

Abstract
Agents capable of preventing the misfolding and sequestration of the microtubule-stabilizing protein tau into insoluble fibrillar aggregates hold considerable promise for the prevention and/or treatment of neurodegenerative tauopathies such as Alzheimer’s disease. Because tauopathies are characterized by amyloidosis that is restricted to the central nervous system (CNS), plausible candidate compounds for in vivo evaluation must both prevent tau fibrillization and achieve significant brain levels. Recently, we reported the discovery of the aminothienopyridazine (ATPZ) class of tau aggregation inhibitors and now describe a series of new analogues that are both effective inhibitors of tau fibrillization and display significant brain-to-plasma exposure ratios after administration to mice. Further, two of the most promising examples, 15 and 16, were found to reach significant brain exposure levels following oral administration. Taken together, these results suggest that examples from the ATPZ class hold promise as candidates for in vivo efficacy studies in animal models of neurodegenerative tauopathies.
Keywords: aminothienopyridazine, tauopathy, Alzheimer’s disease, amyloid, tau-aggregation inhibitor
Introduction
The misfolding and aggregation within neurons of the microtubule (MT)-associated protein (MAP) tau comprises a key pathogenic phenomenon shared by different neurodegenerative conditions, collectively known as tauopathies, which include Alzheimer’s disease (AD), Pick’s disease and certain forms of frontotemporal dementia (FTD) including FTD with Parkinsonism linked to chromosome 17 (FTDP-17).1 The protein tau, expressed predominantly in neurons, stabilizes the MT network within axons, thereby facilitating axonal transport of proteins, trophic factors and other cellular constituents, including neurotransmitters. Tau is in dynamic equilibrium between the MT-bound and -unbound states in the cytosol. This equilibrium is believed to be controlled predominantly by the action of tau kinases and phosphatases, as the phosphorylation state of tau is known to modulate the affinity of this protein for the MTs.2 Under physiological conditions, the vast majority (i.e., ~99%) of tau is associated to MTs.3, 4 Conversely, under pathological conditions, tau is abnormally disengaged from the MTs and as such becomes considerably more prone to misfolding.5 Once tau is misfolded, a self-assembly reaction can ensue that ultimately results in the formation of characteristic structures, such as neurofibrillary tangles (NFTs) and neuropil threads, that constitute the diagnostic signatures of neurodegenerative tauopathies. Although the exact mechanism(s) of tau-mediated neurodegeneration have not been completely defined, aggregation of tau can lead to neuropathology at least in part by causing or contributing to axonal transport deficits arising from the loss of the normal MT-stabilizing function of tau.5 Further, different forms of aggregated tau may also contribute to neuropathology by other less defined mechanisms.6 Agents capable of preventing the self-assembly of tau thus comprise an attractive strategy for the prevention and/or treatment of neurodegenerative tauopathies.7, 8
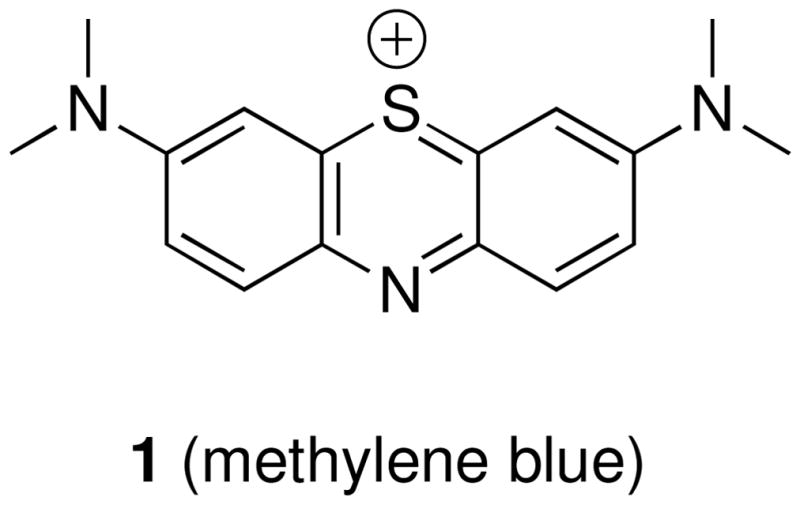
In recent years, several compound classes have been disclosed that can effectively inhibit the assembly of tau into filaments in vitro.9–19 However, with the exception of a member of the phenothiazines, methylene blue (1, Figure 1), which has progressed to human clinical trials, no tau assembly inhibitor has been assessed for efficacy in vivo. Notably, the results from a Phase 2 study, involving 321 patients with mild or moderate AD, revealed a significant reduction in cognitive decline in patients that received methylene blue compared to a placebo. These promising findings, if confirmed, validate the idea that tau aggregation inhibitors can be therapeutically useful for the treatment of neurodegenerative tauopathies. However, since methylene blue is known to modulate a multitude of biological targets,20 whether the outcome observed in the Phase 2 study can be ascribable solely to a direct inhibition of tau aggregation is not yet clear. As a result, further evaluation of the therapeutic potential of tau aggregation inhibitors is likely to require in vivo efficacy studies involving additional candidate compounds. To this end and in light of the fact that tauopathies are characterized by amyloidosis that is restricted to the central nervous system (CNS), candidate compounds for in vivo testing will have to be brain-penetrant. Although numerous classes of tau fibrillization inhibitors have been reported in recent years, including some which exhibit calculated physical-chemical properties potentially appropriate for blood-brain barrier (BBB) permeation,17 to date there are no reports demonstrating brain penetration of any of these candidates.
Figure 1.
Recently, we reported the discovery of a novel class of tau aggregation inhibitors, known as the aminothienopyridazines (ATPZ), which exhibit a promising combination of activity in tau fibrillization assays as well as drug-like physical-chemical properties.21 To evaluate better the potential of the ATPZs as possible candidates for future in vivo efficacy studies, we designed and synthesized a set of derivatives focused on possible BBB-permeability. These compounds were evaluated for efficacy against tau aggregation in vitro, as well as for brain penetration in vivo. Notably, these experiments (vide infra) led to the identification of prototype compounds (i.e., 15, and 16), which are both effective in preventing the fibrillization of tau in vitro and capable of reaching significant brain levels in mice after oral administration.
Compound Design and Synthesis
The design of the ATPZs employed in these studies took into account the structure-activity relationships (SARs) for this class,21 as well as key physical-chemical properties such as lipophilicity (i.e., calculated partition coefficient between n-octanol and water, or ClogP), polar surface area (PSA), the number of hydrogen-bond donors (HBD) and acceptors (HBA) and molecular weight, all of which are known to play an important role in determining the ability of small molecules to permeate biological membranes via passive diffusion.22 Thus, based on our understanding of the SAR summarized in Figure 2, we designed analogues modified in the X and Y fragments (Figure 2) as these regions of the molecule were known to be relatively tolerant to a series of structural variations.
