

Abstract
This report details the invention of a method to enable syntheses of psychotrimine, 1, and the kapakahines F and B, 2 & 3, on a gram scale and in a minimum number of steps. Mechanistic inquiries are presented for the key enabling quaternization of indole at the C3 position by electrophilic attack of an activated aniline species. Excellent chemo-, regio-, and diastereoselectivities are observed for reactions with o-iodoaniline, an indole cation equivalent. Additionally, the scope of this reaction is broad with respect to the tryptamine and aniline components. The anti-cancer profiles of psychotrimine, 1, and kapakahines F and B, 2 & 3, have also been evaluated.
Introduction
Of all the known nitrogen-bearing natural products, those containing an indole heterocycle hold a special place in the annals of organic chemistry. This priority position in “alkaloid space” is due to several factors including the interesting structures of many of their members, their novel bioactivities, and their historical importance in the development of organic chemistry.i Furthermore, certain indole alkaloids are even used as “yardsticks” to measure progress in the art and science of synthesis – strychnine being particularly prominent in this regard.ii
An important subset of indole alkaloids are those made up of more than one indole subunit.iii The vast majority of such structures involve linkages between the carbon atoms of the indoles, such as those shown in Figure 1, which have all succumbed to elegant total syntheses.iv A rarity among polymeric indole alkaloids are those linked by the nitrogen atom of the indole, such as those in Figure 2.
Figure 1.

A sampling of polymeric indole alkaloids with carbon-carbon linkages between indole subunits.
Figure 2.

Representatives of the subgroup of oxidatively conjoined tryptamines containing nitrogen to carbon connectivity.
The first member of this subset was isolated in 1944 and named chaetomin, 12.va,b Its intriguing structure was not fully elucidated until 1978 (then renamed chetomin)5c,d and it was not until the very end of the twentieth century that more natural products with this peculiar connectivity were isolated.vi In 1995 Scheuer and co-workers isolated a family of macrocyclic peptides, the kapakahines, 2 & 3, exhibiting an N1-C3 linkage between two tryptophan subunits.vii A decade later more natural products expressing this motif were isolated, including ones akin to chetominviii and the only reported polymeric cyclotryptamine alkaloids with the N1-C3 linkage, psychotrimine, 1,ix and psychotetramine, 15.x These natural products have both fascinating structures and biological functions that are of considerable interest. For example, chetomin, 12, disrupts binding of a key cofactor in the hypoxia-inducible factor pathway and has been shown to affect dose-dependent tumor reduction in mice.5f The kapakahines, only isolated in minute quantities (0.3 mg of kapakahine B was isolated from 840 g of crude sponge ethanolic extract), have been reported to exhibit promising anti-leukemia activity. Psychotrimine has been shown to possess activity against colon and lung cancers,xi and was equally potent as a currently employed colon cancer therapy, 5-fluorouracil.
The absence of any direct means to synthesize these natural products led us to consider the invention of a method to bridge this gap in the capabilities of chemical synthesis.xii This full account describes in detail the underlying rationale, mechanism, and scope of a method that has made possible the concise gram-scale total syntheses of highly complex natural products such as psychotrimine and those belonging to the kapakahine family.xiii
Biosynthetic Considerations and Initial Forays
Whereas the biogenetic origins of dimeric indole alkaloids linked at carbon are presumed to proceed via a radical dimerization process,xiv the genesis of nitrogen-linked siblings in this family is not well understood. A number of possibilities exist for how Nature might fashion this exotic connectivity. Perhaps the most straightforward possibility would be a radical- or cationic-based, oxidative dimerization to directly form the N1-C3 bond (Figure 3A). A second possibility, reminiscent of the speculated biosynthesis of the chartelline alkaloids, hinges on the propensity of a nitrogen substituent to migrate from C2 to C3 of indole via a [1,5] sigmatropic rearrangement (Figure 3B). Although migration of carbon atoms from C2 to C3 is well precedented, to our knowledge, the sole experimental demonstration of a nitrogen migration in such a context stems from the total synthesis of chartelline C (19 → 20).xv Thus, as illustrated in Figure 3C, an analogous process may be operative in a hypothetical rearrangement of 21 to 24 (via 22 and 23). Such a scenario would require position-selective intermolecular oxidative dimerization of tryptophan units to afford the N1-C2 linked dimer 21. Naturally occurring alkaloids such as 21 are not known, but heteroatom linkages at C2 of tryptophan are precedented, such as celogentin C (vide infra).xvi
Figure 3.

Possible biosynthetic routes to dimeric tryptamines containing the N1-C3 bond.
A third possibility would involve direct nucleophilic displacement of a halide by the indole nitrogen (Figure 3D). Although the work of Rainier has demonstrated that such a process can occur, it requires a strong basexvii and proceeds to give the opposite diastereochemical outcome that is required for the kapakahines via epimerization of the tryptophan α-proton. A final possible biogenesis invokes the “Somei hypothesis” for dimerization via N-hydroxyindoles (26, Figure 3E) with concomitant loss of water.18a Somei has demonstrated that in certain contexts this type of bond formation is facile,xviiib but a quaternary center cannot be formed in more than trace quantities via this process.xix The following series of experiments were designed to interrogate the innate reactivity of molecules relevant to the presumed biogenetic hypothesis in Figures 3A and 3C.
Our studies commenced with the reinvestigation of the sole example of N1-C3 tryptamine dimerization reported in a book chapter in 2000 (Scheme 1).xx The described reaction used a cobalt (II) salen precatalyst in the presence of oxygen, conditions that are well known to operate through an SET process.20b–c Unfortunately, the only melatonin dimer observed was not 28, but rather the constitutionally isomeric structure linked from N1 to C2, 29 (Scheme 1, verified by X-ray crystallography). While disappointing, this reactivity represented an opportunity to explore the C2 to C3 rearrangement proposed in Figure 3C. As depicted in Scheme 2, when dimer 29 was reduced using zinc in acetic acid, the desired tryptamine dimer 30 was isolated in 96% yield. With the necessary oxidation state in place, dozens of thermal and acidic conditions were screened to elicit isomerization (PtCl4, CuOTf, RuCl(H)(CO)(PPh3)3, AgNO3, NiCl2, TiF4, BF3•OEt2, Sc(OTf)3, ZnCl2, etc.), but were unsuccessful. The recalcitrance of this intermediate to isomerize was compounded by its propensity to rapidly undergo oxidation back to 29, along with other oxidation byproducts. During the course of the Lewis acid screen, it was found that AuCl3 acted as an electrophilic chlorine source to cleanly provide the C3-chlorinated indolenine 31a.xxi
Scheme 1.

Structural reassignment of the melatonin oxidative dimerization product.a
aReagents and conditions: (a) Co(salen)2 (0.18 equiv), O2, DCM, 23 °C, 24 h, 35%.
Scheme 2.

Failure of the necessary bond migration.
a Reagents and conditions: (a) Zn (20 equiv), AcOH, 23 °C, 1 h, 96%; (b) N-chlorosuccinimide (1.1 equiv), DCM, 0 → 23 °C, 1 h, 94%; (c) K2CO3 (100 equiv), MeOH, 23 °C, 2 h, 93%.
Isomerization reactions on this chlorinated species, 31a (generated in a simple way using NCS), and the related C3-methoxy indolenine, 31b, (prepared by treatment of 31a with K2CO3/MeOH) were also attempted by subjection to acidic,xxii basic,xxiii or thermal conditions. Additionally, treatment of 31a with silver (I) triflate or 31a or 31b with NaBH4 did not afford the desired indole connectivity (32). Although not thoroughly explored, subjection of 29 to acidic conditions led to significant decomposition and recovery of monomeric melatonin species 27.xxiv Despite these setbacks it was reasoned that a redox neutral isomerization event of the nitrogen substituent from C2 to C3 was still possible. To investigate whether the electron rich nature of melatonin dimer 29 somehow prevented the isomerization reaction, the electron neutral analog, lacking the C5 methoxy groups, was synthesized.
Cobalt-mediated oxidation conditions were attempted with electron neutral tryptamines; however, the N1-C2 product analogous to 29 could not be detected in the reaction mixture. Only monomeric oxidation byproducts were isolated, including the N-formyl kynurenine (oxidative cleavage of the indole 2,3-π bond) and the C3-hydroxy pyrroloindoline.xxv An extensive screen of oxidants did, however, lead to the isolation of trace amounts of an N1-C3 linked tryptamine dimer, using Koser’s reagent (PhI(OH)OTs),xxvi as shown in Figure 4. The continued screen of hypervalent iodine oxidants and other transition metal oxidants did not lead to any improvement of this non-selective reaction nor the detection of the N1-C2 isomer. Even in the case of the hexapeptide 35, macrocyclization was not observed.
Figure 4.

Direct oxidative dimerization of a tryptamine to give N1-C3 bond formation.
A stepwise strategy was therefore pursued as documented in Scheme 3. A Buchwald-Goldberg-Ullmann reactionxxvii was well suited for the task of merging bromotryptamine 37 (available in three steps from tryptamine) and tryptamine 33, furnishing dimer 39 after Boc-removal with TFA (20%, unoptimized). Unfortunately, this compound was also resistant to isomerization and underwent similar oxidative decomposition as the melatonin dimer 30. Exploration of oxidative transformations on this substrate were not pursued due to the lengthy and low-yielding sequence to obtain useful quantities of 39.xxviii
Scheme 3.

Stepwise synthesis of N1-C2 linked tryptamine dimer 39.a
aReagents and conditions: (a) 33 (4.0 equiv), CuI (1.0 equiv), (±)-trans-N,N′-dimethyl-1,2-cyclohexanediamine (0.20 equiv), K3PO4 (11 equiv), 1,4-dioxane, 101 °C, 24 h; (b) TFA/DCM (1:4), 23 °C, 1 h, 21 % (2 steps).
At this juncture the realization and acceptance was made that a purely biogenetic strategy would likely not be feasible in synthetically useful yield, therefore a surrogate for tryptamine was sought out. The work of Renfroe, Bergman and Castle in oxidative heterocoupling was particularly interesting in this regard,xxix wherein the coupling of nitrogen nucleophiles with C3-substituted indoles to afford C2-linked indoles was achieved simply using NCS (Figure 5).
Figure 5.

