


Abstract
A real-time assay for CpG-specific cytosine-C5 methyltransferase activity has been developed. The assay applies a break light oligonucleotide in which the methylation of an unmethylated 5′-CG-3′ site is enzymatically coupled to the development of a fluorescent signal. This sensitive assay can measure rates of DNA methylation down to 0.34 ± 0.06 fmol/s. The assay is reproducible, with a coefficient of variation over six independent measurements of 4.5%. Product concentration was accurately measured from fluorescence signals using a linear calibration curve, which achieved a goodness of fit (R2) above 0.98. The oligonucleotide substrate contains three C5-methylated cytosine residues and one unmethylated 5′-CG-3′ site. Methylation yields an oligonucleotide containing the optimal substrate for the restriction enzyme GlaI. Cleavage of the fully methylated oligonucleotide leads to separation of fluorophore from quencher, giving a proportional increase in fluorescence. This method has been used to assay activity of DNMT1, the principle maintenance methyltransferase in human cells, and for the kinetic characterization of the bacterial cytosine-C5 methyltransferase M.SssI. The assay has been shown to be suitable for the real-time monitoring of DNMT1 activity in a high-throughput format, with low background signal and the ability to obtain linear rates of methylation over long periods, making this a promising method of high-throughput screening for inhibitors.
INTRODUCTION
The methylation of bases in the DNA of both prokaryotic and eukaryotic genomes is the most common of enzymatic base modifications (1). Three methylated bases are known to occur in nature: N6-methyladenine (2), which is found in bacteria and eukaryotes, N4-methylcytosine (3), a minor component of bacterial DNA and C5-methylcytosine (4), which is the most prevalent of the known DNA modifications (5). The methyl group is transferred enzymatically by DNA methyltransferases, which use S-adenosylmethionine (AdoMet) as the methyl donor. In bacteria, adenine-N6 methylation plays a key role in mismatch repair (6), the control of gene expression (7) and regulation of the cell cycle (8). In metazoa, cytosine-C5 methylation is the only DNA methylation which has been identified (9). Cytosine methylation in higher eukaryotes is critical for the control of cellular differentiation and development (10), gene expression, where promoter methylation leads to gene repression (11) and has also been implicated in X-chromosome silencing (12). Cytosine-C5 methylation mainly occurs within the sequence 5′-CG-3′ and within a higher eukaryotic genome, typically 60–90% of cytosine residues within this sequence are methylated (13). The process of cytosine-C5 methylation is of medical interest as variation from normal methylation patterns and the corresponding alteration in gene expression has been shown to be involved in the development of many conditions, such as fragile X chromosome (14) and cancer (15). Cancer development has been linked to hypermethylation of CG-rich sequences (CpG islands), which occur within the promoter regions of tumour suppressor genes, which, in turn, causes gene repression (16). In mammals, three higher-activity cytosine-C5 methyltransferases have been identified: DNA methyltransferase 1 (DNMT1) (17), DNMT3a and DNMT3b (18). A fourth enzyme, DNMT2, has been observed to have a very low methyltransferase activity in vitro (19).
DNMT1 is the most abundant methyltransferase in mammalian cells. It is responsible for the maintenance of methylation patterns in DNA (20) and as such shows a preference for hemimethylated over unmethylated substrates (21,22). DNMT1 has been shown to interact with a wide range of proteins (23). Examples include proliferating cell nuclear antigen (PCNA), which directs DNMT1 to replication sites (24) and UHRF1 (ubiquitin-like, containing PHD and RING finger domains 1), which is thought to target DNMT1 to hemimethylated DNA (25). In cancer cell lines, depletion of DNMT1 with anti-sense inhibitors has been linked with the demethylation of tumour suppressor gene promoters and reactivation of expression (26), indicating that DNMT1 is responsible for this hypermethylation. These results imply that DNMT1 inhibitors may prove to be effective anti-cancer drugs (27,28) and an example of their application in cancer treatment is the DNMT1 inhibitor 5-azacytidine (Vidaza), which has been approved for use in the treatment of myelodysplastic syndrome (29). The ability to assay DNMT1 activity is essential to investigate its substrate specificity in terms of DNA sequence and methylation state, to gain a greater understanding of the allosteric control of its C-terminal catalytic domain by the N-terminal regulatory domain (24,30), the effect of the wide range of proteins with which it is known to interact with upon activity (23) and screening compound libraries for potential inhibitors.
A commonly adopted approach to the kinetic analysis of methyltransferase activity has used the transfer of a 3H labelled methyl group from AdoMet to an oligonucleotide substrate, with the degree of methylation quantified by filter binding and scintillation counting (31,32). This assay has been widely applied, including in the analysis of DNMT1 activity (22), but this assay format is labour intensive and is neither continuous nor appropriate for high-throughput screening. Cytosine-C5 methyltransferase activity has also been measured using radiolabelled AdoMet in an alternative format in which the oligonucleotide substrate was immobilized onto a microplate using a biotin–avidin interaction (33). The oligonucleotide was then cleaved from the plate and scintillation counted. This assay provides the advantages of potential for high-throughput and lower background radioactivity than the filter-binding assay, but involves multiple processes and does not yield real time kinetic data. Alternative assay formats have been described that measure the turnover of AdoMet to S-adenosylhomocysteine via a colorimetric (34) or fluorescence polarization antibody binding assay (35), respectively. However, neither of these are suitable for continuous measurement as the colorimetric assay involves several steps and the polarization assay is lengthy and limits the AdoMet concentration to no higher than 1 µM.
