| (7) |
where NA is the Avogadro number (6.022 × 1023 molecules × mol−1), z is the mass per cell (μgDW/109 cells), and t is the timescale factor (60, in this case).
Formulation of general coupling constraints
Typically, network reconstructions do not stoichiometrically represent reactants that are both substrates and products in the same reactions. Their involvement is implicit and not explicitly represented in the reaction. An example is an enzyme in a metabolic reaction (Fig. 2). However, in the E-matrix, proteins are explicitly included in the reactions they catalyze (Fig. 2). The four explicit reactions (v1–v4) are equivalent to the reaction (v0) in the implicit formulation. It follows that the synthesis of the recycled reactant E is not essential to permit steady-state flux through v1–v4, as it is recycled by the last reaction (v4). Subsequently, the conversion of A + B → C will occur regardless of whether the model is synthesizing E.
Figure 2.
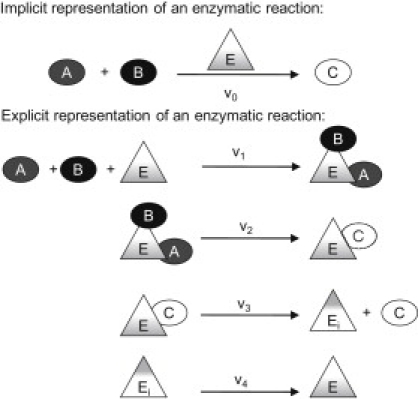
Schematic representation of the participation of tr/tr enzymes in network reactions. In canonical network formulations, enzyme reaction participation is implied but not explicitly modeled. The tr/tr network produces enzymes; hence, the explicit incorporation of enzymes in their catalyzed reactions is desired. The same approach is applied if the reactant E is a tRNA molecule or a protein.
Consequently, additional constraints are needed to enforce the synthesis of E if its set of explicit reactions is active in a particular steady state. We require the condition
| (8) |
where vsynthesis, E is the synthesis reaction rate of reactant E. Furthermore, it would be desirable to relate the flux through reaction v4 and the synthesis of E with some proportionality,
| (9) |
even though the exact proportion factor can only be approximated within bounds (see below). Note that V4,Vsynthesis, E ≥ 0.
The relationships expressed in Eqs. 8 and 9 can be represented in a linear fashion with
| (10) |
| (11) |
where cmin and cmax (0 < cmin ≤ cmax) are the bounds on the proportion factor (termed “coupling coefficients”). Note that Eq. 10 ensures that a higher flux through v4 raises the lower bound on the synthesis reaction vsynthesis, E. Furthermore, s can be used to allow the synthesis of reactant E without being used in the model up to its value. In this study, however, we set s to be zero, because we intended to determine AOS in which all synthesized reactants are used. Linear inequality coupling constraints retain the numerically scalable character of flux balance analysis.
Because reactant E may be required in multiple reactions, the flux through the recycling reaction (v4) will be their sum. Subsequently, choosing v4 for Eqs. 8 and 9 ensures that the synthesis rate of E will be greater than zero if any network reaction that utilizes E is active.
In steady-state condition, the synthesis flux of E is equal to the degradation flux rate of E. Therefore, consider the toy network shown in Fig. 3 A. Node A has an influx (vin) and two outfluxes (vout1 and vout2). The concentration of A (i.e., [A]) depends on the relative outfluxes, i.e., their ratio to each other given that the flux rates are distinct. This is an inherent property of the Jacobian matrix that contains the dynamic metabolite concentration (25) (dynamic metabolite concentration arises from the fact that the outflows from a node are dependent on the concentration of the compound that the node represents). It follows that the coupling constraints are not artificial constraints added to the network, but rather, that they allow the accurate representation of inherent properties of biochemical networks. Because the exact ratio between the outfluxes is unknown in many cases, we bound it by using cmin and cmax (see Eqs. 10 and 11). A geometric representation of the coupling constraints can be found in Fig. 4.
Figure 3.