Oxidative coupling reactions for the introduction of heteroatom substituents to indole C2.
As shown in Scheme 4A, indoline was chosen as a tryptamine surrogate. In the event, tryptamine 45 was treated with NCS in the presence of Et3N followed by addition of indoline to furnish C3-hydroxyindolenine 47. This product presumably arises from rapid autoxidation of the initially formed adduct during workup. While Fmoc removal proceeded smoothly (DBU, C12H25SH) to deliver 48,xxx attempts to reduce the resulting imine and effect isomerization were unsuccessful (Zn/AcOH, NaBH4/CeCl3). It is possible that imine reduction and dehydration did occur, followed by rapid autoxidation to regenerate 48.
Scheme 4.

Synthesis of a C3-N-functionalized pyrroloindoline.a
aReagents and conditions: (a) (i) N-chlorosuccinimide (1.1 equiv), Et3N (1.0 equiv), DCM, 0 → 23 °C, 1.5 h, (ii) indoline (1.4 equiv), 0 → 23 °C, 12 h, 59%; (b) DBU (1.0 equiv), C12H25SH (2.0 equiv), DCM, 23 °C, 2 h; (c) NCS (1.1 equiv), Et3N (1.2 equiv), DCM, 0 → 23 °C, 15 min; indoline (2.0 equiv), 12 h, 18% 49 and 45% 51.
In order to avoid the protection/deprotection sequence, the Fmoc protected secondary amine was simply masked as a methyl carbamate.xxxi The methyl carbamate of tryptamine (33, Scheme 4) was treated with NCS, which to our surprise provided the chloroindolenine (50)xxxii rather than its ring-chain tautomeric C3-chloropyrroloindoline form.xxxiii Not surprisingly the chloroindolenine exhibited noticeable decomposition upon aqueous work-up or concentration. The crude chloroindolenine was then treated with indoline and the N1-C3 linked isomer, 49, was obtained in 18% yield along with 45% of the C2-linked adduct 51. The exact nature of this highly interesting process is certainly debatable and related transformations are discussed in detail in the Mechanistic Considerations section. We were pleased to have rapid access to the key quaternary center expressed in psychotrimine, 1, and the kapakahines, 2 & 3; however, efficiencies were still low and proceeding via an indoline or an unfunctionalized aniline would require a number of extraneous steps.
If o-iodoaniline could be successfully employed as a nitrogen donor, then rapid access to these natural products would be enabled (Figure 6A). However, as was encountered previously (Scheme 4), issues of regio- and chemoselectivity remained (see potential byproducts B – F in Figure 6B). Therefore, an optimization study was carried out with Boc-Trp-OMe and o-iodoaniline as an indole cation equivalent (Table 1). At the outset, our concerns about selective access to 53 were well-founded since several oxidants screened led to either no observed product or trace quantities along with numerous byproducts and polymeric material. Fortunately, hypervalent iodine (III) acted as a competent oxidant for this transformation, with Koser’s reagent (PhI(OH)OTs) providing a local maximum of efficiency (Table 1, Entries 2–4).
Figure 6.

(A) Nature’s presumed biosynthesis leads to the invention of a direct indole-aniline oxidative coupling reaction. (B) Known reactivity suggests issues of chemo- and regioselectivity.
Table 1.
Optimization of direct coupling with o-iodoaniline.
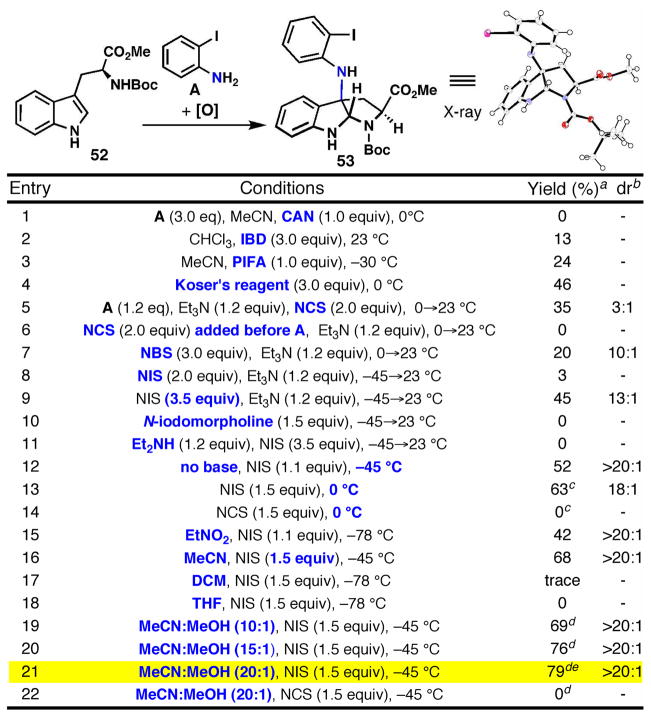 |
Isolated yield of 53.
determined by crude 1H-NMR.
quenched after 10 min.
quenched by addition of Na2S2O3 solution to −45 °C reaction.
gram scale, + 17% 52.
A number of iodine (III)-reagents and reaction conditions failed to improve the yield and conversion to 53 (no recovered starting material). Returning to our initial finding in Scheme 4, a number of different halogenating agents were evaluated (Entries 5–12). To our delight, succinimide-based halogenating agents all delivered 53 with varying amounts of recovered starting material. NIS was found to be optimal in terms of conversion, isolated yield, diastereoselectivity and scalability. If triethylamine is used, the reaction requires ca. 3 equivalents of NIS (Entries 8–9), whereas in the absence of base only 1.1–1.5 equivalents are necessary (Entries 12 and 16). Remarkably, much greater efficiency and diastereoselectivity are obtained in the absence of any amine base, as a result of the lower temperature required for the reaction to occur. This was supported by low temperature quenching experiments, as well as by a slight decrease in diastereoselectivity at higher temperatures (Entry 13). This fact was confirmed by directly injecting an NIS solution at low temperature to a solution of o-iodoaniline and tryptamine while acquiring 1H-NMR data (see Supporting Information for details). All of the data indicates rapid reaction at low temperature only in the absence of base.
Next, a solvent screen revealed that DCM and THF were not competent whereas EtNO2 and MeCN were preferable (Entries 15–18). Finally, the addition of MeOH as cosolvent in a ratio of 20:1 MeCN:MeOH was found to provide the optimal efficiency, affording the product in 79% yield on gram scale (Entry 21). While the minor diastereomer could be detected in minute quantities by crude 1H-NMR (ca. 50:1), removal of this contaminant by column chromatography was simple. No product was observed when NCS was substituted for NIS under either the optimized conditions (Entry 21 vs 22, quenched at −45 °C) or at a higher temperature experiment (Entry 13 vs. 14, quenched after 10 min.). The exact role of MeOH is not entirely clear; however, its addition does increase solubility of the reactants at low temperature. The divergence between NIS and NCS (summarized in Scheme 5), and the role that the oxidant was playing, required further studies varying the substrates and the order of addition in an attempt to uncover the mechanistic basis for the differences in yield and diastereoselectivity.
Scheme 5.

Comparison of order of addition for oxidative cyclization.
Mechanistic Considerations
The following sections present experimental studies to probe the site of oxidation during direct coupling, explain the observed diastereoselectivity, and understand the nature of the active species. It is clear that either initial oxidation of aniline or tryptamine coupling partners can be implicated in the first step of this oxidative coupling reaction, as depicted in Figure 7.
Figure 7.

Some possible mechanisms for N-C bond formation.
Depending on the substrate to be oxidized, several mechanistic pathways can be envisaged and may be classified into two general categories: either initial indole oxidation (Paths A–D) or initial aniline oxidation (Paths E–H). This seemingly simple indole quaternization reaction is mechanistically intriguing and surprisingly complex.
Nature and identity of the oxidized component
If initial indole oxidation were to occur, a structure such as 54 (Figure 7) would be formed; direct interrogation of such an intermediate with aniline was therefore pursued. Specifically, the chlorinated version of 54 (X = Cl) was chosen since it is likely to be more stable (and characterizable) than the corresponding bromo and iodo analogs. This species was generated from 33 using NCS in the presence of Et3N (Scheme 6).
Scheme 6.

Reactions of anilines with the chloroindolenine, 50.a
aReagents and conditions: (a) (i) N-chlorosuccinimide (1.5 equiv), Et3N (1.5 equiv), DCM, 0 → 23 °C, 15 min; (ii) aniline (2.0 equiv), 23 °C, 8 h, 28%; (b) p-iodoaniline (1.5 equiv), N-halosuccinimide (1.5 equiv), Et3N (1.5 equiv), DCM, −45 → 23°C, 3 h.
Addition of aniline to 50 afforded 62 in 28% isolated yield along with unidentified tryptamine byproducts. However, addition of ortho- or para-iodoaniline did not yield coupled product, rather the chloroindolenine 50 and iodoaniline remained unreactive even after a number of hours at room temperature. Apparently, the mildly electron withdrawing iodine substituent is sufficient to inhibit this pathway. In contrast, the addition of NCS to a mixture of tryptamine 33 and p-iodoaniline at 0 °C did provide the pyrroloindoline 63 in 27% yield, along with 47% unreacted starting material. The failure of ortho- or para-iodoaniline to engage the chloroindolenine 50 argues strongly against a pathway for the oxidative coupling reaction that proceeds via such an intermediate with NCS as an oxidant (i.e. Paths A–E, Figure 7). Furthermore, based on these results, it is unlikely that the indolenine 59 (Path F, Figure 7) could act as a competent electrophile at C2 or C3 for an iodoaniline.
Interestingly, the addition of NIS to a mixture of the tryptamine 33 and p-iodoaniline only afforded minute quantities of the product 63 (3%). Instead, the double addition isomer 64 was isolated as the major product (33%), as verified by X-ray crystallography, along with 52% recovered starting material, 33. This curious result can be explained by invoking a pathway originating with aniline oxidation and subsequent reaction of this oxidized species (vide infra) with tryptamine 33 to afford the open-chain tautomeric indolenine of 63. This indolenine can then engage in one of two pathways: (1) ring closure to afford the minor product 63 or (2) a pathway leading to the major product 64 involving first [1,5] sigmatropic rearrangement of the aniline to the indole C2 position. After tautomerization to the 1H indole and reaction with a second equivalent of the oxidized p-iodoaniline species, the product 64 would be delivered. The addition of p-iodoaniline (unactivated) to the ring-chain tautomeric indolenine of 63, while conceivable, is unlikely due to p-iodoaniline’s inability to react with the chloroindolenine 54. It is possible that the difference in product distribution between the NCS and NIS conditions results from the generation of a more reactive oxidized aniline species, derived from NIS as compared to NCS. This study supports the viability of compounds of the type 60 and 61, which most likely arise from initial oxidation of the aniline (Paths G and H). Initial oxidation of the tryptamine (Paths A–D) or a route proceeding via nucleophilic addition of an aniline to an indolenine (Paths E and F) cannot easily account for these observations.
Despite the fact that 50 exists predominantly as the open-chain tautomeric chloroindolenine, the reaction may proceed via the corresponding pyrroloindoline32 (Path A, Figure 7). Thus, the following experiment was devised. When the 3-bromopyrroloindoline, 66, was treated with aniline, production of 67 was only observed at elevated temperature and in the presence of base (Scheme 7). Structure confirmation was then secured by Boc-deprotection to the oxidative coupling product, 62.
Scheme 7.