Figure 2.

Summary of structure tau anti-fibrillization activity of ATPZs.21
With respect to physical-chemical properties, we considered the following criteria: MW ≤450 Da, PSA ≤90 Å2, HBA ≤7, HBD ≤3 and ClogP ≤3. Based on these restrictions in terms of SAR and physical-chemical properties, compounds of general structure F (Scheme 1), bearing a lipophilic para-chloro or para-bromo phenyl (X = Cl or Br) as well as an amide moiety in the Y fragment, were identified as potentially promising (cf., Table 1 for calculated physical-chemical properties). Furthermore, as part of these studies, we included two representative ester derivatives (i.e., 8 and 10) to evaluate the potential of these congeners to be brain-penetrant, possibly acting as lipophilic precursors of the corresponding acid (11). Depending on the rate of esterase-mediated hydrolysis in brain and plasma, we reasoned that such derivatives may potentially lead to an accumulation of the corresponding acid inside the CNS.
Scheme 1. Reagents and Reaction conditions.

a) (i) NaNO2, 37% HCl, ethanol, water, 0 °C, 20 min; (ii) ethyl acetoacetate, sodium acetate, ethanol, water, 0 °C, 2 h; b) X = Cl: ethyl cyanoacetate, ammonium acetate, acetic acid, 170 °C (microwave irradiation), 4 min; c) X = Br: ethyl cyanoacetate, 4-aminobutyric acid, 160 °C, 2.5 h; d) S8, morpholine, ethanol, 150 °C (microwave irradiation), 15 min; e) titanium (IV) isopropoxide, isopropanol, 170 °C (microwave irradiation), 40 min. f) LiOH·H2O, tetrahydrofuran, water, rt, 16 h; g) appropriate amine, BOP reagent, N,N-diisopropylethylamine, dimethylsulfoxide, rt, 4 h.
Table 1.
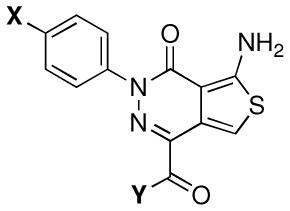 |
Activity Against Tau Fibril Formation | Brain and Plasma Levels at 1 h Time Point After I.P. Administration of 5 mg/kg of Test Compound | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cpd# | X | Y | PCPa | Water Solubilityb | K18PL (ThT)c | K18PL Sed. Assayd | Tau40 Sed. Assaye | Brain (ng/g) | Plasma (ng/ml) | B/Pf |
| 11 | Cl | -OH | MW: 321.7 PSA: 95.9 ClogP: 1.8§ |
>200 μM | 5.4 ± 2.3 μMø (84 ±7 %)* | 79% | 62% | 121 ± 19 | 5226 ± 850 | 0.023 ± 0.003 |
| 8 | Cl | -OEt | MW: 349.8 PSA: 84.9 ClogP: 2.6 |
13 μM | 3.8 ± 1.6 μMø (71 ± 11 %)* | 70% | 48% | 67 ± 53 (8 ± 4.5)† | 20 ± 13 (3558 ± 2000)† | 3.3 ± 0.72 |
| 10 | Cl | OiPr | MW: 363.8 PSA: 84.9 ClogP: 2.9 |
32 μM | 1.6 ± 1.1 μMø (79 ± 1 %)* | 44% | 61% | 436 ± 170 (0)† | 84 ± 14 (1718 ± 650)† | 5.2 ± 1.1 |
| 13 | Cl | -NHMe | MW: 334.8 PSA: 87.8 ClogP: 0.9 |
13 μM | 21.7 ± 16 μMø (78 ± 3 %)* | 68% | 51% | 34 ± 9.5 | 70 ± 16 | 0.49 ± 0.03 |
| 14 | Cl | -NHEt | MW: 348.8 PSA: 87.8 ClogP: 1.4 |
32 μM | 6.9 ± 3.4 μMø (84 ± 6 %)* | 94% | 57% | 886 ± 65 | 500 ± 60 | 1.8 ± 0.14 |
| 15 | Cl | -NHiPr | MW: 362.8 PSA: 87.8 ClogP: 1.8 |
32 μM | 5.9 ± 4.7 μMø (83 ± 4 %)* | 65% | 45% | 1100 ± 160 | 499 ± 130 | 2.2 ± 0.47 |
| 16 | Cl | 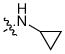 |
MW: 360.8 PSA: 87.8 ClogP: 1.5 |
32 μM | 8.1 ± 5.7 μMø (83 ± 3 %)* | 69% | 58% | 1299 ± 110 | 823 ± 150 | 1.6 ± 0.28 |
| 17 | Cl | -N(Me)2 | MW: 348.8 PSA: 79 ClogP: 0.6 |
32 μM | 20.5 ± 13.6 μMø (75 ± 5 %)* | 69% | 69% | 14 ± 7.5 | 30 ± 7.8 | 0.44 ± 0.15 |
| 18 | Cl | MW: 362.8 PSA: 79 ClogP: 1.1 |
80 μM | 32 ± 4.1 μMø (75 ± 11 %)* | 62% | 58% | 122 ± 41 | 138 ± 45 | 0.89 ± 0.05 | |
| 19 | Br | -NHiPr | MW: 407.3 PSA: 87.8 ClogP: 1.9 |
13 μM | 3.1 ± 0.6 μMø (85 ± 1 %)* | 58% | 55% | 641 ± 210 | 316 ± 56 | 2.0 ± 0.37 |
Physical-chemical properties (MW: molecular weight; PSA: polar surface area; ClogP: calculated partition coefficient between n-octanol and water);
Solubility in the sodium acetate buffer employed in the tau fibrillization assay, as determined by light scattering;
Tau (K18PL) fibrillization assay monitored by thioflavine (ThT) binding and fluorescence (all test compounds tested in triplicate);
Maximal percent inhibition of the K18PL fibrillization as determined by SDS-PAGE analyses of the soluble and insoluble fractions obtained after centrifugation of the fibrillizing K18PL mixtures;
Maximal percent inhibition of the tau40 fibrillization as determined by SDS-PAGE analyses of the soluble and insoluble fractions obtained after centrifugation of the fibrillizing tau40 mixtures;
Ratio between brain/plasma drug level, as determined by LC-MS/MS, 1 h after i.p. administration of 5 mg/Kg of test compound;
ClogP of the neutral form;
Half maximal inhibitory concentration (IC50);
Maximal percent inhibition achieved in the assay;
Detected amount of hydrolysis product (i.e., 11).
