

Abstract
The Torpedo nicotinic acetylcholine receptor (nAChR) is the only member of the Cys-loop superfamily of ligand gated ion channels (LGIC) that is available in high abundance in a native membrane preparation. To study the structure of the other LGICs using biochemical/biophysical techniques, detergent-solubilization, purification, and lipid-reconstitution are usually required. To assess the effects of purification on receptor structure, we used the hydrophobic photoreactive probe 3-trifluoromethyl-3-(m-[125I]iodophenyl)diazirine ([125I]TID) to compare the state-dependent photolabeling of Torpedo nAChR before and after purification and reincorporation into lipid. For purified nAChR, the agonist-sensitive photolabeling within the M2 ion channel domain of positions M2-6, M2-9 and M2-13, the agonist-enhanced labeling of δThr274 (δM2-18) within the δ subunit helix bundle, and the labeling at the lipid-protein interface (αM4) were the same as for nAChR in native membranes. However, addition of agonist did not enhance [125I]TID photolabeling of δIle288 within the δM2-M3 loop. These results indicate that after purification and reconstitution of Torpedo nAChR, the difference in structure between the resting and desensitized states within the M2 ion channel domain was preserved, but not the agonist-dependent change of structure of the δM2-M3 loop. To further characterize the pharmacology of [125I]TID binding sites in the nAChR in the desensitized state, we examined the effect of phencyclidine (PCP) on [125I]TID photolabeling. PCP inhibited [125I]TID labeling of amino acids at the cytoplasmic end of the ion channel (M2-2 and M2-6), while potentiating labeling at M2-9 and M2-13 and allosterically modulating the labeling of amino acids within the δ subunit helix bundle.
The Cys-loop family of ligand gated ion channels (LGIC) includes the nicotinic acetylcholine receptors (nAChR), GABA type A receptor, serotonin type 3 receptor and the glycine receptor. The Torpedo (muscle-type) nAChR has been studied extensively (1) because of its availability in large quantities in native Torpedo membrane preparations, and it is the only eukaryotic LGIC for which there is a three dimensional structure (2;3). Four homologous subunits (2α, β, γ and δ) assemble pseudosymetrically to form the Torpedo nAChR pentamer. The membrane spanning domain of each nAChR subunit consists of a four helix bundle (M1-M4), with the M2 helices from each subunit associating at the central axis to line the lumen of the ion channel and the M1, M3 and M4 helices forming a lipid-exposed outer ring (2).
Due to the low abundance of LGIC receptors in their natural sources (other than the Torpedo nAChR), detergent-solubilization, purification and lipid-reconstitution are necessary to obtain receptor preparations for structural studies using many biophysical and biochemical techniques (4). The type of detergent used to disrupt the native membrane and the composition of the lipid mixture in which the nAChR is reincorporated determine the function of the purified nAChR (5). Solubilization of Torpedo nAChR-rich membranes in cholate, but not other detergents such as octyl glucoside, Triton X-100, or Tween 20, stabilizes nAChR in the resting state (6). While nAChRs reconstituted in lipid vesicles composed of dioleoylphosphatidylcholine (DOPC) were stabilized in the desensitized state, purification of nAChRs in the presence of DOPC, dioleoylphosphatidic acid (DOPA), and cholesterol (CH) has been shown to retain ion-gating activity (7) and agonist-induced state transitions from the resting to the desensitized state, as determined by Fourier Transform Infrared (FTIR) spectroscopy and hydrophobic photolabeling (8;9).
Since its introduction (10), 3-trifluoromethyl-3-(m-[125I]iodophenyl) diazirine ([125I]TID) has been used to study the lipid interface of many membrane proteins (11). As a hydrophobic probe, [125I]TID photolabels amino acids at the nAChR lipid-protein interface (12), within the ion channel, and in the delta subunit helix bundle (13;14). [125I]TID photolabeling within the ion channel reveals changes in nAChR structure between the resting and desensitized states (13), while photolabeling within the δ subunit helix bundle reveals changes in structure between the resting, open, and desensitized states (14).
In this report we use [125I]TID photolabeling to define the structure of Torpedo nAChR after cholate-solubilization, affinity-purification and reconstitution into a lipid environment that preserves nAChR state transitions (purified nAChR hereafter) relative to its structure in its native membrane environment (native nAChR hereafter). While FTIR spectroscopy studies have established that purified and lipid reconstituted Torpedo nAChRs retain similar secondary structure compositions and agonist-induced structural changes (15;16), [125I]TID photolabeling is able to assess at higher resolution the potential differences in structure in the transmembrane domain. To further define [125I]TID as a structural probe of the nAChR in the desensitized state, we use phencyclidine (PCP), an aromatic amine non-competitive antagonist that binds with high affinity to a single site in the nAChR ion channel (Keq = 1 μM, desensitized state) and to additional lower affinity sites (17) to characterize the pharmacological specificity of the [125I]TID binding sites.
EXPERIMENTAL PROCEDURES
Materials
Torpedo californica and their frozen electric organs were obtained from Aquatic Research Consultants (San Pedro, CA). Synthetic lipids and cholesterol were from Avanti Polar Lipids, Inc. (Alabaster, AL). [125I]TID (~10 Ci/mmol) was obtained from Amersham Biosciences (Piscataway, NJ) and stored in ethanol at −4°C. Staphylococcus aureus glutamyl endopeptidase Glu-C (V8 protease) was from ICN Biomedical, and endoproteinase Lys-C (EndoLys-C) was from Roche Applied Sciences. Sodium cholate was from USB Corporation (Cleveland, OH).
Torpedo nAChR Purification and Reconstitution
Torpedo nAChR-rich membranes for affinity purification, isolated from frozen electric organs, and membranes for direct [125I]TID photolabeling, isolated from freshly dissected T. californica electric organs, were prepared, as described previously (18). Torpedo nAChR-rich membranes at 1 mg/ml were solubilized in 1% sodium cholate in vesicle dialysis buffer (VDB, 100 mM NaCl, 0.1 mM EDTA, 0.02% NaN3, 10 mM MOPS, pH 7.5), and the nAChR was affinity-purified on a bromoacetylcholine bromide-derivatized Affi-Gel 10 column (Bio-Rad) and then reconstituted into lipid vesicles composed of DOPC, DOPA, and CH at a molar ratio of 3:1:1, as described (7;9). The lipid to nAChR ratio was adjusted to a molar ratio of 400:1. Based upon SDS-PAGE, the purified nAChR comprised more than 90% of the protein in the preparation. Both the nAChR-rich membranes and the purified nAChR were stored at −80°C.