Nucleophilic displacement of a 3-halopyrroloindoline by an aniline nucleophile.a
aReagents and conditions: (a) N-bromosuccinimide (1.4 equiv), DCM, 23 °C, 1 h, 84%; (b) aniline (6.3 equiv), K3PO4 (1.9 equiv), DMSO, 100 °C, 24 h, 12%; (c) TFA/DCM (1:5), 0 °C, 0.5 h, quant.
While the presence of the Boc functionality prevents a direct comparison of 66 to 55, the harsh conditions required for this transformation suggest that a nucleophilic displacement of a 3-halopyrroloindoline is not operative in the oxidative coupling reaction. In the context of an unprotected 3-halopyrroloindoline, 55, as would be the case in the indole-aniline coupling (Path A, Figure 7), failure to generate the C-N bond is to be expected. Based on the studies of Witkop,33 an aniline or anilide should react more readily as a base towards the indoline N-H, catalyzing the 3-halopyrroloindoline’s decomposition. Indeed, treatment of the methylcarbamate of tryptamine, 33, with tBuOCl, NCS, NBS, or NIS, followed by reaction with o-iodoaniline or its sodium salt, did not result in any product formation. This further reinforces the notion that product formation via such a halogenated tryptamine species (54 or 55, Figure 7) does not occur in the case of o-iodoaniline under these reaction conditions.
Since product formation is not observed for the reaction of 68 (Table 2, X = Cl) with ortho- or para-iodoaniline, the effect of order of addition was analyzed for the reaction of aniline with either pregenerated 68 or 52/NXS. If the diastereoselectivity observed for the chlorination step is conserved in that of the product, that would suggest the intermediacy of a haloindolenine 68 when the oxidant is added last. Thus, Boc-Trp-OMe 52, was reacted with NCS at low temperature and also at room temperature to form two different ratios of chloroindolenines: 1.4:1 and 1.0:1, respectively (Entries 1–2). When aniline was added to the two reaction mixtures at room temperature, the product was obtained with different degrees of diastereoselectivity (7.9:1 and 5.1:1, respectively), albeit in only 23% and 34% yields, respectively. It was found that monitoring the reaction by 1H-NMR confirmed that the minor diastereomer of 68 (Entry 1) reacted faster than the major diastereomer to provide 69, whereas the remaining diastereomer was largely unreactive and decomposed upon aqueous workup.
Table 2.
Diastereoselectivity as a mechanistic footprint.
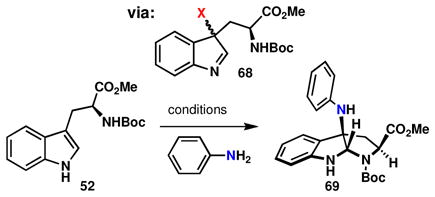 | ||||
|---|---|---|---|---|
| Entry | Conditionsa | dr (68)b | (%)c | dr (69)b |
| 1 | NCS, −78 °C; then aniline, rt | 1.4:1 | 23 | 7.9:1 |
| 2 | NCS, rt; then aniline, rt | 1.0:1 | 34 | 5.1:1 |
| 3 | aniline; then NCS, −78°C | — | 11 | 7.4:1 |
| 4 | aniline; then NCS, rt | — | 10 | 7.6:1 |
| 5 | NBS, rt; then aniline, rt | 1.0:1 | 13 | 10.4:1 |
| 6 | NIS, rt; then aniline, rt | — | 0 | — |
aniline (2.0 equiv), Et3N (1.5 equiv), NXS (1.2 equiv), DCM;
determined by crude 1H-NMR;
isolated yield of 69
We next made a direct comparison to these first two experiments (Entries 1–2) by reversing the order of addition, such that the NCS was added last: either at low temperature or room temperature (Entries 3–4). Interestingly, the diastereoselectivity at low temperature was essentially the same for both of the orders of addition (Entry 1 vs. 3), while the diastereoselectivities were different for the reactions conducted at room temperature (Entry 2 vs. 4, 5.1:1 and 7.6:1 respectively, the diastereomeric ratio for the indolenines 68 would be presumed to be 1.0:1 in both cases). Therefore, a different or additional mechanism may be operative when the order of addition is changed; although with yields ranging from 10–34%, it is difficult to pinpoint the origin of these effects. Importantly, Entry 5 demonstrates that the diastereoselectivity and yield of this transformation are dependent on the oxidant employed even when the reactions are conducted at the same temperature. Initial treatment of 52 with NIS followed by addition of aniline did not lead to formation of the desired product (Entry 6). This difference in reactivity between NCS and NBS is suggestive of either (1) the involvement of the halogen in the stereochemistry-determining step or (2) a change in the relative rates of two pathways. For either scenario, at least two mechanisms must be active for this substrate pair (60 & 61, Figure 7). In light of previous discussions, the simplest interpretation of this study is that the alternative mechanism is one involving attack of tryptamine onto an oxidized aniline species.
Rationalization of the observed diastereoselectivity
The indole-aniline coupling reaction proceeds with strong and often exclusive exo selectivity.xxxiv It is difficult to offer direct experimental evidence that can provide a concrete rationalization for this preference. However, the reaction of tryptophan with a number of electrophiles has shown similar stereochemical outcomes,xxxv including N-phenylselenylphthalimide (N-PSP),xxxvi NBS,xxxvii and DMDO4j–l (Figure 8). Figure 8A shows a mechanistic proposal from Danishefskyxxxviiia and Crich38b to rationalize the observed diastereoselectivity for the phenylselenocyclization of tryptophans. Danishefsky proposes the reversible formation of a selenonium species with the indole 2,3-π bond, followed by an irreversible ring closure to afford a predominance of the kinetically favored exo diastereomer. The origin of this preference is attributed to the greater ease of cyclization of the pre-exo ensemble (72) relative to that of the pre-endo ensemble (71). Specifically, there may be a steric clash between the carbomethoxy functionality and the indole nucleus in the pre-endo ensemble (Figure 8A, 71) that is absent from the pre-exo ensemble, 72. Similarily, Crich posits that reversible electrophilic attack by N-PSP leads to an early transition state, followed by a rate-limiting ring closure. Crich attributes the preferential formation of the exo-diastereomer to greater torsional strain in the transition state for the endo diastereomer in the formation of the pyrrolidine ring.xxxix
Figure 8.

Introduction of various electrophiles to indole C3.
Recently, de Lera has put forth an alternate mechanism for both aminoselenation and bromination chemistry based on DFT calculations (Figure 8B).37 Instead of a thermodynamic preference for the intermediate leading to the exo diastereomer (with equilibration to starting material), de Lera proposes a higher energy transition state in the rate limiting step for formation of the endo diastereomer. The endo isomer arises from activation of the indole 2,3-π bond by the electrophile to form diastereomeric benzylic carbocations (exo diastereomer, 74, Figure 8B), then formation of an azetidine, 75, and finally concerted rearrangement to the pyrroloindoline product, 76. The computed energies for the diastereomeric transition states predicts that the exo product should predominate for both bromination and selenation.
For the case of the DMDO oxidation of tryptophan, an entirely different hypothesis has been suggested (Figure 8C). It is believed that DMDO reacts irreversibly with tryptophans to afford diastereomeric mixtures of the products (78 and 80). The diastereoselectivity is largely dependent upon the substitution at the carboxylic acid and amine functionalities, which supports the hypothesis that a steric interaction determines the stereochemical outcome. Unlike the selenonium and bromonium chemistry, DMDO is known to give 1:1 mixtures of diastereomers for substrates without strong steric demands.xl
Moderate exo-selectivity (69) was observed for oxidation of tryptophan with NCS followed by addition of aniline possibly producing an intermediate such as 81 (Figure 8D). The generally poor ratio for exo-selectivity may be attributed to the ~1:1 d.r. for the chlorination and the irreversibility of the indole oxidation with NCS. Considering the high exo selectivity and yield observed for the oxidative cyclization of o-iodoaniline with NIS (Figure 8E) for substrates without sterically demanding substituents (e.g. Boc-Trp-OMe, 52),xli a similar mechanism to bromination and selenation seems more likely (i.e. reversible formation of diastereomeric intermediates followed by preferential formation of the exo product). In summary, to rationalize the high degree of diastereoselectivity for the C3-quaternization of Boc-Trp-OMe, a reversible process must be invoked.
A difference in rate among diastereomeric transition states, leading to selective formation of the exo diastereomers 53 is reliant upon the reversible formation of intermediates 82 or 83 (similar to 60 or 61 Paths G or H, Figure 7). Reversible bonding interactions have been invoked for the case of phenylnitrenium ions interacting with electron rich arenes, lending credence to this possibility.xlii While the nature of a bonding interaction for indole has not been determined, a σ- or π-complex may be possible. For simplicity the σ-complexes are shown in Figure 7, but π-complexes can also be envisioned. If an equilibrium with reversible bond formation of this type exists, then a subsequent rate limiting ring closure may lead to a predominance of the kinetically favored product – the exo isomer. This explanation can also account for the observed differences in diastereomeric ratio for NCS and NBS (Table 2). For Paths G & H in Figure 7, the halogen remains associated with the complex, stabilizing the cationic charge. The higher atomic number halogens are expected to provide a greater extent of stabilization for either a radical or ionic pathway. This greater stabilization should lead to higher levels of stereoinduction. If π-complex formation occurs, a more favorable bonding interaction would again be predicted for larger halogens, in turn, leading to higher degrees of diastereoselectivity.
What is the nature of the electrophilic aniline species?
For pathways involving an N-haloaniline, the character and extent of bonding between nitrogen and halogen could take a number of forms and may differ between halogens (Figure 9). Gassman’s studies on the reactivity of N-chloroanilines suggest a transient nitrenium and chloride ion pairxliii and it was believed that the introduction of a tryptamine nucleophile to an N-haloaniline would also proceed via this mechanism. While not definitive proof, a number of experiments are consistent with this conclusion. First, the photolysis of o-iodophenylazide (conditions known to generate the nitrene 87 or in acidic solution nitrenium 86)xliv results in some product formation (Scheme 8). The low yield of this process can be attributed to the instability of the product and of this particular nitrenexlv under the reaction conditions. Indeed, resubjection of the product to the reaction conditions led to substantial bond homolysis. However, the poor efficiency of 33 to 90 may also reflect that a nitrenium ion is not a discrete intermediate in the oxidative coupling reaction and N-haloanilines may require assistance from the tryptamine to assume this active form. The low temperature at which N-chloroanilines react is also suggestive of the involvement of tryptamine promoted N-haloaniline decomposition.43
Figure 9.