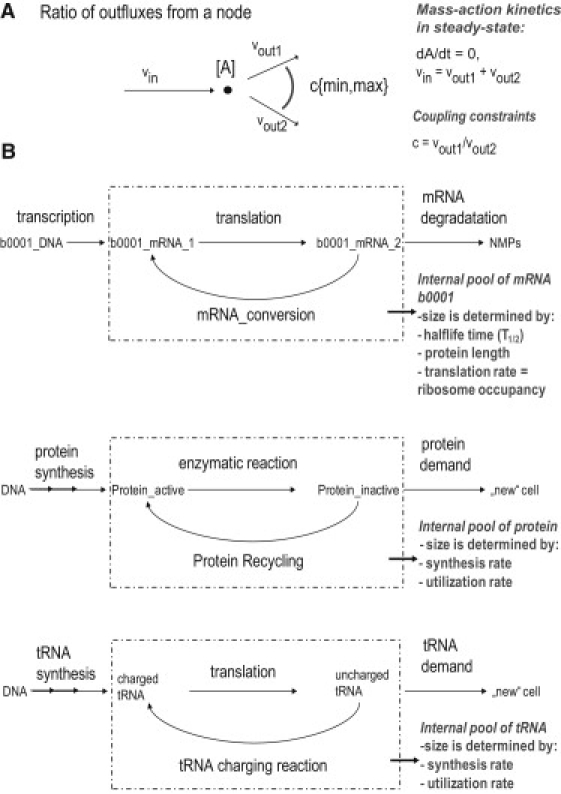
Schematic representations of the mRNA and protein pools present in the E-matrix. (A) Conceptual representation of flux coupling is shown. In steady-state condition, the influx into node A is equal to the sum of outfluxes. Subsequently, there is a ratio describing the relative outfluxes. (B) In contrast to metabolic networks, the tr/tr network requires that component pools are added to ensure that the network functions are similar to known in vivo features. By introducing loops and appropriate constraints, one can represent different pool sizes of the components. NMPs are nucleotide monophosphates.
Figure 4.
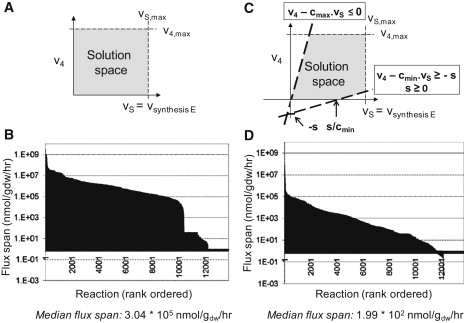
Illustration of geometric interpretation of constraints and the corresponding flux span. (A and B) Uncoupled model. (C and D) Coupled model contains a set of 1056 coupling constraints between 528 network reactions. The flux span corresponds to the variability of each network reaction while producing maximum rate of ribosomes. The simulation condition corresponds to doubling time t = 90 min. See also Fig. S1 for a comparison by cellular subsystems.
Formulation of E-matrix coupling constraints
In the E-matrix, there are three sets of reactions that require coupling:
-
1.
Transcription and translation;
-
2.
Translation and protein utilization; and
-
3.
tRNA synthesis and tRNA utilization (Fig. 3 B).
In each case, the inequalities are the same as Eqs. 10 and 11 but the definition of the coupling coefficients depends on the nature of the coupled reactions.
The following sets of reactions require coupling constraints (see also Fig. 3):
-
1.
Transcription and translation: mRNA degradation reactions (e.g., b0001_mRNA_degr1) were coupled to the corresponding mRNA conversion reactions (e.g., b0001_mRNA_CONV2).
-
2.
Translation and protein utilization: protein demand reactions (e.g., DM_AlaS_mono), which allow the accumulation of proteins in the network, were coupled with the corresponding protein recycling/utilization reactions (e.g., AlaS_mono_RECYCL).
-
3.
tRNA synthesis and tRNA utilization: tRNA charging reactions (e.g., ala1_tRNA_CHARG), representing the tRNA utilization, were coupled with the corresponding tRNA formation reactions (e.g., alaT_ to _ala1).
Coupling transcription and translation
At steady state, the rate of mRNA synthesis vsynthesis, i (transcription) is equal to the rate of mRNA degradation, vdegradation, i, which is given by
| (12) |
where [mRNA]i is the cellular concentration of mRNA i (molecules × cell−1), and T1/2, i is the half-life time of mRNA i (seconds).
Because the E-matrix genes are transcribed in terms of transcription units (16), we will couple the mRNA degradation reaction (vdegradation, i) with the corresponding recycling reaction (vCONV2, i) (Fig. 3). This reaction recycles an mRNA_2 compound released from a translation reaction into an mRNA_1 compound, which is used in translation reactions. This recycling enables the reutilization of a single transcript for multiple translation rounds before degradation. The mRNA recycling reaction forms a cycle together with the translation reactions (Fig. 3). This cycle allows the representation of an internal mRNA pool corresponding to the steady-state concentration of the mRNA, which can be used for quantitative integration of gene expression data on transcript abundance in future studies.
Definition of tr/tr coupling factor
In this section, we derive a meaningful coupling factor (cmin, i, cmax, i) between mRNA degradation reaction (vdegradation, i) with the utilization reaction (vCONV2, i) (Fig. 3),
| (13) |
where vCONV2, i ≡ vtranslation, i.
