

Abstract
The 14 kDa homodimeric N1L protein is a potent vaccinia and variola (smallpox) virulence factor. It is not essential for viral replication, but it causes a strong attenuation of viral production in culture when deleted. The N1L protein is predicted to contain the BH3-like binding domain characteristic of Bcl-2 family proteins, and it is able to bind the BH3 peptides. Its overexpression has been reported to prevent infected cells from committing apoptosis. Therefore, interfering with the N1L apoptotic blockade may be a legitimate therapeutic strategy affecting the viral growth. By using in silico ligand docking and an array of in vitro assays, we have identified sub-micromolar (600 nM) N1L antagonists, belonging to the family of polyphenols. Their affinity is comparable to that of the BH3 peptides (70 nM ÷ 1000 nM). We have also identified the natural polyphenol resveratrol as a moderate N1L inhibitor. Finally, we show that our ligands efficiently inhibit growth of vaccinia virus.
Introduction
The 14 kDa homodimeric N1L protein is a potent vaccinia and variola (smallpox) virulence factor 1–6 and possibly other DNA viruses 7–11. It is not essential for viral replication, however, when deleted, it causes the strongest attenuation of viral production in culture 2, 4. Its deletion also reduces mortality of intracranially infected mice by a factor of 10000 2. Recent determination of the N1L crystal structure showed its structural similarity to Bcl-2 antiapoptotic family members, however it does not share with them significant homology at the level of the amino acid sequence 5, 6, and small molecule inhibitors, antagonizing its activity, have not been identified previously.
Although the exact role of N1L in vaccinia virus life cycle is not fully understood, several recent reports suggest its involvement in modulating cellular apoptotic machinery6, 12. For instance, its overexpression interferes with induced apoptosis6. The strong viral growth attenuation of N1L knockouts suggests that the apoptotic blockade is a rather critical event in the virus life cycle. Therefore, the specific inhibition of the N1L apoptotic blockade should have debilitating effects on virus growth. In addition to poxviruses, several other DNA viruses encode Bcl-2 like anti-apoptotic proteins. For example, these proteins are encoded by gammaherpesviruse Epstein-Barr virus 7, Kaposi’s sarcoma-associated herpesvirus 8, murine gammaherpesvirus 68 11, alphaherpesvirus Marek’s disease virus 9, African swine fever virus10. Thus, N1L antagonists and their derivatives could have rather broad therapeutic application.
The N1L protein is predicted to contain the putative BH3-like binding domain characteristic of Bcl-2 family proteins. However, neither the exact mode of binding of BH3 peptides to N1L has been elucidated, nor have small molecule binders been identified previously. We have addressed the lack of structural information by merging in vitro and in silico methods in the same iterative protocol, in which each round of in silico modeling and ligand screening was followed by comprehensive in vitro testing employing independent biochemical and biophysical methods. In order to model possible conformational changes of N1L upon ligand binding, we have implicitly incorporated the protein flexibility by treating its structure as a collection of conformational subensembles. Although the idea of using multiple receptor conformations for in silico ligand docking is not new 13–19, we look at its application for a given problem from a different perspective. We assume the validity of the energy landscape theory of protein folding, which views a protein as an ensemble of energetically similar conformers on a rugged energy landscape biased toward the native structure 20–27. The protein conformers are in dynamic equilibrium with each other, which can be shifted by such factors as presence of ligands. This ligand-induced conformational equilibrium shift ultimately translates into observable biochemical effect, which helps to identify protein conformers specifically interacting with a ligand. By feeding back this information, we refine the structures of both protein conformers and inhibitor scaffolds.
Iterative application of in silico protein modeling, virtual ligand screening (VLS) and thorough biochemical evaluation of the hits have led to the discovery of submicromolar inhibitors of the N1L BH3-like domain (IC50 = 600 nM ÷ 900 nM) belonging to the family of polyphenols. The leading N1L antagonists are specific to N1L, and possess insignificant affinity toward its cellular homologues Bcl-2 and Bcl-XL. Several of the discovered N1L antagonists efficiently inhibited VacV-GFP virus growth (viral IC50 = 2 μM ÷ 17 μM), while exhibiting minimum non-specific cytotoxicity in control experiments at concentrations up to 150 μM. Additionally, we have identified resveratrol, a natural polyphenol, as a moderate N1L antagonist.
Results
Modeling of the Putative N1L-Bim peptide Complex and In Silico Docking Experiments
As of now, the crystal structure of the complex of N1L with BH3 peptide substrates is not available. Further, no significant amino acid sequence similarity can be observed with other known Bcl-2 family proteins (Supplementary Figure 1). Moreover, the visual appearance of the molecular surface of its putative BH3-binding site bears little resembles to that of Bcl-XL and Bcl-2, which is characterized by a well-defined groove, formed by several helices containing BH regions6 (Supplementary Figure 2B). The equivalent N1L helices are tightly packed together forming closed apo-N1L structure, where the binding groove is buried within (Supplementary Figure 2A). Therefore, in order for N1L to be able to bind BH3 peptides, it must undergo the conformational change. The relatively low affinity of BH3 peptides toward N1L (71 nM ÷ >1000 nM, compare with 2 nM ÷ 10 nM for Bcl-XL) 6 is in agreement with this observation.
The initial approximation of the open N1L conformation was obtained by docking Bim peptide from the human Bim protein into the putative BH3 binding site (see Methods). The peptide docking resulted in the opening of a large groove similar to the one observed in Bcl-XL (Supplementary Figure 2A). The groove is formed by helices α2, α3 and α5. The equivalent helices in Bcl-XL contain important BH regions (Supplementary Figure 2B). We have retained 125 low energy conformations of the holo-N1L structure generated during peptide docking experiments so that the conformational change can be approximately described as a conformational ensemble. All of these conformers were used for in silico ligand docking experiments.