Potential electrophilic aniline species
Scheme 8.

Photochemical generation of a phenylnitrenium ion in the synthesis of 89, along with subsequent decomposition.a
aReagents and conditions: (a) 33 (1.0 equiv), hν, benzene, 23 °C, 24 h, 2%; (b) TFA/DCM (1:10), 23°C, 2 h, 91%.
An anilino radical cation, 85, is not believed to be a discrete intermediate. The addition of agents known to inhibit radical reactions (BHT and O2), including the polymerization of aniline,xlvi did not affect the efficiency of this reaction, suggesting that species such as 85 (Figure 9) are not long lived if involved at all. Also, irradiation with a sunlamp – conditions anticipated to promote a radical pathwayxlvii – did not result in product formation, but instead produced large amounts of insoluble material. The possibility that the N-haloaniline reacts directly via an SN2-like mechanism to form an intermediate indolenine, 58 (Path D, Figure 7), which proceeds to the product, cannot be entirely ruled out.xlviii However, a reaction of this type most likely cannot be considered a reversible process and therefore cannot account for the high levels of stereoinduction.xlix
The failure of many anilines to undergo this transformation and the rapid reaction times have largely impeded other kinetic analyses. A detailed kinetic analysis of this reaction and related transformations will be the subject of a separate disclosure.l Depending on the electronic character of the aniline and nature of the oxidant, initial oxidation of the aniline or tryptamine or both may be involved in product formation.
Mechanism Summary
The oxidation of tryptamine with NCS, followed by the addition of aniline to generate a product of the type 94, provides data supporting the addition of aniline to C2 (93) and subsequent migration (Figure 10A). The diastereoselectivities observed for the oxidative coupling of aniline to tryptophan substrates, requires energetically different diastereomeric transition states resulting in an enhanced ratio of exo products.
Figure 10.

Mechanistic proposal for the indole-aniline oxidative coupling reaction.
For the relevant and special case of o-iodoaniline and a tryptamine reacting with NIS (Figure 10B), we believe a reactive phenylnitrenium and iodide pair, 86, (Figure 9, formed from the reaction of N-iodoaniline with tryptamine) reacts reversibly with tryptamines to form an intermediate, 60 or 61 (Figure 7), that proceeds to a quaternized pyrroloindoline product, 95. This conclusion is supported by order of addition experiments, dependence of product distribution and diastereoselectivity on: reagent, temperature and substrate, generation and reactivity of possible intermediates, and literature precedence. This mechanism can account for all of the experimental observations to date. Furthermore, the trends regarding the substrate scope are consistent with this mechanistic picture.
Scope and limitations of the direct indole-aniline coupling
Analysis of the scope of the reaction revealed distinct trends for substrate tolerance (Figure 11). Consistent with mechanistic considerations in the previous section and consistent with the studies of Gassman and co-workers,43 electron deficient anilines were found to be excellent substrates (97, 99). Although admittedly circumstantial, this point provides further evidence for a pathway proceeding via an electrophilic phenylnitrenium ion,li rather than nucleophilic addition of an aniline to an oxidized tryptamine. If the latter were the case, electron rich anilines would be better participants for N-C bond formation (100 is not formed with NIS). The presence of an ortho substituent is required with only the exception of the electron deficient substrate, para-nitroaniline, which affords useful yields under these conditionslii (p-CF3, p-I, p-Cl, p-H, p-Ph, p-Me, p-OMe, etc. afforded only trace product). Electron deficient tryptamines were more successful than electron rich tryptamines (products 98, 101), but both electronic characters were tolerated. Tryptophan and even dipeptide derivatives participated well in the reaction, providing excellent diastereoselectivities (>20:1) of 53 and 102a (for 102b–c, see Scheme 12).
Figure 11.

Scope of tryptamine and aniline oxidative coupling reaction.
Scheme 12.

Failure to observe direct α-carboline ring system.a
aReagents and conditions: (a) o-iodoaniline (1.2 equiv), N-chlorosuccinimide (2.0 equiv), triethylamine (1.2 equiv), MeCN −45 → 4 °C, 2.5 h, 25%; (b) o-iodoaniline (1.2 equiv), N-iodosuccinimide (1.6 equiv for 102b, 1.55 equiv for 102a, 102c), MeCN −45 → −35 °C, 2.5 h.
Total synthesis of psychotrimine
With a method for direct, scalable quaternization of tryptamines with o-iodoaniline firmly secured, its application in total synthesis was pursued. Our efforts first focused on the synthesis of psychotrimine, 1, which had previously been synthesized by Takayama and co-workers.12d A short model study was first conducted, in order to determine the best method for the synthesis of the N-linked methyltryptamine dimer subunit of psychotrimine, 104 (Scheme 9). Thus, the indole-aniline coupled product, 90, (obtained from the reaction of 33 and o-iodoaniline with NIS in 63% yield) was subjected to Larock indolization to provide the requisite tryptamine dimer 34 in 92% yield.liii It is conceivable that this intermediate could lead to psychotrimine, but that would require a laborious installation of a functional handle at C7 of the indoline. Next, conversion of the methyl carbamates to methyl amines provided access to a methyl tryptamine dimer, 104, which may well be naturally occurring. This constitutional isomer of chimonanthine (Figure 1) has not been isolated to date, but it would not be surprising if non-selective oxidation pathways of methyl tryptamine in Nature would result in the formation of this product analogous to the results from Figure 4.
Scheme 9.

Synthesis of a chimonanthine isomer, 104.a
aReagents and conditions: (a) o-iodoaniline (1.2 equiv), N-iodosuccinimide (1.5 equiv), MeCN/MeOH (20:1), −45 °C, 1 h, 63%; (b) 103 (3.0 equiv), Pd(dppf)Cl2•DCM (0.20 equiv), Cs2CO3 (2.0 equiv), LiCl (1.0 equiv), NMP, 100 °C, 1 h, 92%; (c) sodium bis(2-methoxyethoxy)aluminum hydride (11 equiv), toluene, 110 °C, 30 min, 86%.
Our first route to 1 commenced with the methyl carbamate of 7-bromotryptamine, 105 (Scheme 10). Buchwald-Goldberg-Ullmann coupling with the tryptamine, 33, afforded the dimeric tryptamine 106 in 45% yield. The low yield resulted from a lack of chemoselectivity in the cross-coupling (two indole NH groups present), but this efficiency was significantly greater than with palladium-mediated methods.liv Next, o-iodoaniline was appended to the more reactive indole delivering 98, which was then positioned to participate in a Larock indole ring synthesis to afford the trimeric species 107. The low efficiency of this route to the trimeric structure 107 led to the exploration of a halogen selective Larock route (Scheme 11).
Scheme 10.

Initial route to psychotrimine precursor 107.a
aReagents and conditions: (a) 33 (3.0 equiv), CuI (0.30 equiv), (±)-trans-N,N′-dimethyl-1,2-cyclohexanediamine (0.60 equiv), K2CO3 (7.0 equiv), 1,4-dioxane, 101 °C, 20 h, 45%; (b) o-iodoaniline (1.2 equiv), N-iodosuccinimide (1.5 equiv), MeCN:MeOH 20:1, −45 °C, 1 h, 24%; (c) 103 (7 equiv), Pd(OAc)2 (0.3 equiv), K2CO3 (3.5 equiv), LiCl (1.0 equiv), DMF, 90 °C, 0.5 h, 70%.
Scheme 11.

Total synthesis of psychotrimine (1).a
aReagents and conditions: (a) o-iodoaniline (1.2 equiv), N-iodosuccinimide (3.0 equiv), Et3N (1.2 equiv), MeCN, −45 → 23 °C, 1 h, 61–67%, 25–30% 105; (b) Pd(OAc)2 (0.21 equiv), Na2CO3 (2.6 equiv), LiCl (0.9 equiv), 103 (2.7 equiv), DMF, 102 °C, 20 min, 85%; (c) CuI (0.32 equiv), (±)-trans-N,N′-dimethyl-1,2-cyclohexanediamine (0.60 equiv), K2CO3 (7.0 equiv), 33 (3.0 equiv), 1,4-dioxane, 101 °C, 9 h, 89%; (d) sodium bis(2-methoxyethoxy)aluminum hydride (22 equiv), toluene, 110 °C, 30 min, 89%.
In this sequence, the 7-bromotryptamine methyl carbamate, 105 lv was coupled to o-iodoaniline using NIS and Et3N to afford adduct 101 in 61–67% yield along with 25–30% recovered starting material. Slightly lower efficiencies were encountered without the use of triethylamine or when MeOH was substituted for Et3N. Next, a Larock indole ring synthesis was performed to provide 109 (85% yield), leaving the bromine handle unscathed. Unfortunately, high palladium loadings (20 mol %) were necessary to prevent starting material decomposition under the harsh reaction conditions. However, even with these high loadings, debromination of the starting material (101) or the product (109) was not observed. Next, an N-H selective Buchwald-Goldberg-Ullmann coupling cleanly provided the desired product, 107, in 89% isolated yield. The methyl carbamates of this trimeric species were smoothly converted to the corresponding methyl amines in the natural product, delivering 1 in 89% yield. A large excess of reagent was used to achieve a shorter reaction time with less decomposition of psychotrimine, 1.
The rapid and scalable synthesis of psychotrimine, 1, (4 steps, 41–47% overall from 105, 2.5 g of 1 prepared) is the result of a retrosynthetic analysis tailored to the exact substrate, rather than by applying a preordained strategy or method to its synthesis. While at its conception the retrosynthesis left questions of feasibility, empirical findings led to the invention of a new method, which, as will be described in the following section, has had further applications beyond psychotrimine, 1.
Total syntheses of kapakahines B and F
After successfully synthesizing large quantities of psychotrimine, another even more complex venue for oxidative indole-aniline coupling was explored. In addition to the hallmark N1-C3 linkage between indole subunits in psychotrimine, the kapakahines present a twistedlvi macrocycle, a particularly strained α-carboline ring system, and sensitive imidazolone functionality (Figure 12).7 Thus, the first retrosynthetic simplification of the kapakahines was removal of this imidazolone. Next in the plan was removal of the alanine-leucine residues along with simplification of the N-linked tryptophan to o-iodoaniline, thereby giving rise to the tripeptide alkyne 116 and an indole-aniline oxidative coupling product 117. The α-carboline present in 117 could presumably arise from the direct formation of the six-membered ring, under the standard conditions of aniline-indole coupling (Figure 12).
Figure 12.