[125I]TID Photolabeling
Twenty milligrams of Torpedo nAChR-rich membranes (1.5 nmol [3H]Acetylcholine binding sites/mg protein) or 5 mg of purified Torpedo nAChR were incubated with ~250 μCi of [125I]TID (~ 2.5 μM) in 10 ml of Torpedo physiological saline (TPS: 250 mM NaCl, 5 mM KCl, 3 mM CaCl2, 2 mM MgCl2, and 5 mM sodium phosphate, pH 7.0) and divided into 5 ml aliquots in round bottom flasks. TPS or drug(s) was added to the membrane suspensions, which were stirred for 40 min, then irradiated with a 365 nm UV lamp (Model EN-16, Spectrotonics, Westbury, NY) for 20 minutes at a distance of less than 1 cm. For purified nAChR, labeling was carried out in the absence or presence of 200 μM Carb. For nAChR-rich membranes, labeling was carried out under three different labeling conditions: 1. resting state labeling (no drug added), 2. desensitized state labeling (in the presence of 200 μM Carb); 3. labeling in the presence of 200 μM Carb and 100 μM PCP.
SDS-Polyacrylamide Gel Electrophoresis
[125I]TID-labeled nAChRs were resuspended in electrophoresis sample buffer (12.5 mM Tris-HCL, 2% SDS, 8% sucrose, 1% glycerol, 0.01% bromophenol blue, pH 6.8), and the polypeptides were resolved on 1.5 mm thick gels, with 8% polyacrylamide/0.32% bis-acrylamide in the separating gel (19;20). Following electrophoresis, gels were stained with Gel Code Blue® stain reagent (Pierce), and processed for phosphor imaging to track 125I subunit incorporation, and the stained α, β, γ and δ nAChR subunit bands were excised.
To isolate fragments containing the αM2 or αM4 segments, the α subunit bands were soaked in overlay buffer (5% sucrose, 125 mM Tris-HCl, 0.1% SDS, pH 6.8) for 20 min, transferred to the wells of a 15% acrylamide mapping gel, and overlaid with 200 μg of V8 protease (~1000 u/mg; MP Biomedicals, Solon, OH) in overlay buffer for “in gel” digestion (20;21). This digestion reproducibly generates four non-overlapping peptides, αV8-4 (beginning at αSer1), αV8-18 (αThr52), αV8-20 (αSer173) and αV8-10 (αAsn339) (20). Based on the phosphor image of the mapping gel, the bands were excised that contained the labeled subunit proteolytic fragments αV8-20, which includes the M1, M2 and M3 transmembrane segments, and αV8-10, which includes the M4 segment,.
Alpha subunit fragments and intact β, γ and δ subunits were recovered from gel pieces by passive elution. Eluates were concentrated by centrifugal filtration to a final volume of 300 μl (Vivaspin 15 Mr 5000 concentrators; Vivascience, Stonehouse, UK) and then acetone precipitated (75% acetone at −20°C overnight) to remove SDS. Subunits or subunit fragments were then resuspended in resuspension buffer (15 mM Tris, 0.5 mM EDTA, 0.1% SDS, pH 8.1) for proteolytic digestion.
Isolation of Transmembrane segments
The αM4 segment was generated by digestion of αV8-10 with trypsin. Four volumes of 0.5% Genapol in 50 mM NH4HCO3 buffer (pH 8.1) were added to 1 volume of subunit fragments to dilute the SDS content, and trypsin (1:1 protein to enzyme ratio) in 0.1 volume of 20 mM CaCl2 was added and the digestion was allowed to proceed for 2 days at room temperature (22). The digests were purified by reversed-phase HPLC (rpHPLC, see below), with the fragments containing αM4 eluting as a broad hydrophobic peak. The fragment beginning near the N-terminal of αM2 was generated from endoproteinase Lys-C (EndoLys-C, Roche Diagnostics, Indianapolis, IN) digests of αV8-20. The αV8-20 fragment in 100 μl of resuspension buffer was incubated for 2 weeks with 0.5 unit of EndoLys-C, and the digests were then fractionated by rpHPLC and the 125I peak was pooled for amino acid sequencing.
To isolate fragments beginning near the N-terminal of the βM2, δM1 and δM2 segments, β and δ subunits were digested with trypsin (12–16 h) and EndoLys-C (2 weeks), respectively. The digests were resolved on a 1.5 mm thick, small pore (16.5%T, 6%C) Tricine SDS-PAGE gel (23). For the β subunit digest, the βM2 segment was isolated by rpHPLC purification of the major radioactive band in the Tricine gel which runs with an apparent molecular mass of 8 kDa (13). The fragments beginning near the N-terminal of the δM1 and δM2 were isolated by rpHPLC fractionation of material eluted from the gel band with apparent molecular mass of 10–14 kDa (14).
The fragment beginning near the N-terminal of the δM2-M3 loop and extending through the δM3 was generated by digestion of the δ subunit with V8 protease in solution (22). Delta subunit in resuspension buffer was incubated with 200 μg of V8 protease for 1–2 days at room temperature, and the digest was then fractionated by rpHPLC. While this fractionation does not resolve the M3 fragment from the other transmembrane segments, the 125I incorporated within the δM2-M3 loop was determined from amino acid sequencing by the use of o-phthalaldehyde as described below.
Reversed-Phase HPLC Purification and Sequence Analysis
rpHPLC was performed on an HP 1100 binary system using a Brownlee Aquapore BU-300 column (70μ, 100 × 2.1mm; PerkinElmer #0711-0064) and Brownlee Newguard RP-2 guard column at 40 °C. Solvent A was 0.08% trifluoroacetic acid (TFA) in water, and Solvent B was 0.05% TFA in 60% acetonitrile/40% 2-propanol. A nonlinear elution gradient at 0.2 mL/min was employed (25% to 100% Solvent B in 75 min, shown as dotted line in figures), and fractions were collected every 2.5 min (36 fractions/run). The elution of peptides was monitored by the absorbance at 215 nm, and the 125I in each fraction was determined by γ-counting.
