

Abstract
Sphingosine 1-phosphate (S1P), a potent phospholipid growth and trophic factor, is synthesized in vivo by two sphingosine kinases. Thus these kinases have been proposed as important drug targets for treatment of hyper-proliferative diseases and inflammation. We report here a new class of amidine-based sphingosine analogs that are competitive inhibitors of sphingosine kinases exhibiting varying degrees of enzyme selectivity. These inhibitors display KI values in the submicromolar range for both sphingosine kinases and, in cultured vascular smooth muscle cells, decrease S1P levels and initiate growth arrest.
Introduction
Sphingosine kinases 1 and 2 (SphK1 & 2)a catalyze the phosphorylation of D-erythro sphingosine to form sphingosine 1-phosphate (S1P).1 The sphingosine kinases control, in large part, the equilibrium between the survival factor, S1P, and its pro-apoptotic metabolic precursor, ceramide.2 S1P has been demonstrated to be a potent agonist at five membrane-bound G-protein coupled receptors, known as S1P1-5,3 whose roles in physiologic and pathophysiologic states are currently under investigation. The most well understood receptor subtype, S1P1, is now recognized as the receptor responsible for the anti-apoptotic properties of S1P and is also implicated in the control of lymphocyte trafficking.4 Due to their regulation of S1P production, SphKs1 and 2 have been proposed to be important small molecule drug targets.5
SphK null mice, small interfering RNAs and small molecule inhibitors have provided insight into the physiologic importance of these enzymes. Sphk1-/- and Sphk2-/- mice develop normally while the double null genotype is embryonic lethal at mid-gestation.6 While these data suggest a compensatory mechanism for SphK1 & 2, their unequal distribution in cellular compartments,7 SphK1's high degree of inducibility,8 and SphK2's pro-apoptotic BH3 domain9,10 have led researchers to view the SphK isoforms as unequal with respect to their roles in hyper-proliferative disease states. In support of this longstanding hypothesis, Spiegel and coworkers demonstrated the effectiveness of a recently developed SphK1 selective inhibitor in the treatment of an animal model of leukemia.11 However, another report indicated that manipulation of SphK2 might also be important in preventing neoplastic disease progression.12 Studies such as these demonstrate the importance of both sphingosine kinase types in disease. However, the study of dual SphK inhibitors is underrepresented in the chemical literature. A recent report in this journal documented the effectiveness of a dual inhibitor in U937 cells and its potential as a future mode of therapy.13
We sought to add to this growing pool of inhibitors by synthesizing a novel class of dual sphingosine kinase inhibitors. Turning to examples from the chemical literature, we noticed that conventional methods of kinase inhibition involve the use of adenosine analogs to target the ATP biding site. Although this strategy has been successful,14 the ATP binding site can be similar across a wide array of kinases and such inhibitors are often burdened by a lack of selectivity and off-target effects. In terms of sphingosine kinases, the amino acid sequence of the ATP binding domain of SphK1 and 2 is conserved across a number of diacylglycerol (DAG) kinase family members rendering the established ATP-targeting strategy particularly problematic.
While characterizing previously reported SphK substrates, we discovered a class of sphingosine-like dual SphK1/2 inhibitors. Herein we document the effectiveness of targeting the sphingosine-binding domain of the sphingosine kinases by creating a series of amidine-based, SphK1/2 inhibitors including molecules with affinity constants of less than 1 μM. We also demonstrate an SAR strategy that that was effective in improving potency and selectivity between SphK1 & 2. These inhibitors are effective in depressing S1P levels in cultured cells and initiate growth arrest in proliferating smooth muscle cells.
Results
Initial inhibitor design
The initial design of substrate-based SphK1/2 inhibitors required an understanding of previously evaluated SphK substrates. We and others have documented that the immunomodulatory investigational drug, fingolimod (FTY720), is inactive until phosphorylated by Sphk2.15, 16, 17 FTY720-P is a potent agonist at the S1P receptors, most prominently the S1P1 receptor.13 FTY720 has been shown to be efficacious in clinical trials of remitting relapsing multiple sclerosis.18 We have examined the receptor selectivity and metabolism of a number of classes of FTY720 analogues.19, 20, 21, 22 Despite continuing interest in their role as cell-signaling entities, the SAR associated with sphingosine analogs as SphK substrates (particularly substrates of SphK1) has remained largely undefined. However, a recent study from our laboratories involving the design and evaluation of a series of heterocyclic amino alcohols as SphK substrates provided insight into the structural requirements necessary for phosphorylation. Although synthetic analogs of sphingosine phosphorylated by both SphKs are rare, we previously reported (R)-2-amino-2-(5-(4-octylphenyl)-1,2,4-oxadiazol-3-yl)propan-1-ol (VPC45129) as an amino-alcohol substrate possessing such activity.23 We postulated that if the hydroxyl portion of this substrate were deleted, creating the (S)-deoxy-5-phenyl-1,2,4-oxadiazole (1), an efficient substrate-based dual inhibitor of the SphKs could be created.