Initial retrosynthetic analysis of the kapakahines.
It was quickly discovered that direct synthesis of the α-carboline ring would not be feasible (Scheme 12A). For dipeptide 118, the only discernible product observed was the C2 coupled aniline isomer, 119, which likely arises from the initial oxidative coupling of aniline at indole C3 and subsequent [1,5] shift to the C2 position to give the product 119 after tautomerization. This shift is most likely attributable to the slowed rate of ring-closure for a six-membered ring, relative to that of a five-membered ring. Additionally, the decreased nucleophilicity of the amide in 118, relative to carbamate functionality is expected to diminish the propensity for cyclization. This slowed rate rendered the major pathway to be a steric decompression via a [1,5] shift of the aniline to the C2 position. Alternative conditions or substrates did not give the desired product.lvii
The well-documented challengelviii of forming α-carboline systems such as that found in the kapakahines coupled with our failure to achieve its direct synthesis (Scheme 12A), prompted the design of a radically different approach (Scheme 12B). We reasoned that it might be possible to achieve the conversion of the readily accessible pyrroloindoline architecture (isomer A) into the topologically distinct, yet constitutionally isomeric, α-carboline system (isomer B). Since these two isomers might be in a dynamic equilibrium (favoring isomer A) it would be necessary to achieve an irreversible kinetic trap. While highly speculative, the powerfully simplifying nature of this plan was enticing.
Beginning with dipeptide 120, the pyrroloindoline was obtained by direct coupling with aniline in the absence of base, which led to an increased rate of reaction (Scheme 11C). This oxidative-coupling tolerates a variety of typical carbamate protecting-groups on the phenylalanine amine (102a–c) and remains exclusively exo selective (102). The absence of the endo diastereomer by TLC, LC-MS and crude 1H-NMR is consistent with our studies on Boc-Trp-OMe (vide supra). In line with previous observations (Scheme 11A), direct formation of the α-carboline was not observed.
In order to investigate isomerization strategies, the Larock coupling partner tripeptide (116) was required (Scheme 13). A route to enantiopure alkyne amino-acid (122) was therefore devised. Several methods were available including using Schollköpf’s chiral auxiliarylix–lx or using an asymmetric catalyst in an alkylation strategy.lxi Alternatively, beginning with serine-derived iodoalaninelxii (121), Knochel’s copper/zinclxiii coupling to bromo TES-acetylenelxiv provided large amounts of the alkyne amino acid as a single enantiomer.lxv The copper/zinc mediated coupling proved to be reliable on large scale if the iodoalanine (121) was synthesized and used quickly due to slow decomposition. Methyl ester hydrolysis of the alkyne and peptide coupling to H2N-Ala-Leu-OBnlxvi proceeded with high-efficiency to provide tripeptide 116.
Scheme 13.

Preparation of the tripeptide fragment (116). a
aReagents and conditions: (a) CuCN (0.9 equiv), LiCl (1.8 equiv), Zn (3.6 equiv), TMSCl (0.1 equiv), Br(CH2)2Br (0.2 equiv), DMF, −20 → 23 °C, 11 h, 60%; (b) LiOH (1.2 equiv), THF/H2O 1:1, 0 °C, 0.5 h; H2N-Ala-Leu-OBn (1.1 equiv), EDC (1.2 equiv), HOBt (1.4 equiv), THF, 0 → 23 °C, 10 h, 97% (two steps).
Larock coupling of 102b with 116 furnished 123 (Scheme 14), which contains the necessary framework to complete kapakahine F (2). Importantly, the use of Pd(dppf)Cl2 led to an increase in reaction time in addition to less depreciation of yield with increasing scale.53b Double deprotection of the Cbz and benzyl ester in 123 occurred without event and the macrocyclic peptide coupling was attempted under standard conditions, but isomerization of the pyrroloindoline was not observed. Instead, the structure of the product was firmly established by HMBC correlation to be the pyrroloindoline product, 124. Alternatively, coupling of tripeptide 126lxvii to the secondary amine 125 provided 127, which was prepared for a Larock macrocyclization. However, in this critical step the same undesired macrocyclic intermediate, 124, was obtained. Obtaining this product provided an opportunity to investigate the possibility of achieving an isomerization event, as illustrated in Figure 13.
Scheme 14.

Two routes to the pyrroloindoline-containing macrocycle (124).
Reagents and conditions: (a) 116 (2.0 equiv), Pd(dppf)Cl2•DCM (0.20 equiv), Cs2CO3 (2.0 equiv), LiCl (1.0 equiv), DMF, 100 °C, 1.5 h, 55%; (b) 10% Pd/C (0.20 equiv), H2, MeOH, 1 h (c) HATU (3.0 equiv), HOBt (2.5 equiv), iPr2NEt (2.2 equiv), DMF, 6 h, 41% (2 Steps); (d) Et2NH/DCM, 1:3, 1 h, quant; HATU (2.0 equiv), HOBt (1.5 equiv), Et3N (2.0 equiv), 22% and 125 25% (2 steps); (e) Pd(OAc)2 (0.34 equiv), NaOAc (6 equiv), LiCl (0.96 equiv), DMF, 100 °C, 12 h, 67%.
Figure 13.

Failure to observe isomerization to the desired α-carboline.
With a number of relevant substrates in hand, several approaches for the crucial isomerization of a pyrroloindoline to an α-carboline were evaluated (Figure 13): (A) initial isomerization of differentially substituted pyrroloindolines having an o-iodoaniline linked at C-3 (128), (B) isomerization with an indole at C3 in a macrocyclic context (131), and (C) rearrangement analogous to scenario A with aniline functionality and where both nitrogens of tryptophan and phenylalanine are amides (127). All three approaches outlined above were attempted to give an α-carboline with or without imidazolone formation. It was believed that formation of an imidazolone could act as a point of irreversibility, such that even if the α-carboline were less favored, kinetic trapping could afford the desired product.
To commence these studies, treatment of the pyrroloindoline, 102a (Scheme 12), with a variety of acidic conditions (TFA, AcOH, TMSI, SiO2, etc.) to remove the Boc group and possibly undergo subsequent rearrangement (Figure 13A), caused extensive decomposition accompanied by loss of the newly coupled aniline, analogous to that shown in Scheme 8. Removal of the Cbz group in 102b, avoiding aryliodide reduction or loss of the aniline altogether, proved fruitless (NaBH4, TMSI, BF3•OEt2, Pd(OAc)2/Et3SiH, etc.). The Fmoc removal proceeded cleanly; however, direct isomerization to the α-carboline with or without concomitant imidazolone formation was not observed (BF3•Et2O, Sc(OTf)3, MeOH reflux, etc.). Failure to isomerize these readily obtained intermediates dictated the necessity of a late-stage isomerization strategy (scenario B & C, Figure 13).
Thus, several reactions were attempted to convert the pyrroloindoline, 131, to the α-carboline, 132 or 133, (Figure 13B). Thermal, acidic and basic conditions were attempted, as well as the presence of various Lewis acids (ZrF4, Zr(CF3acac)4, ZrCl4, ZrO2, ZrBr4, Zr(Ot-Bu)4, SiO2, AlMe3, AlCl3, Ti(Oi-Pr)4, In(OTf)3, Zn(OTf)2, etc.), all to no avail. Attempts to convert the corresponding activated carbonyl of 131 (R = OH, Cl, OCH2CF3, OC6F5) to 133 by an isomerization and imidazolone trap were also unsuccessful. The final approach to test isomerization, scenario C (Figure 13), was limited by the presence of the aniline functionality, necessitating relatively mild conditions for substrate manipulation due to the lability of this functionality (vida supra). It was believed that the aniline may impart a different structural bias than the indole at the C3 position due to steric and electronic factors. Nevertheless, a variety of conditions failed to provide isomerization to 134 or imidazolone formation to 135.
With this information in hand the approach involving macrocyclization by peptide-coupling (123, Scheme 15) was reinvestigated. After extensive experimentation it was found that EDC/HOAt-mediated macrolactamization in the absence of baselxviii afforded the desired carboline macrocycle 132 in 64% isolated yield along with 6% of the pyrroloindoline 124 (easily separable by column chromatography).lxix The formation of the α-carboline containing macrocycle 132 may proceed through several mechanisms; three of which are discussed here. The first possibility, which is easily ruled out considering the results in Figure 13B, is that the pyrroloindoline containing macrocycle 124 isomerizes to the product 132. Indeed, resubjection of 124 to the reaction conditions did not afford 132, establishing that the reaction is under kinetic control.
Scheme 15.