The synthesis of these compounds was achieved via the reaction sequence illustrated in Scheme 1, which entails (a) a diazonium coupling reaction to form hydrazones of general structure B; (b) a Knovenagel-type condensation between B and ethyl cyanoacetate to form pyridazines C;23, 24 and (c) a Gewald aminothiophene synthesis25 to furnish the desired ATPZs of general structure D. Compound 8 was then trans-esterified to the corresponding isopropyl ester 10 upon treatment with Ti(iPrO)4 in isopropanol. Alternatively, compounds 8 and 9 were saponified, and the resulting acids (11 and 12, respectively) employed in series of BOP coupling reactions with various primary and secondary amines to furnish compounds 13–19.
Compound Evaluation In Vitro
The solubility of all test compounds in the sodium acetate buffer used for subsequent fibrillization reactions was determined by turbidimetric measurements (Table 1). Compounds were then evaluated in a heparin-induced tau assembly assay, in which the fibrillization of the truncated K18 tau fragment (comprised of four MT-binding repeats) bearing the P301L mutation (K18PL) found in FTDP-17, was monitored by thioflavine-T (ThT) binding and fluorescence (Figure 3A).21
Figure 3.

(A) Representative dose-response curves in the heparin-induced K18PL fibrillization assay; (B) SDS-PAGE analysis of supernatant (S) or pellet (P) samples obtained after centrifugation of fibrillizing mixtures incubated in the presence of DMSO vehicle, or 100 μM of test compound (11 and 16).
The inhibitory activity of test compounds was then confirmed with an orthogonal sedimentation assay in which compound-treated fibrillizing mixtures were centrifuged and quantitation of K18PL tau in the soluble and insoluble fraction was performed by densitometric analyses of SDS-PAGE (Figure 3B). As summarized in Table 1, the ATPZs exhibited IC50 values in the 2–32 μM range in the primary ThT assay, with maximal percent inhibition of 70–85%. The activity against tau fibrillization was confirmed by sedimentation assay, where the majority of compounds exhibited >50% reduction in pelletable material compared to the untreated control. Although many of the ATPZ compounds were found to reach a solubility limit within the upper concentration range used in the biochemical assay, maximal inhibitory effect was typically achieved at compound concentrations below 30 μM, where the majority of compounds appeared to be fully soluble (cf., solubility data on Table 1 with representative dose-response curves shown in Figure 3A). This indicates that the inhibition of tau aggregation is likely to be caused by the ATPZs present in solution. However, to further ensure that the observed inhibitory activity of these ATPZs may not be artifactually generated by the presence of insoluble or aggregated test compound,26 we conducted additional experiments in which both DMSO- and compound-treated fibrillizing mixtures were initially centrifuged for 30 min prior to incubation (i.e., at time zero of the fibrillization reaction) to remove any pelletable material. As shown in Figure 4A, no significant differences in compound-mediated inhibition of tau assembly were observed with and without the pre-centrifugation step. Furthermore, the IC50 values of the centrifuged and control samples did not appear to differ appreciably (Figure 4C). Moreover, SDS-PAGE analysis of the soluble and pelletable material after the initial centrifugation step revealed no significant difference between compound- and vehicle- treated mixtures; in all cases K18PL remained largely in the soluble fraction (Figure 4B) and thus was not precipitated by the test compound. Collectively, the activity and SDS-PAGE data confirm that the inhibition of K18PL fibrillization produced by the ATPZ analogues is not caused by insoluble or aggregated material.
Figure 4.

(A) Comparison of maximal percent inhibition of K18PL fibrillization caused by test compounds in which the fibrillizing mixtures did (sup) or did not (tot) undergo centrifugation at time zero to remove insoluble material. Compounds were added at 50 μM to the tau fibrillization mixture and incubated for 30 min. Samples were then divided in two halves, one of which was incubated without further treatment (“tot”), and one that was centrifuged at 186,000g for 30 min. After centrifugation of the latter sample, the supernatants (“sup”) was collected and incubated as per the fibrillization protocol.21 (B) Representative examples of SDS-PAGE analysis of supernatant (S) or pellet (P) samples obtained after centrifugation of fibrillizing mixtures prior to incubation; (C) Representative dose-response curves with (“sup”) and without (“tot”) initial centrifugation.
All test compounds also inhibited the aggregation of full-length tau (tau40) as determined by sedimentation assay, which revealed efficacy values in the 50–60% range (cf., Table 1). Both K18PL and tau40 activity data are in keeping with prior SAR which suggested that the presence of halogens in the para position of the phenyl ring, as well as carboxylic acid, esters and amides in the Y fragment, would be generally well tolerated.21 Also consistent with our previous studies is the observation that all ATPZs tested (i.e., 8, 10, 11, 13–19) did not appear to interfere with the normal MT-stabilizing function of tau in a MT-polymerization assay (Figure 5).
Figure 5.

MT-assembly assay. Tubulin (30 μM) and tau40 (20 μM) were incubated with the test compound (50 μM; e.g., 16 [green triangles]) or DMSO (open and black squares) to evaluate possible interference in the tau-promoted MT-polymerization that may be caused by the ATPZ inhibitors of tau aggregation. As control experiment, MT-polymerization assay was also conducted in the absence of tau40 (open blue circles). Additional results are presented in the Supporting Information.
Finally, to evaluate possible major toxicities associated with the ATPZs, all test compounds were evaluated in a cytotoxicity assay that employs rapidly dividing HEK-293 cells. All compounds were found to be non-toxic at 100 μM concentration.
Pharmacokinetic Studies
Test compounds underwent preliminary evaluations of brain penetration, in which 5 mg/Kg of each compound was administered intraperitoneally (i.p.) to a group of three normal mice; drug levels in brain and plasma were determined at a single time-point (1 h) by LC-MS/MS using pre-validated calibration curves. The results of these experiments, summarized in Table 1, revealed that with the exception of 11, all other test compounds displayed significant brain uptake as demonstrated by the brain-to-plasma exposure ratios (B/P) above 0.3. The lack of brain penetration of 11 was not unexpected given that the carboxylic acid moiety of this compound would be mostly negatively charged at physiological pH, and thus likely result in limited passive diffusion of the compound across the BBB. Conversely, the more lipophilic ester derivatives (8 and 10) exhibited comparatively higher B/P ratio. In particular, 10 was found to reach significantly higher brain concentrations compared to the corresponding acid 11. However, both esters appeared to have relatively short half-lives in plasma, as indicated by the limited amount of parent drug detected after 1 h from administration of the compounds. Moreover, monitoring for the hydrolyzed metabolite (i.e., 11) in both brain and plasma revealed a considerable amount of acid 11 in plasma, but not in the brain 1 h after administration of either ester. This observation suggests that while lipophilic esters may be able to gain access to the CNS (e.g., 10), these derivatives may not produce an accumulation of the corresponding ATPZ acid in the CNS at the rate observed in plasma due to significantly less esterase-mediated hydrolysis in the brain. Finally, among the amide derivatives, 14, 15 and 16 appeared to be particularly interesting due to favorable B/P ratios combined with promising indications of good metabolic stability. Compound 16 was selected for full PK analysis in which brain and plasma drug levels were determined at six time points (i.e., 30 min, 1 h, 2 h, 4 h, 8 h, and 16 h) after intravenous (i.v.) administration of 2 mg/kg of the test compound to a group of eighteen normal mice (three mice per time point). The results from this experiment, illustrated in Figure 6A, confirmed that the total B/P exposure ratio over 16 h is ~1.6. Furthermore, these data reveal that 16 exhibits good metabolic stability as indicated by an elimination half-life ≥ 2.5 h in plasma.