The therapeutic potential of DNMT1 inhibitors has provided the drive to develop a new assay suitable for the kinetic analysis of cytosine-C5 methyltransferase activity in a format applicable to high-throughput screening. Recently, two fluorescence-based methyltransferase assays have been developed. The first general strategy relies upon protection (by methylation) of a restriction endonuclease cleavage site, coupled to an enzyme-linked immunosorbent assay (ELISA) detection format (36). This method is compatible with high-throughput screening and is suitable for assaying the activity of a broad range of methyltransferases, but it requires a series of lengthy processes and does not provide real-time data. An alternative approach couples the use of a methylation sensitive restriction endonuclease with the protection or promotion of cleavage of a break light oligonucleotide. A molecular break light is a single-stranded self-complementary oligonucleotide, which spontaneously forms a hairpin loop structure with a fluorophore at the 5′ terminus and a quencher dye at the 3′ terminus (37). In this structure, the fluorophore and quencher are locked close in space, promoting efficient quenching. Cleavage of the stem leads to the melting of the short cleaved sequence and separation of fluorophore and quencher resulting in an increase in fluorescence. This assay format has been applied to DNA adenine-N6 methyltransferase activity (Dam) (38–40), in all cases the oligonucleotide stem contains the 5′-GATC-3′ Dam recognition sequence. The most recent assay coupled a hemimethylated substrate with a restriction enzyme (DpnI), which will preferentially cleave the fully methylated substrate, so that one methylation event by Dam is reported by a proportional increase in fluorescence because of DpnI cleavage. This convenient assay format has the advantage of providing real-time data, which allows kinetic analysis of methylation.
We have developed a molecular break light assay for the continuous measurement of cytosine-C5 methyltransferase activity in which a partially methylated oligonucleotide acts as a substrate for cytosine-C5 methylation. The resultant fully methylated oligonucleotide is the optimal substrate (41) for the cytosine-C5 methylation-specific restriction endonuclease GlaI (42). Cleavage of the break light stem results in separation of the fluorophore and quencher, resulting in an increase in fluorescence. Using this assay, we have measured DNMT1 activity and determined the 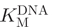 and
and 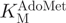 for the bacterial cytosine-C5 methyltransferase M.SssI.
for the bacterial cytosine-C5 methyltransferase M.SssI.
MATERIALS AND METHODS
Materials
Restriction endonuclease GlaI was purchased from SE Technologies (Zweibrücken, Germany), M.SssI and human DNMT1 were obtained from New England Biolabs (Herts., UK). Oligonucleotides 1 and 2 for fluorescence assays were purchased from ATDBio (Southampton, UK). AdoMet and mineral oil (sterile filtered, mouse embryo tested) were purchased from Sigma–Aldrich (Poole, UK). GelRed Stain was purchased from Cambridge Bioscience Limited. Fluorescence measurements were carried out in either 96-well half-area black flat-bottomed, or 384-well low-volume, hibase flat-bottomed microplates from Greiner Bio-One Ltd (Stonehouse, UK). Measurements were taken using a Tecan Safire 2 microplate reader (Reading, UK). Assays in 384-well plates were prepared and initiated using a Beckman Coulter BioMek 3000 liquid handling system, equipped with a 200-µl single-channel and 20-µl eight-channel pipette head from Beckman Coulter (High Wycombe, UK). Data were analysed with the commercially available software SigmaPlot 10 (Systat Inc., San Jose, CA, USA).
Cytosine-C5 methyltransferase activity assays
The break light oligonucleotide sequences used in methyltransferase assays were; oligonucleotide 1: 5′ (F)CCTATGCGCMATCAGTTTTCTGATGCMGCMATAGG(Q) 3′; oligonucleotide 2: 5′ (F)CCTATGCMGCMATCAGTTTTCTGATGCMGCMATAGG(Q) 3′, where the cytosine-C5 methyltransferase recognition sequence is shown in bold and the GlaI recognition sequence is underlined, (F) is Cy3 and (Q) is a dabcyl quencher.
Cytosine-C5 methyltransferase activity was measured in triplicate, in Greiner 384-well plates in a total assay volume of 20 µl at 37°C unless otherwise stated. All assays were prepared in buffer A, which contained 10 mM Tris/HCl pH 7.5, 5 mM MgCl2, 1 mM DTT, 5% glycerol, 0.1 mg/ml BSA, 20–100 mM NaCl and 0–1 mM AdoMet. The 384-well plate assays were prepared by making an oligonucleotide solution in buffer A containing 1.33 × the required oligonucleotide concentration. Fifteen microlitres of this solution was then added to the microplate per well and the plate equilibrated at 37°C for 15 min. An enzyme solution was prepared on ice, containing GlaI and either additional DNMT1 or M.SssI in buffer A. The assays were initiated by the addition of 5 µl of enzyme solution. Using the same method, 96-well plate assays were prepared, scaling the volumes up to a total of 100 µl per well. Cy3 emission was then measured over time and for M.SssI methylation reactions the initial rate of reaction was calculated over the first 500 s of steady-state methylation or for DNMT1 experiments, steady-state rates were measured over the first 1000 s after the lag phase. Background fluorescence increase due to non-specific oligonucleotide 1 cleavage by GlaI was accounted for by subtracting a negative control, which contained GlaI but lacked both DNMT1 and M.SssI. The rate of fluorescence increase could be converted into a rate of reaction using a fluorescence calibration curve for kinetic analysis and the data were fitted to the Michaelis–Menten equation (43).
Fluorescence was monitored using a Tecan Safire2 microplate reader, using 10 readings per well for each measurement and a 0.1 s pause between each movement and reading. The excitation wavelength was 543 nm and the emission wavelength 563 nm, with a bandwidth of 5 nm for measurements in a 96-well plate, or 7 nm for a 384-well plate. The gain setting was 190 for 96-well-plate measurements and 165 or 133 for 384-well-plate measurements as stated. The Z-position was 9000 µm for 96-well plate and 12 500 µm for 384-well-plate measurements, with an integration time of 40 µs.
Oligonucleotide 2 calibration curve
Assays containing buffer A supplemented with 0 mM AdoMet, 100 mM NaCl, 3.56, 4.75, 6.33, 8.43, 11.3, 15.0 and 20 nM oligonucleotide 2 and either 0.2 or 1.0 U/well GlaI for 20 (384-well) or 100 µl (96-well) assays, respectively. For 100 µl assays, fluorescence was measured at a gain of 190 and for 20 µl assays, at gains of 133 and 165. The endpoint fluorescence values were measured at each concentration.
Analysis of GlaI activity against oligonucleotides 1 and 2
To confirm the selectivity of GlaI cleavage between the 5′-GCGC-3′ sequences of partially methylated oligonucleotide 1 and fully methylated oligonucleotide 2 followed the pattern reported by Tarasova et al. (41), oligonucleotides 1 or 2 (10 μM) were incubated with GlaI (8 U) for 1 h and then analysed by urea-PAGE. The effect of oligonucleotide 1 concentration upon GlaI activity was measured in a series of activity assays in buffer A supplemented with 50 µM AdoMet, 100 mM NaCl, 2.4 U/well GlaI and 0.028, 0.047, 0.078, 0.13, 0.22, 0.36, 0.6 and 1 µM oligonucleotide 1. The dependence of rate upon concentration of the fully methylated oligonucleotide 2 was measured in a series of activity assays containing 50 µM AdoMet, 100 mM NaCl, 0.05 U/well GlaI and 0.028, 0.047, 0.078, 0.13, 0.22, 0.36 and 0.6 µM oligonucleotide 2.