Successful isomerization to the desired α-carboline (132).a
aReagents and conditions: (a) 10% Pd/C (0.20 equiv), H2, MeOH, 1 h; (b) EDC (3.0 equiv), HOAt (6.0 equiv), DCM/DMF (20:1), 12 h, 70% (124:132, 1:11).
The second possibility involves amide bond formation with intermediate 137. Macrocyclization of intermediate 137 may be more favorable than of 138 due to the greater number of degrees of freedom in the absence of the α-carboline. Amide bond formation of intermediate 137 would generate an indolenine (not depicted) that would then be engaged by the phenylalanine amide, rather than the tryptophan amide, providing the α-carboline 132. For this pathway, it is difficult to rationalize the selectivity observed based on examination of the substrate, as both the tryptophan and phenylalanine amides are sterically and electronically similar. If this pathway does proceed, the preferential formation of a six-membered ring could only be the result of subtle conformational effects. Not surprisingly, such imine intermediates are neither isolated nor observed, as is frequently the case for indolenine species due to their relative instability.
A third mechanistic postulate would involve ring closure of 137 to afford intermediate 138, which then participates in macrocyclization. This pathway allows for a clear explanation for the observed selectivity via a Curtin-Hammett scenario – an equilibrium between 136 and 138 and rapid amidation of the primary amine 138 over the secondary amine 136 results in a predominance of the desired product. Both the second and third mechanistic pathways rely on the well-documented rate difference for acylation of a primary versus a secondary amine.lxx
The preferential formation of the undesired macrocycle 124 using HATU, HOBt, and Hünig’s base in DMF (Scheme 14, 123 → 124) is not surprising in light of literature on amide-bond formation with secondary amines such as proline.lxxi Our optimized conditions, avoiding the use of base and using a minimal amount of DMF for solubility, results in a reduced rate of peptide bond formation and is therefore expected to be more discriminating for a primary amine (137 or 138).
The exceedingly mild conditions utilized to achieve the topological shift of pyrroloindoline 123 to α-carboline 132 is remarkable given the well-documented stability of pyrroloindolines and their widespread occurrence in nature. As alluded to above, this transform dramatically simplified the structural challenge posed by the kapakahines.
Macrocycle 132 was found to decompose slowly if left at room temperature for extended periods, underscoring the necessity for mild conditions for its production or manipulation. Therefore, once the material was isolated, it was immediately carried forward to the construction of the last ring. The imidazolone formation was all that remained to synthesize kapakahine F, 2. It was reasoned that once the imidazolone ring was formed and the Boc group removed, the natural product, 2, should be a stable entity. Hydrolysis of the ester proceeded uneventfully (Scheme 15), but the resulting acid could not be stored for extended periods and was immediately processed. Several different conditions were attempted, (EDC and HOBt, HBTU, HATU, etc.), that were successful at ring-closure with typical conversion of 85–100%. However, 5 – 40% of the uncoupled acid 139 was regenerated after treatment with TFA, which was difficult to separate and detrimental to producing the kapakahines on large scale. Other conditions to remove the Boc group were attempted (TMSOTf, TBSOTf, AcOH, etc.), however, an unacceptable amount of hydrolysis still occurred. It was found that the presence of trace HOBt or other coupling agents during the Boc removal with TFA was causing an increase in the amount of hydrolyzed product.
After several aqueous washes, and checking the crude intermediate for coupling agents by LC-MS and 1H-NMR, the trace impurities isolated together with 133 still caused unacceptable levels of hydrolysis. The only solution was to exclude nucleophilic peptide-coupling reagents during imidazolone formation. Thus, treatment with oxalyl chloride and triethylamine led to clean conversion to the imidazolone with starting material absent by 1H-NMR and LC-MS (Scheme 16). Concentration of the reaction in vacuo to drive off excess oxalyl chloride and several washes with NaHCO3 and NaOH to remove salts gave large quantities of Boc-kapakahine F (133). Exposure of Boc-kapakahine F, 133, to TFA provided kapakahine F, 2, with excellent efficiency and purity (64% isolated yield, 3 steps, > 1g synthesized) and without imidazolone hydrolysis (139). HPLC co-injection of natural and synthetic kapakahine F co-eluted perfectly; however, the kapakahine F 1H-NMR spectra obtained did not precisely match the literature spectra.lxxii The conversion of kapakahine F (2) to B (3) was undertaken to secure our structural assignment. The overall yield of 2 is 13.1% over 12 steps from serine.
Scheme 16.