Synthesis of 1
Envisioning the stereochemistry of the amine to be readily available from L-alanine, the synthesis of 1 commenced the N-Cbz protection of the amino acid. Aqueous ammonium hydroxide in methanol were used to covert the methyl ester to the corresponding amide, although a reaction time of 72 hours was necessary for moderate yields. The primary amide was then converted to the nitrile 2 through a trifluoroacetic anhydride mediated dehydration. Hydroxylamine hydrochloride and triethylamine in refluxing ethanol then provided the amide-oxime 3. In our previously cited study on heterocyclic amino alcohols as SphK substrates, we showed that amide-oximes such as 3 are excellent substrates for PyBOP mediated couplings, providing an efficient route to precursors of phenyl-1,2,4-oxadiazole systems.23 Utilizing this methodology, 4 was synthesized, which upon heating in DMF in the presence of molecular sieves, afforded 5 in more than 80% yield.
The final deprotection of 5 could be achieved using standard palladium hydrogenolysis conditions, yielding 1 as the free amine. Surprisingly, after 15 hours under these conditions, several products were present by TLC analysis. Isolation of the major product proved to be more difficult than anticipated. Thus, only small amounts of material were recovered from the deprotection of 5.
Reductive cleavage of 1,2,4-oxadiazole yielding a primary amidine
Based on analytical LCMS spectra, the mass of 1 was inconsistent with that of the desired product, indicating a reduction beyond that of the N-Cbz group had taken place. After a search through the chemical literature, an example was found showing catalytic palladium hydrogenolysis conditions, similar to those used in the deprotection of 5, catalyzed the N-O bond cleavage of phenyl-substituted 1,2,4-oxadiazoles.24 Applying this model to our system, we hypothesized that a reduction of 1 would lead to a rapid tautomerization to the acyl-amidine 7. After bond isomerization to the E isomer, 8, a rearrangement yielding 9 was possible.
Primary amidine synthesis verifies rearrangement
To verify the rearrangement, the primary amidine 9 was independently synthesized via a separate synthetic sequence for comparison of LCMS and NMR spectra. Since amidines can be easily generated from a nitrile precursor, 25,26 synthesis of the desired product would be quite similar to that of 1. Stirring the protected alanine methyl ester in ammonium hydroxide for 72 hours proved an inefficient means of generating the amide precursor necessary in the synthesis of 2. To circumvent this process, the N-Boc protection of L-alaninamide hydrochloride initiated the synthesis of the primary amidine.
Deprotection and coupling of 10 proved challenging as alkylation of the nitrile by the tert-butyl cation generated in the deprotection was a consistent problem. Limiting the formation of the alkylated-nitrile side product was achieved by closely monitoring reaction times of the trifluoroacetic acid mediated deprotection of 10 and immediately co-evaporating excess solvent with diethyl ether. Coupling of the TFA salt of 10 to 4-octylbenzoic acid proceeded in relatively low yields to form the amide 11. Limited amide formation was most likely the result of steric hindrance about the α-carbon of the amine.
As mentioned, an existing literature procedure for the formation of amidines from nitriles made amidine products very accessible. However, slow formation of the transient imidate precursor made mild heating necessary to ensure moderate product formation. After addition of ammonium chloride, the amidinium salt 9 was recovered as a white solid on precipitation with ethyl acetate.
The target-directed synthesis of amidine 9, verified the cleavage and rearrangement of the 5-phenyl-1,2,4-oxadiazole based on NMR, LCMS and HRMS analysis. When this compound was evaluated at the SphKs (Table 1), it was found to be a moderately potent dual inhibitor, displaying KI values in the mid-micromolar range.
Table 1.