The translation flux is the product of translation rate and mRNA concentration:
| (14) |
Using the derivation described in the Supporting Material, we obtain
| (15) |
where rtl is translation rate of a ribosome (in aa × s−1 × ribosome−1), rspace is the minimum spacing of two ribosomes on a transcript, and vtranslation, i is in nmol × × h−1.
To obtain the vdegradation, i in the same unit, Eq. 12 needs to be converted,
| (16) |
where vdegradation, i is in nmol × gDW−1 × h−1.
Under the steady-state assumption, we can equate Eqs. 15 and 16. Furthermore, because the recycling reaction rate (vCONV2, i) is equal to the translation reaction rate for mRNA i in the network, it follows that
| (17) |
Subsequently, the coupling factor cmax, i between the degradation and translation rate is
| (18) |
The minimum coupling factor cmin, i was determined assuming one ribosome bound per transcript,
| (19) |
where LP, i is the length of the protein i (in amino acids).
Why are the coupling constraints valid?
As mentioned above, the flux through mRNA synthesis/degradation is independent of mRNA translation/recycling flux in steady-state condition. I.e., no constraint on synthesis/degradation reactions would affect the translation/recycling reactions. Subsequently, a set of constraints had to be included that would define possible ratios the reaction fluxes of synthesis/degradation and translation/recycling can take—i.e., the coupling constraints. These constraints do not enforce the identity of degradation and translation fluxes but rather their correlation (Fig. 4 C). Such correlation can be readily justified by the fact that high ribosome occupancy on a transcript (i.e., high translation rate) protects the transcript from degradation. In addition, if the maximal possible ribosome occupancy is achieved, the translation rate can only be increased by augmenting the mRNA synthesis flux—which is equal to increasing the mRNA degradation flux in steady state.
Coupling protein synthesis and utilization and tRNA synthesis and utilization
The protein and tRNA synthesis reactions were coupled to their utilizing reactions in a similar fashion. However, an arbitrary number of 105 was chosen for the coupling factor (cmax, i), because the interpretation of this factor is quite different from the mRNA recycling. As most proteins and tRNAs are assumed to be stable in the timescale of an average cell's doubling time, protein and tRNA degradation were ignored. The turnover rate of a protein or tRNA is limited and depends on the individual species. The coupling factor represents such turnover limitation as it enforces the synthesis of more protein/tRNA if they are highly used in the network. The value for cmax, i represents the largest possible value in terms of numerical stability, meaning that all other feasible solutions resulting from smaller coupling factors lay within the analyzed set. cmin, i was set to be one.
In total, 1056 additional inequality constraints (628 on mRNA, 120 on tRNA, and 308 on protein synthesis) were added to the E-matrix, resulting in a problem size of 13,047 equality and inequality constraints and 13,726 variables (reactions). This additionally constrained E-matrix (Ecoupled-matrix) was used throughout this work unless stated differently.
Flux variability analysis and flux span
Given a set of constraints, flux variability analysis (FVA) (18) can be used to assess the network flexibility and network redundancy. In this study, we fixed the ribosome production rate to its maximal value (νDM_rib 50, max = νDM_rib50, max = max, based on Table S2). Then, every network reaction i was minimized and maximized. The flux span of a network reaction i is given by |vi, max – vi, min| = spani.
Alternate optimal solutions
Alternate optimal solutions (AOS) were determined using FVA, which was carried out as described above. All solution vectors were stored and used for subsequent analysis.
Principal component analysis of alternate optimal solutions
To identify reactions that account for the greatest variance in flux between different simulation conditions, we used principal component analysis (PCA) (see Supporting Material for detailed description). We used the set of flux vectors corresponding to AOS, P ∈ Rn, N, in the nullspace of the stoichiometric matrix, S ⋅ P ≡ 0, which lay in an n-dimensional flux vector space, but used PCA to reveal the intrinsically significant axes, which account for the variation within this set. First, we calculate the flux covariance matrix, C ∈ Rn, n, where the covariance between two fluxes is given by
with denoting the average flux of reaction i over all N flux vectors. Singular value decomposition of the covariance matrix gives
where U = V as C is a square diagonally symmetric matrix. Each row of V contains components, or singular vectors, of the covariance matrix. Each singular vector gives the direction of an intrinsic axis, which is linearly independent from all other intrinsic axes. The standard deviation for each principal component may be calculated by taking the square root of the singular values, the diagonal entries in Σ (26). PCA of the covariance matrix is mathematically equivalent to PCA of the AOS themselves, but the former is computationally more efficient (27).