Completion of kapakahines F, 2, and B, 3.
Reagents and conditions: (a) LiOH, THF/H2O/MeOH (40:2:1), 1 h; HATU (4.0 equiv); TFA/DCM (1:6), 0.5 h; (d) LiOH, THF/H2O/MeOH (40:2:1), 1 h; (COCl)2 (4.0 equiv), Et3N (1.0 equiv), DCM, 1 h; TFA/DCM (1:10), 1 h, 64% (three steps); (f) Boc-Phe-OH (1.2 equiv), EDC (2.0 equiv), HOBt (1.8 equiv), Et3N (3.0 equiv), DCM 1 h; TFA/DCM (1:10), 1 h, 81% (two steps).
This conversion proved uneventful and surprisingly the use of EDC and HOBt followed by Boc deprotection with TFA did not hydrolyze the imidazolone. The basis for the difference in stability between 133 and Boc-3 remains elusive. A comparison of NMR spectra (1H and 13C) for natural and synthetic 3 as well as HPLC co-injection confirmed the structure of the natural product and proved that 2 was also correctly synthesized.
Preliminary Biological Evaluation
With access to large quantities of these rare natural products (1–3), biological testing could be undertaken. Synthetic kapakahines B and F did not possess significant cytotoxicity in the NCI 60-cell line. However, kapakahine B had an IC50 of 11.7 μM for breast cancer cell line DU4475.lxxiii Psychotrimine did show activity in the NCI screen and was submitted to a five-dose GI50 study and had a broad spectrum of activity between 1.3 and 40 μM for the 60-cell linelxxiv (see Supporting Information for details). In addition to the NCI, large quantities of 1–3 have been donated to Bristol-Myers Squibb and samples are freely available from our lab on request.
Conclusion
This full account has traced the inspiration, design, development, mechanistic study, and application of a new reaction for the direct merger of anilines with tryptamines and related species. This oxidative coupling reaction is believed to proceed via a nitrenium ion generated from an aniline followed by nucleophilic capture at C3 of the tryptamine. This powerfully simplifying reaction has enabled the gram-scale synthesis of the three complex alkaloids: psychotrimine and kapakahine F and B. It is worth mentioning that this work culminated from an adherence to a set of guidelines at the outset of the planning stages of this work.lxxv Aside from the invention of the direct coupling of anilines to tryptamines, notable aspects of the total syntheses of 1–3 include: (1) absence of protecting group chemistry and excellent redox economy in the synthesis of 1, (2) chemoselective indole arylation using a Buchwald-Goldberg-Ullmann reaction (109→107), (3) chemoselective Larock indole syntheses in highly complex settings (98→107, 101→109, 102b→123, 127→124), and (4) a remarkable late-stage architectural shift from a pyrroloindoline to α–carboline ring system (123→132).lxxvi
Supplementary Material
Acknowledgments
Dedicated to Professor Albert Eschenmoser on the occasion of his 85th birthday. We are grateful to the NCI and Bristol-Myers Squibb (Dr. Percy Carter) for biological testing of 1, 2 and 3 and to Yoichi Nakao for providing HPLC samples of 2 and 3. Drs. D. H. Huang and L. Pasternack are acknowledged for NMR spectroscopic assistance, particularly with the direct injection experiments. Drs. Arnold L. Rheingold and Antonio DiPasquale are thanked for X-ray crystallographic analysis. Funding for this work was provided by the NIH/NCI (CA134785), the Skaggs Institute for Chemical Biology, Bristol-Myers Squibb (unrestricted grant and graduate fellowship to T.N.), and the Canadian Institutes of Health Research (postdoctoral fellowship to C. A. L.).
Footnotes
Supporting Information Available: Detailed experimental procedures, copies of all spectral data, and full characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- i.For numerous examples see: Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. 1. Wiley-VCH; New York: 1996. and Nicolaou KC, Snyder SA. Classics in Total Synthesis II. 1. Wiley-VCH; Weinheim: 2003.
- ii.Bonjoch J, Solé D. Chem Rev. 2000;100:3455. doi: 10.1021/cr9902547. [DOI] [PubMed] [Google Scholar]
- iii.For reviews see: May JA, Stoltz B. Tetrahedron. 2006;62:5262.Steven A, Overman LE. Angew Chem, Int Ed. 2007;46:5488. doi: 10.1002/anie.200700612.Mulzer J, Gaich T, Siengalewicz P. Angew Chem, Int Ed. 2008;47:8170. doi: 10.1002/anie.200801735.Kam TS, Choo YM. In: The Alkaloids. Cordell GA, editor. Vol. 63. Academic Press; San Diego: 2006. pp. 181–337.
- iv.Vinblastine: Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b.Yokoshima S, Ueda T, Kobayashi S, Sato A, Kuboyama T, Tokuyama H, Fukuyama T. J Am Chem Soc. 2002;124:2137. doi: 10.1021/ja0177049.Mangeney P, Andriamialisoa RZ, Langlois N, Langlois Y, Potier P. J Am Chem Soc. 1979;101:2243. doi: 10.1021/jo01336a006.Kutney JP, Choi LSL, Nakano J, Tsukamoto H, McHugh M, Boulet CA. Heterocycles. 1988;27:1845.Kuehne ME, Matson PA, Bornmann WG. J Org Chem. 1991;56:513.Magnus P, Mendoza JS, Stamford A, Ladlow M, Willis P. J Am Chem Soc. 1992;114:10232.Asperazine: Govek SP, Overman LE. J Am Chem Soc. 2001;123:9468. doi: 10.1016/j.tet.2007.05.127.Govek SP, Overman LE. Tetrahedron. 2007;63:8499.Quadrigemine C: Lebsack AD, Link JT, Overman LE, Stearns BA. J Am Chem Soc. 2002;124:9008. doi: 10.1021/ja0267425.Himastatin: Kamenecka TM, Danishefsky SJ. Angew Chem, Int Ed. 1998;37:2993. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2993::AID-ANIE2993>3.0.CO;2-I.Kamenecka TM, Danishefsky SJ. Angew Chem, Int Ed. 1998;37:2995. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2995::AID-ANIE2995>3.0.CO;2-6.Kamenecka TM, Danishefsky SJ. Chem Eur J. 2001;7:41. doi: 10.1002/1521-3765(20010105)7:1<41::aid-chem41>3.0.co;2-d.Perophoramidine: Fuchs JR, Funk RL. J Am Chem Soc. 2004;126:5068. doi: 10.1021/ja049569g.Chimonanthine: Hendrickson JB, Göschke R, Rees R. Tetrahedron. 1964;20:565.Scott AI, McCapra F, Hall ES. J Am Chem Soc. 1964;86:302. doi: 10.1021/ja01017a041.Link JT, Overman LE. J Am Chem Soc. 1996;118:8166.Movassaghi M, Schmidt MA. Angew Chem, Int Ed. 2007;46:3725. doi: 10.1002/anie.200700705.Haplophytine: Ueda H, Satoh H, Matsumoto K, Sugimoto K, Fukuyama T, Tokuyama H. Angew Chem, Int Ed. 2009;48:7600. doi: 10.1002/anie.200902192.Nicolaou KC, Dalby SM, Li S, Suzuki T, Chen DYK. Angew Chem, Int Ed. 2009;41:7480. doi: 10.1002/anie.200904588.Communesin F: Yang J, Wu H, Shen L, Qin Y. J Am Chem Soc. 2007;129:13794. doi: 10.1021/ja075705g.
- v.(a) Waksman SA, Bugie E. J Bacteriology. 1944;48:527. doi: 10.1128/jb.48.5.527-530.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Geiger WB, Conn JE, Waksman SA. J Bacteriology. 1944;48:531. doi: 10.1128/jb.48.5.531-536.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McInnes AG, Taylor A, Walter JA. J Am Chem Soc. 1976;98:6741. doi: 10.1021/ja00437a074. [DOI] [PubMed] [Google Scholar]; (d) Brewer D, McInnes AG, Smith DG, Taylor A, Walter JA, Loosli HR, Kis ZL. J Chem Soc, Perkin Trans 1. 1978;10:1248. [Google Scholar]; (e) Kikuchi T, Kadota S, Nakamura K, Nishi A, Taga T, Kaji T, Osaki K, Tubaki K. Chem Pharm Bull. 1982;30:3846. [Google Scholar]; (f) Kung AL, et al. Cancer Cell. 2004;6:33. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- vi.For the isolation of alkaloid 13, see the following conference report: Masashi H, Mitsuyoshi S, Hiroshi M, Yuji S. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu. 1994;36:557.
- vii.(a) Nakao Y, Yeung BKS, Yoshida WY, Scheuer PJ, Kelly-Borges M. J Am Chem Soc. 1995;117:8271. [Google Scholar]; (b) Yeung BKS, Nakao Y, Kinnel RB, Carney JR, Yoshida WY, Scheuer PJ, Kelly-Borges M. J Org Chem. 1996;61:7168. doi: 10.1021/jo960725e. [DOI] [PubMed] [Google Scholar]; (c) Nakao Y, Kuo J, Yoshida WY, Kelly M, Scheuer PJ. Org Lett. 2003;5:1387. doi: 10.1021/ol026830u. [DOI] [PubMed] [Google Scholar]
- viii.a) Li GY, Li BG, Yang T, Yan JF, Liu GY, Zhang GL. J Nat Prod. 2006;69:1374. doi: 10.1021/np0602970. [DOI] [PubMed] [Google Scholar]; b) Ding G, Jiang L, Guo L, Chen X, Zhang H, Che Y. J Nat Prod. 2008;71:1861. doi: 10.1021/np800357g. [DOI] [PubMed] [Google Scholar]
- ix.Takayama H, Mori I, Kitajima M, Aimi N, Lajis NH. Org Lett. 2004;6:2945. doi: 10.1021/ol048971x. [DOI] [PubMed] [Google Scholar]
- x.The relative configuration of psychotetramine is not known, see: Mori I. M S Thesis. Chiba University; Japan: 2004.
- xi.Yohei M. Ph D Thesis. Chiba University; Japan: 2008. [Google Scholar]
- xii.Marsden SP, Watson EL, Raw SA. Org Lett. 2008;10:2905. doi: 10.1021/ol801028e.Toumi M, Couty F, Marrot J, Evano G. Org Lett. 2008;10:5027. doi: 10.1021/ol802155n.Espejo VR, Rainier JD. J Am Chem Soc. 2008;130:12894. doi: 10.1021/ja8061908.For a 16 linear step synthesis of 1 (13.2% overall yield), see: Matsuda Y, Kitajima M, Takayama H. Org Lett. 2008;10:125. doi: 10.1021/ol702637r.Takahashi N, Ito T, Matsuda Y, Kogure N, Kitajima M, Takayama H. Chem Commun. 2010;46:2501. doi: 10.1039/b923918a.Beaumont S, Pons V, Retailleau P, Dodd RH, Dauban P. Angew Chem Int Ed. 2010;49:1634. doi: 10.1002/anie.200906650.
- xiii.(a) Newhouse T, Baran PS. J Am Chem Soc. 2008;130:10886. doi: 10.1021/ja8042307. [DOI] [PubMed] [Google Scholar]; (b) Newhouse T, Lewis CA, Baran PS. J Am Chem Soc. 2009;131:6360. doi: 10.1021/ja901573x. [DOI] [PubMed] [Google Scholar]
- xiv.Dimerization at carbon has extensive precedent, for example: Cheek GT, Nelson RF. J Org Chem. 1978;43:1230.Balogh-Hergovich E, Speier G. J Chem Soc Perkin Trans 1. 1986:2305.Shen X, Lind J, Eriksen TE, Merenyi G. J Chem Soc Perkin Trans 2. 1990:597.Dryhurst G. Chem Rev. 1990;90:795.Verotta L, Orsini F, Sbacchi M, Scheildler MA, Amador TA, Elisabetsky E. Bioorg Med Chem. 2002;10:2133. doi: 10.1016/s0968-0896(02)00078-0.Ishikawa H, Takayama H, Aimi N. Tetrahedron Lett. 2002;43:5637.Ishikawa H, Kitajima M, Takayama H. Heterocycles. 2004;63:2597.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238. doi: 10.1126/science.1170777. and references cited therein.
- xv.(a) Baran PS, Shenvi RA, Mitsos CA. Angew Chem, Int Ed. 2005;44:3714. doi: 10.1002/anie.200500522. [DOI] [PubMed] [Google Scholar]; (b) Baran PS, Shenvi RA. J Am Chem Soc. 2006;128:14028. doi: 10.1021/ja0659673. [DOI] [PubMed] [Google Scholar]
- xvi.(a) Ma B, Litvinov DN, He L, Banerjee B, Castle SL. Angew Chem, Int Ed. 2009;121:6220. doi: 10.1002/anie.200902425. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Feng Y, Chen G. Angew Chem, Int Ed. 2010;49:958. doi: 10.1002/anie.200905134. [DOI] [PubMed] [Google Scholar]; (c) Hu W, Zhang F, Xu Z, Liu Q, Cui Y, Jia Y. Org Lett. 2010;12:956. doi: 10.1021/ol902944f. [DOI] [PubMed] [Google Scholar]
- xvii.The perturbation of pH in a restricted environment, such as an enzyme pocket, is well-known; however, the necessity for strong base reduces the likelihood of such a transformation.
- xviii.(a) Somei M. Adv Het Chem. 2002;82:101. [Google Scholar]; (b) Somei M. Heterocycles. 1999;50:1157. [Google Scholar]
- xix.Somei has reported a 1% yield for N1-C3 bond formation when N-hydroxy melatonin and indole (10 equiv) were treated with MsCl: Hayashi T, Peng W, Nakai Y-y, Yamada K, Somei M. Heterocycles. 2002;57:421.
- xx.Adam W, Deutsche F. Peroxide Chemistry: Mechanistic and Preparative Aspects of Oxygen Transfer. Wiley-VCH; DFG, Weinheim: 2000. p. 542.For the use of cobalt (II) in a related transformation see: Nishinaga A. Chem Lett. 1975:273.Mori K, Goto M, Sakai T. Chem Pharm Bull. 1985;33:4167.
- xxi.For a related CuCl2 chlorination, see: Baloghhergovich E, Speier G. J Chem Soc, Perkin Trans 1. 1986:2305.