Figure 6.

(A) Brain and plasma level of 16 after i.v. administration of 2 mg/Kg; (B) Brain and plasma level of 16 after oral (p.o.) administration of 5 mg/Kg; (C) Brain and plasma level of 15 after oral administration of 5 mg/Kg.
Finally, in order to investigate the potential for ATPZ congeners to be orally administered, we evaluated brain and plasma level of 16 and 15 after oral administration (oral gavage) of 5 mg/Kg. In both cases, significant concentrations of each test compound was achieved in the brain after oral administration (cf., Figure 6B and 6C). Comparison of the integrated area under the curve (AUC) in plasma after oral and i.v. administration of 16 revealed an oral bioavailability (F) of ~70% (cf., Figure 6A and 6B).
Discussion
Despite the hypothesis that tau aggregation inhibitors may be therapeutically useful for AD and related tauopathies, to date only one compound of this type, methylene blue, has entered clinical trials. Numerous additional classes of compounds have been shown to inhibit tau assembly in vitro, but there are no reports of these molecules being evaluated in vivo for pharmacokinetic (PK) properties or efficacy in models of tauopathy. Because BBB permeability is known to be a major bottleneck that hampers the development of new CNS-active drugs,27 an early evaluation of the brain penetration of candidate compounds is important, as such studies would permit focus on the most promising compound type. For these reasons and to assess the potential of ATPZ inhibitors as possible candidate compounds for in vivo evaluation of efficacy, we conducted a study in which selected analogues, designed for improved BBB-permeability, were evaluated for in vitro activity as well as for brain penetration. Results from the in vitro efficacy studies appeared to be fully consistent with our previous results and confirmed that the ATPZs are most effective in preventing tau fibrillization when present in ~1:1 molar ratio with tau (i.e., 15 μM in the fibrillization assay).21 Assuming that similar stoichiometric requirements also exist in vivo, any ATPZ candidate for in vivo efficacy study will have to reach free brain concentrations that are comparable to that of the unbound fraction of tau. The total intraneuronal tau concentration (i.e., MT-bound and MT-unbound tau) is estimated to be in the low μM range3 and, as previously noted, >99% of the protein is bound to MTs under physiological conditions. 3, 4 Although, the concentration of the MT-unbound fraction of tau is likely to increase in diseased neurons, this may be in the sub μM range. Thus, only compounds that readily cross the BBB would be viable candidates forin vivo evaluations of efficacy. Interestingly, preliminary evaluation of brain exposures of the ATPZ test compounds revealed that with the exception of the acid derivative 11, all other ATPZ congeners exhibited B/P ratios above 0.3. Considering that most CNS-active drugs typically exhibit B/P >0.3–0.5,28 these results indicate that ATPZs have the potential to achieve appreciable brain concentrations. Furthermore, selected amide derivatives, such as 14, 15 and 16, were found to reach brain concentrations above 800 ng/g (i.e., >2 μM) 1h after i.p. administration of 5 mg/Kg. These results were further confirmed by complete PK studies on 16, which displayed an average brain-to-plasma AUC exposure ratio of ~1.6. Equally important, significant brain concentrations were also observed after oral administration of 15 or 16, with the oral bioavailability of the latter compound being ~70%. Collectively, the results from the PK studies combined with the promising in vitro activity and safety data suggest that the ATPZ class of tau aggregation inhibitors hold considerable promise as candidate compounds for efficacy testing in transgenic mouse models of tauopathies.
Conclusions
Although the preliminary Phase 2 clinical data obtained with methylene blue suggest that tau aggregation inhibitors may be therapeutically useful, further validation of the therapeutic potential of compounds of this type for the treatment and/or prevention of neurodegenerative tauopathies is likely to require additional in vivo efficacy studies involving different candidate compounds. To this end, the ATPZs presented here appear to be very promising candidates due to a favorable combination of biological activity in vitro and desirable PK properties, including excellent brain penetration and oral bioavailability.
Experimental Section
Materials and methods
All solvents were reagent grade. All reagents were purchased from Aldrich or Acros and used as received. Thin layer chromatography (TLC) was performed with 0.25 mm E. Merck pre-coated silica gel plates. Flash chromatography was performed with silica gel 60 (particle size 0.040 – 0.062 mm) supplied by Silicycle and Sorbent Technologies. TLC spots were detected by viewing under a UV light. Infrared (IR) spectra were recorded on a Jasco Model FT/IR-480 Plus spectrometer. All melting points were obtained on a Thomas-Hoover apparatus. Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker AMX-500 spectrometer. Chemical shifts were reported relative to solvents. High-resolution mass spectra were measured at the University of Pennsylvania Mass Spectrometry Center on either a VG Micromass 70/70H or VG ZAB-E spectrometer. Single-crystal X-ray structure determinations were performed at the University of Pennsylvania with an Enraf Nonius CAD-4 automated diffractometer. Analytical reverse-phased (Sunfire™ C18; 4.6×50 mm, 5 mL) high-performance liquid chromatography (HPLC) was performed with a Waters binary gradient module 2525 equipped with Waters 2996 PDA and Waters micromass ZQ. All samples were analyzed employing a linear gradient from 10% to 90% of acetonitrile in water over 8 minutes and flow rate of 1 mL/min, and unless otherwise stated, the purity level was >95%. Preparative reverse phase HPLC purifications were performed on a Gilson instrument (i.e., Gilson 333 pumps, a 215 liquid handler, 845Z injection module, and PDA detector) employing Waters SunFire™ preparative C18 OBD™ columns (5 μm 19×50 or 19×100 mm). Purifications were carried out employing a linear gradient from 10% to 90% of acetonitrile in water for 15 minutes with a flow rate of 20 mL/min. Yields refer to chromatographically and spectroscopically pure compounds.