For sequence analysis, the rpHPLC fractions containing the αM4 and δM1 segments were loaded onto PVDF filters using Prosorb® Sample Preparation Cartridges (Applied Biosystems #401959) and the filters were treated with Biobrene as recommended by the manufacturer. For other samples, rpHPLC fractions were drop-loaded onto Biobrene-treated Micro TFA filters (Applied Biosystems #401111) at 45°C. Amino acid sequencing was performed on an Applied Biosystems PROCISE 492 protein sequencer. One-sixth of the eluate of each Edman degradation cycle was used for amino acid identification/quantification, and five-sixths were collected for 125I counting. For each peptide detected, the amount of amino acid (f(x), in pmol) in cycle x, determined from the peak height(x) − peak height(x−1), was fit to the equation f(x) = I0Rx to determine the initial amount of the peptide (I0) and the sequencing repetitive yield (R). Ser, His, Trp, and Cys were not included in the fits due to known problems with their quantification. For some samples, sequencing was interrupted at a specific cycle and the filter was treated with o-phthalaldehyde (OPA) before resuming sequencing (indicated by an arrow in figures). OPA reacts efficiently with primary amino acids but not secondary amines (proline), and treatment with OPA prevents further sequencing of fragment not containing a proline at that cycle (24;25). The efficiency of amino acid photolabeling (cpm/pmol) was calculated as (cpmx−cpm(x−1))/5IoRx.
RESULTS and DISCUSSION
[125I]TID labeling of purified Torpedo nAChR
[125I]TID photolabeling of affinity purified Torpedo nAChR reconstituted into DOPC, DOPA, and CH was characterized by the same relative incorporation into subunits in the absence of agonist as seen for Torpedo nAChR in its native membrane and by the same ~90% reduction in subunit photolabeling upon desensitization by agonist (6;8). To extend these studies, we photolabeled purified and native Torpedo nAChRs on a preparative scale and isolated for sequence analysis the δ subunit fragments containing δM1, δM2 and δM2-M3 loop/δM3 (Supplementary Figures S1 and S2). As reported previously (13), for native nAChR in the absence of agonist (resting state), within δM2 (Figure 1A) [125I]TID photolabeled two amino acids in the middle of the ion channel domain: δM2-9 (δLeu265, 40 cpm/pmol) and δM2-13(δVal269, 130 cpm/pmol). In the presence of agonist (desensitized state) (Figure 1A and B), δM2-18 (δThr274), which projects into the pocket formed by the δ subunit helix bundle (see summary Figure 6), was the most efficiently labeled amino acid (15 cpm/pmol), and the labeling at δM2-9 and δM2-13 was reduced by >95%. δM2-6 (δSer262), near the cytoplasmic end of the ion channel, and δM2-22 (δLeu278), which projects into the δ subunit helix bundle, were also labeled in the presence of agonist. Similar state-dependent photolabeling of amino acids was found within δM2 segments isolated from [125I]TID-labeled purified nAChR (Figures 1C and D). In the presence of agonist, the major peak of 125I release was associated with δM2-18 (5 cpm/pmol), and labeling at δM2-9 and δM2-13, which was predominant in the absence of agonist (17 cpm/pmol and 44 cpm/pmol, respectively), was reduced to less than 1 cpm/pmol. In addition, δM2-6 was labeled at ~ 1 cpm/pmol (Table 1). While in the absence of agonist in the native nAChR there was no detectable labeling of δM2-6 or δM2-18, in the purified nAChR those positions were labeled at ~30% the efficiency seen in the presence of agonist. This labeling of δM2-6 and δM2-18 in the absence of agonist provides evidence that a higher fraction of the purified nAChRs are in the desensitized state than in the native membranes (~20 % (26)).
Figure 1. The effect of affinity-purification and lipid-reconstitution on [125I]TID photoincorporation into δM2 in nAChRs in the resting and desensitized states.

125I (○, ●) and PTH-amino acids (□, ■) released during sequence analysis through δM2 from native (A and B) or purified (C and D) nAChRs. Native Torpedo nAChR-rich membranes or affinity-purified and lipid-reconstituted nAChR were photolabeled with [125I]TID in the absence (○, □) or presence (●, ■) of Carb, and EndoLys-C digests of δ subunits were fractionated by Tricine SDS-PAGE and by rpHPLC (Supplemental Figure S1) to isolate fragments beginning at δMet257. The primary sequence began at δMet257 for native nAChR (A and B, Io, control (□), 31 pmol,+ Carb (■), 37 pmol) and for purified nAChR (C and D, Io, −Carb (□), 35 pmol, +Carb (■), 30 pmol), with a fragment beginning at δAsn437 present at ~ 1 pmol in all samples. For both the native and purified nAChRs, in the absence of Carb the peaks of 125I release in cycles 9 and 13 reflected labeling of δLeu265 and δVal269, which was reduced by >95% for the +Carb samples which had the major peak of 125I release in cycle 18, consistent with labeling of δThr-274, and in cycles 6 (δSer262). B and D, 125I release profiles from panels A and C are replotted on an expanded scale to show clearly the peaks of release for the +Carb samples. The amino acid sequence quantified is shown above each panel, with the solid bar indicating the span of the M2.
Figure 6. Residues photolabeled by [125I]TID within the transmembrane domain of native and reconstituted nAChRs.

Views of the membrane-spanning helices (shown as cylinders) of the Torpedo nAChR structure (PDB # 2BG9): A, looking down the channel from the base of the extracellular domain; and B, (A) looking parallel to the membrane with 2 subunits removed for clarity, rotated 90° from (A). Subunits are color-coded: α, gold; β blue; γ, green; and δ, magenta. Residues photolabeled by TID are included in stick format, color coded by domain and conformation: ion channel, resting state (red); ion channel, desensitized state (PCP inhibitable) (yellow); δ subunit helix bundle, desensitized state (cyan); lipid-protein interface (green). A Connolly surface model of TID is included in A for scale.
TABLE 1. The efficiency of [125I]TID photoincorporation in amino acids within the δ subunit (cpm/pmol of PTH-derivative).