- xxii.Movassaghi M, Schmidt MA, Ashenhurst JA. Org Lett. 2008;10:4009. doi: 10.1021/ol8015176. [DOI] [PubMed] [Google Scholar]
- xxiii.Richter JM, Ishihara Y, Masuda T, Whitefield BW, Llamas T, Pohjakallio A, Baran PS. J Am Chem Soc. 2008;130:17938. doi: 10.1021/ja806981k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxiv.(a) Hugel G, Royer D, Sigaut F, Lévy J. J Org Chem. 1991;56:4631. [Google Scholar]; (b) Güller R, Borschberg HJ. Tetrahedron Lett. 1994;35:865. [Google Scholar]
- xxv.Nakagawa M, Okajima H, Hino T. J Am Chem Soc. 1977;99:4424. doi: 10.1021/ja00455a035. [DOI] [PubMed] [Google Scholar]
- xxvi.(a) Moriarty RM, Vaid RK, Koser GF. Synlett. 1990:365. [Google Scholar]; (b) Koser GF. Aldrichimica Acta. 2001;34:89. [Google Scholar]
- xxvii.Antilla JC, Klapars A, Buchwald SL. J Am Chem Soc. 2002;124:11684. doi: 10.1021/ja027433h. [DOI] [PubMed] [Google Scholar]
- xxviii.Burns NZ, Baran PS, Hoffmann RW. Angew Chem, Int Ed. 2009;48:2854. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- xxix.For introduction of imidazole nucleophiles to haloindolenines, see: Renfroe, H. B. 3-Substituted-2-(Heteroaryl) Indoles. 4,511,573. US Patent. 1985 Apr 16;Anilines: Bergman J, Engqvist R, Stålhandske C, Wallberg H. Tetrahedron. 2003;59:1033.Imidazoles: He L, Yang L, Castle SL. Org Lett. 2006;8:1165. doi: 10.1021/ol060016f.Ma B, Banerjee B, Litvinov DN, He L, Castle SL. J Am Chem Soc. 2010;132:1159. doi: 10.1021/ja909870g.
- xxx.Sheppeckll JE, Kar H, Hong H. Tetrahedron Lett. 2000;41:5329. [Google Scholar]
- xxxi.For recent reviews on protecting group free synthesis, see: Hoffmann RW. Synthesis. 2006:3531.Young IS, Baran PS. Nat Chem. 2009;1:193. doi: 10.1038/nchem.216.
- xxxii.Species 50 was characterized by conducting the reaction in deuterated solvent (1H, 13C, and HSQC NMR experiments support this structural assignment).
- xxxiii.Ohno M, Spande T, Witkop B. J Am Chem Soc. 1968;90:6521. doi: 10.1021/ja01025a055. [DOI] [PubMed] [Google Scholar]
- xxxiv.See Figure 11 for excellent diastereoselectivities in the oxidative coupling reaction.
- xxxv.In the case of acid-mediated ring closure, the more stabile endo diastereomer is obtained due to the reversibility of product formation. See reference 38b for further explanation.
- xxxvi.Marsden SP, Depew KM, Danishefsky SJ. J Am Chem Soc. 1994;116:11143. [Google Scholar]
- xxxvii.López CS, Pérez-Balado C, Rodríguez-Graña P, de Lera ÁR. Org Lett. 2008;10:77. doi: 10.1021/ol702732j. [DOI] [PubMed] [Google Scholar]
- xxxviii.(a) Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ. J Am Chem Soc. 1999;121:11953. [Google Scholar]; (b) Crich D, Huang X. J Org Chem. 1999;64:7218. doi: 10.1021/jo982385y. [DOI] [PubMed] [Google Scholar]
- xxxix.The postulated difference between torsional strains of the transition states, as compared to that of the products, is suggested to arise from differing hybridization of the carbamate nitrogen lone pair. See reference 38b for further discussion.
- xl.(a) Kawahara M, Nishida A, Nakagawa M. Org Lett. 2000;2:675. doi: 10.1021/ol005513p. [DOI] [PubMed] [Google Scholar]; (b) May JP, Fournier P, Pellielli J, Patrick BO, Perrin DM. J Org Chem. 2005;70:8424. doi: 10.1021/jo051105t. [DOI] [PubMed] [Google Scholar]; (c) May JP, Patrick BO, Perrin DM. Synlett. 2006:3403. [Google Scholar]; (d) González-Vera JA, García-López MT, Herranz R. Tetrahedron. 2007;63:9229. [Google Scholar]
- xli.It is also instructive to compare the high diastereoselectivity in production of 102a–c with the poor selectivity for formation of compound 8 in reference 40c.
- xlii.For reversible interactions of phenyl nitreniums with arenes, see: Robbins RJ, Laman DM, Falvey DE. J Am Chem Soc. 1996;118:8127.Kung AC, Falvey DE. J Org Chem. 2005;70:3127. doi: 10.1021/jo0477749.Chiapperino D, Mcllroy S, Falvey DE. J Am Chem Soc. 2002;124:3567. doi: 10.1021/ja011049n.For recent studies on reversible reactions of electrophiles with olefins, see: Denmark SE, Collins WR, Cullen MD. J Am Chem Soc. 2009;131:3490. doi: 10.1021/ja900187y.Denmark SE, Burk MT, Hoover AJ. J Am Chem Soc. 2010;132:1232. doi: 10.1021/ja909965h.
- xliii.(a) Gassman PG, Campbell GA. J Am Chem Soc. 1971;93:2567. doi: 10.1021/ja00733a701. [DOI] [PubMed] [Google Scholar]; (b) Gassman PG, Campbell GA, Frederick RC. J Am Chem Soc. 1972;94:3884. [Google Scholar]; (c) Gassman PG, Campbell GA. J Am Chem Soc. 1972;94:3891. [Google Scholar]
- xliv.(a) Leyva E, Platz MS, Persy G, Wirz J. J Am Chem Soc. 1986;108:3783. doi: 10.1021/ja00284a049. [DOI] [PubMed] [Google Scholar]; (b) McClelland RA, Kahley MJ, Davidse PA, Hadzialic G. J Am Chem Soc. 1996;118:4794. [Google Scholar]
- xlv.Watt DS, Kawada K, Leyva E, Platz MS. Tetrahedron Lett. 1989;30:899. [Google Scholar]
- xlvi.(a) Ding Y, Padias AB, Hall HK., Jr J Polymer Sci: Part A: Polymer Chemistry. 1999;37:2569. [Google Scholar]; (b) Wojciechowski PM, Zierkiewicz W, Michalska D, Hobza P. J Chem Phys. 2003;118:10900. [Google Scholar]; (c) Kato K, Yamanouchi K. Chemical Physics Letters. 2004;397:237. [Google Scholar]; (d) Sapurina I, Stejskal J. Polym Int. 2008;57:1295. [Google Scholar]
- xlvii.Tanner DD. J Am Chem Soc. 1964;86:4674. [Google Scholar]
- xlviii.For N-C bond formation that follows second order kinetics (not necessarily SN2) between arenes and activated esters of N-phenylhydroxylamine, see: Shudo K, Ohta T, Okamoto T. J Am Chem Soc. 1981;103:645.Hamel P, Girard Y, Atkinson JG. J Chem Soc, Chem Commun. 1989:63.Novak M, Martin KA, Heinrich JL. J Org Chem. 1989;54:5430.Helmick JS, Martin KA, Heinrich JL, Novak M. J Am Chem Soc. 1991;113:3459.Novak M, Rangappa KS, Manitsas RK. J Org Chem. 1993;58:7813.
- xlix.While a reversible π-stacking interaction could be invoked, it is difficult to envision a stereochemical preference for the exo diastereomer in this scenario in light of the known thermodynamic bias for the endo diastereomer: Crich D, Banerjee A. Acc Chem Res. 2007;40:151. doi: 10.1021/ar050175j. Qualitatively, a solution of o-iodoaniline and 33 led to a yellow solution (both materials are initially white crystalline solids). Unfortunately, UV-vis and 1H or 13C-NMR experiments did not allow observation of changes indicative of π-stacking.
- l.(a) Mathew JS, Klussmann M, Iwamura H, Valera F, Futan A, Emanuelsson EAC, Blackmond DG. J Org Chem. 2006;71:4711. doi: 10.1021/jo052409i. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG. Angew Chem Int Ed. 2005;44:4302. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- li.Takeuchi H, Takano K. J Chem Soc, Chem Commun. 1983:447. [Google Scholar]
- lii.The increased yield for para-iodoaniline to afford 63 (27%) and 47% recovered 33 with NCS (unoptimized) suggests substrates can be optimized when standard conditions fail (Scheme 6).
- liii.(a) Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652. [Google Scholar]; (b) Ujjainwalla F, Warner D. Tetrahedron Lett. 1998;39:5355. [Google Scholar]; (c) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689. [Google Scholar]
- liv.(a) Wolfe JP, Tomori H, Sadighi JP, Yin J, Buchwald SL. J Org Chem. 2000;65:1158. doi: 10.1021/jo991699y. [DOI] [PubMed] [Google Scholar]; (b) Harris MC, Buchwald SL. J Org Chem. 2000;65:5327. doi: 10.1021/jo000674s. [DOI] [PubMed] [Google Scholar]; (c) Altman RA, Anderson KW, Buchwald SL. J Org Chem. 2008;73:5167. doi: 10.1021/jo8008676. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Coste A, Toumi M, Wright K, Razafimahaléo V, Couty F, Marrot J, Evano G. Org Lett. 2008;10:3841. doi: 10.1021/ol8015513. [DOI] [PubMed] [Google Scholar]
- lv.The methyl carbamate was available in quantitative yield from the commercial 7-bromotryptamine, whose preparation can be found in reference 13a. An alternative preparation may be found in the Supporting Information.
- lvi.Kapakahi is the Hawaiian word for “twisted.”
- lvii.Varying the substrate or oxidant led to unproductive oxidation, double addition or no reaction at all.
- lviii.Chaetominine isolation Jiao RH, Xu S, Liu JY, Ge HM, Ding H, Xu C, Zhu HL, Tan RX. Org Lett. 2006;8:5709. doi: 10.1021/ol062257t.Synthesis of chaetominine Snider BB, Wu X. Org Lett. 2007;9:4913. doi: 10.1021/ol7022483.Malgesini B, Forte B, Toumi M, Couty F, Marrot J, Evano G. Org Lett. 2008;10:5027. doi: 10.1021/ol802155n.Borghi D, Quartieri F, Gennari C, Papeo G. Chem Eur J. 2009;15:7922. doi: 10.1002/chem.200900793.Coste A, Karthikeyan G, Couty F, Evano G. Synthesis. 2009;17:2927. doi: 10.1002/anie.200901099.
- lix.Schöllkopf U, Groth U, Deng C. Angew Chem Int Ed Engl. 1981;20:798–799. [Google Scholar]
- lx.(a) Ma J, Yin W, Zhou H, Cook JM. Org Lett. 2007;9:3491. doi: 10.1021/ol071220l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou H, Liao X, Yin W, Ma J, Cook JM. J Org Chem. 2006;71:251. doi: 10.1021/jo052081t. [DOI] [PubMed] [Google Scholar]; (c) Yu J, Wearing XZ, Cook JM. J Org Chem. 2005;70:3963. doi: 10.1021/jo040282b. [DOI] [PubMed] [Google Scholar]; (d) Zhou H, Liao K, Cook JM. Org Lett. 2004;6:249. doi: 10.1021/ol0362212. [DOI] [PubMed] [Google Scholar]; (e) Liu X, Deschamp JR, Cook JR. Org Lett. 2002;4:3339. doi: 10.1021/ol020101x. [DOI] [PubMed] [Google Scholar]; (f) Ma C, Liu X, Li X, Flippen-Anderson J, Yu S, Cook JM. J Org Chem. 2001;55:4525. doi: 10.1021/jo001679s. [DOI] [PubMed] [Google Scholar]
- lxi.Castle SL, Srikanth GSC. Org Lett. 2003;5:3611. doi: 10.1021/ol035236x. [DOI] [PubMed] [Google Scholar]
- lxii.Trost BM, Rudd MT. Org Lett. 2003;5:4599. doi: 10.1021/ol035752n. [DOI] [PubMed] [Google Scholar]
- lxiii.Yeh MCP, Knochel P. Tetrahedron Lett. 1989;30:4799. [Google Scholar]
- lxiv.Nie X, Wang G. J Org Chem. 2006;71:4734. doi: 10.1021/jo052317t. [DOI] [PubMed] [Google Scholar]
- lxv.Jsselstijn MI, Kaiser J, van Delft FL, Schoemaker HE, Rutjes FPJT. Amino Acids. 2003;24:263. doi: 10.1007/s00726-002-0408-3. [DOI] [PubMed] [Google Scholar]
- lxvi.Boggs NT, Bruton HD, Craig DH, Helpern JA, Marsh HC, Pegram MD, Vandenbergh DJ, Koehler KA, Hiskey RG. J Org Chem. 1982;47:1812. [Google Scholar]
- lxvii.Garfunkle J, Kimball FS, Trzupek JD, Takizawa S, Shimamura H, Tomishima M, Boger DL. J Am Chem Soc. 2009;131:16036. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxviii.Although no base is added, either DMF or trace dimethylamine may act as a base in this peptide coupling reaction.
- lxix.Products 124 and 132had Rf’s of 0.58 and 0.43 respectively (4:1 EtOAc:hexanes).
- lxx.Bodanszky M, Bodanszky A. The Practice of Peptide Synthesis. Springer-Verlag; Berlin: 1993. [Google Scholar]
- lxxi.Carpino LA, El-Faham A. J Org Chem. 1994;59:695. [Google Scholar]
- lxxii.For a comparison of synthetic kapakahine F (2) 1H-NMR to the natural source, please see reference 13b. For HMQC, HMBC and ROESY NMR data for kapakahine F (2), please see the Supporting Information.
- lxxiii.Assays performed by Bristol-Myers Squibb.
- lxxiv.Assays performed by the NCI.
- lxxv.(a) Baran PS, Maimone TJ, Richter JM. Nature. 2007;446:404. doi: 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]; (b) Shenvi RA, O’Malley DP, Baran PS. Acc Chem Res. 2009;42:530. doi: 10.1021/ar800182r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hendrickson JB. J Am Chem Soc. 1975;97:5784. [Google Scholar]
- lxxvi.During the review of this manuscript, a synthesis of kapakahine F was reported that proceeded in 19 linear steps and 6.8% overall yield (23 mg produced) from tryptophan, see: Espejo VR, Rainier JD. Org Lett. 2010;12 doi: 10.1021/ol100672z.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.