9 activity at SphK1 and SphK2
| KI at SphK1 | KI at SphK2 | |
|---|---|---|
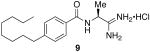 |
55 μM | 20 μM |
Structure-activity relationship of 9
Realizing the potential of a dual SphK inhibitor, we focused on optimizing 9. With this in mind, our lead compound was divided into four distinct regions for SAR evaluation (figure 2).
Figure 2.

SAR regions of 9
Head group and linker region analogs
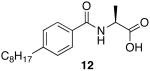
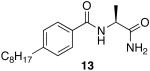
The first region studied was the head group of 9. To determine if the unique chemistry of the amidine was critical to the function of 9, the moiety was replaced with a carboxylic acid to yield 12 and a primary amide 13 (Scheme 4).
Scheme 4.

synthesis of head group analogs 12 and 13.
A small set of analogs was envisioned to probe the steric and stereochemical requirements for potent SphK inhibition by amidine based inhibitors. To better understand the steric requirements for sphingosine kinase inhibition, 14 was constructed through a PyBOP mediated amide coupling and converted to the corresponding amidine, 15 (Scheme 5).
Scheme 5.

Synthesis of linker region analog 15.
Inversion of the stereocenter in the linker region of 9 was easily achieved by starting with D-alanine. Isobutylchloroformate (IBCF) followed by ammonia in methanol was found to be a facile route for the conversion of the carboxylic acid to the primary amide en route to the nitrile 16.27 Subsequent deprotection and coupling afforded the benzamide 17, which was easily converted to the amidine resulting in the R stereoisomer of 9, 18 (Scheme 6).
Scheme 6.

Synthesis of linker region analog 18.
Finally, to increase steric bulk in the linker region, a cyclopropyl ring was installed in place of the methyl group on 9. Synthesis of this derivative was achieved through the conversion of 4-octylbenzoic acid to the corresponding acid chloride which was immediately coupled to 1-amino-1-cyclopropanecarbonitrile hydrochloride to provide the benzamide 19. Despite increased activation of the acyl chloride, amide formation in the presence of the cyclopropyl ring was extremely sluggish and yields of 30 – 40% were common. On the use of standard conditions, 20 was isolated as a white solid (Scheme 7).
Scheme 7.

Synthesis of Linker Region Analog 20.
Tail region and amide region analogs
To determine the optimal tail length for analogs of 9, a number of cyclopropyl derivatives with differing tail lengths were evaluated. Amide coupling conditions in the synthesis of these derivatives were identical to that of 20 (Scheme 8A).
Scheme 8.


Scheme 8A: Synthesis of tail region analogs 22, 23, 24 and 25.
Scheme 8B: Synthesis of inverse amide tail region analogs 27, 28, 29, 30.
Finally, the amide region of 9 was inverted to determine the importance of this portion of the lead molecule. Starting with an aniline derivative and 1-cyano-1-cyclopropane, PyBOP was used to synthesize the corresponding aryl-amide. Amide coupling reactions to form the reversed amide cyclopropyl scaffold were significantly improved over the previously described amide series most likely due to a decrease in steric bulk about the nucleophilic amine. After isolation of the amide, standard conditions yielded the corresponding amidines. (Scheme 6B)
Direct in vitro evaluation of sphingosine kinase inhibitors at SphK 1 & 2
We determined the KI values of each inhibitor at recombinant SphK1 and SphK2 (see Experimental section for details). As documented in the Table, the KI values ranged from 0.2 to greater than 100 μM (i.e., no inhibition observed when inhibitor was included at 50 μM). The inhibitors appeared to be competitive, that is, inclusion of the inhibitor increased the KM, but did not lower the Vmax, values for D-erythro-sphingosine. Data are displayed in tabular format to show the experimental KI values for the amidine-based inhibitors at SphK1 and SphK2. We also show the experimental KI values as a ratio to the experimental KM of sphingosine to indicate inhibitor potency and selectivity at the sphingosine kinases (Table 2).
Table 2.