Analysis of reproducibility and non-specific cleavage by GlaI in coupled M. SssI assays
To analyse reproducibility, a coupled M.SssI assay was repeated six times under standard assay conditions at the following final concentrations: 1 µM oligonucleotide 1, 50 µM Adomet, 10 nM M.SssI and 2.4 U/well GlaI in buffer A in two independent experiments. The background fluorescence increase due to non-specific cleavage of oligonucleotide 1 by GlaI was determined by comparing complete assays containing M.SssI and GlaI (in the column labelled ‘assay’ in Table 2) with a negative control containing only GlaI (Table 2, ‘negative control’). Background cleavage of partially methylated oligonucleotide 1 by GlaI was calculated as a percentage of the complete assay signal (i.e. negative control signal divided by assay signal).
Table 2.
Background cleavage by GlaI in a coupled M.SssI assay
| Concentration of oligonucleotide 1 (µM) | Rate of fluorescence change (AU/s) |
Background cleavage (%) | |
|---|---|---|---|
| Assay | Negative control | ||
| 0.03 | 3.33 | 0.47 | 14.1 |
| 0.05 | 4.07 | 0.38 | 9.34 |
| 0.08 | 5.36 | 0.59 | 11.0 |
| 0.13 | 7.53 | 0.88 | 11.7 |
| 0.22 | 10.4 | 1.25 | 12.0 |
| 0.36 | 14.3 | 1.48 | 10.4 |
| 0.60 | 18.4 | 1.64 | 8.91 |
| 1.00 | 21.8 | 0.30 | 1.38 |
Assays contained oligonucleotide 1, AdoMet, M.SssI and GlaI. Negative control assays were prepared identically except that they lacked M.SssI. Background cleavage was calculated as the percentage of assay signal caused by non-specific cleavage of partially methylated oligonucleotide 1 by GlaI.
Comparison of stepwise methylation and restriction reactions with the coupled activity assay
Stepwise assays comprised of 20 μl of buffer A supplemented with 250 nM oligonucleotide 1 and 10 nM M.SssI and 100 mM NaCl. Triplicate methylation reactions were initiated with 10 µM AdoMet at eight time points and incubated at 37°C for 0, 1, 2, 4, 8, 16, 32 and 64 min. Triplicate control assays were also prepared containing no AdoMet and incubated under the same conditions. Reactions were stopped by heat denaturation and then cooled on ice. The resultant solutions were then equilibrated at 37°C for 5 min in a black, flat-bottomed, low-volume, 384-well Greiner microplate and the cleavage reactions were then initiated by the addition of 2.4 U/well GlaI. Subsequent fluorescence increases due to separation of fluorophore and quencher were monitored in a Tecan Safire2 microplate reader until an endpoint was reached. Coupled activity assays containing M.SssI and GlaI were prepared in triplicate as described previously and contained the same substrate and enzyme concentrations as the stepwise assays. The fluorescence of the coupled assays was monitored over 64 min. Negative control rates (containing no AdoMet) were subtracted from the complete assays and fluorescence changes for both the stepwise and the coupled assays were converted to fractional turnover, calculated for each time point as (fluorescence signal/maximum change in fluorescence) to facilitate comparison. After GlaI-mediated cleavage of methylated oligonucleotide 1, assays were analysed by 20% Urea-PAGE. The resultant gel was visualized on a Bio-Rad Molecular Imager Gel Doc XR+ system using the trans-UV mode before and after staining with GelRed stain.
Catalytic DNMT1 activity assay
DNMT1 activity was assayed in duplicate in 96-well half-area black flat-bottomed microplates. The assay volume was 100 µl; assays were incubated at 37°C for 8 h and measurements were taken every 145 s. Assays contained 30 nM oligonucleotide 1, 25 mM NaCl, 1 mM AdoMet, 2.8 nM DNMT1 and 0.8 U/well GlaI in buffer A. Wells were covered with 5 µl mineral oil to minimize evaporation.
Effect of increasing oligonucleotide concentration upon DNMT1 activity
The effect of the variation of oligonucleotide concentration upon DNMT1 activity was assayed in duplicate at a gain of 165, in buffer A containing 50 mM NaCl, 1 mM AdoMet and 30, 60, 120 and 240 nM oligonucleotide 1. The DNMT1 concentration was 2.8 nM and the GlaI concentration was 0.2 U/well.
Determination of the optimal GlaI concentration for the kinetic analysis of M.SssI activity
The concentration of GlaI required for kinetic analysis of M.SssI was determined in experiments containing buffer A supplemented with 1 µM oligonucleotide 1, 50 µM AdoMet, 100 mM NaCl, 10 nM M.SssI and GlaI concentrations of 0, 0.2, 0.4, 0.8, 1.2, 1.6, 2.4 and 3.2 U/well at a gain setting of 133.
Kinetic analysis of M.SssI activity
For kinetic analysis, M.SssI was assayed at a gain setting of 133 in buffer A in each case containing 100 mM NaCl, 10 nM M.SssI and 2.4 U/well GlaI. The dependence of M.SssI activity upon the concentration of oligonucleotide 1 was measured in a series of assays additionally containing 50 µM AdoMet and 0.028, 0.047, 0.078, 0.13, 0.22, 0.36, 0.6 and 1 µM concentrations of oligonucleotide 1. The effect of AdoMet concentration upon M.SssI activity was measured in assays additionally containing 1 µM oligonucleotide 1 and AdoMet concentrations of 0.28, 0.47, 0.78, 1.3, 2.16, 3.6, 6 and 10 µM.