The docking site was defined in the center of mostly positively charged cavity formed at the Bim peptide-binding site. This cavity is completely concealed within the closed N1L conformation (Supplementary Figure 2A). The molecular surface region with similar charge distribution can be observed in the Bcl-XL peptide-binding groove, and is formed by BH regions (Supplementary Figure 2B). During iterative rounds of ligand optimization, the chosen docking site was further refined to accommodate improved ligand structures.
The predicted binding mode of a select ligand is shown on Supplementary Figure 2A. In a given binding mode, the ligand is predicted to strongly interact with N1L helices α2 and α5, which are equivalent to Bcl-XL helices containing BH1 and BH3 regions. Note, that during optimization rounds hundreds of N1L conformers were accumulated. The conformers were organized into subensembles depending of the docking scores of validated ligands. Within a conformational subensemble, associated with a particular ligand, multiple predicted binding modes of a ligand could be observed.
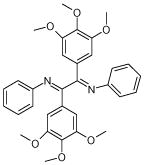
From the ligand structures presented in Table 1, it is easy to recognize that they belong to a family of polyphenols, an important class of biologically active compounds. The predicted multiple mode binding of the discovered ligands is consistent with their chemical nature and certain structural symmetry. Polyphenols are negatively charged under the physiological conditions and form strong electrostatic interactions with mostly positively charged binding site cavity.
Table 1.
Best N1L antagonists obtained at each scaffold optimization round.
| 1 (ID2/I) | 2 (ID9/I) | 3 (ID10/I) | 4 trans-Resveratrol* |
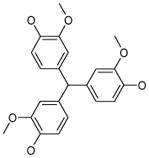 |
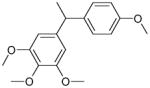 |
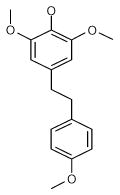 |
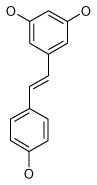 |
| IC50 = 8.8 μM | IC50 = 5.6 μM | IC50 = 5.0 μM | IC50 = 21.9 μM |
| XLogP = 4.1 | XLogP = 4.1 | XLogP = 3.7 | XLogP = 3.1 |
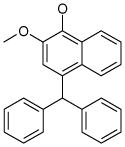
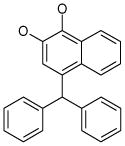
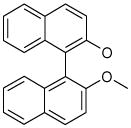
| 5 (ID13/II) | 6 (ID15/II) | 7 (ID23/III) | 8 (ID26/III) |
 |
 |
 |
 |
| IC50 = 2.4 μM | IC50 = 1.8 μM | IC50 = 0.6 μM | IC50 = 1.5 μM |
| XLogP = 6.7 | XLogP = 6.2 | XLogP = 5.8 | XLogP = 6.0 |
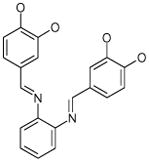
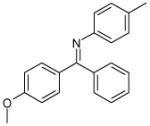
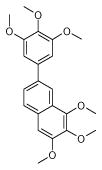
| 9 (ID29/III) | 10 (ID30/III) | 11 (ID31/III) | 12 (ID41/III) |
 |
 |
 |
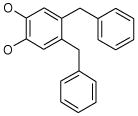 |
| IC50 = 1.2 μM | IC50 = 1.7 μM | IC50 = 0.9 μM | IC50 = 0.9 μM |
| XLogP = 2.6 | XLogP = 5.4 | XLogP = 4.6 | XLogP = 5.1 |
In parentheses, ligand IDs correspond to those in the Supplementary Table 1. Optimization round is indicated by a roman numeral next to ligands ID. Best IC50 values shown.
Resveratrol was added to the compound set because of its structural similarity to ligand 3. The octanol-water partitioning XLogP coefficients of the compounds were obtained from the PubChem database (http://pubchem.ncbi.nlm.nih.gov).
Recently, such polyphenol as gossypol and its derivatives have been reported to antagonize Bcl-2 family of proteins28, 29. Importantly, the ligand 8 (Table 1), discovered on the third round of the optimization protocol, is the superstructure of gossypol, and its close derivatives, apogossypol and apogossypolone28, 29.
Determination of IC50 Values for N1L Ligands
The top hits, predicted in in silico ligand docking experiments, were obtained from NCI and tested for interference with the BH3 peptide binding to N1L. The competitive IC50 values were determined by fluorescence polarization assay (FPA), as described in Methods (Table 1, Supplementary Table 1). Three rounds of ligand scaffolds optimization have lead to over 8 fold improvement of their affinity. The best obtained IC50 values are in submicromolar range (600 nM and 900 nM, Table 1), which is comparable with the reported affinity of BH3 peptides (71 nM ÷ over 1000 nM)6. For several ligands, IC50 values were also determined at multiple N1L concentrations to demonstrate IC50 dependence on protein concentration (Supplementary Table 1), and to obtain better IC50 values at low N1L concentration (IC50 asymptotically approaches true KD with decreasing receptor concentration). Unfortunately, we were not able to decrease N1L concentration below 500 nM due to the assay sensitivity limitations.
The obtained IC50 values were used to rationalize the hits selection for the subsequent round of in silico ligand scaffold optimization. Two main criteria were applied while selecting hits for optimization: 1) improved IC50; 2) insignificant cross reactivity with Bcl-XL and Bcl-2 proteins. The hits, selected at each round of optimization, are highlighted in the Supplementary Table 1.