The 125I incorporation in each residue was calculated from the observed 125I release as described under “Experimental Procedures”, and the mass was calculated from the initial and repetitive yield.
| Native Torpedo nAChR | Purified Torpedo nAChR | ||||||
|---|---|---|---|---|---|---|---|
| Experiment I | Experiment II | Experiment III | |||||
| Control | Carb | Carb | Carb and PCP | Control | Carb | ||
| δM2-6 | Ser262 | < 0.3 | 0.6 | 1.7 | <0.1 | 0.5 | 0.9 |
| δM2-9 | Leu265 | 43 | 1.4 | 2.5 | 5.2 | 17 | 0.7 |
| δM2-13 | Val269 | 130 | 2.4 | 3.3 | 3.9 | 44 | 1.2 |
| δM2-18 | Thr274 | < 0.3 | 15 | 23 | 16 | 1.3 | 4.6 |
| δ M2-M3 | Ile288 | 1.0 | 6.5 | 11 | 6.4 | 0.8 | 0.8 |
| δ M1 | Phe232 | < 0.5 | 5.0 | 8.3 | 4.4 | 1.0 | 1.6 |
| Cys236 | 1.0 | 42 | 38 | 15 | 2.3 | 4.6 | |
| αM4 | Cys412 | 7.7 | 13 | 38 | 41 | 100 | 80 |
To further characterize the structure of the δ subunit transmembrane domain in the purified nAChR, subunit fragments beginning at δPhe209, containing δM1, and at δThr281, containing the δM2-M3 loop and extending through δM3, were isolated from [125I]TID-labeled native and purified nAChRs and sequenced (Figure 2). For native nAChRs, [125I]TID labeled δPhe232 (5 cpm/pmol) and δCys236 (42 cpm/pmol) within δM1 (Figure 2A) and δIle288 (7 cpm/pmol) in the δM2-M3 loop (Figure 2B) only in the presence of agonist (desensitized state). For purified nAChR, [125I]TID photolabeled δPhe232 and δCys236 within the δM1 (Figure 2C) in the absence (1 and 2 cpm/pmol, respectively) and the presence of agonist (2 and 5 cpm/pmol, respectively). The efficiency of [125I]TID photoincorporation into δIle288 within the δM2-M3 loop was 1 cpm/pmol in the absence or presence of agonist (Figure 2D).
Figure 2. The effect of affinity-purification and lipid-reconstitution on [125I]TID photoincorporation in δM1 (A and C) and in the δM2-δM3 loop (B and D) in the absence or presence of agonist.

125I (○, ●) and PTH-amino acids (□, ■) released during sequencing of fragments isolated from EndoLys-C digests (A and C) and V8 protease digests (B and D) of δ subunits isolated from [125I]TID-labeled native (A, B) or purified Torpedo nAChR (C, D) as described in “Experimental Procedures” and in Supplemental Figures S1 and S2. During sequencing the filters were treated with OPA at cycle 20 to chemically isolate δM1 (A & C) or in cycle 6 to chemically isolate M3 (B & D) by preventing further sequencing of fragments not containing a proline in those cycles. A & C, Before treatment with OPA the primary amino acid sequence began at δPhe206 for native (A, Io, 4 pmol, − and +Carb) and purified nAChRs (B, Io, 14 pmol, − and +Carb), and sequencing of that fragment continued after OPA treatment. For the native membranes, before OPA treatment the fragment beginning at δMet257 was present as a secondary sequence (+Carb (◆), Io, 2 pmol; −Carb, 1 pmol (not shown)), and it is this fragment that is the source of the peaks of 125I release in cycles 9 and 13 (−Carb, ○) and in cycle 18 (●, +Carb). After treatment with OPA in cycle 20, for the native nAChR there were peaks of 125I release in cycles 27 (δPhe232), and 31 (δCys236) +Carb, with no detectable 125I release in the −Carb sample. For the purified nAChR, there were peaks of 125I release in cycles 27 and 31 in the absence and presence of Carb. B & D, Before OPA treatment in cycle 6, fragments were present beginning at δIle192, δVal443, and δThr281, each at ~ 12 pmol (B), while after OPA treatment, sequencing continued only for the δThr281 fragment from native (B, Io, 12 pmol − or +Carb) and purified nAChRs (D, Io, 8 pmol − or +Carb). The amino acid sequence quantified is shown above each panel, with the solid bar indicating the span of the M1 (A &C) and M3 (B & D) segments.
Since the agonist-sensitivity of [125I]TID photolabeling within the ion channel was similar for the purified nAChR as for the native Torpedo nAChR, these results indicate that the orientation of the M2 helices in the closed state is retained after purification as well as the differences in structure between the resting (closed channel) and desensitized states. [125I]TID photolabeling also provided evidence of state-dependent changes in the structure of the δ subunit helix bundle after purification, but not in the δM2-M3 loop. In contrast to the native nAChR, [125I]TID photolabeled δPhe232 and δCys236 within δM1 in the purified nAChR in the absence of agonist, consistent with a higher fraction of purified nAChRs in the desensitized state, as was also evidenced by the labeling of δM2-6 and δM2-18. The lack of agonist-enhanced photolabeling of δIle288, which in the purified nAChR in the absence (or presence) of agonist is labeled at the same low level as in the native nAChR in the absence of agonist, indicates that either the structure of the δM2-M3 loop was perturbed after solubilization, purification and reconstitution or that its structure was retained but not its orientation in the desensitized state relative to the δ subunit helix bundle. Further studies are required to distinguish between these two possibilities.
Another structural component that might be affected by detergent-solubilization and reconstitution is the lipid-protein interface. To test this, we examined the [125I]TID labeling within αM4. For both native and purified nAChR (Figure 3A and B, respectively), [125I]TID photoincorporated mainly at αCys412 within αM4, with similar labeling efficiency in the absence and presence of agonist. Thus the surface of the αM4 helix most exposed to lipid remains the same in the purified nAChR as in the native nAChR. Amino acids within each M3 segment are also positioned at the lipid interface (12), but we were unable to identifying the photolabeled amino acids within δM3 in native (Figure 2B) or purified nAChRs (Figure 2D) because of the increasing background 125I release seen beyond sequencing cycle 10, which probably originated from 125I release due to random internal cleavage of other labeled fragments blocked by OPA. Successful identification of the amino acids within the M3 helices photolabeled by [125I]TID in the native nAChR required sequence analysis, without OPA block, of highly purified M3 fragments (12).
Figure 3. Affinity-purification and lipid-reconstitution has no effect on [125I]TID photolabeling within αM4.