PCA was carried out on the AOS for simulations corresponding to t = 90 min doubling time. A control point in our model is a reaction, or component, that, when alternated, leads to significant changes of the functional states of the model. For example, a control point in gene expression is therefore a gene that, when repressed, alters the transcription of many other genes and thus the function of the cell. The key control points of gene expression were determined by collecting flux values from the AOS for all mRNA degradation reactions (which are equivalent to the flux values of mRNA synthesis reactions in steady state). PCA was carried out on the resulting matrix (with dimensions of 314 × 27, 452) as described above. Additionally, we tilted the eigen-vectors to obtain a clearer picture of the eigen-reactions. The procedure used was described in Barrett et al. (28).
Results
Comparison of flux span with and without flux coupling
We expected a significant reduction in the size of the steady-state solution space in the Ecoupled-matrix. To assess the change in solution space size, we determined the flux span of the E-matrix reactions and of the Ecoupled-matrix (Fig. 4). For this comparison, we used the same simulation condition, corresponding to a doubling time of 90 min, with the exception that the Ecoupled-matrix contained the additional coupling constraints as described above. We found that the coupling constraints reduced the mean flux span by two orders of magnitude (from 1.1 × 107 ± 9.2 × 107 nmol × gDW−1 × h−1 in the E-matrix to 6.76 × 104 ± 1.38 × 106 nmol × gDW−1 × h−1 in the Ecoupled-matrix) (Fig. 4). The small change in standard deviation of the flux span indicates that the coupling constraint's effect was not limited to reactions with very large fluxes (Fig. 4). The same trend was observed when the median flux span was compared (from 3.04 × 105 nmol × gDW−1 × h−1 to 1.99 × 102 nmol × gDW−1 × h−1). In other words, if the feasible, steady-state solution space of the E-matrix had a certain volume, then the addition of coupling constraints led to a reduction in solution space volume by a factor of (1/160)n, where n is the number of dimensions. This shrinkage in size of a steady-state feasible set is substantial, and shows the benefit of the coupling constraints in the assessment of physiological relevant flux states.
AOS for maximal ribosome production
First, we tested whether the additional constraints altered the Ecoupled-matrix ribosome production capabilities. We found that the computed ribosome values were in good agreement with the published experimental data (24) and the in silico production capabilities of the E-matrix (16) (data not shown). Subsequently, we used FVA to enumerate all AOS that produced ribosomes at maximal rate and have an optimal (minimal or maximal) value for at least one other network reaction. This FVA-derived subset of AOS thus corresponded to extreme (or boundary) AOS. The characteristics of the AOS of four different models, corresponding to doubling times of t = 24, t = 60, t = 90, and t = 100 min, were determined.
Average distance of alternate optima solutions
Because the FVA-derived AOS represent only a subset of all possible AOS, we computed the average Euclidean distance between the AOS. The distance between two AOS also represents a measure of how evenly they are distributed in the solution space. We compared the distance of 106 pairs of AOS (Fig. 5). As expected, the AOS were not evenly distributed; however, the average distance between the AOS was 9.2 × 106 nmol × gDW−1 × h−1 ± 1.3 × 107 nmol × gDW−1 × h−1.
Figure 5.
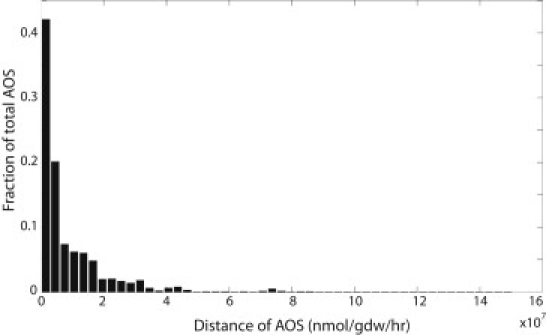
Distance between AOS in the Ecoupled-matrix. To assess the overall distance between the set of AOS, we computed the distance between 106 randomly chosen AOS pairs (doubling time t = 90).
Principal component analysis of alternate optimal solutions
Principal component analysis (PCA) is an objective, nonparametric, analytical method in wide use for a variety of applications, including signal processing (29) and mRNA expression analysis (30–32). Furthermore, singular value decomposition has been used to study the topology and structure of metabolic networks (33) and to analyze the key reactions that are regulated within the human red blood cell (26). In the latter, singular value decomposition was applied on uniformly sampled points in the steady-state solution space to identify the eigen-reactions, which themselves correspond to the modes that represent the key branch points and thus, the key control points (reactions) in the network (26,34). We used PCA to 1), investigate the effective dimensionality of the Ecoupled-matrix; and 2), to determine the number of branch points, or control points, in the gene expression system of E. coli's tr/tr machinery.