In parallel to N1L, the ligands were although assayed against structurally related cellular antiapoptotic proteins Bcl-2 and Bcl-XL (Figure 1). These proteins were assayed together with N1L on the same plate and at the same time. Note, that incomplete titration curves are shown for Bcl-2 and Bcl-XL (ligand saturation was not achieved in an indicated concentration ranges) solely to demonstrate that given ligands poorly interact with these proteins. These experiments allowed addressing issues of ligand specificity. The ligands, determined to bind well to all three proteins, were discarded from further scaffold optimization rounds. Additionally, the current database of N1L conformers was updated by discarding those conformers where the cross-reacting ligands scored the best in in silico docking experiments.
Figure 1. Determination of IC50 values of select N1L antagonists by fluorescence polarization experiments.

A. Ligand 7; IC50(N1L) = 0.6 μM; IC50(F1L) = 1.4 μM. B. Ligand 11; IC50(N1L) = 0.9 μM; IC50(F1L) = 1.7 μM. C. Ligand 12; IC50(N1L) = 0.9 μM; IC50(F1L) = 0.6 μM; IC50(Bcl-2) = 4.7 μM; IC50(Bcl-XL) = 8.5 μM. FPA, normalized fluorescence polarization; Log([L], M), decimal logarithm of the ligand concentration in M. Data were fit to the variable slope dose-response equation. FPA assay was performed as described in Methods. Incomplete titration curves (ligand saturation was not achieved in a given concentration range) are shown for Bcl-2 and Bcl-XL in panels A and B solely to demonstrate poor ligand binding to these proteins.
Equally importantly, the cross-reactivity testing allowed the implicit identification of the non-specific promiscuous compounds. The FPA assay is sensitive to such experimental artifacts as a ligand fluorescence quenching, and the ligand-induced peptide or protein aggregation because of the limited compound solubility or its strong non-specific interactions with peptide and/or protein. Thus, a compound, inducing the decrease of the fluorescence polarization equally well in the experiments with all three proteins, is potentially a non-specific promiscuous ligand.
The vaccinia virus contains another Bcl-2-like antiapoptotic protein closely related to N1L, F1L12. At the last round of optimization, we have screened several of our ligands against recombinant F1L protein, and determined that our best N1L antagonists also efficiently bind to F1L (Figure 1).
Differential Scanning Calorimetry Experiments
In addition to FPA experiments, we have assessed the thermodynamics of ligand interaction with N1L by differential scanning calorimetry (DSC) (Figure 2) at each round of scaffold optimization. DSC measures the effects of ligand binding on protein melting temperature and enthalpy of unfolding. The melting temperature is the measure of quality of packing of protein tertiary structure, while the enthalpy of unfolding (area under DSC peak) is proportional to tertiary structure content of a protein.
Figure 2. Thermodynamics of ligands interaction with N1L as determined by differential scanning calorimetry.

A (−), N1L, 71μM, DMSO only, Tm=58.6 °C, ΔH=13.1 kcal/mol, ligand 3: 214 μM, Tm= 54.4 °C, ΔH=11.0 kcal/mol; ligand 6: 71 μM, Tm= 57.5 °C, ΔH=8.7 kcal/mol; resveratrol: 214 μM, Tm= 54.2 °C, ΔH=10.3 kcal/mol. B. (−), DMSO only, Tm=57.2 °C, ΔH=16.1 kcal/mol, ligand 7: 106 μM, Tm= 54.9 °C, ΔH=12.3 kcal/mol; ligand 9: 106 μM, Tm= 52.3 °C, ΔH=17.8 kcal/mol; ligand 11: 106 μM, Tm= 56.3 °C, ΔH=20.3 kcal/mol; ligand 12: 106 μM, Tm= 48.6 °C, ΔH=11.2 kcal/mol. C. N1L, 71 μM protein, DMSO, Tm=58.6 °C, ΔH=13.1 kcal/mol; N1L-Resv, 71 μM protein, 214 μM resveratrol, Tm=54.2 °C, ΔH=10.3 kcal/mol; N1L I6K, 71 μM protein, DMSO, Tm=61.5 °C, ΔH=15.8 kcal/mol; N1L I6K – Resv, 71 μM protein, 214 μM resveratrol, Tm=61.1 °C, ΔH=15.9 kcal/mol.
All of the tested ligands induced decrease of the N1L melting temperature, suggesting that N1L assumes more flexible and open conformational state upon ligand binding, which also might influence its homodimeric state. The broadening of the DSC peaks also indicated presence of multiple protein species in the system. Interestingly, in spite of decrease of melting temperature, the binding of the majority of ligands had little or no effect of enthalpy of unfolding (except for ligand 12). In the case of ligand 11, the increase of enthalpy of unfolding and slight decrease of melting temperature were observed (Figure 2B). Collectively, the DSC data are consistent with an assumption that the binding of ligands must induce strong conformational changes of N1L.
The same experiments were conducted with stable monomeric I6K N1L mutant (Figure 2C). As expected, the monomeric N1L mutant is at least as stable as wild type dimer. The presence of select ligands also did not have any dramatic effects on its unfolding, indicating that these ligands do not significantly interact with stable monomeric form of N1L.
Analytical Ultracentrifugation Studies
Modeling, and DSC studies suggested that the binding of a small molecule to N1L is possible only after a significant rearrangement of its tertiary structure. This rearrangement might alter the subunit interface and affect the stability of a dimer. To determine if the ligand binding has any effect on the homodimeric state of N1L, the analytical ultracentrifugation (AUC) experiments were conducted for select ligands (Supplementary Figure 4). In the absence of ligands, the equilibrium constant of N1L dimerization (KD) was 6 nM. The ligands 7 (100 μM) and 11 (100 μM) clearly destabilized the N1L dimer by increasing KD to 3.3 μM and 8 μM, respectively.
Antiviral Properties of N1L Antagonists
The antiviral properties of N1L antagonists were studied in CV1 and HT1080 cells using a Green Fluorescent Protein (GFP)-expressing recombinant vaccinia virus (Figure 3, Supplementary Figure 5). The Supplementary Figure 5 presents raw assay data collected for certain ligands. Additionally, it provides examples of both toxic and non-toxic ligands.