RESULTS
Assay design
To enable the continuous measurement of cytosine-C5 methyltransferase activity, an oligonucleotide (oligonucleotide 1) was used which lacks cytosine methylation at a single site out of a possible four within the sequence. We have termed this the (n-1) strategy, where n is the number of methylated bases required for the oligonucleotide to become the optimal substrate for a methylation-sensitive restriction endonuclease. It allows the coupling of a single methylation event by a methyltransferase to a proportional increase in fluorescence. In this case, the optimal restriction site for GlaI, a 5-methylcytosine-specific endonuclease is the palindromic sequence 5′-GCMGCM-3′ and so n = 4. Oligonucleotide 1 therefore contains three 5-methylcytosines, leaving a single site available for methylation. In the assay, methylation of oligonucleotide 1 forms the tetra methylated product oligonucleotide 2. In situ GlaI restriction of fully methylated 2 leads to the separation of fluorophore and quencher with a resultant proportional increase in fluorescence (Figure 1).
Figure 1.

Break light cytosine-C5 methyltransferase activity assay. The fluorescence of the hemimethylated oligonucleotide 1 is quenched by the dabcyl group. It is a substrate for cytosine-C5 methyltransferases such as DNMT1 or M.SssI and upon methylation yields the fully methylated oligonucleotide 2, which is cleaved by GlaI, separating the fluorophore from the quencher and resulting in an increase in fluorescence, which is proportional to the concentration of oligonucleotide 3.
Calibration of oligonucleotide fluorescence
Calibration curves were used to convert fluorescence measurements from real-time assays into a concentration of cleaved fully methylated oligonucleotide 2. The concentration of oligonucleotide 2 was varied between 3.56 and 20 nM and endpoint fluorescence values were fitted to a linear function (Supplementary Figure S1). The gradient of each fit and the measures of goodness of fit to a linear function (R2) are shown in Table 1.
Table 1.
Oligonucleotide 2 fluorescence calibration curve data
| Assay volume (µl) | Gain | Gradient (AU/nM) | R2 |
|---|---|---|---|
| 20 | 133 | 183 ± 7.1 | 0.990 |
| 20 | 165 | 1347 ± 29 | 0.997 |
| 100 | 190 | 962 ± 50 | 0.982 |
R2 is a measure of the goodness of fit to a linear function.
Analysis of assay reproducibility and non-specific cleavage by GlaI
Partially and fully methylated oligonucleotides 1 and 2 were incubated with GlaI and then analysed by urea-PAGE (Figure 2). The experiment confirmed the selectivity of GlaI for the tetramethylated substrate as originally reported by Tarasova et al. (41). In a further experiment, the signal from background cleavage of partially methylated oligonucleotide 1 by GlaI over a concentration range of 0.03–1 μM was calculated as a percentage of the complete assay signal (Table 2). The data show that non-specific cleavage by GlaI accounts for ∼10–15% of the overall assay signal and thus does not have a substantial effect on the quality of the results.
Figure 2.

Urea-PAGE analysis of partially and fully methylated oligonucleotides incubated with GlaI. Partially methylated oligonucelotide 1 or fully methylated oligonucleotide 2 (10 μM) were incubated with GlaI (8 U) for 1 h. (A) Fluorescence image. (B) White light image. Lanes: M, oligonucleotide ladder (bases); lane 1, oligonucleotide 1; lane 2, GlaI treated oligonucleotide 1; lane 3, oligonucleotide 2; lane 4, GlaI-treated oligonucleotide 2.
The reproducibility of the assay was investigated by determining the standard deviation and the coefficient of variation (CV) in rate of fluorescence change for six identical assays and negative control assays (lacking M.SssI) under standard conditions. The average negative control signal was found to be very low at 2.7 ± 2.7% of the assay signal. The assay signal was found to be highly reproducible with an average of 21.2 ± 1.2 AU/s, giving a CV of 4.5%.
Kinetic analysis of GlaI activity
The rate of cleavage of the partially methylated substrate oligonucleotide 1 by GlaI must be sufficiently slow to allow satisfactory differentiation between the fluorescence signal observed due to background cleavage of partially methylated oligonucleotide 1 and that caused by cleavage of fully methylated oligonucleotide 2. The optimal NaCl and MgCl2 concentrations for GlaI selectivity were determined in a simple experiment in which the concentrations of NaCl (0–200 mM) and MgCl2 (1–20 mM) were varied (Supplementary Figure S2) and this was identified as 100 mM NaCl and 5 mM MgCl2. The effect of the NaCl concentration on DNMT1 activity was analysed to ensure these conditions were appropriate (Supplementary Figure S3). The break light assay was used to determine  of GlaI for oligonucleotide 1 (Figure 3A) and oligonucleotide 2 (Figure 3B). The dependence of GlaI activity upon oligonucleotide concentration was fitted to the Michaelis–Menten equation. The
of GlaI for oligonucleotide 1 (Figure 3A) and oligonucleotide 2 (Figure 3B). The dependence of GlaI activity upon oligonucleotide concentration was fitted to the Michaelis–Menten equation. The 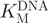 for oligonucleotide 1 was observed to be 236 ± 23 nM and the Vmax to be 0.100 ± 0.004 fmol/U/s; in comparison, for oligonucleotide 2, the
for oligonucleotide 1 was observed to be 236 ± 23 nM and the Vmax to be 0.100 ± 0.004 fmol/U/s; in comparison, for oligonucleotide 2, the 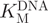 was 44.8 ± 11 nM and the Vmax was 28.5 ± 1.8 fmol/U/s. The relative specificity constant [defined as Vmax/KM (44)] for oligonucleotide 1 is 4.2 × 10–10 U–1 s–1 and for oligonucleotide 2 is 6.4 × 10–7 U–1 s–1, which indicates that GlaI shows a considerable preference for the fully methylated oligonucleotide 2. This preference is highly advantageous in our coupled assay format, as the restriction enzyme must selectively cleave the fully methylated oligonucleotide 2 in the presence of a relatively high concentration of the partially methylated methyltransferase oligonucleotide 1.
was 44.8 ± 11 nM and the Vmax was 28.5 ± 1.8 fmol/U/s. The relative specificity constant [defined as Vmax/KM (44)] for oligonucleotide 1 is 4.2 × 10–10 U–1 s–1 and for oligonucleotide 2 is 6.4 × 10–7 U–1 s–1, which indicates that GlaI shows a considerable preference for the fully methylated oligonucleotide 2. This preference is highly advantageous in our coupled assay format, as the restriction enzyme must selectively cleave the fully methylated oligonucleotide 2 in the presence of a relatively high concentration of the partially methylated methyltransferase oligonucleotide 1.