125I (○, ●) and PTH-amino acids (□, ■) released during sequence analysis of the fragment beginning at αTyr401, which was isolated by rpHPLC from trypsin digests of αV8-10 from [125I]TID-labeled native (A) or purified nAChR (B) photolabeled with [125I]TID in the absence (□, ○) or presence of Carb (■, ●). In each condition, the primary amino acid sequence began at αTyr401 (A, Io, 15 pmol, − or +Carb; B, 3 pmol − or +Carb) and the major peak of 125I release was in cycle 12 (αCys412). The amino acid sequence detected is shown above the panel, with the solid bar indicating the span of the M4 segment.
Table 1 compares the efficiencies of [125I]TID photolabeling of amino acids (cpm/pmol) in the δ subunit transmembrane domain and in αM4 for native and purified nAChRs in the absence and presence of agonist. For the native nAChR in the desensitized state, comparison of the results for two independent labeling experiments establishes that the efficiency of labeling at an individual position within the δ subunit can vary by 30% from the average value, but the relative efficiencies of photolabeling of individual amino acids within each experiment is quite reproducible (for example, the ratios of labeling at δThr274 to δIle288 and δThr274 to δPh232 were 2.2±0.1 and 2.9±0.1, respectively). Comparison of labeling efficiencies of positions in the native and purified nAChRs reveals that the most striking difference is the enhanced labeling of the purified nAChR at the lipid interface (αCys412) relative to the protein interior (ion channel or helix bundle). In the native nAChR in the resting state, the ratio of labeling of αCys412 to δM2-13 was 0.06, while it was 2.2 for the purified nAChR. For native nAChR in the desensitized state, the ratio of labeling of αCys412 to δM2-18 was ~1.3, while it was 16 for the purified nAChR. This difference results both from an increase in the labeling efficiency at the lipid interface and a reduced labeling efficiency in the protein interior. Although the 400 to 1 molar ratio of lipid to purified nAChR is similar to the average value in the Torpedo nAChR-rich membrane preparation, neither the specific lipid composition nor the bilayer asymmetry of the Torpedo membranes (5;27) is maintained, which will likely contribute to differences in TID partitioning between bulk lipid and the nAChR in the two environments (28). In addition, the highly unsaturated fatty acids in the Torpedo lipids (27) may function as a scavenger competing for photoactivated TID at the nAChR-lipid interface. While differential partitioning may account for the relatively reduced labeling within the protein interior (ion channel, δ helix bundle), it can not account for the difference we observe in the state dependence of TID labeling in the δM2-M3 loop.
The effects of PCP on [125I]TID photolabeling of nAChR in the desensitized state
For nAChR in the desensitized state, PCP binds to a single high affinity binding site per receptor which is assumed to be within the ion channel (29). To examine the effect of PCP on [125I]TID photolabeling within the ion channel and the δ subunit helix bundle of Torpedo nAChR in the desensitized state, transmembrane fragments were isolated and sequenced from Torpedo nAChR-rich membranes photolabeled with [125I]TID in the presence of agonist or agonist and PCP. Within the ion channel, PCP completely inhibited [125I]TID photoincorporation into αM2-6, βM2-2, βM2-6, and δM2-6, but it enhanced photolabeling at βM2-13, δM2-9 and δM2-13 (Figure 4). Within the δ subunit helix bundle, PCP reduced by ~30% the efficiency of labeling of δM2-18 (Figure 4C) and by ~50% the labeling δPhe232 and δCys236 within δM1 segment (Figure 5A) or δIle288 in the δM2-M3 loop (Figure 5B). At the lipid-protein interface PCP did not inhibit labeling of αCys412 within αM4 (Table 1).
Figure 4. For nAChRs in the desensitized state, PCP inhibits [125I]TID photoincorporation into amino acids only at the cytoplasmic end of the M2 segments.
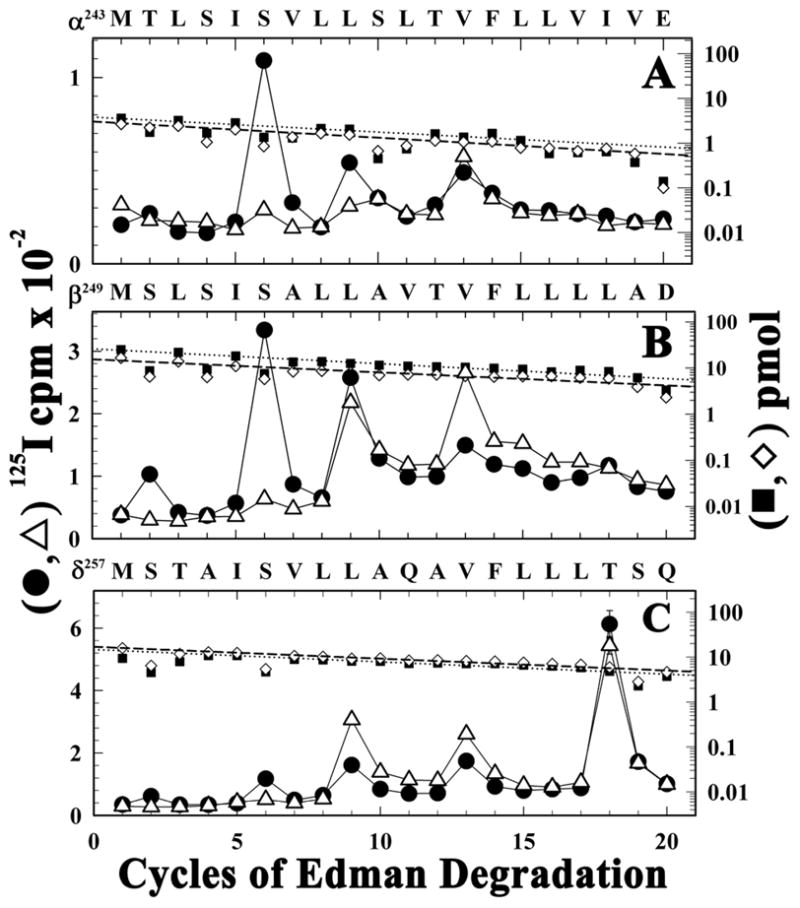
125I (●, △) and PTH-amino acids (■, ◇) released during sequence analysis of the fragments beginning at the N-termini of αM2 (A), βM2 (B), and δM2 (C) that were isolated from native Torpedo nAChR labeled with [125I]TID in the presence of Carb (●, ■) or Carb and PCP (△, ◇). A, The primary amino acid sequence began at αMet243 (Io, 4 pmol each condition). The major peak of 125I release in cycle 6 +Carb (αSer248, 90 cpm) was reduced to 10 cpm +Carb +PCP. B, The primary amino acid sequence began at βMet249 (Io, +Carb, 20 pmol; +Carb+PCP, 16 pmol) with a secondary sequence beginning at βLys216 (Io, < 1 pmol). In the presence of PCP, the peaks of 125I release in cycles 2 (βSer250) and 6 (βSer254) were reduced by >90%, the release in cycle 9 (βLeu257) was reduced by <15%, and the release in cycle 13 (βVal261) was increased by 200%. C, The primary amino acid sequence began at δMet257 (Io, 16 pmol, both conditions). In the presence of Carb, the major peak of release in cycle 18 indicated labeling of δThr274 at 22 cpm/pmol which was reduced by 30% in the presence of PCP. The peaks of 125I release in cycles 2 and 6 indicated labeling of δSer258 and δSer262 at 0.4 and 1.7 cpm/pmol which was reduced by 90% +PCP. δLeu265 (cycle 9) and δVal269 (cycle 13) were labeled at 2.5 and 3.3 cpm/pmol (+Carb), and that labeling was increased by 100% and 20%, respectively, in the presence of PCP.