Figure 3. Inhibition of vaccinia virus growth by N1L antagonists.

A. VacV-GFP growth inhibition EC50 values obtained in CV1 and HT1080 cell lines. B. Ligand cytotoxicity in the absence of VacV-GFP. The indicated ligands were added to the cells at 150 μM. The ligand numbering corresponds to that of the Table 1.
The viral growth inhibition potency of certain ligands achieved 2 ÷ 18 μM (cellular EC50), while no significant non-specific cytotoxicity was observed at compounds concentrations above 100 μM (ligands 2, 9 and 11). Thus, even our worst ligand (9, EC50 = 18 μM) has a maximum working concentration to EC50 ratio of 5.6 (best being 50 for ligand 2), which is sufficient for complete and specific inhibition of viral growth.
Purification and Characterization of N1L Monomeric Mutants
The following mutations were chosen based on the calculated decrease of the free binding energy (see Methods) of the dimer subunits relative to the wild type N1L: I6K, I6H, E92R and E92C. The mutations are located on the well-defined hydrophobic interface of the N1L dimer, opposite of the putative BH3-like domain. The mutants were purified and their oligomeric state was assessed by both FPLC and AUC. From the above mutants, the I6K mutant did not exhibit detectable oligomerization as determined by both methods, and was chosen for further experiments.
Discussion and Conclusions
The eukaryotic viruses usually require several days to achieve maximum production of progeny viral particles within the infected cells. The massive virus multiplication allows the virus to spread to other cells. Higher eukaryotic cells have evolved mechanisms aimed at slowing or stopping the virus replication cycle. The most important of these mechanisms is the induction of self-destruction in the process of apoptosis. The triggering of apoptosis results in destruction of mitochondrial function by recruiting Bak and Bax proteins. The Blc-2 proteins act as signal transduction checkpoint regulators of mitochondrial destruction by modulating the activity of Bak and Bax proteins through heterodimerization.
During their evolution viruses have developed mechanisms to block apoptosis, permitting completion of their replication cycle, by expressing proteins mimicking Bcl-2 function. The presence of Bcl-2 like proteins in the genome of several DNA viruses, including vaccinia and smallpox, suggests an importance of the apoptotic blockade for their life cycle. Therefore, targeting viral proteins, which manipulate host cell apoptotic signaling pathways, could be a promising point of the therapeutic intervention.
Designing small molecule inhibitors, interfering with protein-protein interactions, has long been recognized to be the most challenging problem of the modern rational drug discovery. The strong binding of a small molecule to a protein-protein interface must be accompanied by a free energy gain comparable to that of the binding of a significantly larger molecule (binding of a peptide or a protein takes place across much larger interface). Thus, in order for a small molecule to be able to compete with a protein or peptide, its binding must induce such a receptor conformational change that would significantly contribute to the decrease of overall free energy of a system. The bottleneck of the problem is an inability of available in silico methods to reliably model the ligand-induced protein conformational changes.
Previously, it has been demonstrated that an implicit treatment of an “induced-fit” of a receptor by using multiple representative conformers is sufficiently accurate for purposes of drug discovery30–32. In this work, we although treat a receptor as an ensemble of multiple representative conformers. However, we look at the application of this method from a different perspective.
Specifically, we assume the validity of the energy landscape theory of protein folding, which describes a folded protein as a dynamic ensemble of energetically close conformers biased toward the native structure20, 25, 33–40. We use the framework of this theory to formulate the logistics of hit discovery and optimization, and to test it in a real life application.
As was pointed out by Friere 36 and Ma et el. 37, the ligand binding could be viewed as a “lock-and-key” binding to an “induced-fit-like” states already present within a conformational ensemble representing the folded state of a protein. If a ligand binds to one of the “induced-fit-like” states with sufficient free energy, then this particular state becomes the most populated, and the overall protein folded state is changed. The change of the receptor folded state ultimately translates into an observable biological effect.
There could be many “induced-fit-like” states, and potentially there are many ligands that could bind and stabilize these states. The binding of a non-specific ligand can be pictured as multiple modes binding to an array of receptor conformers. Such treatment of a ligand binding simplifies the task of finding the initial hits: a diverse set of receptor conformers, roughly approximating the putative induced-fit-like state, should lead to a discovery of weak, non-specific binders. This conformer set was constructed from subensembles generated during the peptide docking experiments. The application of VLS (see Supplemental Materials and Methods) using this conformational set resulted in the identification of several low affinity binders (Table 1, Supplementary Table 1, Supplementary Figure 3, Round 1). The affinity of these binders was significantly improved after two iterations of optimization protocol.
It is easy to see, that the majority of the discovered ligands belongs to the family of polyphenols, an important class of biologically active compounds. Polyphenols, in spite of high aromatic content, are rather soluble in aqueous solutions due to the labile protons of hydroxyl groups attached to the phenyl rings. The predicted octanol-water partitioning coefficients (XLogP41, http://pubchem.ncbi.nlm.nih.gov) of approximately half of the compounds are well within the range, acceptable for drug-like small molecules (< 5.0, although tamoxifene XLogP = 8.3). Moreover, the ligand 8 is a superstructure of gossypol29, apogossypol28, apogossypolone42 and their numerous derivatives, which are well-characterized Bcl-XL/Bcl-2 ligands. It is not surprising that the ligand 8 is a poor antiviral - due to its cross-reactivity with Bcl-XL and Bcl-2, resulting in cytotoxicity in uninfected cells (Figure 3B). Interestingly, we have discovered that natural polyphenol trans-resveratrol is a moderate N1L inhibitor and a weak Bcl-XL inhibitor (IC50 = 190 μM). These findings are consistent with several reports of its antiviral activity43–45.