Effective dimensionality of Ecoupled-matrix
First, when considering the entire network, we found that the first 10 modes (Z scores) could reconstruct 90% of the variance between AOS that corresponds to maximal ribosome production (Fig. 6, left panel, blue line). The first four corresponding eigen-reactions consist of
-
1.
Diphosphate, proton, and water exchange;
-
2.
Diphosphate, proton, Pi, and water exchange;
-
3.
GTP, GDP, and water exchange; and
-
4.
ATP, Pi, and water exchange.
Figure 6.
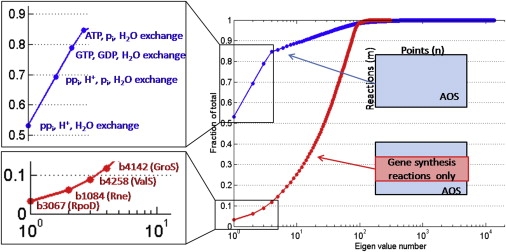
Principal component analyses (PCA). Z scores of the entire Ecoupled-matrix network (A) and of the gene expression reactions (B). The PCA analysis was performed on the set of alternate optimal solutions (AOS) (doubling time t = 90 min). Note that there are m reactions in the network and the number of AOS (points) is n = 2m.
Consequently, changing the flux rate for any of these reactions will have a significant effect on which functional states can be achieved by the network. These results indicate thus that at maximal ribosome production rate, the energy state of the cell mainly controls the achievable cellular states (e.g., mRNA production, protein synthesis). An integrative model of tr/tr and metabolism will be of great value for further investigation of the role of energy metabolism in macromolecular synthesis. Corresponding efforts have been recently initiated by the authors.
Synthesis of key genes for maximal ribosome production
To investigate the set of tr/tr genes that are likely to correspond to key control points (26), we performed the PCA on the subset of mRNA synthesis reactions. We found that 75 modes were necessary to recover 90% of the information content in the AOS for the 314 protein coding genes (Fig. 6, left panel, red line). This result was quite different from the PCA analysis of the entire network where 10 modes were sufficient to recover the majority of information content in the AOS. The first eigen-reaction was dominated by the expression of σ70 (b3067, RpoD), the primary σfactor during exponential growth targeting a wide range of promoters, and thus genes, essential for normal growth (35). The second eigen-reaction consisted of the gene synthesis reaction for b1084 (Rne), a component of the multiprotein complex degradosome, which is responsible for mRNA degradation in E. coli. The third eigen-reaction is dominated by the synthesis of the valyl-tRNA synthetase (b4258, ValS), which is responsible for charging valyl-tRNA molecules. Valine is the third most frequent amino acid in E. coli's genome. The fourth eigen-reaction consists of the synthesis of b2794 (QueF) and b1084 (Rne). QueF is a protein involved in the synthesis of pre_Q0, a precursor to queuosine that is an important modified nucleotide in E. coli's tRNA. The next two eigen-reactions are dominated by genes involved in protein folding, namely, b4142 (GroS), b0014 (DnaK), and b0015 (DnaJ). GroS is part of the protein-folding complex GroEL/S, which helps to fold larger proteins (36). DnaK and DnaJ are components of the second protein folding system in E. coli, DnaKJ/GrpE.
Taken together, the first six modes of the genes expression reactions recovered ∼35% of the information content and the corresponding eigen-reactions consisted of the main players involved in transcription, translation, mRNA degradation, and protein folding. Based on the proposed interpretation of the eigen-reactions as key control points (26), it is to be expected that the gene expression of these seven genes is highly regulated in E. coli. In fact, preliminary analysis of the regulatory rules for E. coli genes indicate that there are at least 30 transcriptional regulators involved in controlling the synthesis of tr/tr genes under different environmental conditions (I. Thiele, unpublished results).
Length and reaction participation of alternate optima solutions
Metabolic networks are known for their redundancy, which increases the flexibility and fitness of the cell to sudden environmental changes (37,38). For the E-matrix, a certain rigidity is expected, because the majority of the associated functions have only one coding gene in the genome. When optimizing for the ribosome synthesis rate in the Ecoupled-matrix, the number of active reactions in the AOS can be used as a measure of network flexibility. We found that, on average, ∼6500 reactions (≈50%) were active per AOS, i.e., they had a nonzero flux value. Three-thousand-eight-hundred of these 6500 reactions were active in all AOS in a simulation condition. Overall, a set of 3616 reactions was active in all AOS under all simulated conditions. An additional 1048 reactions were active in 95% of the AOS under all simulation conditions.
