

Abstract
Ligand-mediated gene induction by steroid receptors is a multistep process characterized by a dose–response curve for gene product that follows a first-order Hill equation. This behavior has classically been explained by steroid binding to receptor being the rate-limiting step. However, this predicts a constant potency of gene induction (EC50) for a given receptor-steroid complex, which is challenged by the findings that various cofactors/reagents can alter this parameter in a gene-specific manner. These properties put strong constraints on the mechanisms of gene induction and raise two questions: How can a first-order Hill dose–response curve (FHDC) arise from a multistep reaction sequence, and how do cofactors modify potency? Here we introduce a theoretical framework in which a sequence of steps yields an FHDC for the final product as a function of the initial agonist concentration. An exact determination of all constants is not required to describe the final FHDC. The theory predicts mechanisms for cofactor/reagent effects on gene-induction potency and maximal activity and it assigns a relative order to cofactors in the sequence of steps. The theory is supported by several observations from glucocorticoid receptor-mediated gene induction. It identifies the mechanism and matches the measured dose–response curves for different concentrations of the combination of cofactor Ubc9 and receptor. It also predicts that an FHDC cannot involve the DNA binding of preformed receptor dimers, which is validated experimentally. The theory is general and can be applied to any biochemical reaction that shows an FHDC.
Keywords: dose-response, Michaelis-Menten, gene expression, steroid receptors, glucocorticoids, pharmacology
In ligand-mediated gene induction, the amount of gene expressed depends on the amount of ligand present. Thus, the specific shape and properties of the dose–response curve of gene induction, which is of crucial importance for development, differentiation, and homeostasis in many biological systems, provide a quantitative means for probing the gene-induction process. In many cases, the dose–response curve in gene induction obeys a sigmoidal curve, but not all sigmoidal curves have the same shape. For example, a dose–response curve obeying a first-order Hill equation or function (Hill coefficient equal to 1) goes from 10 to 90% of maximum activity over an 81-fold change in ligand concentration, whereas only a 9-fold change is required in a second-order Hill function, which thus has a different shape (Fig. S1). (A first-order Hill function is sometimes called a Michaelis–Menten function.) Depending upon the shape of the dose–response curve, the responsiveness of gene induction to the same variation in ligand concentration will differ greatly. In addition to the shape, the position or potency [i.e., concentration required for 50% of maximal response (EC50)] and maximum activity (Amax) of the dose–response curve are required to specify the amount of gene expressed for a given amount of ligand. Despite the vital role that the dose–response curve plays in biological systems, the mechanisms that determine its shape and position remain poorly understood. Therefore, a better understanding of the control and shape of the dose–response curve may provide insights into the gene-induction process.
Steroid-mediated gene induction provides an excellent system for investigating basic questions about gene induction and dose–response curves. The current model is that steroids enter the cell by passive diffusion and bind to intracellular receptors that can be predominantly cytoplasmic, as for glucocorticoid receptors (GRs), or nuclear, as for estrogen receptors. After a poorly understood step called activation, the receptor-steroid complex binds as a dimer with high affinity to biologically active DNA sequences [called hormone response elements (HREs)] to recruit additional transcriptional cofactors and modify the rates of transcription of nearby genes by the RNA polymerase complex. Glucocorticoids are an important class of steroids because they affect almost every cell and tissue in the body and are used to treat a variety of conditions, including asthma, autoimmune diseases, and cancer (1). In most experiments, the observed dose–response curves closely match a first-order Hill function. This is the dose response for the actual amount of protein product measured, not the receptor occupancy or an initial reaction rate. The addition of various cofactors can shift the EC50 and Amax yet preserve the shape of the dose–response curve (2–4). The fact that the multiple steps between ligand-receptor binding and translation to protein can yield a first-order Hill dose–response curve (FHDC) that can change position is remarkable. Here we develop a theoretical framework for a ligand-regulated gene-induction process that predicts the shape, position, and maximum activity of these dose–response curves. The theory is based on modeling the gene-induction process as an arbitrarily long sequence of complex-forming steps or reactions. A set of stringent but biologically plausible constraints that yield an FHDC arises from the theory. The theory is mathematically tractable in that it manages the parameter explosion of mathematical modeling by generating formulas for the FHDC with a small number of parameters that can be applied quantitatively to data. Although here it is applied specifically to glucocorticoid-induced gene expression, our paradigm is applicable to any general biochemical pathways that exhibit FHDCs.
Model
Obtaining FHDCs.
The basic principles of our theory can be demonstrated in some simple examples. In steroid-receptor-regulated gene induction, the ligand binds to an intracellular receptor. The receptor-ligand complex then associates with DNA to initiate a series of transcriptional and translational processes (see Fig. S2 for an example of a reaction sequence). The classic explanation for an FHDC is that receptor-ligand binding is the rate-limiting step (5). For example, consider the reaction 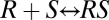 , for a receptor R and a ligand S. If the reaction obeys mass-action kinetics, then in equilibrium the concentrations obey
, for a receptor R and a ligand S. If the reaction obeys mass-action kinetics, then in equilibrium the concentrations obey 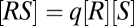 , where q is the affinity or association constant and square brackets indicate concentration. By mass conservation,
, where q is the affinity or association constant and square brackets indicate concentration. By mass conservation, 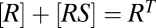 , where RT is the total receptor concentration. Solving the concentration and conservation equations gives the classic FHDC relationship between the complex and the free-ligand concentration,
, where RT is the total receptor concentration. Solving the concentration and conservation equations gives the classic FHDC relationship between the complex and the free-ligand concentration,
 |
where Amax = RT and EC50 = 1/q are both constants. If this receptor-ligand reaction is the rate-limiting step for gene induction, then the concentration of gene product can be assumed to be proportional to [RS]. However, experiments show that EC50 and Amax are not constant and can be changed by cofactors that act downstream of ligand-receptor binding (6), thereby ruling out the classic explanation. Thus, we are left with two questions: How can an FHDC arise for multiple reaction steps, and how can Amax and EC50 be controlled by cofactors?