Ethyl 2-(2-(4-chlorophenyl)hydrazono)-3-oxobutanoate (4)
Prepared as previously reported.29 Yield: 83%, mp: 84–85 °C (from ethanol, Lit.29 81–84 °C). 1H NMR (CDCl3): δ 1.41 (t, J = 7.0 Hz, 3H), 2.60 (s, 3H), 4.34 (q, J = 7.2 Hz, 2H), 7.34–7.36 ppm (m, 4H). 13C NMR (CDCl3): δ 14.5, 30.9, 61.2, 117.6, 126.5, 129.8, 131.0, 140.4, 164.9, 197.4 ppm. IR: ν 3359, 1706, 1617 cm−1. HRMS (ESI+): calculated for C12H13ClN2NaO3+ 291.0518 found 291.0512.
Ethyl 2-(2-(4-bromophenyl)hydrazono)-3-oxobutanoate (5)
4-Bromoaniline (2.0 g, 11.63 mmol) was dissolved in 37% hydrochloric acid (2.94 mL), ethanol (1.50 mL) and water (1.50 mL). The reaction mixture was cooled to 0 °C in an ice water bath before a solution of sodium nitrite (0.88 g, 12.75 mmol) in water (2.0 mL) was added dropwise. The resulting mixture was stirred at 0 °C for 20 min. Sodium acetate (3.72 g, 45.3 mmol) in water (5.9 mL) and ethyl acetoacetate (1.51 g, 1.43 mL, 11.63 mmol) were added and the reaction mixture was stirred at 0 °C for 2 h. The precipitated solid was then filtered, washed with water, and dried under high vacuum for 16 h to provide 5 as yellow solid that was crystallized in ethanol and used without further purification in the next step. Yield: 98%.
Ethyl 1-(4-chlorophenyl)-5-cyano-4-methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate (6)
Prepared as previously reported.30 Yield: 47%, mp: 160–161 °C (from ethanol, Lit.30 190 °C). 1H NMR (CDCl3): δ 1.41 (t, J = 7.0 Hz, 3H), 2.76 (s, 3H), 4.43 (q, J = 7.2 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H). 7.62 ppm (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3): δ 14.2, 19.4, 63.0, 112.4, 116.0, 126.3, 129.4, 135.5, 137.5, 138.5, 150.8, 155.8, 162.0 ppm. IR: ν 2235, 1726, 1685 cm−1. MS (ESI+): calculated for C15H13ClN3O3+ 318.06 found 318.07.
Ethyl 1-(4-bromophenyl)-5-cyano-4-methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate (7)
A mixture of 5 (2.0 g, 6.39 mmol), 4-aminobutyric acid (1.33 g, 12.99 mmol), and ethyl cyanoacetate (1.1 g, 1.0 mL, 9.44 mmol) was stirred neat at 160 °C for 2.5 h. After cooling the residue was purified by silica gel flash chromatography (using an ethyl acetate/n-hexane gradient of 5% to 80% of ethyl acetate) to provide 7 as white solid. Yield: 76%, mp: 174–176 °C (from ethanol). 1H NMR (CDCl3): δ 1.41 (t, J = 7.0 Hz, 3H), 2.76 (s, 3H), 4.43 (q, J = 4.43 Hz, 7.2 Hz, 2H), 7.56 (d, J = 9.0 Hz, 2H), 7.64 ppm (d, J = 9.0 Hz, 2H). 13C NMR (CDCl3): δ 14.2, 19.4, 62.9, 112.4, 116.0, 123.5, 126.6, 132.3, 137.6, 139.1, 150.8, 155.7, 162.0 ppm. IR: ν 2230, 1727, 1681 cm−1. MS (ESI+): calculated for C15H13BrN3O3+ 362.02 found 362.01.
Synthesis of thienyl derivatives with general structure D (Gewald reaction) Representative Example: Ethyl 5-amino-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (8)
A mixture of 6 (0.100 g, 0.31 mmol), sulfur (0.015 g, 0.47 mmol), and morpholine (0.55 g, 0.55 mL, 0.63 mmol) was heated to 150 °C using microwave irradiation for 15 min. After cooling, the precipitate which formed was collected and purified by preparative HPLC to give 8 as yellow solid. HPLC-MS retention time 8.20 min. Yield: 64%, mp: 189–191 °C (from ethanol). 1H NMR (CDCl3): δ 1.43 (t, J = 7.3 Hz, 3H), 4.44 (q, J = 7.2 Hz, 2H), 6.20 (broad s, 2H), 7.25 (s, 1H), 7.42 (d, J = 8.5 Hz, 2H), 7.56 ppm (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3): δ 14.4, 62.2, 104.9, 106.9, 127.1, 127.2, 128.9, 133.3, 134.0, 139.2, 159.4, 161.8, 163.0 ppm. IR: ν 3411, 3303, 1724, 1709, 1660 cm−1. HRMS (ESI+): calculated for C15H13ClN3O3S+ 350.0366 found 350.0355.
Ethyl 5-amino-3-(4-bromophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (9)
Was synthesized in the same manner as 8 starting with 7. Yield: 55%, mp: 198–200 °C (from ethanol). 1H NMR (CDCl3): δ 1.42 (t, J = 7.0 Hz, 3H), 4.44 (q, J = 7.2 Hz, 2H), 6.12 (broad s, 2H), 7.25 (s, 1H), 7.51 (d, J = 8.5 Hz, 2H), 7.58 ppm (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3): δ 14.1, 62.1, 105.9, 106.7, 121.3, 127.1, 127.5, 131.8, 134.0, 139.7, 159.4, 161.9, 163.0 ppm. IR: ν 3407, 3310, 1709, 1657 cm−1. HRMS (ESI+): calculated for C15H13BrN3O3S+ 393.9861 found 393.9879.
Isopropyl 5-amino-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylate (10)
A mixture of 8 (0.050 g, 0.143 mmol) and titanium (IV) isopropoxide (0.002 g, 2.1 μL, 7 μmol) in isopropanol (0.5 mL) was heated to 170 °C using microwave irradiation for 40 min. After cooling, the solvent was evaporated and the residue was purified by preparative HPLC to provide 10 as yellow solid. HPLC-MS retention time 8.73 min. Yield: 77%, mp: 118–120 °C (from ethanol). 1H NMR (CDCl3): δ 1.41 (d, J = 6.5 Hz, 6H), 5.26–5.33 (m, 1H), 6.25 (broad s, 2H), 7.17 (s, 1H), 7.38 (d, J = 8.5 Hz, 2H), 7.57 ppm (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3): δ 22.0, 70.1, 104.7, 106.8, 127.1, 127.2, 128.8, 133.1, 134.4, 139.2, 159.4, 161.9, 162.5 ppm. IR: ν 3408, 3294, 3145, 1716, 1663, 1585 cm−1. HRMS (ESI+): calculated for C16H14ClN3O3NaS+ 386.0342 found 386.0331.