Figure 3.

Dependence of GlaI activity on oligonucleotide concentration. (A) Using partially methylated oligonucleotide 1 as the substrate and (B) using fully methylated oligonucleotide 2 as the substrate.
Comparison of stepwise methylation and restriction reactions with the coupled activity assay
To ensure the fluorescence signal from the coupled assay was accurately reflecting the degree of substrate methylation, the coupled assay was compared with a time course in which the methylation and restriction reactions were carried out by independent steps. The results of this experiment are shown in Figure 4. For the time course, the methylation reactions were stopped by heat denaturation of the methyltransferase at selected time points, then in a second step, the cooled samples were restricted with GlaI and the fluorescent signal monitored until an endpoint was reached. These endpoint fluorescence readings were converted to fractional turnover (fluorescence/maximum fluorescence) and plotted against the average of three coupled assays (Figure 4A). The time course of stepwise activity assays, a sample of the coupled assay at the endpoint (64 min) and appropriate controls were analysed by urea PAGE (Figure 4B and C). The results of this gel analysis show the cleaved, fluorescent product (7 bp) increasing with time and no cleavage in the negative controls (no AdoMet) and are in good agreement with the fluorescence measurements shown in Figure 4A.
Figure 4.

Analysis of time courses for coupled and stepwise enzymatic reactions. (A) Comparison of time courses of product formation in stepwise (filled circles) and coupled (solid line) assays expressed as fractional turnover. (B) Gel analysis of coupled and stepwise enzymatic reactions. The fluorescence image is of a 20% urea-polyacrylamide gel of stepwise and coupled assays and was recorded before staining. Lanes were as follows: 1, coupled assay negative control (no AdoMet); 2, stepwise assay negative control (no AdoMet) incubated for 0 min; 3, stepwise assay negative control (no AdoMet) incubated for 64 min; 4-11, stepwise assay incubated for 0, 1, 2, 4, 8, 16, 32 and 64 min respectively; 12, coupled assay. (C) As in (B) but after staining with GelRed stain.
Analysis of DNMT1 activity using the molecular break light assay
DNMT1 activity was measured over a time period of 8 h using the 14 bp stem oligonucleotide 1 at concentration of 30 nM as a substrate and a DNMT1 concentration of 2.8 nM. A plot of the real-time fluorescence data against time for the assay and negative control (containing no DNMT1) shows that, whilst the fluorescence due to background cleavage decreases slowly over the course of the assay, the fluorescence due to cleavage of the DNMT1 methylation product oligonucleotide 2 continues to increase (Figure 5A). During this assay, DNMT1 was observed to maintain a roughly linear activity for 8 h, with no apparent burst phase (Figure 5A). After subtraction of the background signal (Figure 5B), the fluorescence increase can be fitted to a linear function (goodness of fit, R2 = 0.99) allowing the steady-state rate to be determined as 0.83 ± 0.07 pmol/mg/s from a set of duplicate assays. The total change in fluorescence during this assay corresponds to the turnover of 1.18 ± 0.06 pmol of substrate oligonucleotide, which is 39.3% of the initial substrate concentration and equates to 4.2 ± 0.2 turnovers of DNMT1.
Figure 5.

Activity of DNMT1 with oligonucleotide 1. (A) Comparison of fluorescence changes in a full assay (solid black line) and a negative control assay lacking DNMT1 (dotted line). (B) Plot of change in the concentration of oligonucleotide 3 after background subtraction.
The dependence of DNMT1 activity upon oligonucleotide concentration
The effect of increasing oligonucleotide concentration upon the observed steady state rate was investigated in a series of assays containing 30–240 nM oligonucleotide 1 and 2.8 nM DNMT1. At each DNA concentration, a short lag phase was observed (Figure 6A). Such lag phases are commonly observed in coupled enzyme assays (45) and in this case may correspond to the time taken for the concentration of fully methylated oligonucleotide 2 to reach a steady-state concentration. Plotting the steady-state rates against concentration of oligonucleotide 1 gives a roughly proportional linear increase in rate (Figure 6B). The maximum observed rate of oligonucleotide 1 turnover by DNMT1 was 2.97 ± 0.27 h–1 at a substrate DNA concentration of 240 nM. The rate of background cleavage of oligonucleotide 1 by GlaI at each concentration was low, but increases slightly with increasing oligonucleotide concentration. This experiment demonstrates the advantage of GlaI having a high specificity for the oligonucleotide 2 over oligonucleotide 1, as the background cleavage is very slow even at relatively high substrate concentrations.
Figure 6.

Effect of increasing oligonucleotide 1 concentration on DNMT1 activity. (A) Real-time data averaged from duplicate assays. Assays contained oligonucleotide 1 at concentrations of 30 nM (black line), 60 nM (orange line), 120 nM (green line) and 240 nM (blue line) with associated negative controls (lacking DNMT1) shown as dotted lines. (B) Plot of steady-state rate against concentration of oligonucleotide 1.
Determination of the optimal GlaI concentration for the kinetic analysis of M.SssI activity
M.SssI was utilized as a model methyltransferase to test the application of the break light assay for kinetic analysis of cytosine-C5 methylation. To ensure that the rate of fluorescence increase was not limited by GlaI concentration at high (1 µM) DNA concentrations, a set of assays were prepared in which GlaI concentration was varied (0–3.2 U/well) and oligonucleotide 1 (1 µM), M.SssI (10 nM) and AdoMet (50 µM) concentrations were fixed. The concentration of DNA was over twice that required to ensure M.SssI was saturated and therefore functioning at Vmax. The observed rate of methylation was then plotted against GlaI concentration (Figure 7). Increasing concentrations of GlaI resulted in an increase in the observed initial rate of product formation up to a concentration of 1.6 U/well. Further increases in GlaI concentration did not result in a significant increase in the reaction rate and a concentration of 2.4 U/well GlaI was selected for the kinetic analysis of M.SssI activity, to ensure that the observed rate of fluorescence increase reflects the rate of methylation.
Figure 7.

Effect of GlaI concentration on the observed activity of M.SssI using oligonucleotide 1 as a substrate.