In the gene-induction process, the receptor-steroid complex undergoes an activation step (or steps) culminating in binding to DNA, which is followed by multiple other steps such as the binding of transcription factors and the recruitment of polymerase (Fig. S2). These steps are envisaged as the building of larger and larger complexes (7). However, considering just two steps of such a sequence, for example, 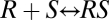 and
and 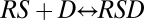 (where D represents DNA), does not generally lead to an FHDC for the product [RSD] in terms of [S], which can be seen as follows. The dose–response curve is computed by combining the concentration equations [RS] = q1[R][S] and [RSD] = q2[RS][D] with the mass-conservation equations [R] + [RS] + [RSD] = RT, [D] + [RSD] = DT, to eliminate [R], [D], and [RS], and obtain an equation for [RSD] and [S]. The resulting equation is quadratic [RSD], ruling out an FHDC (SI Section 1). Including additional complex-forming reactions does not alleviate this problem but makes the resulting equation even more complicated. An FHDC can exist if the resulting equation is linear in [RSD] (bilinear in [S] and [RSD]).
(where D represents DNA), does not generally lead to an FHDC for the product [RSD] in terms of [S], which can be seen as follows. The dose–response curve is computed by combining the concentration equations [RS] = q1[R][S] and [RSD] = q2[RS][D] with the mass-conservation equations [R] + [RS] + [RSD] = RT, [D] + [RSD] = DT, to eliminate [R], [D], and [RS], and obtain an equation for [RSD] and [S]. The resulting equation is quadratic [RSD], ruling out an FHDC (SI Section 1). Including additional complex-forming reactions does not alleviate this problem but makes the resulting equation even more complicated. An FHDC can exist if the resulting equation is linear in [RSD] (bilinear in [S] and [RSD]).
Hence, a sequence of complex-building reactions does not yield an FHDC in general because of the presence of quadratic and other nonlinear terms. However, the quadratic term does not exist if [RSD] can be disregarded or is of negligible value in the conservation equation for [R] (i.e., if [R] + [RS] + [RSD] = RT can be replaced by [R] + [RS] = RT). In this case, the two reactions 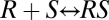 and
and 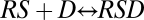 “decouple” and can be solved independently to yield Eq. 1 (with q = q1) and
“decouple” and can be solved independently to yield Eq. 1 (with q = q1) and 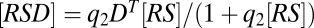 . Combining the two equations then yields the FHDC
. Combining the two equations then yields the FHDC
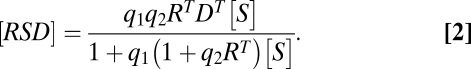 |
This property that the combination of two FHDCs results in another FHDC has been used previously (8, 9). Here we show how this property, which arises because the FHDC is in the family of Möbius or fractional linear transforms and forms a group under function composition (SI Section 2), can be generalized to any set of complex-forming biochemical reactions and allows us to derive a formula for the dose–response curve for an arbitrarily long reaction sequence. We consider the biological conditions under which this can occur below. We also note that the expressions for 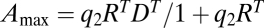 and
and 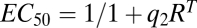 indicate that they can now be altered by factors downstream of receptor-steroid binding.
indicate that they can now be altered by factors downstream of receptor-steroid binding.
This FHDC property can also be exploited in diverse ways, which is seen by adding a third reaction,  , where U is a cofactor such as Ubc9. The concentration equations for the three reactions are now
, where U is a cofactor such as Ubc9. The concentration equations for the three reactions are now 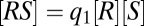 ,
, 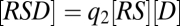 , and
, and 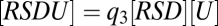 , and the mass-conservation equations are
, and the mass-conservation equations are 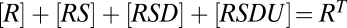 ,
, 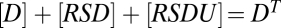 , and
, and 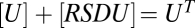 . The equation giving [RSDU] as a function of [S] is quartic (SI Section 1) and hence does not yield an FHDC. However, if downstream products in the conservation equations can be disregarded as before, such that
. The equation giving [RSDU] as a function of [S] is quartic (SI Section 1) and hence does not yield an FHDC. However, if downstream products in the conservation equations can be disregarded as before, such that 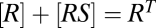 ,
, 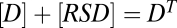 , and
, and 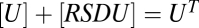 , then the three reactions decouple and [RSDU] has an FHDC with respect to [S]. Alternatively, if the conservation equations obey
, then the three reactions decouple and [RSDU] has an FHDC with respect to [S]. Alternatively, if the conservation equations obey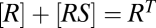 ,
, 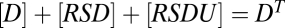 , and
, and 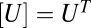 , then this yields a different FHDC. This second scenario could occur, for example, if the concentration of factor D were much smaller or limited compared with the other factors, and thus the reaction
, then this yields a different FHDC. This second scenario could occur, for example, if the concentration of factor D were much smaller or limited compared with the other factors, and thus the reaction 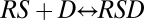 acts like a steady-state analog of a rate-limiting step, which we term the concentration-limiting step (CLS).
acts like a steady-state analog of a rate-limiting step, which we term the concentration-limiting step (CLS).
Biological Conditions Leading to an FHDC.