Synthesis of compounds with general structure E (esters hydrolysis). Representative Example: 5-Amino-3-(4-chlorophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (11)
Lithium hydroxide monohydrate (0.063 g, 1.29 mmol) was added to a solution of 8 (0.150 g, 0.43 mmol) in tetrahydrofuran (3 mL) and water (2 mL). The reaction mixture was stirred at room temperature for 16 h. 1 N HCl was added (pH ~ 2), the formed precipitate was filtered and purified by preparative HPLC to afford 11 as yellow solid. HPLC-MS retention time 6.63 min. Yield: 67%, mp: > 300 °C (from ethanol). 1H NMR (CD3OD): δ 7.19 (s, 1H), 7.44 (d, J = 9.0 Hz, 2H), 7.58 ppm (d, J = 9.0 Hz, 2H). 13C NMR (DMSO-d6): δ 104.4, 104.5, 126.8, 128.3, 128.9, 132.0, 134.5, 140.2, 158.9, 163.9, 164.6 ppm. IR: ν 3439, 3322, 3065, 1712, 1646, 1590 cm−1. HRMS (ESI+): calculated for C13H9ClN3O3S+ 322.0053 found 322.0062.
5-Amino-3-(4-bromophenyl)-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxylic acid (12)
Prepared in the same manner as 11 starting with 9. HPLC-MS retention time 6.76 min. Yield: 77%, mp: > 300 °C (from ethanol). 1H NMR (DMSO-d6): δ 7.12 (s, 1H), 7.52 (d, J = 9.0 Hz, 2H), 7.63 (broad s, 2H), 7.66 ppm (d, J = 8.5 HZ, 2H). 13C NMR (DMSO-d6): δ 104.4, 104.5, 120.4, 126.8, 128.6, 131.9, 134.5, 140.6, 158.8, 163.9, 164.5 ppm. IR: ν 3439, 3322, 3065, 1712, 1646 cm−1. HRMS (ESI+): calculated for C13H7BrN3NaO3S+ 387.9367 found 387.9378.
Synthesis of compounds with general structure F. Representative Example: 5-Amino-3-(4-chlorophenyl)-N-methyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (13)
DIPEA (0.028 g, 38 μL, 0.23 mmol) was added to a mixture of 11 (0.050 g, 0.15 mmol), methylamine (2.0 M solution in THF, 110 μL, 0.22 mmol), and BOP reagent (0.097 g, 0.22 mmol) in anhydrous DMSO (2 mL). The reaction mixture was stirred at room temperature for 4 h. Water was added and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered and evaporated. The residue was purified by preparative HPLC to give the 13 as yellow solid. HPLC-MS retention time: 6.83 min. Yield: 52%, mp: 192–194 °C (from ethanol). 1H NMR (DMSO-d6): δ 2.75 (d, J = 5.0 Hz, 3H), 7.24 (s, 1H), 7.51 (d, J = 9.0 Hz, 2H), 7.60 (broad s, 2H), 7.69 (d, J = 9 Hz, 2H), 8.27 ppm (broad s, 1H). 13C NMR (DMSO-d6): δ 25.7, 103.9, 104.6, 126.0, 127.5, 128.1, 131.0, 131.6, 136.1, 139.5, 158.3, 162.9, 163.0 ppm. IR: ν 3418, 3302, 1656, 1593 cm−1. HRMS (ESI+): calculated for C14H10ClN4O2S+ 333.0213 found 333.0210.
5-Amino-3-(4-chlorophenyl)-N-ethyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (14)
Prepared in the same manner as 13 starting with 11 using ethylamine hydrochloride. HPLC-MS retention time: 7.43 min. Yield: 58%, mp: 203–205 °C (from ethanol). 1H NMR (CDCl3): δ 1.23 (t, J = 7.3 Hz, 3H), 3.41–3.47 (m, 2H), 6.18 (broad s, 2H), 7.07 (s, 1H), 7.45 (d, J = 9.0 Hz, 2H), 7.51 (d, J = 9.0 Hz, 2H), 7.60 ppm (broad s, 1H). 13C NMR (CDCl3): δ 14.9, 34.3, 106.8, 106.9, 126.7, 127.3, 129.0, 133.3, 135.6, 139.2, 159.5, 161.4, 162.6 ppm. IR: ν 3405, 3297, 1652, 1600 cm−1. HRMS (ESI+): calculated for C15H13ClN4NaO2S+ 371.0345 found 371.0341.
5-Amino-3-(4-chlorophenyl)-N-isopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (15)
Prepared in the same manner as 13 starting with 11 using isopropylamine. HPLC-MS retention time: 6.58 min. Yield: 89%, mp: 197–199 °C (from ethanol). 1H NMR (CD3OD): δ 1.22 (d, J = 6.5 Hz, 6H), 4.09–4.19 (m, 1 H), 7.25 (s, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.59 ppm (d, J = 8.5 Hz, 2H). 13C NMR (CD3OD): δ 19.6, 39.9, 102.9, 103.3, 124.9, 126.1, 126.9, 131.2, 135.1, 138.1, 158.0, 161.4, 162.1 ppm. IR: ν 3404, 3294, 3178, 1651, 1590 cm−1. HRMS (ESI+): calculated for C16H15ClN4O2NaS+ 385.0502 found 385.0492.
5-Amino-3-(4-chlorophenyl)-N-cyclopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (16)
Prepared in the same manner as 13 starting with 11 using cyclopropylamine. HPLC- MS retention time: 7.43 min. Yield: 41%, mp: 188–190 °C (from ethanol). 1H NMR (DMSO-d6): δ 0.56–0.60 (m, 2H), 0.66–0.70 (m, 2H), 2.76–2.81 (m, 1H), 7.19 (s, 1H), 7.50, (d, J = 9.0 Hz, 2H), 7.67 (d, J = 9.0 Hz, 2H), 8.28 ppm (broad s, 1H). 13C NMR (DMSO-d6): δ 5.7, 22.6, 103.8, 104.4, 125.9, 127.5, 128.1, 131.0, 136.3, 139.4, 158.3, 163.0, 163.7 ppm. IR: ν 3408, 3302, 1654, 1593 cm−1. HRMS (ESI+): calculated for C16H13Cl4O2N4NaS+ 383.0345 found 383.0333.