The inspection of the Supplementary Table 1 reveals that the discovered ligands lack consistency in both structure and potency. The severity of structural inconsistency actually prevents the R-group analysis or any other attempt to correlate ligand structures with their affinities. However, the overall improvement of affinity and specificity is observed from one round of the optimization to another. The ligand structure and potency inconsistencies are the side effects of the receptor-centered ligand scaffold optimization strategy employed in this work. The lock-and-key paradigm, which follows from the energy landscape theory of protein folding, implies that multiple ligand scaffolds target multiple receptor conformers with possibly different affinities. To structurally diversify the ligand set in the course of in silico hits derivatization, we deliberately used chemical fingerprints as a chemical similarity measure (see Supplementary Materials and Methods) to maximize the scaffold hopping. This strategy has lead to the broadening of the ligand optimization “tree” (Supplementary Figure 3B) and increasing of the chances to discover cell-permeable, non-toxic and specific ligands.
As expected, the binding of ligands to N1L induces major protein conformational changes, manifested in decrease of N1L melting temperature (Figure 2). The decrease of melting temperature upon ligand binding is often considered to be an alarming sign of non-specific nature of its interactions with a protein. However, in many cases the decrease of melting temperature indicates that upon ligand binding protein adapts more open/flexible conformation. For instance, cofilin binding to F-actin decreased F-actin melting temperature by ~10 degrees46, while F-actin binding to plectin ABD decreased plectin ABD melting temperature by ~5 degrees47. In the case of N1L, the decrease of melting temperature is mostly due to its monomerization, as was confirmed by AUC experiments. The monomerization of native homodimeric form of N1L (KD = 6 nM vs. KD = 8 μM in the presence of ligand 11) is thermodynamically possible only if individual subunits are driven by a ligand binding into an energetically similar to, or more favorable than corresponding conformational state within a homodimer. The increase of enthalpy of unfolding of N1L and insignificant decrease of its melting temperature in the presence of the ligand 11 indicates this transition (Figure 2B).
The effects of ligand binding onto oligomeric state of N1L indicated possible biological importance of the homodimeric state of N1L. To further investigate this phenomenon and to validate that the observed monomerization of wild type N1L caused by specific interactions with ligands, we have designed, expressed and purified stable monomeric N1L mutant I6K (N1L I6K). The DSC experiments performed with select ligands and I6K, suggested that these ligands do not significantly interact with N1L I6K (Figure 2C). Importantly, the mutation has caused an increase of the enthalpy of unfolding and a slight increase of the melting temperature indicating thermodynamic stabilization of the individual subunits. These results are consistent with the binding of the ligand 11 to the wild type N1L (Figure 2B), which manifested in enthalpy increase and insignificant decrease of melting temperature.
Finally, we have investigated the antiviral properties of the most potent and specific N1L antagonists obtained at different rounds of ligand optimization (Figure 3, Supplementary Figure 5). Several ligands exhibited potent antiviral properties by efficiently inhibiting production of VacV-GFP virus, while showing little non-specific cytotoxicity in control experiments. The key to success of these studies was that the ligand specificity issue was addressed by testing the N1L ligands for cross-reactivity against other proteins from Bcl-2 family such as Bcl-XL and Bcl-2. This testing was routinely performed at each round of optimization. The non-specific ligands, interacting with all three proteins, were discarded from following rounds of scaffold optimization together with their associated receptor conformer subensembles (see Supplementary Materials and Methods). This step eliminated the majority of non-specific ligand scaffolds, which bind to induced-fit-like states energetically favorable for all three structurally related proteins, and, are cytotoxic to cells because of their suppression of cellular Bcl-2 family proteins.
Interestingly, vaccinia virus contains another Bcl-2-like antiapoptotic protein, F1L12. We have screened several of our ligands against recombinant F1L protein, and determined that their affinity to F1L is comparable to that of N1L (Figure 1). This unplanned off-target activity of the N1L ligands might strengthen their antiviral potency.
In summary, we have discovered several potent and specific N1L antagonists, which possess strong vaccinia virus growth attenuation properties. The discovered ligands belong to a family of polyphenols, a class of compounds attracting significant attention as drug candidates28, 42. We have although demonstrated that, in those cases, where inhibition of close receptor homologues is undesirable, the ligand specificity can be achieved by targeting receptor “induced-fit-like” conformations that are energetically less favorable for its homologues.
Experimental Section
Protein Expression and Purification
Recombinant N1L protein and its monomeric mutants were overexpressed and purified as previously described 6. Recombinant human Bcl-XL and Bcl-2 proteins were prepared as previously described 48. Recombinant F1L protein, lacking both N-terminal and C-terminal transmembrane regions was purified as follows. The F1L DNA sequence coding for residues 35–190 was amplified from vaccinia Western Reserve (WR) by PCR and subcloned into pGEX plasmid (Novagen) as an N-terminal GST-fusion. The GST-tagged F1L was expressed in Escherichia coli (E. coli) BL21(DE3) cells. The protein expression was induced with 500 μM IPTG at 20 °C for 18 hrs. The cell pellets were homogenized in PBS. The cell lysate containing GST-F1L was loaded to a glutathione Sepharose 4B column (GE Healthcare) and washed with 5 column volumes of PBS. The GST tag was removed by incubating beads with bound protein with thrombin (0.5 μg/ml) at room temperature overnight. Then F1L was eluted with PBS. The protein was further purified with a Superdex 200 column in 20 mM Tris pH 8.0, 100 mM NaCl, 5 mM β-mercaptoethanol.