The theory hinges on having much smaller concentrations of downstream complexes than either upstream complexes or downstream cofactors. Small downstream concentrations of complexes could arise if cofactors and complexes only act transiently but produce a lasting response. For example, in the second scenario, if the second and third reactions were of the form  and D′ + U ⇔ D* + U, where D′ and D* are different states of D, then the same mass-conservation equations would result, leading to an FHDC for final product [D*] with respect to [S]. Possible biological mechanisms are that a cofactor could bind transiently to DNA but affect the DNA state (e.g., methylation, ubiquitination, uncoiling, untwisting, etc.), facilitate the binding of another cofactor, or alter the mRNA state during translation (6, 10, 11). Considerable experimental evidence has been advanced in support of transient binding (dubbed “hit and run”) of GR to endogenous genes (12, 13). We show below how these concepts can be applied to a sequence of complex-forming reactions in a general gene-induction process.
and D′ + U ⇔ D* + U, where D′ and D* are different states of D, then the same mass-conservation equations would result, leading to an FHDC for final product [D*] with respect to [S]. Possible biological mechanisms are that a cofactor could bind transiently to DNA but affect the DNA state (e.g., methylation, ubiquitination, uncoiling, untwisting, etc.), facilitate the binding of another cofactor, or alter the mRNA state during translation (6, 10, 11). Considerable experimental evidence has been advanced in support of transient binding (dubbed “hit and run”) of GR to endogenous genes (12, 13). We show below how these concepts can be applied to a sequence of complex-forming reactions in a general gene-induction process.
General Theory.
Consider a sequence of n binary reactions of the form 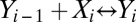 , where i = 1, 2,…, n, is an index for a reaction. Let Y0 be the ligand, X1 be the receptor, and Y1 be the receptor-ligand complex. We call the subsequent X variables activators or cofactors and the Y variables products. The reactions need not be reversible; they only should reach a stationary or a steady state. Hence, any number of the reactions could be irreversible with decay, that is,
, where i = 1, 2,…, n, is an index for a reaction. Let Y0 be the ligand, X1 be the receptor, and Y1 be the receptor-ligand complex. We call the subsequent X variables activators or cofactors and the Y variables products. The reactions need not be reversible; they only should reach a stationary or a steady state. Hence, any number of the reactions could be irreversible with decay, that is, 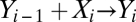 and
and 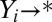 , or be transient (hit and run), e.g.
, or be transient (hit and run), e.g. 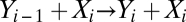 . For mass action, the steady-state concentrations obey
. For mass action, the steady-state concentrations obey 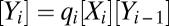 and mass conservation implies
and mass conservation implies 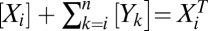 for i = 1, 2,…, n. The n association constants
for i = 1, 2,…, n. The n association constants 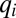 and the n total concentrations XiT are free parameters. The concentration and conservation equations consist of 2n equations in 2n + 1 unknowns. The equation governing [Yn] as a function of [Y0] is a high-degree polynomial, for which [Yn] is not in general a first-order Hill function of [Y0].
and the n total concentrations XiT are free parameters. The concentration and conservation equations consist of 2n equations in 2n + 1 unknowns. The equation governing [Yn] as a function of [Y0] is a high-degree polynomial, for which [Yn] is not in general a first-order Hill function of [Y0].
As shown in the examples above, an FHDC can arise if some of the downstream terms in the conservation equations are zero or very small (automatically satisfied for hit and run). It can be shown mathematically that the general form for the mass-conservation equations to ensure an FHDC has the form
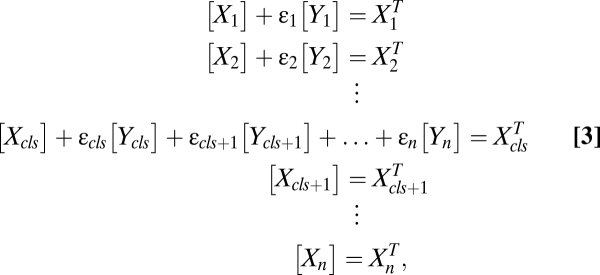 |
where 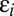 is 1 or 0, indicating the presence or absence of [Yi] in the equations. Step cls is the general definition of the CLS (introduced above). In this step, the concentration of factor Xcls is small compared with the concentration of the other activators. Hence, all terms appear in its conservation condition. For all steps after the CLS, the free concentration of any activator is equal to its total concentration. Hence, the location of the CLS can move by adding or removing varying amounts of activators. The CLS also need not exist for all situations.
is 1 or 0, indicating the presence or absence of [Yi] in the equations. Step cls is the general definition of the CLS (introduced above). In this step, the concentration of factor Xcls is small compared with the concentration of the other activators. Hence, all terms appear in its conservation condition. For all steps after the CLS, the free concentration of any activator is equal to its total concentration. Hence, the location of the CLS can move by adding or removing varying amounts of activators. The CLS also need not exist for all situations.
For reactions obeying the steady-state concentration equations and mass conservation (Eq. 3), the equations can be solved exactly using the group property of the fractional-linear transformation (SI Section 2) to obtain a formula for the dose–response curve,
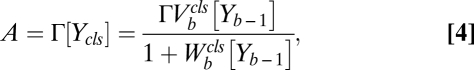 |
where A is the activity of the gene and 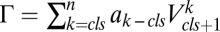 , where
, where 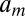 are positive constants,
are positive constants, 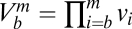 ,
, 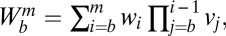 and
and 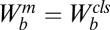 , for
, for 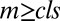 . We use the convention
. We use the convention 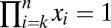 if
if 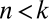 . The factors vi and wi have explicit formulas listed in Table 1. These factors specify the action of the cofactor at step i and take different forms depending on location with respect to the CLS. As is evident from comparison with Eq. 1,
. The factors vi and wi have explicit formulas listed in Table 1. These factors specify the action of the cofactor at step i and take different forms depending on location with respect to the CLS. As is evident from comparison with Eq. 1, 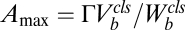 and EC50 = 1/Wbm. If the sequence of reactions does not have a CLS then A has the same formula as Eq. 4, except that cls is the index for the last reaction of interest and
and EC50 = 1/Wbm. If the sequence of reactions does not have a CLS then A has the same formula as Eq. 4, except that cls is the index for the last reaction of interest and 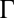 is a positive constant.
is a positive constant.