The binding constants for D-erythro-sphingosine were measured in the presence (K′M) or absence (KM) of a fixed concentration (0.1 – 50 μM) of a VPC compound (I). KI values were calculated using the equation: KI = [I] / (K′M/KM − 1).
| KI (μM) SphK1 | KI (μM) SphK2 | KI/KM (SphK1) | KI/KM (SphK2) | |
|---|---|---|---|---|
| Sphingosine (KM) | 10 | 5 | — | — |
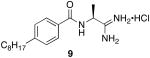 |
55 | 20 | 5.5 | 4 |
 |
>100 | >100 | >10 | >20 |
 |
>100 | >100 | >10 | >20 |
 |
> 100 | 60 | > 10 | 12 |
 |
> 100 | 25 | > 10 | 5 |
 |
25 | 23 | 2.5 | 4.6 |
 |
5 | 4 | 0.5 | 0.8 |
 |
0.2 | 0.5 | 0.02 | 0.1 |
 |
10 | 32 | 1 | 6.4 |
 |
36 | 10 | 3.6 | 2 |
 |
3 | 4 | 0.3 | 0.8 |
 |
0.3 | 6 | 0.03 | 1.2 |
 |
8.4 | > 100 | 0.8 | > 20 |
 |
30 | > 100 | 3 | > 20 |
To test for specificity, the two most potent inhibitors (23, 28) were tested at several recombinant diacylglycerol kinases (types γ, δ1, ζ) and no inhibition was observed at concentrations up to 100 μM (not shown). Many sphingosine analogs, such as DMS, have been shown to act as inhibitors of members of the protein kinase C family of enzymes. Thus, we evaluated the same two inhibitors at PKCα and observed no inhibition at concentrations up to 10 μM.
In vitro evaluation of SphK inhibitors in smooth muscle cells
SphK1 has been described as an immediate-early response gene in many cell types. In vascular smooth muscle cells, activation of the S1P1 receptor by S1P can result in smooth muscle cell proliferation and growth. Indeed, previous studies with compounds developed by our group to target S1P receptors documented that selective inhibition of the S1P1 receptor prevented S1P-induced vascular smooth muscle cell proliferation/growth in vitro and in vivo.28 In Figure 3A and 3B respectively, we show that serum (10% FBS) induces vascular smooth muscle cell Sphk1 mRNA and SphK1 activity while SphK2 mRNA and activity was unchanged. Serum stimulation also increases smooth muscle cell proliferation and growth. We tested the hypothesis that serum-induced smooth muscle cell proliferation was regulated in part by Sphk1. Cells were pre-treated with 9 and the enantiomer 18 for 30 min and then stimulated with 10% serum for 24 hours. 9 blocked serum-induced changes in S1P intracellular concentration (Figure 3C). 9, not 18, also blocked smooth muscle cell proliferation (EC50 ∼10uM, Figure 3D). As documented in Figure 3E, 9 was not cytotoxic to the cells on visual microscopic inspection. A cell viability assay was performed; this revealed that 9 did not induce cytotoxicity/death (Figure 3F).
Figure 3. Inhibition of Sphk1 with 9 prevents serum-induced smooth muscle cell proliferation.
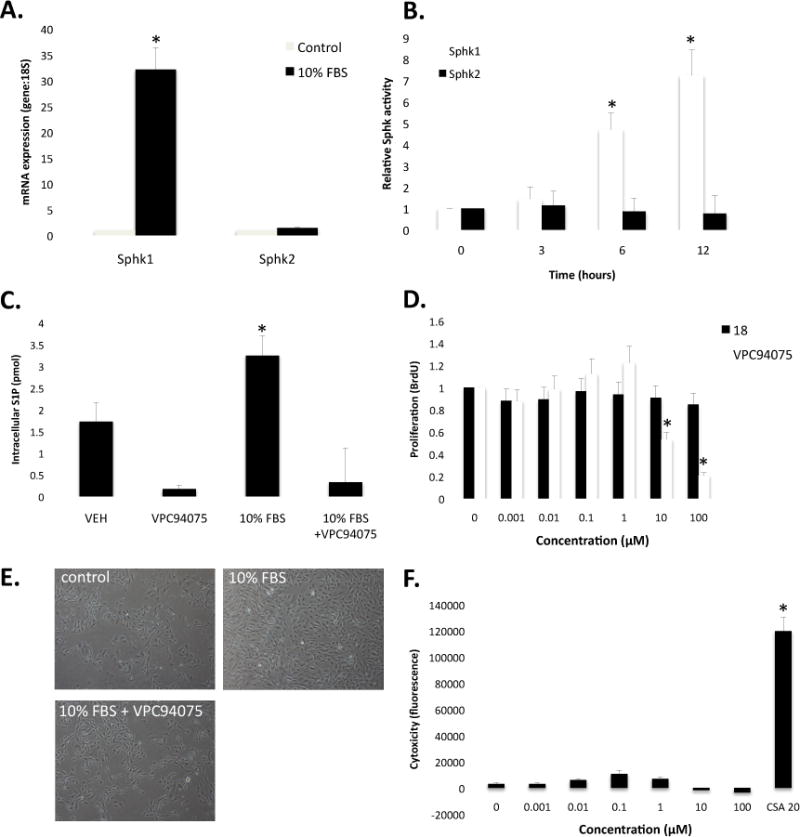
A. Quantitative real-time PCR analysis of Sphk1 and Sphk2 mRNA normalized to 18S mRNA following 4 hours of serum (10% FBS) stimulation (N=3). B. Sphk1 and Sphk2 activity ([32P]-S1P) following 0, 3, 6, and 12 hrs of 10% FBS stimulation (N=3). C. Changes in intracellular S1P concentration (C17 S1P) measured by LC/MS following 12 hrs of 10% FBS stimulation (N=2). D. Dose-response of 18 and 9 on smooth muscle cell incorporation of BrdU in response to 24 hr 10% FBS stimulation (N=6). E. Light microscopy images (20× magnification) in control, 10% FBS-treated and 10% FBS + 9 (10 μM). F. Cytotoxic response to 9 (0.001 μM – 100 μM) and 20 μM cyclosporine A (CSA) as measure by the Promega MultiTox-Fluor Assay (N=3). * denotes P < 0.05, N denotes biological replicate; mean ± st dev.
Discussion