5-Amino-3-(4-chlorophenyl)-N,N-dimethyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (17)
Prepared in the same manner as 13 starting with 11 using dimethylamine hydrochloride. HPLC-MS retention time: 6.43 min. Yield: 62%, mp: 242–244 °C (from ethanol). 1H NMR (CDCl3): δ 3.14 (s, 3H), 3.16 (s, 3H), 6.18 (broad s, 2H), 6.73 (s, 1H), 7.41 (d, J = 9.0 Hz, 2H), 7.52 ppm (d, J = 9.0 Hz, 2H). 13C NMR (CDCl3): δ 35.7, 39.0, 104.0, 106.9, 127.0, 128.1, 128.8, 132.9, 139.2, 139.3, 159.1, 161.7, 164.3 ppm. IR: ν 3414, 3300, 1646, 1598 cm−1. HRMS (ESI+): calculated for C14H13ClN4NaO2S+ 371.0345 found 371.0366.
5-Amino-3-(4-chlorophenyl)-N-ethyl-N-methyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (18)
Prepared in the same manner as 13 starting with 11 using ethylmethylamine. HPLC-MS retention time 6.85 min. Yield: 47%, mp: 148–150 °C (from ethanol). 1H NMR (CDCl3): δ 1.19–1.26 (m, 3H), 3.46–3.63 (m, 2H), 6.16 (broad s, 2H), 6.70 (s, 1H), 7.40–7.42 (m, 2H), 7.51–7.54 ppm (m, 2H). 13C NMR (CDCl3): δ 12.2, 14.0, 32.9, 36.3, 42.9, 46.1, 103.5, 106.4, 111.3, 117.0, 126.7, 126.8, 127.0, 127.1, 127.8, 127.9, 128.8 (x2), 132.8, 132.9, 159.1 (x2), 162.2, 164.0, 164.5 ppm. IR: ν 3409, 3290, 3175, 1635 cm−1. HRMS (ESI+): calculated for C16H15ClN4NaO2S+ 385.0502 found 385.0491.
5-Amino-3-(4-bromophenyl)-N-isopropyl-4-oxo-3,4-dihydrothieno[3,4-d]pyridazine-1-carboxamide (19)
Prepared in the same manner as 13 starting with 12. HPLC-MS retention time 8.20 min. Yield: 68%, mp: 222 °C dec (from ethanol). 1H NMR (DMSO-d6): δ 1.15 (d, J = 6.6 Hz, 6H), 4.02–4.10 (m, 1H), 7.20 (s, 1H), 7.60 (broad s, 2H), 7.62 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 8.9 Hz, 2H), 7.99 ppm (broad d, 1H). 13C NMR (DMSO-d6): δ 22.0, 40.1, 103.8, 104.5, 119.4, 125.9, 127.9, 131.1, 136.5, 139.9, 158.3, 161.6, 162.9 ppm. IR: ν 3412, 3305, 1655, 1598 cm−1. HRMS (ESI+): calculated for C16H15BrN4NaO2S+ 428.9997 found 428.9986.
Solubility Studies
Compounds were serially diluted in DMSO to achieve a 2.5 fold dilution series from 20 mM down 8 points to 3 μM on a 384-well master plate (Costar 3672). Aliquots (2 μL) were transferred to a clear 96-well assay plate (Fisher 12-565-501) containing 198 μL fibrillization buffer (100 mM NaOAc pH 7.0) for a 100-fold dilution (200 μM down to 0.03 μM). The plate was incubated with agitation for 2.5 h, and the absorbance at 550, 600, 650 and 700 nm was measured on a Spectramax M5 spectrophotometer. DMSO (1%) in fibrillization buffer served as a control that was subtracted from each data point. The absorbances were averaged and the concentration at which the average absorbance rose above 0.03 AU was an indication of insoluble compound, with solubility limits reported as the previous concentration in the dilution series. Absorbance scans of compounds in DMSO indicated that the compounds tested here did not have any absorbance peaks in the above range, therefore all absorbance was due to light scattering by insoluble material.
Tau K18PL Fibrillization Assay
Activity evaluations of test compounds in the K18PL fibrillization assay was performed as previously described.21
Secondary Sedimentation Assay (K18PL and tau40)
The determination of compound-induced effects on the amount of tau (K18PL or tau40) that remains soluble upon centrifugation following incubation with test compounds was conducted as previously described.9
MT-Assembly Assay
The MT assembly assay was conducted in a 384 well plate essentially as described in Hong et al..31 Wild-type full-length human brain tau (tau40, 2N4R) was employed in this assay. Lyophilized bovine brain tubulin (Cytoskeleton Inc., TL238) was reconstituted in 1x RAB (100 mM MES pH 6.9, 1mM EDTA, 0.5 mM MgSO4) pH 6.9 to a concentration of 10 mg/mL. ATPZ compounds (62.5 μM) were added to tau (25 μM) in RAB pH 6.9 and pre-incubated at room temperature for 120 min. To initiate the MT assembly reaction, 8.25 μL per well of 10 mg/mL tubulin was dispensed on a UV-clear 384-well plate (NUNC 265196) followed by 1.0 μL of 100 mM GTP (1x RAB pH 6.9) and 40 μL of the ATPZ:tau mixture. The final reaction mixture was 30 μM tubulin, 20 μM tau, 50 μM ATPZ and 2 mM GTP. The plate was then incubated in a Spectramax M5 plate reader at 37 °C and the absorbance at 340 nm was measured every minute for 45 min.
Cytotoxicity Assay
Cells were plated in each well of a 96-well plate in 100 μL of media (20,000 cells; DMEM plus 10% fetal calf serum with antibiotics). After 24 h, half-log serial dilutions of the test compounds, starting at 200 μM, were added in a further 100 μL of media. The cells were cultured for an additional 72 h and then Alamar blue (Biosource, Camarillo, CA) was added, with cell viability at each drug concentration measured using a SpectraMax M5 fluorescence plate-reader (Molecular Devices, Sunnyvale, CA). CC50 values were generated using Prism software.