Kinetic analysis of M.SssI activity
The break light assay was used to determine 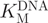 (oligonucleotide 1) and
(oligonucleotide 1) and 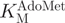 for M.SssI. In the activity assays (Figure 8A), a slowing of the rate of fluorescence change were observed in the initial phase (up to ∼100 s), but this was followed by a stable steady-state phase (up to 600 s), which was used to determine the reaction rates. The source of the variation in rate during the initial phase is at present unknown: some of the possible factors might include temperature equilibration of the plate, mixing of the wells or rate-limiting product release, resulting in a burst phase. For both substrates, the dependence of M.SssI activity upon substrate concentration was fitted to the Michaelis–Menten equation.
for M.SssI. In the activity assays (Figure 8A), a slowing of the rate of fluorescence change were observed in the initial phase (up to ∼100 s), but this was followed by a stable steady-state phase (up to 600 s), which was used to determine the reaction rates. The source of the variation in rate during the initial phase is at present unknown: some of the possible factors might include temperature equilibration of the plate, mixing of the wells or rate-limiting product release, resulting in a burst phase. For both substrates, the dependence of M.SssI activity upon substrate concentration was fitted to the Michaelis–Menten equation. 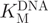 was found to be 234 ± 36 nM (Figure 8B), which compares well with the value of 180 nM reported previously (46) for a 30-bp hemimethylated oligonucleotide, although for an alternative sequence of the same length, M.SssI showed a significantly higher
was found to be 234 ± 36 nM (Figure 8B), which compares well with the value of 180 nM reported previously (46) for a 30-bp hemimethylated oligonucleotide, although for an alternative sequence of the same length, M.SssI showed a significantly higher 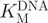 of 1.6 µM (47). Using our assay,
of 1.6 µM (47). Using our assay, 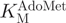 for M.SssI was measured to be 675 ± 110 nM (Figure 8C). The literature values for
for M.SssI was measured to be 675 ± 110 nM (Figure 8C). The literature values for  are 50 nM (46) and 2.2 µM (48) and may depend on the precise assay conditions used; however, the value determined using our assay falls between these two. The range of values calculated for kcat, from data shown in Figures 7, 8B and C are 0.98 ± 0.02, 0.73 ± 0.03 and 0.49 ± 0.02 turnovers/min, respectively. These values are similar to the reported kcat for the protein of 0.16 turnovers/min (46).
are 50 nM (46) and 2.2 µM (48) and may depend on the precise assay conditions used; however, the value determined using our assay falls between these two. The range of values calculated for kcat, from data shown in Figures 7, 8B and C are 0.98 ± 0.02, 0.73 ± 0.03 and 0.49 ± 0.02 turnovers/min, respectively. These values are similar to the reported kcat for the protein of 0.16 turnovers/min (46).
Figure 8.

Kinetic analysis of M.SssI activity. (A) Background-subtracted raw data showing the first 600 s for methylation of oligonucleotide 1 by M.SssI. The concentration of oligonucleotide 1 increases from lowest to highest as follows: 28, 47, 78, 130, 220, 360, 600 and 1000 nM. (B) Dependence of M.SssI activity on oligonucleotide concentration. (C) Dependence of M.SssI activity on AdoMet concentration.
DISCUSSION
The cytosine-C5 methyltransferase assay reported herein is the first to permit continuous monitoring of cytosine methylation. This has been achieved using an (n-1) molecular break light assay, in which a single methylation event forms the optimal GlaI restriction endonuclease substrate, which is cleaved in situ resulting in a proportional increase in fluorescence. The reproducibility, sensitivity and relative background cleavage were all within the range that indicates the assay is suitable for the analysis of cytosine-C5 methyltranferases. Validation of the coupled assay format has been achieved by comparing the continuous coupled assay to a stopped time-course experiment, in which the methylation and cleavage reactions were carried out in separate steps. This experiment shows that both methods give comparable results whilst also demonstrating the greater level of information it is possible to obtain from the real-time assay. An appropriate concentration of magnesium chloride and a suitable ionic strength have been identified to permit the normal functioning of both the methyltransferase and the restriction enzyme.
We have utilized this assay format for the analysis of DNMT1 activity. In an assay containing 30 nM oligonucleotide substrate a linear increase in methylation was observed over time, which remained steady over 8 h. The ability to sustain methylation over an extended period depends upon the NaCl concentration; below 50 mM, a linear rate over extended periods of up to 8 h is observed, whereas at higher concentrations of up to 100 mM, the length of this linear phase is reduced. This observation is consistent with the reported reduction of DNMT1 activity at salt concentrations over 50 mM (49). There is a slight decrease in selectivity of GlaI for oligonucleotide substrate 2 over 1 at lower NaCl concentration; however, even under these conditions, the rate of background cleavage of 1 remained very low.
The observation of a constant rate of methylation of partially methylated oligonucleotide 1 over four turnovers implies that in our assay, DNMT1 does not exhibit burst phase kinetics as reported for other hemimethylated 5′-CG-3′ oligonucleotides (47), but the observed activity more closely mirrors the de novo DNMT1 activity observed when unmethylated DNA is the substrate (50). It may be that the short 14 bp stem present in oligonucleotide substrate 1 is not sufficiently long to interact with the N-terminal regulatory domain and may not be subject to the complex allosteric control that has been proposed for DNMT1 (50). The activity assay appears to function with hairpin oligonucleotides containing a complimentary stem as short as 14 bp, but the oligonucleotides previously used as DNMT1 substrates in kinetic analysis were 30–36 bp in length (50,51) and extension of oligonucelotide 1 may yield a better model substrate. At a concentration of 240 nM oligonucleotide 1, the observed rate of DNMT1 turnover was estimated to be 2.97 ± 0.27 h–1, which is comparable to other reported activities against 5′-CG-3′ hemimethylated substrates of 3.0 h–1 (52).
A linear relationship was observed between DNMT1 activity and oligonucleotide concentration; however, at all concentrations of DNA a lag phase was observed before the linear steady-state rate of reaction was reached (Figure 6A). A lag phase has previously been reported in experiments with DNMT1 and an unmethylated substrate and is observed to increase with the concentration of DNA, although it is not clear that the lag phases are the result of identical processes. It may be that the lag phase observed in our assay arises from the time taken for the concentration of fully methylated oligonucleotide 2 to reach a steady-state concentration. If this were the case, then the addition of higher concentrations of GlaI would minimize the lag phase. However, the non-specific cleavage of oligonucleotide 1 is minimized by adding the smallest possible amount of GlaI. Therefore, a compromise concentration of GlaI was required, which gave an acceptable lag phase and a tolerable level of GlaI-dependent background cleavage of oligonucleotide 1.