The sphingosine kinases control, in part, the cellular equilibrium of the pro-survival lipid S1P and its pro-apoptotic precursor ceramide and the ratio of these two contrasting metabolites has been proposed to be critical to the proliferation, survival and death of cells.29 This hypothesis predicts a role for the SphKs in hyper-proliferative disease models. The highly inducible nature of SphK1 mRNA and enzyme activity has focused most attention on this SphK type,8 but a recent literature report indicates that SphK2 is the key kinase isoform in macrophage polarization to an anti-cancer phenotype.12 Such contrasting data suggests that dual and selective SphK inhibitors are needed to better characterize the relative role of these lipid kinase types.
Perhaps the most distinct structural aspect of 9 was the amidine head group. We immediately wished to understand the role this unique functional group played in sphingosine kinase inhibition. Realizing that basic amines are critical for substrate recognition by the SphKs,30 and that a well-known alkyl amidine, Distamycin A, was reported to have a pKa of 11.6 in water and DMSO,31 we postulated that a highly basic amidines was critical to SphK inhibition. To test this hypothesis, a carboxylic acid (12) and primary amide (13) derivatives were synthesized. Both are similar in size and shape to the amidine but negatively charged or neutral at a physiological pH. After evaluation at the SphKs, there was no inhibition observed in either case, at concentrations up to 100 μM. We concluded that the unique electrostatic properties associated with amidines in an aqueous environment are critical to the function of 9 and its ilk.
In the initial design of 1, we considered a previous study of the SphKs30 that identified aspartic acid residues in the substrate-binding domain that are critical to recognition of the basic amine in sphingosine. As a result, there is a high degree of stereoselectivity associated with substrates of the sphingosine kinases including analogs of the pro-drug FTY720, which are prominent SphK2 substrates.16 Interestingly, D-(+)-threo-sphingosine, wherein the amine displays stereochemistry opposite that of the natural substrate, has been found to be a weak inhibitor of the SphKs,32 Indeed, in our own study of heterocyclic amino-alcohol substrates, R-enantiomers of the imidazole analogs were found to be the most active kinase substrates, reinforcing this concept of steroselective kinase recognition.23 Presuming the same stereochemical preference existed for the other racemic amino-alcohols studied, this relative stereochemistry influenced the stereochemistry of our initial target 1.
After the unexpected palladium catalyzed reductive ring opening and rearrangement of 1 to yield the primary amidine, 9, we directed our efforts to the elucidation of the stereochemical preference of kinase inhibition. Evaluation of the enantiomer, 18, at both kinases revealed that inversion of stereochemistry in the linker region significantly influenced the activity profile, building in selectivity at SphK2 over SphK1. Although previously demonstrated to be important in neoplastic disease,12 SphK2 has been described as being pro-apoptotic. However, a lack of potent inhibitors of this SphK isoform prevents further understanding of this enzyme in such disease models. The unique activity associated with 18 makes this ligand a candidate for future scaffolds of SphK2 selective inhibitors to elucidate the role of this kinase in disease.