Fluorescence Polarization Assay
Binding of N1L, F1L, Bcl-XL and Bcl-2 to the BH3 domains of several Bcl-2 family proteins was quantified using fluorescence polarization anisotropy-based peptide binding assays 48. Fluorescein isothiocyanate (FITC)-conjugated synthetic peptides comprising the BH3 domains of pro-apoptotic Bcl-2 proteins (BH3-Bim, FITC-aminohexanoyl (Ahx)-DMRPEIWIAQELRRIGDEFNAYYAR; BH3-Bak, FITC-Ahx-PSSTMGQVGRQLAIIGDDINRRYDS) were prepared at the Burnham Institute’s medicinal chemistry core facility. Varying concentrations of proteins were incubated with 10 nM of the corresponding peptides: N1L, Bcl-XL were incubated with BH3-Bak, while F1L, Bcl-2 with BH3-Bim. The resulting change of fluorescence polarization was measured on Analyst TM AD assay system (LJL Biosystems) and used to calculate IC50 values.
Differential Scanning Calorimetry
DSC experiments were performed at a scanning rate of 1 K/min under 3.0 atm of pressure using N-DSC II differential scanning calorimeter (Calorimetry Sciences Corp, Provo, UT). DSC samples contained PBS buffer, 1.0 mg/ml N1L, 2 or 5% dmso and 107 or 214 μM compounds.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were performed in ProteomeLab XL-I (Beckman Coulter) analytical ultracentrifuge. N1L samples in PBS at concentrations 0.45, 0.15 and 0.05 mg/ml were loaded in 6-channel equilibrium cells and span in An-50 Ti 8-place rotor at 20,000 rpm, 20 °C for 24 hours. AUC samples also contained 2% dmso and between 50μM and 100μM compounds. Data were analyzed using Hetero Analysis software (by J.L. Cole and J.W. Lary, University of Connecticut). The best fit of the sedimentation equilibrium data for N1L alone and in the presence of ligand 12 was achieved using monomer-dimer-tetramer equilibrium model, and in the presence of ligands 7 and 11 – using monomer – dimer equilibrium model.
Modeling of Bim Peptide Binding and Construction of Subensemble Collection
The complex of N1L with Bim peptide was modeled by in silico docking of peptide at the putative BH3-like domain. The starting complex conformation was built by superimposition of N1L crystal structure (2I39, 6) with the crystal structure of Bcl-XL in complex with Bim peptide (1PQ1, 49). The conformational change of single N1L monomer upon ligand binding was modeled by globally optimizing the initial model of the complex with biased probability Monte Carlo (BPMC) 50. The BPMC was performed in internal coordinates space as implemented in the ICM program 51. The docking simulation by BPMC was set up as follows. The flexible region of the N1L monomer was defined as an amino acid stretch encompassing residues from L29 to I66. This sequence stretch is composed from three clearly defined loops (L29 - D35, L50 - G53, Q61 - I66) and two α-helices (D36 - T49, P54 - N60). For the residues comprising the loops, both the side chain χ and backbone Phi, Psi angles were unfixed; while for the residues comprising α-helices only side chain χ angles were unfixed. In addition to these angles, the side chain χ angles of amino acid residues within 10 A of the Bim peptide were unfixed. The rest of the N1L monomer structure was held constant. The Bim peptide was fixed at its initial position by weighed harmonic restraints and its side chain χ angles were unfixed. During the minimization run, the weight of harmonic restraints was gradually diminished, allowing the peptide to move. At the end of the minimization, top 25 low energy complex conformations were retained. Then, the peptide was removed from the ICM complex object. For each of the N1L conformations, the side chain χ of amino acid residues within 10 ⇒ of the Bim peptide were unfixed, the conformations were “relaxed” by BPMC, and top 4 conformations together with the initial “bound” conformation were retained. The outlined procedure resulted in the construction of conformational stack containing 25 subensembles each composed of 5 conformers (125 conformers total).
Virus inhibition and cytotoxicity assays
CV-1, HeLa, and HT1080 cells (ATCC # CCL-70, CCL-2, and CRL-12011 respectively) were grown in the Dulbecco’s Modified Eagle Media (DMEM) (Mediatech) containing 10% fetal calf serum (FCS) (HiClone), 4.5 g/L glucose, L-glutamate, pyruvate, and 1000 U/ml Pen-Strep (Omega) in a 37C incubator with 5% CO2. The vaccinia virus strain WR (VacV-WR) (ATCC # VR-119) and the recombinant virus VacV-NP-GFP expressing green fluorescent protein (GFP) fused to the influenza virus nucleoprotein 52 (provided by Shane Crotty, La Jolla Institute for Allergy & Immunology) were grown in the media containing 2.5% FCS. The confluent monolayers of cells in 96-well plates (seeded at 1-2*10E4 cells/well 12 h before use) were infected with VacV-NP-GFP at multiplicity of infection (MOI) of 0.05–0.1 for 1 hour before addition of the compounds. All experiments were conducted in duplicates. Controls included rifampicin or no compound. Compounds were added in three fold dilutions. After 2.5 day incubation the media was replaced with phosphate-buffered saline (PBS), and the GFP fluorescence (Ex/Em=485nm/535nm) of VacV-NP-GFP-infected cells was measured using PHERAstar (BMG Labtech). The cell viability was measured in uninfected cells under the same conditions using the ATPLite assay following the manufacturer protocol (PerkinElmer). The inhibitory concentrations (IC50) and toxicity concentrations (TC50) were calculated using Prism-5 (GraphPad). TC50s of fairly nontoxic compounds were estimated using their max concentration (150 uM). The inhibition of VacV-WR was validated by incubation 130 uM of compounds 16 and 50 (or 100 ug/ml of rifampicin as a control). The virus titer was measured by the plaque assay after a 1.7-day incubation of a compound with CV-1 cells infected at MOI=0.05 and 1.0.