Table 1.
Values for vi and wi for activator and inhibitor at position i before, at, or after CLS
| Position | Activator | Activator with inhibitor |
Before CLS 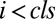
|
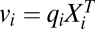 |
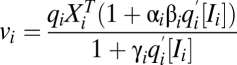 |
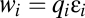 |
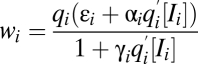 |
|
At CLS 
|
vi same as before CLS | vi same as before CLS |
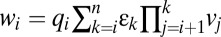 |
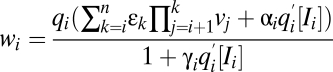 |
|
After CLS 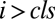
|
vi same as before CLS | 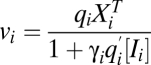 |
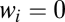 |
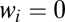 |
Eq. 4 depends on 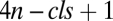 free parameters, namely the
free parameters, namely the 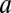 ,
, 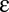 , q, and
, q, and 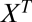 parameters, and the choice of these parameters specifies a model for the data. In general, all of these parameters and even the number of steps will be unknown. However, the formula is useful because the number of effective parameters necessary to specify a model can be reduced because any sequence of steps collapses or “telescopes” into a single first-order Hill function with an effective Amax and EC50. The result is that there are now only three effective “steps” to consider (before, at, or after the CLS), which reduces the possible parametric models that can explain a given experiment to a small number involving a finite set of cofactors. One also does not need to know a priori whether there even is a CLS. By applying the different possible models to the data, the existence of a CLS can be inferred. The CLS could be located experimentally by finding the cofactor that affects the dose–response curve in the appropriate manner for the CLS, as predicted by the theory. The reaction sequence also has a “modular” structure in that reactions, or a sequence of reactions, can be inserted, deleted, or combined without affecting the FHDC. Arbitrarily complex reaction sequences that maintain first-order Hill form can be constructed in this way. Thus, different genes could mix and match different pieces of the reaction sequence and all have FHDCs but with different Amax and EC50.
parameters, and the choice of these parameters specifies a model for the data. In general, all of these parameters and even the number of steps will be unknown. However, the formula is useful because the number of effective parameters necessary to specify a model can be reduced because any sequence of steps collapses or “telescopes” into a single first-order Hill function with an effective Amax and EC50. The result is that there are now only three effective “steps” to consider (before, at, or after the CLS), which reduces the possible parametric models that can explain a given experiment to a small number involving a finite set of cofactors. One also does not need to know a priori whether there even is a CLS. By applying the different possible models to the data, the existence of a CLS can be inferred. The CLS could be located experimentally by finding the cofactor that affects the dose–response curve in the appropriate manner for the CLS, as predicted by the theory. The reaction sequence also has a “modular” structure in that reactions, or a sequence of reactions, can be inserted, deleted, or combined without affecting the FHDC. Arbitrarily complex reaction sequences that maintain first-order Hill form can be constructed in this way. Thus, different genes could mix and match different pieces of the reaction sequence and all have FHDCs but with different Amax and EC50.
Incorporation of Inhibitors.
We have only considered activators so far. However, it is known that inhibitors, which act by reducing the concentration or efficacy of activators or products, can affect gene induction (10, 11). The reaction scheme for an inhibitor Ii acting at step i that preserves FHDC form is
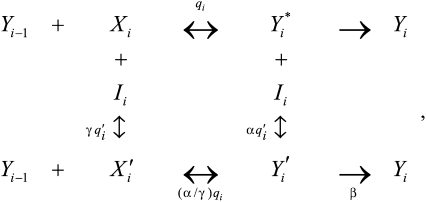
where the parameters obey 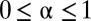 ,
,  , and
, and  . The case of
. The case of 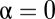 is called competitive inhibition,
is called competitive inhibition, 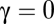 is called uncompetitive inhibition,
is called uncompetitive inhibition, 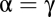 is called noncompetitive inhibition,
is called noncompetitive inhibition, 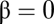 is called linear inhibition, and
is called linear inhibition, and 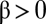 is called partial inhibition. Although I is called an inhibitor, its actions need not be inhibitory or repressive on the final activity. For example, if
is called partial inhibition. Although I is called an inhibitor, its actions need not be inhibitory or repressive on the final activity. For example, if 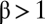 , I can be activating because it diverts the output from a lower-yield product to a higher-yield one. The dependence of
, I can be activating because it diverts the output from a lower-yield product to a higher-yield one. The dependence of 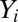 and
and 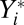 on Yi−1 is derived in SI Section 3 (following refs. 10 and 11) by solving the concentration and mass-conservation equations to obtain formulas for factors vi and wi, which specify the actions of the inhibitor on the activator at step i and are listed in Table 1. Inhibitory steps can be inserted anywhere in the reaction sequence before the CLS, but only competitive inhibitors can occur after the CLS.
on Yi−1 is derived in SI Section 3 (following refs. 10 and 11) by solving the concentration and mass-conservation equations to obtain formulas for factors vi and wi, which specify the actions of the inhibitor on the activator at step i and are listed in Table 1. Inhibitory steps can be inserted anywhere in the reaction sequence before the CLS, but only competitive inhibitors can occur after the CLS.
Results
Application to Steroid-Mediated Gene Induction.