The suitability of the assay for the kinetic analysis of cytosine-C5 methylation was tested by the kinetic characterization of the bacterial cytosine-C5 methyltransferase M.SssI. The kinetic constants measured for M.SssI using our assay were comparable to those reported in the literature (46,47) confirming the validity of the assay for the kinetic analysis of cytosine-C5 methylation. The underlying cause of the decrease in apparent reaction rates observed in the first ∼100 s of the M.SssI assays is not known, but reaction rates were determined using data from the steady-state region, which occurred between ∼100 and 600 s.
In the case of the DNMT1 methylation reaction, even at the highest DNA concentration (240 nM), the reaction had progressed to 0.18% of completion in 1000 s and in the fastest observed M.SssI reaction only 5.5% conversion had been achieved after 600 s. This relatively low level of turnover allowed the reaction rates observed to be treated purely as initial rates of reaction, if it is assumed that the GlaI substrate concentration remains essentially constant for both methyltransferase and restriction enzyme a direct background subtraction can be made. It is therefore relatively simple to account for the slight increase in fluorescence observed due to non-specific GlaI activity and obtain accurate rate data.
This strategy for the analysis of cytosine-C5 methyltransferase activity has a number of practical advantages. It provides the first real-time assay for cytosine-C5 methyltransferase activity, which results in a fluorescence increase that is directly proportional to substrate methylation, greatly facilitating kinetic analysis. This has been demonstrated using M.SssI and DNMT1 and provides a convenient alternative to the radioactive filter-binding assay for this purpose. This simple and practically robust assay is suitable for high-throughput analysis of DNMT1 activity and is likely to provide an efficient method for screening large compound libraries for DNMT1 inhibitors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The 6th Framework Programme of the European Commission under the contract NMP4-CT-2005-017114 ‘RECEPTRONICS’ (to R.J.W.); Engineering and Physical Sciences Research Council (to M.D.M.S.); DSTL Porton Down (to J.C.M.); the Royal Society via a University Research Fellowship (to P.L.R.). Funding for open access charge: Research donation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Professor T. Brown (Southampton University) for advice and helpful discussions.
REFERENCES
- 1.Colot V, Rossignol JL. Eukaryotic DNA methylation as an evolutionary device. Bioessays. 1999;21:402–411. doi: 10.1002/(SICI)1521-1878(199905)21:5<402::AID-BIES7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Dunn DB, Smith JD. The occurrence of 6-methylaminopurine in deoxyribonucleic acids. Biochem. J. 1957;68:627–636. doi: 10.1042/bj0680627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrlich M, Wilson GG, Kuo KC, Gehrke CW. N4-methylcytosine as a minor base in bacterial DNA. J. Bacteriol. 1987;169:939–943. doi: 10.1128/jb.169.3.939-943.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson TB, Coghill RD. Researches on pyrimidines C111. The discovery of 5-methylcytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus. J. Am. Chem. Soc. 1925;47:2838–2844. [Google Scholar]
- 5.Adams RLP. DNA methylation - the effect of minor bases on DNA protein interactions. Biochem. J. 1990;265:309–320. doi: 10.1042/bj2650309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Modrich P. Methyl-directed DNA mismatch correction. J. Biol. Chem. 1989;264:6597–6600. [PubMed] [Google Scholar]
- 7.Low DA, Weyand NJ, Mahan MJ. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect. Immun. 2001;69:7197–7204. doi: 10.1128/IAI.69.12.7197-7204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Løbner-Olesen A, Skovgaard O, Marinus MG. Dam methylation: coordinating cellular processes. Curr. Opin. Microbiol. 2005;8:154–160. doi: 10.1016/j.mib.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 10.Ehrlich M. Expression of various genes is controlled by DNA methylation during mammalian development. J. Cell. Biochem. 2003;88:899–910. doi: 10.1002/jcb.10464. [DOI] [PubMed] [Google Scholar]
- 11.Eden S, Cedar H. Role of DNA methylation in the regulation of transcription. Curr. Opin. Genet. Dev. 1994;4:255–259. doi: 10.1016/s0959-437x(05)80052-8. [DOI] [PubMed] [Google Scholar]
- 12.Chow JC, Brown CJ. Forming facultative heterochromatin: silencing of an X chromosome in mammalian females. Cell. Mol. Life Sci. 2003;60:2586–2603. doi: 10.1007/s00018-003-3121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 14.Stoger R, Kajimura TM, Brown WT, Laird CD. Epigenetic variation illustrated by DNA methylation patterns of the fragile-X gene FMR1. Hum. Mol. Genet. 1997;6:1791–1801. doi: 10.1093/hmg/6.11.1791. [DOI] [PubMed] [Google Scholar]
- 15.Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 17.Roy PH, Weissbach A. DNA methylase from HeLa cell nuclei. Nucleic Acids Res. 1975;2:1669–1684. doi: 10.1093/nar/2.10.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okano M, Xie SP, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 19.Hermann A, Schmitt S, Jeltsch A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. J. Biol. Chem. 2003;278:31717–31721. doi: 10.1074/jbc.M305448200. [DOI] [PubMed] [Google Scholar]
- 20.Pradhan S, Esteve PO. Mammalian DNA (cytosine-5) methyltransferases and their expression. Clin. Immunol. 2003;109:6–16. doi: 10.1016/s1521-6616(03)00204-3. [DOI] [PubMed] [Google Scholar]
- 21.Hermann A, Goyal R, Jeltsch A. The DNMT1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 22.Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- 23.Spada F, Rothbauer U, Zolghadr K, Schermelleh L, Leonhardt H. Adv. Enzyme Regul. 2006;46:224–234. doi: 10.1016/j.advenzreg.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Chuang LSH, Ian HI, Koh TW, Ng HH, Xu GL, Li B.FL. Human DNA (cytosine-5) methyltransferase PCNA complex as a target for p21(WAF1) Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 25.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 26.Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, MacLeod AR. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003;33:61–65. doi: 10.1038/ng1068. [DOI] [PubMed] [Google Scholar]