Ligand Source and Compound Databases
All ligands used in this work and corresponding databases in SDF format were obtained from “The NCI/DTP Open Chemical Repository” at http://dtp.nci.nih.gov. The compounds were all greater than 95% pure as certified by the supplier (NCI DTP Discovery Services). The database for discovery of initial low affinity hits was constructed by further clustering by chemical fingerprint similarity the NCI drug-like database. The ligand NCI/DTP accession numbers (NSC) are provided in the Supplementary Table 1. The ligands were dissolved in 100% DMSO and stored at −20 °C until further use.
Design of Monomeric N1L Mutants
The necessary mutations were identified as follows. The set of potential mutation sites was compiled upon visual inspection of the N1L dimer interface. The amino acid mutations were introduced into the N1L structure using simple charge/property inversion logic. The mutations were introduced into the N1L amino acid sequence, the side chains of the introduced amino acids were locally minimized, and the resulting structures were submitted to DCOMPLEX server53 at http://sparks.informatics.iupui.edu/czhang/complex.html for the binding energy evaluation. Four highest energy mutations were chosen, based on the results of the predictions: E92R, E92C, I6K and I6H.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants AI-078048 and CA-113318 to RJC, GM071872 and GM074832 to AR, 5 P01 AI055789-05 to LRC.
Abbreviations
- FPA
fluorescence polarization assay
- DSC
differential scanning calorimetry
- AUC
analytical ultracentrifugation
- GFP
green fluorescent protein
- VLS
virtual ligand screening
Footnotes
Supporting Information Available: Additional computational and biological data, and in silico modeling protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kotwal GJ, Moss B. Vaccinia virus encodes a secretory polypeptide structurally related to complement control proteins. Nature. 1988;335:176–178. doi: 10.1038/335176a0. [DOI] [PubMed] [Google Scholar]
- 2.Kotwal GJ, Hugin AW, Moss B. Mapping and insertional mutagenesis of a vaccinia virus gene encoding a 13,800-Da secreted protein. Virology. 1989;171:579–587. doi: 10.1016/0042-6822(89)90627-2. [DOI] [PubMed] [Google Scholar]
- 3.Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244:365–396. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett N, Symons JA, Tscharke DC, Smith GL. The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence. J Gen Virol. 2002;83:1965–1976. doi: 10.1099/0022-1317-83-8-1965. [DOI] [PubMed] [Google Scholar]
- 5.Cooray S, Bahar MW, Abrescia NG, McVey CE, Bartlett NW, Chen RA, Stuart DI, Grimes JM, Smith GL. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gen Virol. 2007;88:1656–1666. doi: 10.1099/vir.0.82772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoyagi M, Zhai D, Jin C, Aleshin AE, Stec B, Reed JC, Liddington RC. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 2007;16:118–124. doi: 10.1110/ps.062454707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hickish T, Robertson D, Clarke P, Hill M, di Stefano F, Clarke C, Cunningham D. Ultrastructural localization of BHRF1: an Epstein-Barr virus gene product which has homology with bcl-2. Cancer Res. 1994;54:2808–2811. [PubMed] [Google Scholar]
- 8.Sarid R, Sato T, Bohenzky RA, Russo JJ, Chang Y. Kaposi’s sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat Med. 1997;3:293–298. doi: 10.1038/nm0397-293. [DOI] [PubMed] [Google Scholar]
- 9.Afonso CL, Tulman ER, Lu Z, Zsak L, Rock DL, Kutish GF. The genome of turkey herpesvirus. J Virol. 2001;75:971–978. doi: 10.1128/JVI.75.2.971-978.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neilan JG, Lu Z, Afonso CL, Kutish GF, Sussman MD, Rock DL. An African swine fever virus gene with similarity to the proto-oncogene bcl-2 and the Epstein-Barr virus gene BHRF1. J Virol. 1993;67:4391–4394. doi: 10.1128/jvi.67.7.4391-4394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Virgin HWt, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA, Huang DC, Colman PM. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 2008;15:1564–1571. doi: 10.1038/cdd.2008.83. [DOI] [PubMed] [Google Scholar]
- 13.Shoichet BK, Kuntz ID. Matching chemistry and shape in molecular docking. Protein Eng. 1993;6:723–732. doi: 10.1093/protein/6.7.723. [DOI] [PubMed] [Google Scholar]
- 14.Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 15.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 16.Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins. 1997;(Suppl 1):215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 17.Knegtel RM, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. J Mol Biol. 1997;266:424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 18.Bouzida D, Rejto PA, Arthurs S, Colson AB, Freer ST, Gehlhaar DK, Larson V, Luty BA, Rose PW, Verkhivker GM. Computer simulations of ligand-protein binding with ensembles of protein conformations: a monte carlo study of HIV-1 protease binding energy landscapes. Int J Quant Chem. 1999;72:73–84. [Google Scholar]
- 19.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 21.Weinkam P, Zong C, Wolynes PG. A funneled energy landscape for cytochrome c directly predicts the sequential folding route inferred from hydrogen exchange experiments. Proc Natl Acad Sci U S A. 2005;102:12401–12406. doi: 10.1073/pnas.0505274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavez LL, Gosavi S, Jennings PA, Onuchic JN. Multiple routes lead to the native state in the energy landscape of the beta-trefoil family. Proc Natl Acad Sci. 2006;103:10254–10258. doi: 10.1073/pnas.0510110103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finke JM, Onuchic JN. Equilibrium and kinetic folding pathways of a TIM barrel with a funneled energy landscape. Biophysical Journal. 2005;89:488–505. doi: 10.1529/biophysj.105.059147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy M, Chavez LL, Finke JM, Heidary DK, Onuchic JN, Jennings PA. The native energy landscape for interleukin-1beta. Modulation of the population ensemble through native-state topology. J Mol Biol. 2005;348:335–347. doi: 10.1016/j.jmb.2005.02.059. [DOI] [PubMed] [Google Scholar]