Figure 5. [125I]TID photoincorporation within δM1 and the δM2-M3 loop in the presence of PCP.
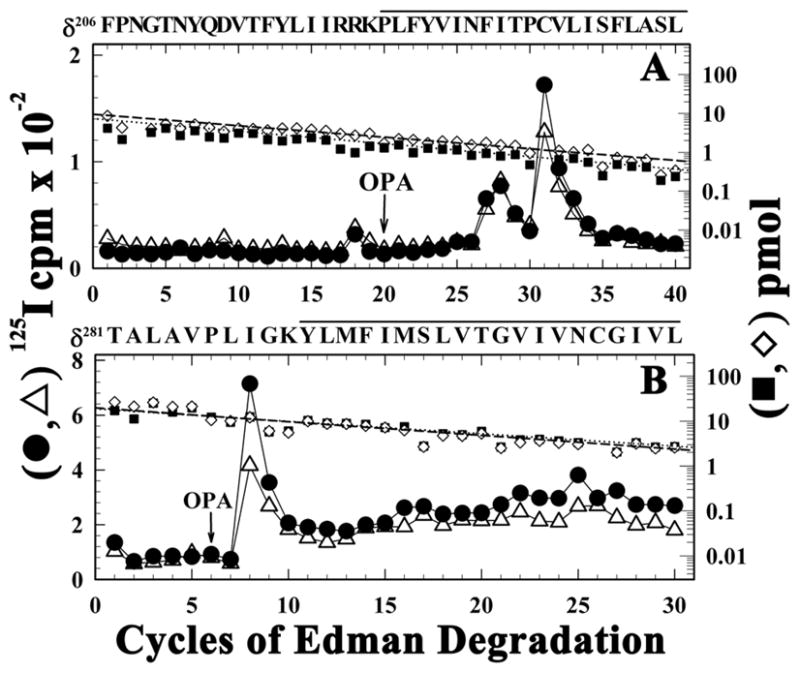
125I (●, △) and PTH-amino acids (■, ◇) released during sequence analysis of the fragments beginning at δPhe206 before δM1 (A) and δThr281 in the δM2-M3 loop (B) that were isolated from native Torpedo nAChR labeled with [125I]TID in the presence of Carb (●, ■) or Carb and PCP (△, ◇) as described under the Experimental Procedures and the legend of Figure 2. During sequencing the filters treated with OPA before cycle 20 (A) or 6 (B) to prevent sequencing of fragments not containing a proline in those cycles. A, The primary amino acid sequence began at δPhe206 (+Carb (■), I0, 7 pmol; +Carb + PCP (◇), 10 pmol). In the presence of Carb, the peaks of 125I release in cycles 27 and 31 indicated labeling of δPhe232 and δCys236 at 8 and 38 cpm/pmol, respectively, which was reduced by 50% in the presence of PCP. B, The fragment beginning at δThr281 was present at 20 pmol (both conditions). In the presence of Carb, the major peak of 125I release in cycle 8 indicated labeling of δIle288 at 11 cpm/pmol, which was reduced to 6 cpm/pmol in the presence of PCP. The amino acid sequences of the fragments are shown above each panel, with solid bars indicating the span of the δM1 (A) and δM3 (B) segments.
For nAChRs in the desensitized state, sequence analysis provided no evidence of [125I]TID photolabeling of αM2-18 (αIle260) or βM2-18 (βLeu266) (Figures 4A and B), in contrast to the labeling of δM2-18 (δThr274). However, aliphatic side chains have lower intrinsic reactivity with TID than a threonine (30). We also found no evidence of [125I]TID photolabeling of βIle280, the position corresponding to δIle288, or of other amino acids in the βM2-M3 loop or within βM1 (i.e., labeling, if it occurred was at <15% the efficiency of the labeling of δIle288 or δPhe232). Similarly, we found no evidence of labeling within the γM2-M3 loop in nAChRs in the desensitized state. (data not shown). Thus for nAChRs in the desensitized state, [125I]TID appears to bind selectively within the pocket formed by the δ subunit helix bundle and not in the pockets formed by transmembrane helices of the α, β, or γ subunit transmembrane domains.
For nAChRs in the resting state, TID binding in the ion channel at the level of M2-9 and -13 is inhibited competitively by tetracaine, a closed channel blocker, and allosterically by PCP (31–33). For nAChRs in the desensitized state, our results, along with the fact that PCP inhibited [3H]chlorpromazine photolabeling at positions M2-6 (and -2 or -9, depending on the subunit) (34), indicate that PCP and [125I]TID bind in a mutually exclusive manner at the cytoplasmic end of the ion channel in the desensitized state, and that PCP binding at that site may cause a subtle perturbation of the structure of the δ subunit helix bundle as evidenced by the reduced efficiency of labeling. Since [125I]TID photolabeling of M2-9 and M2-13 in the nAChRs equilibrated with agonist was less than 5% of that for nAChR in the resting state, further studies are required to determine whether the labeling of those positions in the presence of agonist results from a small fraction of nAChRs remaining in the resting state, which would be consistent with the observed effects of PCP on the labeling.