Understanding that inversion of stereochemistry affected kinase selectivity, we next directed our attention to determining how the deletion of stereochemistry affected the potency and selectivity of our inhibitors. Such an approach would invariably impact the steric bulk in the linker region of this class of SphK inhibitors. The achiral 15 was devoid of steric bulk in the linker region and displayed a substantial loss of kinase inhibition at SphK1, although weak inhibition at SphK2 was retained. Eliminating chirality and increasing steric bulk in the case of the 20, resulted in enhanced potency at SphK1 with no change in SphK2, thus improving activity as a dual inhibitor. After establishing the influence of sterics in the linker region on SphK inhibition, this scaffold was carried forward in the analysis of tail region SAR.
Realizing the tail region of our lead compound was significantly smaller than that of the natural substrate, sphingosine, we postulated that increasing linear size in this region of the molecule would optimize kinase inhibition. The amidine 23, having a tail region 12 carbon atoms with the cyclopropyl head group, displayed nanomolar potencies at both kinases. Comparing the KI/KM ratios of 23 at SphK1 & 2 shows only slight selectivity toward SphK1. The amidines 24 and 25 demonstrated the limits to alkyl chain length, as these analogs having 14 and 16 carbon atoms displayed a steep decrease in inhibitory activity. Such a result is most likely due to the size of the hydrophobic pocket in the substrate-binding domain of the kinases.
Inversion of the benzamide region of the molecule provided the most compelling results in terms of kinase selectivity. Overall, a similar trend of alkyl group length and potency was observed with the inverted benzamides – potency at the SphKs was increased at a length of 12 carbons, but decreased with 14 and 16 carbons. Most intriguing was the selectivity this series displayed to SphK1. The amidine 28, the 12-carbon analog, displayed a dramatic degree of selectivity for SphK1 with potencies of 0.3 μM at SphK1 and 6 μM at SphK2. Comparing the KI/KM ratios of 28 at SphK1 & 2 showed that the analog displayed 40-fold selectivity for SphK1 over SphK2. Much like 18, 28 displayed unforeseen selectivity that could serve as a template for future classes of selective, amidine-based SphK inhibitors. Such compounds, if active in vivo, could be useful in testing hypotheses of the relative roles of SphK types in hyper-proliferative diseases.
Plasma levels of S1P are reported as being in the high nanomolar range, typically between 200 and 800 nM.33 While cellular levels of sphingolipids, a group that exists in a myriad of forms, are typically considered to be approximately an order of magnitude higher.34 Not surprisingly, the observed KM of the sphingosine kinases for sphingosine is approximately 10 μM and 5 μM for SphK135 and SphK2,36 respectively. Consequently, previously described selective SphK inhibitors exhibiting seemingly poor KI values have been effective at lowering cellular S1P production and proliferation. While such inhibitors have proven quite effective at elucidating the role of the sphingosine kinases in various disease states, the relatively high dosages required for such inhibitors renders their therapeutic index unfavorable. At the outset of this study, we hoped to identify a new class of sphingosine kinase inhibitors that would be effective both in cells and in vivo with a therapeutic index that would eliminate the likelihood of drug toxicity. While the amidine-containing compounds identified in this study have improved potency over reported sphingosine kinase inhibitors, they are largely charged at physiologic pH, leading us to consider whether such molecules could penetrate cells. Evaluation of our lead compound 9 at vascular smooth muscle cells has demonstrated that amidine based sphingosine kinase inhibitors are effective at lowering overall S1P levels in these cells, correlating with a decrease in cell proliferation. Currently, efforts are underway to better characterize the more potent amidine-based inhibitors in a number of disease models.
Conclusions
At the outset of this study we sought to synthesize a deoxy 5-phenyl-1,2,4-oxadiazole derivative of a previously described SphK1 and SphK2 dual substrate. After an unexpected palladium catalyzed ring-opening and rearrangement, a new amidine based sphingosine kinase inhibitor, 9, was developed that displayed moderate potencies as an inhibitor of SphK1 and SphK2. Demonstration of the effectiveness of a moderate inhibitor in a smooth muscle cell based hyper-proliferative disease model illustrates the potential of this new class of sphingosine kinase inhibitors to answer long-standing questions on the pharmacology of the sphingosine kinases. We have identified a potent dual SphK inhibitor (23), a potent selective SphK1 inhibitor (28) and a moderately potent SphK2 selective inhibitor (18). Most importantly, we have begun to identify the pharmacophore associated with amidine-based sphingosine kinase inhibitors that we hope to exploit in future SphK inhibitors active in vivo.