- 25.Onuchic JNaW, PG Theory of protein folding. Curr Opin Struct Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Freer ST, Rose PW. Complexity and simplicity of ligand-macromolecule interactions: the energy landscape perspective. Curr Opin Struct Biol. 2002;12:197–203. doi: 10.1016/s0959-440x(02)00310-x. [DOI] [PubMed] [Google Scholar]
- 27.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Freer ST, Rose PW. Simulating disorder-order transitions in molecular recognition of unstructured proteins: where folding meets binding. Proc Natl Acad Sci U S A. 2003;100:5148–5153. doi: 10.1073/pnas.0531373100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei J, Kitada S, Rega MF, Emdadi A, Yuan H, Cellitti J, Stebbins JL, Zhai D, Sun J, Yang L, Dahl R, Zhang Z, Wu B, Wang S, Reed TA, Lawrence N, Sebti S, Reed JC, Pellecchia M. Apogossypol derivatives as antagonists of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2009;8:904–913. doi: 10.1158/1535-7163.MCT-08-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azmi AS, Mohammad RM. Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol. 2009;218:13–21. doi: 10.1002/jcp.21567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 32.Cheltsov AV, Bisson WH, Bruey-Sedano N, Lin B, Chen J, Goldberger N, May LT, Christopoulos A, Dalton JT, Sexton PM, Zhang XK, Abagyan R. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc Natl Acad Sci USA. 2007;104:11927–11932. doi: 10.1073/pnas.0609752104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rejto PA, Freer ST. Protein conformational substates from X-ray crystallography. Progress in Biophysics & Molecular Biology. 1996;66:167–196. doi: 10.1016/s0079-6107(97)85629-5. [DOI] [PubMed] [Google Scholar]
- 34.Onuchic JN. Contacting the protein folding funnel with NMR. Proc Natl Acad Sci USA. 1997;94:7129–7131. doi: 10.1073/pnas.94.14.7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoemaker BA, Wang J, Wolynes PG. Structural correlations in protein folding funnels. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freire E. Statistical thermodynamic linkage between conformational and binding equilibria. Adv Prot Chem. 1998;51:255–279. doi: 10.1016/s0065-3233(08)60654-3. [DOI] [PubMed] [Google Scholar]
- 37.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Engineering. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 38.Kristjansdottir S, Lindorff-Larsen K, Fieber W, Dobson CM, Vendruscolo M, Poulsen FM. Formation of native and non-native interactions in ensembles of denatured ACBP molecules from paramagnetic relaxation enhancement studies. J Mol Biol. 2005;347:1053–1062. doi: 10.1016/j.jmb.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Lindorff-Larsen K, Rogen P, Paci E, Vendruscolo M, Dobson CM. Protein folding and the organization of the protein topology universe. Trends Biochem Sci. 2005;30:13–19. doi: 10.1016/j.tibs.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433:128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 41.Wang R, Fu Y, Lai L. A new atom-additive method for calculating partition coefficients. J Chem Inf Comput Sci. 1997;37:615–621. [Google Scholar]
- 42.Arnold AA, Aboukameel A, Chen J, Yang D, Wang S, Al-Katib A, Mohammad RM. Preclinical studies of Apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in Follicular Small Cleaved Cell Lymphoma model. Mol Cancer. 2008;7:20. doi: 10.1186/1476-4598-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Docherty JJ, Fu MM, Stiffler BS, Limperos RJ, Pokabla CM, DeLucia AL. Resveratrol inhibition of herpes simplex virus replication. Antiviral Res. 1999;43:145–155. doi: 10.1016/s0166-3542(99)00042-x. [DOI] [PubMed] [Google Scholar]
- 44.Docherty JJ, Fu MM, Hah JM, Sweet TJ, Faith SA, Booth T. Effect of resveratrol on herpes simplex virus vaginal infection in the mouse. Antiviral Res. 2005;67:155–162. doi: 10.1016/j.antiviral.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 45.Docherty JJ, Smith JS, Fu MM, Stoner T, Booth T. Effect of topically applied resveratrol on cutaneous herpes simplex virus infections in hairless mice. Antiviral Res. 2004;61:19–26. doi: 10.1016/j.antiviral.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Bobkov AA, Muhlrad A, Pavlov DA, Kokabi K, Yilmaz A, Reisler E. Cooperative effects of cofilin (ADF) on actin structure suggest allosteric mechanism of cofilin function. J Mol Biol. 2006;356:325–334. doi: 10.1016/j.jmb.2005.11.072. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Alvarez B, Bobkov A, Sonnenberg A, de Pereda JM. Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin beta 4. Structure. 2003;11:615–625. doi: 10.1016/s0969-2126(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 48.Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity. 2003;19:341–352. doi: 10.1016/s1074-7613(03)00234-6. [DOI] [PubMed] [Google Scholar]
- 50.Abagyan R, Totrov M. Biased Probability Monte-Carlo Conformational Searches and Electrostatic Calculations For Peptides and Proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 51.Abagyan R, Totrov M, Kuznetsov D. ICM: a new method for structure modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]
- 52.Anton LC, Schubert U, Bacik I, Princiotta MF, Wearsch PA, Gibbs J, Day PM, Realini C, Rechsteiner MC, Bennink JR, Yewdell JW. Intracellular localization of proteasomal degradation of a viral antigen. J Cell Biol. 1999;146:113–124. doi: 10.1083/jcb.146.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H, Zhang C, Liu S, Zhou Y. Web-based toolkits for topology prediction of transmembrane helical proteins, fold recognition, structure and binding scoring, folding-kinetics analysis and comparative analysis of domain combinations. Nucleic Acids Res. 2005;33:193–197. doi: 10.1093/nar/gki360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.