CONCLUSIONS
In this report, we used hydrophobic photolabeling with [125I]TID to assess the effect of detergent solubilization, affinity-purification and lipid-reconstitution on the structure of the well-characterized Torpedo nAChR. In the nAChR transmembrane segments, the amino acids photolabeled by [125I]TID are shown in Figure 6 in views of the transmembrane domain from the base of the extracellular domain (Figure 6A) and from the lumen of the channel toward β-δ-α (Figure 6B). We found that after cholate-solubilization and purification in the presence of DOPC, DOPA and CH, the structure of the nAChR ion channel domain in the resting state and the change in structure of the ion channel domain between the resting and desensitized states were retained. However, the loss of agonist-enhanced labeling in the δM2-M3 loop indicates that this region in the purified nAChR at the interface between the extracellular and transmembrane domains does not undergo the expected change in structure between the resting and desensitized states. Since [125I]TID photolabels in the δ helix bundle and δM2-M3 at least ten times more efficiently in the open state than in the equilibrium desensitized state (14), it will be important in future studies to use [125I]TID photolabeling in conjunction with rapid-mixing and freeze-quench techniques to probe the structure of that pocket and the δM2-M3 loop in the purified nAChR after transient exposure to agonist. We have characterized the purified nAChR after reconstitution in a PC/PA/CH environment that supports channel gating and receptor state transitions. In the future it will be important to examine the effect of CH on the structure of the nAChR transmembrane domain, since CH has been shown to be essential for nAChR gating, while the transition from the open to fast desensitized state is independent of CH concentration (7;35;36). When the human α4β2 nAChR was purified by the same protocol (37), [125I]TID photolabeling established that the amino acids exposed at the lipid interface were consistent with homology models based upon the Torpedo nAChR structure. Based upon our studies of the purified, reconstituted Torpedo nAChR, it is likely that other structural features of the transmembrane domains in purified neuronal nAChRs will be the same as in their native membrane environment.
Supplementary Material
Abbreviations
- nAChR
nicotinic acetylcholine receptor
- LGIC
ligand gated ion channel
- Carb
carbamylcholine
- rpHPLC
reversed-phase high-performance liquid chromatography
- OPA
o-phthalaldehyde
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TFA
trifluoroacetic acid
- PTH
phenylthiohydantoin
- [125I]TID
3-trifluoromethyl-3-(m-[125I]iodophenyl) diazirine
- Tricine
N-tris(hydroxymethyl)methylglycine
- VDB
vesicle dialysis buffer
- TPS
Torpedo physiological saline
- MOPS
4-morpholinopropanesulfonic acid
- EndoLys-C
endoproteinase Lys-C
- V8 protease
S. aureus glutamyl endopeptidase
- DOPC
dioleoylphosphatidylcholine
- DOPA
dioleoylphosphatidic acid
- CH
cholesterol
- PCP
phencyclidine
- FTIR
Fourier Transform Infrared
Footnotes
This research was supported in part by United States Public Health services Grant GM-58448 (J.B.C.), by an award to Harvard Medical School from the Howard Hughes Biomedical Research Support Program for Medical Schools (J.B.C.), by American Heart Association Texas Affiliate Grant-In-Aid 0755029Y (M.P.B.), and by the South Plains Foundation (M.P.B.).
Supporting Information Available: Two figures describing the purification by SDS-PAGE and/or rpHPLC of [125I]TID-photolabeled fragments from EndoLys-C and V8 protease digests of nAChR δ subunits. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Changeux J-P, Edelstein SJ. Nicotinic Acetylcholine Receptors: From Molecular Biology to Cognition. Odile Jacob Publishing; New York, NY: 2005. [Google Scholar]
- 2.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–958. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 3.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Ä resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 4.Cascio M. Structure and function of the glycine receptor and related nicotinicoid receptors. J Biol Chem. 2004;279:19383–19386. doi: 10.1074/jbc.R300035200. [DOI] [PubMed] [Google Scholar]
- 5.Barrantes FJ. Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res Rev. 2004;47:71–95. doi: 10.1016/j.brainresrev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy MP, Moore MA. Effects of lipids and detergents on the conformation of the nicotinic acetylcholine receptor from Torpedo californica. J Biol Chem. 1992;267:7655–7663. [PubMed] [Google Scholar]
- 7.Fong TM, McNamee MG. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry. 1986;25:830–840. doi: 10.1021/bi00352a015. [DOI] [PubMed] [Google Scholar]
- 8.daCosta CJB, Ogrel AA, McCardy EA, Blanton MP, Baenziger JE. Lipid-protein interactions at the nicotinic acetylcholine receptor - A functional coupling between nicotinic receptors and phosphatidic acid-containing lipid bilayers. J Biol Chem. 2002;277:201–208. doi: 10.1074/jbc.M108341200. [DOI] [PubMed] [Google Scholar]
- 9.Hamouda AK, Sanghvi M, Sauls D, Machu TK, Blanton MP. Assessing the lipid requirements of the Torpedo californica nicotinic acetylcholine receptor. Biochemistry. 2006;45:4327–4337. doi: 10.1021/bi052281z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunner J, Semenza G. Selective labeling of the hydrophobic core of membranes with 3-(trifluoromethyl)-3-m-([125I]iodophenyl)diazirine, a carbene-generating reagent. Biochemistry. 1981;20:7174–7182. doi: 10.1021/bi00528a019. [DOI] [PubMed] [Google Scholar]
- 11.Brunner J. New photolabeling and crosslinking methods. Ann Rev Biochem. 1993;62:483–514. doi: 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- 12.Blanton MP, Cohen JB. Identifying the lipid-protein interface of the Torpedo nicotinic acetylcholine receptor: secondary structure implications. Biochemistry. 1994;33:2859–2872. doi: 10.1021/bi00176a016. [DOI] [PubMed] [Google Scholar]