This high number of active reactions is a consequence of the linear structure of the transcriptional and translational network (16): A gene is transcribed into mRNA; its mRNA is then either degraded or used as a template for translation into a protein, which catalyzes one or more biochemical transformation along this path. In contrast, metabolic networks have more interconnections with numerous alternative (redundant) pathways. Subsequently, an average of ∼30% of the reactions present in E. coli's metabolic network were found to be active per AOS (19). This observation was quite different from our observation of active reactions in the Ecoupled-matrix. These results illustrate the fundamental differences in topology and redundancy found between the networks of these two important cellular functions.
Essential genes are expressed in all AOS
The Ecoupled-matrix accounts for a total of 314 protein-coding genes, many of which are directly involved in processes of the macromolecular machinery (16). First, we analyzed how many genes were expressed in all AOS. We found that at a doubling time t = 90 min, 227 genes (73%) were expressed in all AOS (termed, required genes), and only two genes were not expressed in any AOS. These two genes, b4292 (fecR) and b4293 (fecI, σ19), are part of the same operon and hence coexpressed in the network. The transcription factor σ19 was not expressed in any AOS, as none of the included genes has σ19-dependent transcription (16,39). In fact, σ19 seems to have few genomic binding sites in E. coli (B. K. Cho, University of California, San Diego, personal communications, 2009). Eighty-five of 314 (27%) genes were transcribed in many but not all AOS. We compared the required genes with in vitro essentiality data (23). E. coli has 303 essential genes (in rich medium) (23), 99 of these genes were present in the E-matrix network, and 91 of these essential genes were required genes in all simulated conditions (doubling times of 24, 60, 90, and 100 min).
Only eight in vitro essential genes were not active in all AOS (Table S3). Four of these essential genes were metabolic genes that were coexpressed with genes involved in the synthesis machinery. As the E-matrix does not account for metabolism, no gene essentiality was expected and this disagreement can be neglected. The remaining four genes were involved in different processes of the synthesis machinery (Table S3). RpoE (b2573) is the minor σfactor (σE) in E. coli, which responds to heat shock and other stress situations. In the E-matrix, only four transcription units are dependent on σE transcription. However, as σE has only ∼70 binding sites on the E. coli's genome, it is very likely that the E-matrix did not account for essential functions dependent on σE transcription. In contrast, GroS (4142) is the smaller subunit of the GroEL/ES chaperone that is responsible for correct folding of larger proteins. Many of the E-matrix proteins can be folded spontaneously, in a DnaK/J-GrpE chaperone-dependent, and/or in a GroEL/ES-dependent manner. The information was included in the E-matrix based on two large-scale experimental studies identifying targets specific for these chaperones (40,41). The overlapping action of DnaK/J-GrpE chaperone and GroEL/S chaperone explains the missing essentiality of GroS and of GrpE (b2614) in the Ecoupled-matrix. The last false-negative predictions included proteins for a tRNA modification, TilS (b0188), which modifies the nucleotide at position 34 in ileX and ileY-tRNA (conversion of cytidine into lysidine) (42). These two tRNA recognize the same codon (ATA), which was less frequently used in the E-matrix associated genes compared to the genome (I. Thiele, unpublished data), which may explain why TilS is not essential to our calculations.
Conclusions
In this study, we investigated the properties of E. coli's transcriptional and translational machinery when optimized for maximal ribosome production. This objective seems in agreement with experimental observations reporting direct correlation between achieved growth rate and cellular ribosome content (24). We introduced what we believe to be a new type of constraints to the network, which coupled outfluxes of a node to certain ratios (see Fig. 3). These coupling constraints represent inherent properties of biochemical reaction networks (43). The use of these constraints, in addition to mass-balance and flux rate constraints, led to further refinement of the physiological feasible set of flux states. In fact, we found that the coupling constraints led to a reduction to (1/160)n of the original, constrained, steady-state solution space. These additional constraints represent thus a significant advance in constraint-based modeling techniques.