To demonstrate the utility of our theory, we have applied it to steroid-receptor-mediated gene induction, where an FHDC has been observed experimentally. When a cofactor is added, Amax and EC50 of the dose–response curve can increase, decrease, or not change (4). Mechanisms for these various combinations can be predicted using Eq. 4 expressed in a form so that the inhibitor [Ii] or activator 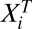 concentrations are visible (SI Section 4). Leaving out the combination of both not changing, we have experimentally observed seven of the eight remaining scenarios that are listed in Table 2 along with the predicted mechanisms. Although these suggested mechanisms are not unique (some of the scenarios are consistent with multiple mechanisms; see also SI Section 5), the predictions are consistent with previous results (6), as described below.
concentrations are visible (SI Section 4). Leaving out the combination of both not changing, we have experimentally observed seven of the eight remaining scenarios that are listed in Table 2 along with the predicted mechanisms. Although these suggested mechanisms are not unique (some of the scenarios are consistent with multiple mechanisms; see also SI Section 5), the predictions are consistent with previous results (6), as described below.
Table 2.
Effect of cofactors on Amax and EC50
| Amax | EC50 | Cofactors and context | Mechanism and position |
| Decrease | Decrease | CBP (with GR) (3), NCoR (with PR) (17) | (L and U) or (A after CLS) |
| Decrease | Increase | GMEB2, NCoR (with GR) (43), CPT, H8, DRB (with high GR and Ubc9) (6) | (C or L) before or at CLS |
| Decrease | No change | DRB, H8 (with high GR), VPA (with high GR and Ubc9) (6) | (P and U) before CLS |
| Increase | Decrease | TIF2 (with GR) (2, 3), Ubc9 (with high GR) (6, 14) | A before or after CLS |
| Increase | Increase | Not observed but predicted | C after CLS |
| Increase | No change | TSA, VPA, Ubc9 (with low GR) (6, 14) | A at or after CLS |
| No change | Decrease | SRC-1 (with GR) (3) | A after CLS |
| No change | Increase | TIF2 siRNA (with GR) (30) | L or C anywhere |
A, activator; I, inhibitor; L, linear inhibitor; P, partial inhibitor; C, competitive inhibitor; U, uncompetitive inhibitor.
Quantitative Predictions for Ubc9.
To uniquely identify a mechanism, the set of parameters specifying a model must be estimated directly from the data. We show this for the unusual capacity of the comodulator Ubc9 to cause different responses in transiently transfected cells depending upon the concentration of steroid receptor (6, 14, 15). (Experimental methods are in SI Section 8.) To generate numerous data points for an exacting test of our model, we used six different steroid (Dex) concentrations to define the dose–response curves for four levels of GR with three amounts of Ubc9 (Fig. 1). With low levels of GR, Ubc9 increases the Amax of luciferase activity while perturbing EC50 marginally. At higher receptor concentrations, there is a less proportional increase in Amax and a greater decrease in EC50. In all cases, an FHDC is always seen. In addition to extracting parameters to specify a mechanism, applying Eq. 4 to the data of Fig. 1 also gives an independent test of the model. We first must reexpress Eq. 4 so that three factor concentrations (steroid, receptor, and Ubc9) are visible in the formula (SI Section 4). However, whereas the steroid and receptor are assumed to appear in the first reaction, it is not known where Ubc9 appears. Hence, we must consider the possibility that it is before, at, or after a possible CLS. Recall that the CLS represents the step for which the cofactor has the smallest concentration compared with the other cofactors. The case of no CLS has the same formula as Ubc9 appearing before the CLS. The applicable formula has the form (SI Section 4)
Fig. 1.

Theoretical versus observed dose–response curves with different amounts of GR and Ubc9. The induction by Dex of transiently transfected GREtkLUC reporter and different amounts of GR (0.1, 2, 10, and 25 ng GR plasmid) ± Ubc9 plasmid in CV-1 cells was determined. Solid circle, 0 ng Ubc9; open square, 135 ng Ubc9; solid triangle, 175 ng Ubc9 (error bars, SD for triplicates). The vertical line indicates the EC50 for the dose–response curve with the same line style.
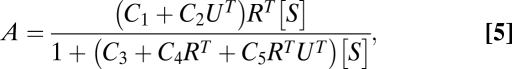 |
where A represents the luciferase activity, UT denotes the total Ubc9 concentration, RT equals the total steroid-receptor concentration, [S] is the free-steroid concentration, which we assume is approximated by the total steroid concentration, and the C parameters (given in Table S1) differ depending on where Ubc9 acts. Table S1 shows that if Ubc9 acts at the CLS (i.e., it is the concentration-limiting step), then it cannot affect EC50, so we can rule that possibility out. We fit Eq. 5 to the complete dataset of multiple doses of S, R, and Ubc9. Fig. 1 shows that the model captures the data fairly well, thereby reinforcing the validity of the model. The fitting algorithm and estimated parameter values are in SI Section 6. Given the parameters, we can predict where Ubc9 acts. The model fit finds the ratio of C1/C2UT ∼ 1. If Ubc9 acts before the CLS, then this ratio implies that the endogenous to exogenously applied Ubc9 ratio in the cell is near 1 (UT = 0 implies there is no exogeneous Ubc9). However, quantitative reverse transcription–PCR measurements give a ratio of ∼100, which rules out this possibility. Thus, we conclude that Ubc9 is an activator that acts after the CLS, as was hypothesized previously (14, 15).
GR Monomer Versus Dimer Binding to DNA.