- 27.Reid GK, Besterman JM, MacLeod AR. Selective inhibition of DNA methyltransferase enzymes as a novel strategy for cancer treatment. Curr. Opin. Mol. Ther. 2002;4:130–137. [PubMed] [Google Scholar]
- 28.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R, et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin. Cancer Res. 2005;11:3604–3608. doi: 10.1158/1078-0432.CCR-04-2135. [DOI] [PubMed] [Google Scholar]
- 30.Fatemi M, Hermann A, Pradhan S, Jeltsch A. The activity of the murine DNA methyltransferase DNMT1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J. Mol. Biol. 2001;309:1189–1199. doi: 10.1006/jmbi.2001.4709. [DOI] [PubMed] [Google Scholar]
- 31.Geier GE, Modrich P. Recognition sequence of the Dam methylase of Escherichia coli K12 and mode of cleavage of DpnI endonuclease. J. Biol. Chem. 1979;254:1408–1413. [PubMed] [Google Scholar]
- 32.Reich NO, Mashhoon N. Inhibition of EcoRI DNA methylase with cofactor analogs. J. Biol. Chem. 1990;265:8966–8970. [PubMed] [Google Scholar]
- 33.Roth M, Jeltsch A. Biotin-avidin microplate assay for the quantitative analysis of enzymatic methylation of DNA by DNA methyltransferases. Biol. Chem. 2000;381:269–272. doi: 10.1515/BC.2000.035. [DOI] [PubMed] [Google Scholar]
- 34.Hendricks CL, Ross JR, Pichersky E, Noel JP, Zhou ZS. An enzyme-coupled colorimetric assay for S-adenosylmethionine-dependent methyltransferases. Anal. Biochem. 2004;326:100–105. doi: 10.1016/j.ab.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 35.Graves TL, Zhang Y, Scott JE. A universal competitive fluorescence polarization activity assay for S-adenosylmethionine utilizing methyltransferases. Anal. Biochem. 2008;373:296–306. doi: 10.1016/j.ab.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woo YH, Rajagopalan P.TR, Benkovic SJ. A nonradioactive DNA methyltransferase assay adaptable to high-throughput screening. Anal. Biochem. 2005;340:336–340. doi: 10.1016/j.ab.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 37.Biggins JB, Prudent JR, Marshall DJ, Ruppen M, Thorson JS. A continuous assay for DNA cleavage: the application of “break lights” to enediynes, iron-dependent agents, and nucleases. Proc. Natl Acad. Sci. USA. 2000;97:13537–13542. doi: 10.1073/pnas.240460997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mashhoon N, Pruss C, Carroll M, Johnson PH, Reich NO. Selective inhibitors of bacterial DNA adenine methyltransferases. J. Biomol. Screen. 2006;11:497–510. doi: 10.1177/1087057106287933. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Yan H, Wang K, Tan W, Zhou X. Hairpin fluorescence DNA probe for real-time monitoring of DNA methylation. Anal. Chem. 2007;79:1050–1056. doi: 10.1021/ac061694i. [DOI] [PubMed] [Google Scholar]
- 40.Wood RJ, Maynard-Smith MD, Robinson VL, Oyston P.CF, Titball RW, Roach PL. Kinetic analysis of Yersinia pestis DNA adenine methyltransferase activity using a hemimethylated molecular break light oligonucleotide. PLoS ONE. 2007;2:E801. doi: 10.1371/journal.pone.0000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarasova GV, Nayakshina TN, Degtyarev S.KH. Substrate specificity of new methyl-directed DNA endonuclease GlaI. BMC Mol. Biol. 2008;9:7. doi: 10.1186/1471-2199-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chernukhin VA, Najakshina TN, Abdurashitov MA, Tomilova JE, Mezentzeva NV, Dedkov VS, Mikhnenkova NA, Gonchar DA, Degtyarev SK. A novel restriction endonuclease GlaI recognizes methylated sequence 5′-G(5mC) GC-3′. Biotechnologia. 2006;4:31–35. [Google Scholar]
- 43.Michaelis L, Menten ML. Die kinetik der invertinwirkung. Biochem. Z. 1913;49:334−336. [Google Scholar]
- 44.Koshland DE. The application and usefulness of the ratio kcat/KM. Bioorg. Chem. 2002;30:211–213. doi: 10.1006/bioo.2002.1246. [DOI] [PubMed] [Google Scholar]
- 45.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: Wiley Interscience; 1993. pp. 85–87. [Google Scholar]
- 46.Rathert P, Rasko T, Roth M, Slaska-Kiss K, Pingoud A, Kiss A, Jeltsch A. Reversible inactivation of the CG specific Sssl DNA (cytosine-C5)-methyltransferase with a photocleavable protecting group. Chembiochem. 2007;8:202–207. doi: 10.1002/cbic.200600358. [DOI] [PubMed] [Google Scholar]
- 47.Flynn J, Glickman JF, Reich NO. Murine DNA cytosine-C-5 methyltransferase: pre-steady- and steady-state kinetic analysis with regulatory DNA sequences. Biochemistry. 1996;35:7308–7315. doi: 10.1021/bi9600512. [DOI] [PubMed] [Google Scholar]
- 48.Lee WJ, Zhu BT. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis. 2006;27:269–277. doi: 10.1093/carcin/bgi206. [DOI] [PubMed] [Google Scholar]
- 49.Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells – purification, sequence specificity, and mode of interaction with DNA. Proc. Natl Acad. Sci. Biol. 1983;80:5559–5563. doi: 10.1073/pnas.80.18.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svedruzic ZM, Reich NO. DNA cytosine C-5 methyltransferase DNMT1: catalysis-dependent release of allosteric inhibition. Biochemistry. 2005;44:9472–9485. doi: 10.1021/bi050295z. [DOI] [PubMed] [Google Scholar]
- 51.Pradhan S, Esteve PO. Allosteric activator domain of maintenance human DNA (cytosine-5) methyltransferase and its role in methylation spreading. Biochemistry. 2003;42:5321–5332. doi: 10.1021/bi034160+. [DOI] [PubMed] [Google Scholar]
- 52.Bacolla A, Pradhan S, Roberts RJ, Wells RD. Recombinant human DNA (cytosine-5) methyltransferase II. Steady-state kinetics reveal allosteric activation by methylated DNA. J. Biol. Chem. 1999;274:33011–33019. doi: 10.1074/jbc.274.46.33011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.