We determined AOS consistent with optimal ribosome production using flux variability analysis. These AOS correspond to the extreme points of the bounded, convex polytope meaning that all feasible, steady-state solutions, consistent with the applied constraints, lay within the set of AOS. Principal component analysis of these AOS revealed that metabolic coupling is dominant in furnishing capability for determining the expression of model genes. In particular, the energy currency exchange was found to be crucial. These results are consistent with experimental data indicating that the overarching goal of growing cells is energy (ATP) synthesis. Analysis of key control points of in silico gene expression suggested that the expression state is determined by genes involved in transcription, mRNA degradation, protein folding and active tRNA availability. This is a systems biology result describing the systemic properties of E. coli's protein synthesis machinery. Lastly, analysis of in silico gene expression revealed that the majority of in vitro essential genes were expressed in all AOS, i.e., they need to be expressed in any functional network state leading to optimal ribosome production. This is the first time to our knowledge that a gene essentiality study has been carried out in silico for a nonmetabolic network. Furthermore, overlapping essentiality with in vitro data suggest that optimal ribosome production is indeed a driving force of growing cells. In comparison to previous, experimental studies, we derived supporting evidence from a systems biology approach, in which all known information was collected into a consistent format. The systematic analysis of the collective information revealed inherent properties consistent with experimental data. None of the available models of macromolecular synthesis is currently able to accurately represent and determine these inherent properties, which renders this study a milestone in molecular systems biology. As a next step, one could imagine integration of the protein synthesis machinery with a metabolic network of E. coli, to enable further in silico studies into the relationships among ribosome production, the energy state of the cell, and environmental growth conditions.
Supporting Material
Seventeen equations, one figure, and three tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00225-0.
Supporting Material
Acknowledgments
The authors are thankful to N. Jamshidi for valuable discussion.
This study was supported by the National Institutes of Health (grant No. R0157089) and the Department of Energy (award No. DE-SC00092009).
References
- 1.Laffend L., Shuler M.L. Ribosomal protein limitations in Escherichia coli under conditions of high translational activity. Biotechnol. Bioeng. 1994;43:388–398. doi: 10.1002/bit.260430507. [DOI] [PubMed] [Google Scholar]
- 2.Bremer H., Dennis P., Ehrenberg M. Free RNA polymerase and modeling global transcription in Escherichia coli. Biochimie. 2003;85:597–609. doi: 10.1016/s0300-9084(03)00105-6. [DOI] [PubMed] [Google Scholar]
- 3.Mehra A., Hatzimanikatis V. An algorithmic framework for genome-wide modeling and analysis of translation networks. Biophys. J. 2006;90:1136–1146. doi: 10.1529/biophysj.105.062521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alberghina L., Mariani L. Analysis of a cell cycle model for Escherichia coli. J. Math. Biol. 1980;9:389–398. doi: 10.1007/BF00276501. [DOI] [PubMed] [Google Scholar]
- 5.Santillan M., Mackey M.C. Dynamic regulation of the Tryptophan operon: a modeling study and comparison with experimental data. Proc. Natl. Acad. Sci. USA. 2001;98:1364–1369. doi: 10.1073/pnas.98.4.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong P., Gladney S., Keasling J.D. Mathematical model of the Lac operon: inducer exclusion, catabolite repression, and diauxic growth on glucose and lactose. Biotechnol. Prog. 1997;13:132–143. doi: 10.1021/bp970003o. [DOI] [PubMed] [Google Scholar]
- 7.Palsson B.Ø. In silico biotechnology. Era of reconstruction and interrogation. Curr. Opin. Biotechnol. 2004;15:50–51. doi: 10.1016/j.copbio.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Feist A.M., Herrgård M.J., Palsson B.Ø. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reed J.L., Famili I., Palsson B.O. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 10.Durot M., Bourguignon P.Y., Schachter V. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol. Rev. 2009;33:164–190. doi: 10.1111/j.1574-6976.2008.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price N.D., Reed J.L., Palsson B.Ø. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2004;2:886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 12.Thiele I., Palsson B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papin J.A., Palsson B.Ø. Topological analysis of mass-balanced signaling networks: a framework to obtain network properties including crosstalk. J. Theor. Biol. 2004;227:283–297. doi: 10.1016/j.jtbi.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 14.Li F., Thiele I., Palsson B.Ø. Functional assessment of the TLR receptor network. PLOS Comput. Biol. 2009;5:e1000292. doi: 10.1371/journal.pcbi.1000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gianchandani E., Joyce A.R., Papin J.A. Functional states of the Escherichia coli transcriptional regulatory system at the genome-scale. PLOS Comput. Biol. 2009;5:e1000403. doi: 10.1371/journal.pcbi.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiele I., Jamshidi N., Palsson B.Ø. Genome-scale reconstruction of Escherichia coli's transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLOS Comput. Biol. 2009;5:e1000312. doi: 10.1371/journal.pcbi.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K., Berthiaume F., Yarmush M.L. Metabolic flux analysis of postburn hepatic hypermetabolism. Metab. Eng. 2000;2:312–327. doi: 10.1006/mben.2000.0160. [DOI] [PubMed] [Google Scholar]