In the prevailing model of steroid hormone action, preformed dimers of receptor-steroid complex bind to the biologically active HREs of target genes (7, 16). However, our theory predicts that such a cooperative step will destroy the FHDC. Thus, in those cases where a steroid produces an FHDC, our theory predicts that GR dimerization cannot be a necessary step for gene induction and vice versa (see SI Section 7 for a theoretical justification). In support of our theory, recent NMR studies show conformational changes in DNA-bound dimers occurring after the DNA binding of monomeric receptor (18). Furthermore, experiments with both steroid receptors (19–22) and helix-loop-helix zipper transcription factors (23, 24) strongly suggest that the majority of receptor binding to HREs occurs not by preformed dimers but by monomers that then form dimers on the DNA. To further examine this issue, we performed new experiments. We looked at the activity of several known dimerization-defective mutants (GR A477T, R479D, and D481R) (21, 25), plus a new double mutant (GR A477T/I646A) with a mutation in the dimerization domains of both the DNA-binding domain and ligand-binding domain (26). Each mutant is expressed at equivalent levels (Fig. S3), is active, and yields an FHDC (Fig. 2A). We then find that Ubc9 increases the Amax (while decreasing the fold induction) and decreases the EC50 of each mutant, similar to that seen with the wild-type GR (Fig. 2B and Fig. S4). We therefore conclude that defects in GR dimerization do not prevent GR transactivation or the modulatory activity of Ubc9, as predicted by our theory.
Fig. 2.

Modulatory activity of Ubc9 is maintained with dimerization-defective GR mutants. (A) Dose–response curves of wild type (wt GR; circles), double mutant (GRmm; squares), and empty vector (no GR; triangles) with added hSA or Ubc9 (open and closed symbols, respectively). (B) The induction properties with Dex of CV-1 cells transiently transfected with GREtkLUC reporter and wild-type or double mutant GR (A477T/I646A) plasmids ± Ubc9 plasmid were determined as in Fig. 1. The average values (n = 5; ±SEM) were plotted. *P < 0.05, **P ≤ 0.005 versus no Ubc9.
Discussion
We have described a theoretical framework for ligand-mediated gene induction that was applied specifically to glucocorticoid hormone action but is applicable to any biochemical pathway that exhibits an FHDC. The theory produces a formula for the FHDC describing how the Amax and the EC50 can change and yields mechanistic insights that are not obtainable with the common practice of examining just Amax (Table 1). The theory shows that any number of first-order steps of arbitrary complexity telescope into a single expression, with an effective Amax and EC50, consisting of just three steps that are before, at, or after a CLS. The steps involved need not be reversible as long as they reach steady state or equilibrium. An FHDC-preserving reaction sequence has a modular structure in that reactions can be inserted, deleted, or combined. In fact, different genes could possibly mix and match different pieces of the reaction sequence and all have FHDCs but with different Amax and EC50.
Our theory yields valuable information and testable hypotheses even when the precise steps of action are unknown. Despite minimal initial experimental constraints, the theory accounted for all of the different combinations of changing Amax and EC50 that have been observed experimentally. The validity of the theory is further supported by its successes in explaining how steps well downstream of ligand binding can alter the Amax and EC50 (6, 27). The theory defines a CLS, which is an equilibrium/steady-state analog of the rate-limiting step. One property of the CLS is that products from all steps after it can be summed directly and still preserve an FHDC. As shown in Table 1, the theory makes specific predictions of both biochemical mechanism of cofactor action and site of action relative to the CLS. For example, Table 1 predicts that the changes in Amax and EC50 of gene induction by high concentrations of GR upon addition of Ubc9 result from Ubc9 functioning as an activator before or after the CLS. Other experiments then restricted the possible mechanisms to Ubc9 acting as an activator downstream of the CLS, providing confirmation for our previous hypothesis of Ubc9 acting downstream of GR and a rate-limiting step (14). One also does not need to know a priori whether there is a CLS. The CLS could be located by finding a cofactor that affects the dose–response curve, as predicted by the theory.
A stringent test for application of our theory is the invariance of an 81-fold change between 0.1 and 0.9 of maximal induction under all conditions. More generally, the fold change between a fraction p and 1 − p of maximal induction is (1 − p)2/p2. This does not hold for a general complex-forming sequence of reactions. Additionally, we note that the first-order Hill function is the only Hill function that preserves its form when substituted into itself (i.e., forms a group under function composition). Higher-order Hill functions will be driven to a “switch-like” activation curve with the inclusion of more such steps, whereas lower-order ones will be driven to a flat activation curve (Fig. S1). Thus, any reaction scheme that is not described by our theory would be unlikely to produce the 81-fold change.
Another critical test of the model came with its prediction that appreciable DNA binding of preformed receptor dimers, which is widely believed to occur, cannot be necessary for steroid-induced gene expression in all situations. This does not mean that steroid receptors never bind to HREs as preformed dimers. Indeed, examples of non-FHDCs could indicate steps like receptor dimers or other cooperative behaviors that break the FHDC. Instead, our model says that the observation of an FHDC uniquely restricts the mechanism for receptor regulation of the observed gene to one that does not require preformed dimers. We show that three GR mutants with documented low to negligible binding as dimers to DNA, plus a combination double mutant that would be expected to be even less capable of dimer formation, are all still active with Amax and EC50 values that are modulated by exogenous Ubc9, as described by the model (Fig. 2 and Figs. S3 and S4). These and previous results of others suggest that the initial binding to biologically active DNA sequences involves predominantly GR monomers, as opposed to preexisting dimers, when the dose–response curve obeys first-order Hill form.