- 13.White BH, Cohen JB. Agonist-induced changes in the structure of the acetylcholine receptor M2 regions revealed by photoincorporation of an uncharged nicotinic non-competitive antagonist. J Biol Chem. 1992;267:15770–15783. [PubMed] [Google Scholar]
- 14.Arevalo E, Chiara DC, Forman SA, Cohen JB, Miller KW. Gating-enhanced accessibility of hydrophobic sites within the transmembrane region of the nicotinic acetylcholine receptor’s delta- subunit - A time-resolved photolabeling study. J Biol Chem. 2005;280:13631–13640. doi: 10.1074/jbc.M413911200. [DOI] [PubMed] [Google Scholar]
- 15.Methot N, McCarthy MP, Baenziger JE. Secondary structure of the nicotinic acetylcholine receptor: implications for structural models of a ligand-gated ion channel. Biochemistry. 1994;33:7709–7717. doi: 10.1021/bi00190a026. [DOI] [PubMed] [Google Scholar]
- 16.Ryan SE, Demers CN, Chew JP, Baenziger JE. Structural effects of neutral and anionic lipids on the nicotinic acetylcholine receptor. J Biol Chem. 1996;271:24590–24597. doi: 10.1074/jbc.271.40.24590. [DOI] [PubMed] [Google Scholar]
- 17.Heidmann T, Oswald RE, Changeux J-P. Multiple sites of action for noncompetitive blockers on acetylcholine receptor rich membrane fragments from Torpedo marmorata. Biochemistry. 1983;22:3112–3127. doi: 10.1021/bi00282a014. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen SE, Dreyer EB, Cohen JB. Location of ligand binding sites on the nicotinic acetylcholine receptor α-subunit. J Biol Chem. 1986;261:13735–13743. [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.White BH, Cohen JB. Photolabeling of membrane-bound Torpedo nicotinic acetylcholine receptor with the hydrophobic probe 3-trifluoromethyl-3-(m[125I]iodophenyl)diazirine. Biochemistry. 1988;27:8741–8751. doi: 10.1021/bi00424a009. [DOI] [PubMed] [Google Scholar]
- 21.Cleveland DW, Fischer SG, Kirschner MW, Laemmli UK. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 22.Garcia GI, Chiara DC, Nirthanan S, Hamouda AK, Stewart DS, Cohen JB. [3H]Benzophenone photolabeling identifies state-dependent changesin nicotinic acetylcholine receptor structure. Biochemistry. 2007;46:10296–10307. doi: 10.1021/bi7008163. [DOI] [PubMed] [Google Scholar]
- 23.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 24.Brauer AW, Oman CL, Margolies MN. Use of ophthalaldehyde to reduce background during automated Edman degradation. Anal Biochem. 1984;137:134–142. doi: 10.1016/0003-2697(84)90359-2. [DOI] [PubMed] [Google Scholar]
- 25.Middleton RE, Cohen JB. Mapping of the acetylcholine binding site of the nicotinic acetylcholine receptor: [3H]-nicotine as an agonist photoaffinity label. Biochemistry. 1991;30:6987–6997. doi: 10.1021/bi00242a026. [DOI] [PubMed] [Google Scholar]
- 26.Boyd ND, Cohen JB. Desensitization of membrane-bound Torpedo acetylcholine receptor by amine noncompetitive antagonists and aliphatic alcohols: studies of [3H]acetylcholine binding and 22Na+ ion fluxes. Biochemistry. 1984;23:4023–4033. doi: 10.1021/bi00313a003. [DOI] [PubMed] [Google Scholar]
- 27.Rotstein NP, Arias HR, Barrantes FJ, Aveldano MI. Composition of lipids in elasmbranch electric organ and acetylcholine receptor membranes. J Neurochem. 1987;49:1333–1347. doi: 10.1111/j.1471-4159.1987.tb00996.x. [DOI] [PubMed] [Google Scholar]
- 28.Baenziger JE, Ryan SE, Goodreid MM, Vuong NQ, Sturgeon RM, daCosta CJB. Lipid composition alters drug action at the nicotinic acetylcholine receptor. Mol Pharmacol. 2008;73:880–890. doi: 10.1124/mol.107.039008. [DOI] [PubMed] [Google Scholar]
- 29.Arias HR, Bhumireddy P, Bouzat C. Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int J Biochem Cell Biol. 2006;38:1254–1276. doi: 10.1016/j.biocel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Sigrist H, Mühlemann M, Dolder M. Philicity of amino acid side-chains for photogenerated carbenes. J Photochem Photobiol B-Biol. 1990;7:277–287. [Google Scholar]
- 31.Gallagher MJ, Cohen JB. Identification of amino acids of the torpedo nicotinic acetylcholine receptor contributing to the binding site for the noncompetitive antagonist [3H]tetracaine. Mol Pharmacol. 1999;56:300–307. doi: 10.1124/mol.56.2.300. [DOI] [PubMed] [Google Scholar]
- 32.Gallagher MJ, Chiara DC, Cohen JB. Interactions between 3-(trifluoromethyl)-3-m-[125I]iodophenyl)diazirine and tetracaine, phencyclidine, or histrionicotoxin in the Torpedo nicotinic acetylcholine receptor ion channel. Mol Pharmacol. 2001;59:1514–1522. doi: 10.1124/mol.59.6.1514. [DOI] [PubMed] [Google Scholar]
- 33.Arias HR, McCardy EA, Bayer EZ, Gallagher MJ, Blanton MP. Allosterically linked noncompetitive antagonist binding sites in the resting nicotinic acetylcholine receptor ion channel. Arch Biochem Biophys. 2002;403:121–131. doi: 10.1016/S0003-9861(02)00214-X. [DOI] [PubMed] [Google Scholar]
- 34.Revah F, Galzi JL, Giraudat J, Haumont PY, Lederer F, Changeux JP. The noncompetitive blocker [3H]chlorpromazine labels three amino acids of the acetylcholine receptor γ subunit: Implications for the α-helical organization of regions MII and for the structure of the ion channel. Proc Natl Acad Sci USA. 1990;87:4675–4679. doi: 10.1073/pnas.87.12.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Ballester G, Castresana J, Fernandez AM, Arrondo JLR, Ferragut JA, Gonzalez-Ros JM. A Role for Cholesterol as a Structural Effector of the Nicotinic Acetylcholine Receptor. Biochemistry. 1994;33:4065–4071. doi: 10.1021/bi00179a035. [DOI] [PubMed] [Google Scholar]
- 36.Rankin SE, Addona GH, Kloczewiak MA, Bugge B, Miller KW. The cholesterol dependence of activation and fast desensitization of the nicotinic acetylcholine receptor. Biophys J. 1997;73:2446–2455. doi: 10.1016/S0006-3495(97)78273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamouda AK, Sanghvi M, Chiara DC, Cohen JB, Blanton MP. Identifying the lipid--protein interface of the α4 β2 neuronal nicotinic acetylcholine receptor: Hydrophobic photolabeling studies with 3-(Trifluoromethyl)-3-(m-[125I]iodophenyl)diazirine. Biochemistry. 2008;46:13837–13846. doi: 10.1021/bi701705r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.