- 18.Mahadevan R., Schilling C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003;5:264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Reed J.L., Palsson B.Ø. Genome-scale in silico models of E. coli have multiple equivalent phenotypic states: assessment of correlated reaction subsets that comprise network states. Genome Res. 2004;14:1797–1805. doi: 10.1101/gr.2546004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varma A., Boesch B.W., Palsson B.Ø. Biochemical production capabilities of Escherichia coli. Biotechnol. Bioeng. 1993;42:59–73. doi: 10.1002/bit.260420109. [DOI] [PubMed] [Google Scholar]
- 21.Duarte N.C., Herrgård M.J., Palsson B.Ø. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 2004;14:1298–1309. doi: 10.1101/gr.2250904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nomura M. Regulation of ribosome biosynthesis in Escherichia coli and Saccharomyces cerevisiae: diversity and common principles. J. Bacteriol. 1999;181:6857–6864. doi: 10.1128/jb.181.22.6857-6864.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba T., Ara T., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Sys. Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neidhardt F.C., editor. Chemical Composition of Escherichia coli. 2nd ed. Vol. 2. ASM Press; Washington, DC: 1996. [Google Scholar]
- 25.Jamshidi N., Palsson B.Ø. Using in silico models to simulate dual perturbation experiments: procedure development and interpretation of outcomes. BMC Syst. Biol. 2009;3:44. doi: 10.1186/1752-0509-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrett C.L., Price N.D., Palsson B.Ø. Network-level analysis of metabolic regulation in the human red blood cell using random sampling and singular value decomposition. BMC Bioinformatics. 2006;7:132. doi: 10.1186/1471-2105-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jolliffe I.T. 2nd ed. Springer; New York: 2002. Principal Component Analysis. Springer Series in Statistics. [Google Scholar]
- 28.Barrett C.L., Herrgard M.J., Palsson B.Ø. Decomposing complex reaction networks using random sampling, principal component analysis and basis rotation. BMC Syst. Biol. 2009;3:30. doi: 10.1186/1752-0509-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moon T.K., Stirling W.C. Prentice Hall; Upper Saddle River, NJ: 2000. Mathematical Methods and Algorithms for Signal Processing. [Google Scholar]
- 30.Alter O., Brown P.O., Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holter N.S., Mitra M., Fedoroff N.V. Fundamental patterns underlying gene expression profiles: simplicity from complexity. Proc. Natl. Acad. Sci. USA. 2000;97:8409–8414. doi: 10.1073/pnas.150242097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holter N.S., Maritan A., Banavar J.R. Dynamic modeling of gene expression data. Proc. Natl. Acad. Sci. USA. 2001;98:1693–1698. doi: 10.1073/pnas.98.4.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Famili I., Forster J., Palsson B.O. Saccharomyces cerevisiae phenotypes can be predicted by using constraint-based analysis of a genome-scale reconstructed metabolic network. Proc. Natl. Acad. Sci. USA. 2003;100:13134–13139. doi: 10.1073/pnas.2235812100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price N.D., Papin J.A., Palsson B.O. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21:162–169. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 35.Jishage M., Iwata A., Ishihama A. Regulation of RNA polymerase σ-subunit synthesis in Escherichia coli: intracellular levels of four species of σ-subunit under various growth conditions. J. Bacteriol. 1996;178:5447–5451. doi: 10.1128/jb.178.18.5447-5451.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartl F.U., Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 37.Price N.D., Famili I., Palsson B.Ø. Extreme pathways and Kirchhoff's second law. Biophys. J. 2002;83:2879–2882. doi: 10.1016/S0006-3495(02)75297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thiele I., Price N.D., Palsson B.Ø. Candidate metabolic network states in human mitochondria. Impact of diabetes, ischemia, and diet. J. Biol. Chem. 2005;280:11683–11695. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- 39.Keseler I.M., Collado-Vides J., Karp P.D. EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res. 2005;33(Database issue):D334–D337. doi: 10.1093/nar/gki108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerner M.J., Naylor D.J., Hartl F.U. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122:209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 41.Deuerling E., Patzelt H., Bukau B. Trigger factor and DnaK possess overlapping substrate pools and binding specificities. Mol. Microbiol. 2003;47:1317–1328. doi: 10.1046/j.1365-2958.2003.03370.x. [DOI] [PubMed] [Google Scholar]
- 42.Ikeuchi Y., Soma A., Suzuki T. molecular mechanism of lysidine synthesis that determines tRNA identity and codon recognition. Mol. Cell. 2005;19:235–246. doi: 10.1016/j.molcel.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Jamshidi N., Palsson B.Ø. Flux-concentration duality in dynamic nonequilibrium biological networks. Biophys. J. 2009;97:L11–L13. doi: 10.1016/j.bpj.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.