Although the studies of the current report are all with the transiently transfected reporter GREtkLUC, we have observed FHDCs with several endogenous genes for GR-mediated induction and repression (27–30). However, dose–response curves with other endogenous genes and/or other conditions (28) indicate non-FHDCs, which could serve as a valuable screen for systems where additional factors/processes contribute to the overall control of gene expression in a manner that violates the conditions of the theory. Our theory also does not include the effects of biomolecular fluctuations due to small numbers of molecules. We hypothesize that the mean concentrations should not be strongly affected because the regime under which our theory operates is near linear, implying that correlations arising from fluctuations should not be too deleterious. However, fluctuations will imply that the variance of the gene product could be nonnegligible, and this could affect downstream systems that depend on the expressed protein. An analysis with fluctuations could be formulated with the chemical master equation. Our theory could also potentially be useful in synthetic biology (31, 32), where precise control of the amount of gene product is desired. Our theory demonstrates that an exact determination of all rate constants of synthetic reactions is not necessary for an accurate description of the final dose–response curve, because there are only a few effective parameters that need to be tuned and these parameters can compensate for other possibly unknown parameters. The modularity also implies that tuning of the dose–response curve could be achieved by inserting or deleting a small number of synthetic modules to adjust EC50 or Amax as required.
Considerations of EC50 are of major importance in intact organisms, as seen with ecdysone regulation of Drosophila embryo development (33), GR concentration in glucocorticoid-induced apoptosis of mouse thymocytes (34), the role of coactivators in the pituitary resistance to thyroid hormone syndrome (35), and gene-selective responses in human peripheral blood mononuclear cells (30). An FHDC implies that changes in factor concentration act like a rheostat to give a continuum of responses (2, 4, 36, 37), which suggests that gene expression by steroid receptors during development, differentiation, and homeostasis employs differential control, as seen with other transcription factors and morphogens (38–41). This rheostat model is fully compatible with the many observations that the binding of steroid receptors and transcription factors is rapid and readily reversible (12, 13, 42). Indeed, the rapid binding of steroid receptors is a consequence of FHDC and a prediction of our theoretical framework.
Supplementary Material
Acknowledgments
We thank Jessica Lee for excellent technical assistance and Yunguan Sun for some preliminary experiments. We thank Sankar Adhya, Gordon Hager, Terence Hwa, David Levens, and Ty Voss for helpful comments. This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911095107/DCSupplemental.
References
- 1.Stahn C, Löwenberg M, Hommes DW, Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol. 2007;275:71–78. doi: 10.1016/j.mce.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Szapary D, Xu M, Simons SS., Jr Induction properties of a transiently transfected glucocorticoid-responsive gene vary with glucocorticoid receptor concentration. J Biol Chem. 1996;271:30576–30582. doi: 10.1074/jbc.271.48.30576. [DOI] [PubMed] [Google Scholar]
- 3.Szapary D, Huang Y, Simons SS., Jr Opposing effects of corepressor and coactivators in determining the dose-response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor-regulated gene expression. Mol Endocrinol. 1999;13:2108–2121. doi: 10.1210/mend.13.12.0384. [DOI] [PubMed] [Google Scholar]
- 4.Simons SS., Jr What goes on behind closed doors: Physiological versus pharmacological steroid hormone actions. Bioessays. 2008;30:744–756. doi: 10.1002/bies.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baxter JD, Tomkins GM. Specific cytoplasmic glucocorticoid hormone receptors in hepatoma tissue culture cells. Proc Natl Acad Sci USA. 1971;68:932–937. doi: 10.1073/pnas.68.5.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim Y, Sun Y, Chow C, Pommier YG, Simons SS., Jr Effects of acetylation, polymerase phosphorylation, and DNA unwinding in glucocorticoid receptor transactivation. J Steroid Biochem Mol Biol. 2006;100:3–17. doi: 10.1016/j.jsbmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 8.Strickland S, Loeb JN. Obligatory separation of hormone binding and biological response curves in systems dependent upon secondary mediators of hormone action. Proc Natl Acad Sci USA. 1981;78:1366–1370. doi: 10.1073/pnas.78.3.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loeb JN, Strickland S. Hormone binding and coupled response relationships in systems dependent on the generation of secondary mediators. Mol Endocrinol. 1987;1:75–82. doi: 10.1210/mend-1-1-75. [DOI] [PubMed] [Google Scholar]
- 10.Fromm HJ. Initial Rate Enzyme Kinetics. New York: Springer; 1975. [Google Scholar]
- 11.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems (Wiley Classics Library) New York: Wiley-Interscience; 1993. [Google Scholar]
- 12.Nagaich AK, Walker DA, Wolford R, Hager GL. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell. 2004;14:163–174. doi: 10.1016/s1097-2765(04)00178-9. [DOI] [PubMed] [Google Scholar]
- 13.Stavreva DA, Müller WG, Hager GL, Smith CL, McNally JG. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol Cell Biol. 2004;24:2682–2697. doi: 10.1128/MCB.24.7.2682-2697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaul S, Blackford JA, Jr, Cho S, Simons SS., Jr Ubc9 is a novel modulator of the induction properties of glucocorticoid receptors. J Biol Chem. 2002;277:12541–12549. doi: 10.1074/jbc.M112330200. [DOI] [PubMed] [Google Scholar]