Determination of Plasma and Brain Drug Concentrations
Test compounds were administered to two month old female B6C3F1 mice (20 mg average body weight). Whole brain hemispheres, obtained from mice euthanized according to protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee, were homogenized in 10 mM ammonium acetate, pH 5.7 (1:2; w/v) using a handheld sonic homogenizer. Plasma was obtained from blood that was collected into a 1.5 mL tube containing 0.5M EDTA solution and subjected to centrifugation for 10 min at 4500g at 4°C. Aliquots (50 μL) of brain homogenate or plasma were mixed with 0.2 mL of acetonitrile, centrifuged at 15,000g, and the resulting supernatants were used for subsequent LC-MS/MS analysis. The LC-MS/MS system was comprised of an Aquity UPLC and a TQ MS that was controlled using MassLynx software (Waters Corporation, Milford, MA, USA). Compounds were detected using multiple reaction monitoring (MRM) of their specific collision-induced ion transitions. Samples (5 μL) were separated on an Aquity BEH C18 column (1.7 μm, 2.1 × 50 mm) at 35 °C. Operation was in positive electrospray ionization mode, with mobile phase A of 0.1% (v/v) formic acid, and B of either acetonitrile or methanol with 0.1% (v/v) formic acid at a flow rate of 0.6 mL/min using a gradient from 5% to 95% B over two min, followed by wash and re-equilibration steps. The MS was operated with a desolvation temperature of 450 °C and a source temperature of 150 °C. Desolvation and source nitrogen gas flows were 900 L/hr and 50 L/hr, respectively. Source and MS/MS voltages were optimized for each compound using the MassLynx auto tune utility. Standard curves were generated for each compound from brain homogenate and plasma samples that had compound added at 4, 40, 400 and 4000 ng/mL and extracted as above. Peak areas were plotted against concentration and a 1/x weighted linear regression curve was used to quantify the tissue-derived samples using the average peak area from triplicate injections. Drug half-life was calculated using the elimination rate constant determined from the slope of a line plotted through natural log plasma concentration versus time for the 4, 8 and 16 h time points using Microsoft Excel. Area under the brain or plasma concentration curve from 0.5 to 16 h was calculated using the trapezoid rule with GraphPad Prism.
Supplementary Material
List of Abbreviations
- CNS
central nervous system
- ATPZ
aminothienopyridazine
- MT
microtubule
- AD
Alzheimer’s disease
- FTD
frontotemporal dementia
- FTDP-17
frontotemporal dementia with parkinsonism linked to chromosome 17
- NFT
neurofibrillary tangle
- BBB
blood-brain barrier
- SAR
structure-activity relationship
- PSA
polar surface area
- HBD
hydrogen-bond donor
- HBA
hydrogen-bond acceptor
- ThT
thioflavine-T
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- I.P
intraperitoneal
- I.V
intravenous
- PK
pharmacokinetic
- AUC
area under the curve
Footnotes
Supporting Information Available: HPLC chromatograms, mass, IR, 1H NMR, 13C NMR spectra (compounds 8, 10, 11, 13–19), X-ray crystal structures (compounds 8, and 16) and MT-assembly assay results for compounds of 8, 10, 11, 13–19. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative Tauopathies. Ann Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 2.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 3.Congdon EE, Kim S, Bonchak J, Songrug T, Matzavinos A, Kuret J. Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J Biol Chem. 2008;283:13806–13816. doi: 10.1074/jbc.M800247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–1807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 6.Brunden KR, Trojanowski JQ, Lee VMY. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis. 2008;14:393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunden KR, Ballatore C, Crowe A, Smith AB, III, Lee VM-Y, Trojanowski JQ. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: A focus on tau assembly inhibitors. Exp Neurol. 2009 doi: 10.1016/j.expneurol.2009.08.031. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunden KR, Trojanowski JQ, Lee VMY. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VMY. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophy Res Comm. 2007;358:1–6. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickhardt M, von Bergen M, Gazova Z, Hascher A, Biernat J, Mandelkow EM, Mandelkow E. Screening for inhibitors of tau polymerization. Curr Alzheimer Res. 2005;2:219–226. doi: 10.2174/1567205053585891. [DOI] [PubMed] [Google Scholar]
- 11.Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolyl-Hydrazide and Its Derivatives Are Potent Inhibitors of tau Aggregation and Toxicity in Vitro and in Cells. Biochemistry. 2007;46:10016–10023. doi: 10.1021/bi700878g. [DOI] [PubMed] [Google Scholar]
- 12.Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 13.Larbig G, Pickhardt M, Lloyd DG, Schmidt B, Mandelkow E. Screening for inhibitors of tau protein aggregation into Alzheimer paired helical filaments: a ligand based approach results in successful scaffold hopping. Curr Alzheimer Res. 2007;4:315–323. doi: 10.2174/156720507781077250. [DOI] [PubMed] [Google Scholar]
- 14.Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible Expression of Tau Repeat Domain in Cell Models of Tauopathy: Aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- 15.Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, Waldmann H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed Engl. 2007;46:9215–9219. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 17.Chang E, Congdon EE, Honson NS, Duff KE, Kuret J. Structure-activity relationship of cyanine tau aggregation inhibitors. J Med Chem. 2009;52:3539–3547. doi: 10.1021/jm900116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chirita C, Necula M, Kuret J. Ligand-dependent inhibition and reversal of tau filament formation. Biochemistry. 2004;43:2879–2887. doi: 10.1021/bi036094h. [DOI] [PubMed] [Google Scholar]
- 19.Wischik CM, Edwards PC, Lai RYK, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wainwright M, Crossley KB. Methylene Blue - a therapeutic dye for all seasons? J Chemother. 2002;14:431–443. doi: 10.1179/joc.2002.14.5.431. [DOI] [PubMed] [Google Scholar]
- 21.Crowe A, Huang W, Ballatore C, Johnson RL, Hogan AM, Huang R, Wichterman J, McCoy J, Huryn D, Auld DS, Smith AB, III, Inglese J, Trojanowski JQ, Austin CP, Brunden KR, Lee VMY. Identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry. 2009;48:7732–7745. doi: 10.1021/bi9006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson GN, Valant C, Horne J, Figler H, Flynn BL, Linden J, Chalmers DK, Sexton PM, Christopoulos A, Scammells PJ. 2-aminothienopyridazines as novel adenosine A1 receptor allosteric modulators and antagonists. J Med Chem. 2008;51:6165–6172. doi: 10.1021/jm800557d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelhamid IA, Ghozlan SAS, Elnagdi MH. Alkyl-substituted heteroaromatics as precursors to polycyclic heteroaromatics: recent developments. ARKIVOC. 2008;2008:54–84. [Google Scholar]
- 25.Gewald K. Heterocycles from CH-acidic nitriles. VII. 2-Aminothiophene from alpha-Oxo Mercaptans and Methylene-active Nitriles. Chem Ber. 1965;98:3571–3577. [Google Scholar]
- 26.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 27.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reichel A. The role of blood-brain barrier studies in the pharmaceutical industry. Curr Drug Metab. 2006;7:183–203. doi: 10.2174/138920006775541525. [DOI] [PubMed] [Google Scholar]
- 29.Nagakura M, Ota T, Shimidzu N, Kawamura K, Eto Y, Wada Y. Syntheses and antiinflammatory actions of 4,5,6,7-tetrahydroindazole-5-carboxylic acids. J Med Chem. 1979;22:48–52. doi: 10.1021/jm00187a012. [DOI] [PubMed] [Google Scholar]
- 30.Sotelo E, Mocelo R, Suarez M, Loupy A. Synthesis of Polyfunctional Pyridazine Derivatives Using a Solvent-Free Microwave Assisted Method. Synth Commun. 1997;27:2419–2423. [Google Scholar]
- 31.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VMY. Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.