- 15.Cho S, Kagan BL, Blackford JA, Jr, Szapary D, Simons SS., Jr Glucocorticoid receptor ligand binding domain is sufficient for the modulation of glucocorticoid induction properties by homologous receptors, coactivator transcription intermediary factor 2, and Ubc9. Mol Endocrinol. 2005;19:290–311. doi: 10.1210/me.2004-0134. [DOI] [PubMed] [Google Scholar]
- 16.Tsai M-J, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 17.Song L-N, Huse B, Rusconi S, Simons SS., Jr Transactivation specificity of glucocorticoid vs. progesterone receptors: role of functionally different interactions of transcription factors with amino- and carboxyl-terminal receptor domains. J Biol Chem. 2001;276:24806–24816. doi: 10.1074/jbc.M102610200. [DOI] [PubMed] [Google Scholar]
- 18.Holmbeck SMA, Dyson HJ, Wright PE. DNA-induced conformational changes are the basis for cooperative dimerization by the DNA binding domain of the retinoid X receptor. J Mol Biol. 1998;284:533–539. doi: 10.1006/jmbi.1998.2207. [DOI] [PubMed] [Google Scholar]
- 19.Dahlman-Wright K, Siltala-Roos H, Carlstedt-Duke J, Gustafsson J-A. Protein-protein interactions facilitate DNA binding by the glucocorticoid receptor DNA-binding domain. J Biol Chem. 1990;265:14030–14035. [PubMed] [Google Scholar]
- 20.Furlow JD, Murdoch FE, Gorski J. High affinity binding of the estrogen receptor to a DNA response element does not require homodimer formation or estrogen. J Biol Chem. 1993;268:12519–12525. [PubMed] [Google Scholar]
- 21.Liu W, Wang J, Yu G, Pearce D. Steroid receptor transcriptional synergy is potentiated by disruption of the DNA-binding domain dimer interface. Mol Endocrinol. 1996;10:1399–1406. doi: 10.1210/mend.10.11.8923466. [DOI] [PubMed] [Google Scholar]
- 22.Connaghan-Jones KD, Heneghan AF, Miura MT, Bain DL. Thermodynamic dissection of progesterone receptor interactions at the mouse mammary tumor virus promoter: Monomer binding and strong cooperativity dominate the assembly reaction. J Mol Biol. 2008;377:1144–1160. doi: 10.1016/j.jmb.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohler JJ, Metallo SJ, Schneider TL, Schepartz A. DNA specificity enhanced by sequential binding of protein monomers. Proc Natl Acad Sci USA. 1999;96:11735–11739. doi: 10.1073/pnas.96.21.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rentzeperis D, Jonsson T, Sauer RT. Acceleration of the refolding of Arc repressor by nucleic acids and other polyanions. Nat Struct Biol. 1999;6:569–573. doi: 10.1038/9353. [DOI] [PubMed] [Google Scholar]
- 25.Heck S, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bledsoe RK, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 27.Tao Y-G, Xu Y, Xu HE, Simons SS., Jr Mutations of glucocorticoid receptor differentially affect AF2 domain activity in a steroid-selective manner to alter the potency and efficacy of gene induction and repression. Biochemistry. 2008;47:7648–7662. doi: 10.1021/bi800472w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Y, Simons SS., Jr STAMP, a novel predicted factor assisting TIF2 actions in glucocorticoid receptor-mediated induction and repression. Mol Cell Biol. 2007;27:1467–1485. doi: 10.1128/MCB.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y, Tao YG, Kagan BL, He Y, Simons SS., Jr Modulation of transcription parameters in glucocorticoid receptor-mediated repression. Mol Cell Endocrinol. 2008;295:59–69. doi: 10.1016/j.mce.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo M, Simons SS., Jr Modulation of glucocorticoid receptor induction properties by cofactors in peripheral blood mononuclear cells. Hum Immunol. 2009;70:785–789. doi: 10.1016/j.humimm.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 32.Deans TL, Cantor CR, Collins JJ. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell. 2007;130:363–372. doi: 10.1016/j.cell.2007.05.045. [DOI] [PubMed] [Google Scholar]
- 33.Karim FD, Thummel CS. Temporal coordination of regulatory gene expression by the steroid hormone ecdysone. EMBO J. 1992;11:4083–4093. doi: 10.1002/j.1460-2075.1992.tb05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichardt HM, Umland T, Bauer A, Kretz O, Schütz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol. 2000;20:9009–9017. doi: 10.1128/mcb.20.23.9009-9017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan W, Farboud B, Privalsky ML. Pituitary resistance to thyroid hormone syndrome is associated with T3 receptor mutants that selectively impair β2 isoform function. Mol Endocrinol. 2005;19:1529–1542. doi: 10.1210/me.2005-0014. [DOI] [PubMed] [Google Scholar]
- 36.Simons SS., Jr The importance of being varied in steroid receptor transactivation. Trends Pharmacol Sci. 2003;24:253–259. doi: 10.1016/S0165-6147(03)00101-9. [DOI] [PubMed] [Google Scholar]
- 37.Simons SS., Jr How much is enough? Modulation of dose-response curve for steroid receptor-regulated gene expression by changing concentrations of transcription factor. Curr Top Med Chem. 2006;6:271–285. doi: 10.2174/156802606776173465. [DOI] [PubMed] [Google Scholar]
- 38.Gregor T, Tank DW, Wieschaus EF, Bialek W. Probing the limits to positional information. Cell. 2007;130:153–164. doi: 10.1016/j.cell.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc Natl Acad Sci USA. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurdon JB, Bourillot PY. Morphogen gradient interpretation. Nature. 2001;413:797–803. doi: 10.1038/35101500. [DOI] [PubMed] [Google Scholar]
- 41.Dubrulle J, Pourquié O. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature. 2004;427:419–422. doi: 10.1038/nature02216. [DOI] [PubMed] [Google Scholar]
- 42.Gorski SA, Snyder SK, John S, Grummt I, Misteli T. Modulation of RNA polymerase assembly dynamics in transcriptional regulation. Mol Cell. 2008;30:486–497. doi: 10.1016/j.molcel.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaul S, Blackford JA, Jr, Chen J, Ogryzko VV, Simons SS., Jr Properties of the glucocorticoid modulatory element binding proteins GMEB-1 and -2: potential new modifiers of glucocorticoid receptor transactivation and members of the family of KDWK proteins. Mol Endocrinol. 2000;14:1010–1027. doi: 10.1210/mend.14.7.0494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.