Experimental Section
General synthetic materials and methods
All non-aqueous reactions were carried out in oven or flame-dried glassware under an argon or nitrogen atmosphere with dry solvents and magnetic stirring, unless otherwise stated. The argon and nitrogen were dried by passing through a tube of Drierite. Anhydrous diethyl ether (Et2O), toluene, dichloromethane (CH2Cl2), methanol (MeOH), and tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) were purchased from Aldrich or VMR Chemicals and used as received. THF was dried over activated molecular sieves (4 Å) prior to use. All other reagents were purchased from Acros chemicals and Aldrich chemicals. Except as indicated otherwise, reactions were monitored by thin layer chromatography (TLC) using 0.25 mm Whatman precoated silica gel plates. Flash chromatography was performed with the indicated solvents and Dynamic Adsorbents silica gel (particle size 0.023 – 0.040 mm). Proton (1H) and carbon (13C) NMR spectra were recorded on a Varian UnityInova 500/51 or Varian UnityInova 300/54 at 300 K unless otherwise noted. Chemical shifts are reported in ppm (δ) values relative to the solvent as follows: CDCl3 (δ 7.24 for proton and δ 77.0 for carbon NMR), DMSO-d6 (δ 2.50 for proton and δ 39.5 for carbon NMR) CD3OD (δ 3.31 for proton and δ 47.6 for carbon NMR). All high-resolution mass spectrometry was carried out by the High-Resolution Mass Spectrometry Facility in the School of Chemical Sciences at the University of Illinois Urbana-Champagne (Urbana, Il).
General procedure A: conversion of nitriles to amidines
A nitrile (0.21 mmol) was taken up in 2.1 mL of anhydrous methanol; 42 μL of a solution of 0.5 M sodium methoxide in methanol was immediately added. This mixture was allowed to stir for 15 hours at 43 °C at which time 0.231 mmol of ammonium chloride was added to the reaction, still stirring at this elevated temperature. When the reaction appeared complete by TLC, the mixture was allowed to cool to ambient temperature and the solution was evaporated to dryness. The crude material was next taken up in chloroform and filtered through a fine fritted funnel. The eluent was collected and thoroughly dried under vacuum. Finally, the reaction mixture was taken up in ethyl acetate and filtered again through a fine fritted funnel; the product was recovered as a white or off-white crystalline solid.
General procedure B: PyBOP mediated couplings of amines, anilines and amide oximes to carboxylic acids
To a suspension of an amine (0.43 mmol) in anhydrous DCM (4.3 mL) was added the acid (0.43 mmol) and PyBOP (0.43 mmol). Finally, diisopropylethylamine (1.72 mmol) was added and the reaction was allowed to stir for 15 hours. At this time, the reaction mixture was evaporated to dryness and reconstituted in 100 mL of EtOAc. The solution was extracted with four 10 mL portions of 1N HCl followed by one 10 mL portion of brine. The organic layer was dried with MgSO4 and evaporated to dryness. The crude organic material was purified by flash chromatography.
General procedure C: conversion of carboxylic acids to acyl chlorides
A carboxylic acid (0.28 mmol) and a catalytic amount of dimethylformamide were dissolved in dichloromethane (2.8 mL) and the solution was cooled to 0 °C. Oxalyl chloride (0.84 mmol) was added dropwise and the reaction mixture was warmed to room temperature. After stirring for 2 hours, the solution was evaporated to dryness; the crude material was taken up in 3 one mL portions of diethyl ether and immediately evaporated. The oil was dried under vacuum for an additional 30 minutes and carried on for alkylation by an amine.
General procedure D: alkylation of acyl chlorides by amines
To a stirring solution of a primary amine (1.68 mmol) in DCM (8.5 mL) was added an acyl chloride (2.184 mmol) as a solution in an additional volume of DCM (8.5 mL). The mixture was then cooled to 0 °C and diisopropylethyl amine (5.04 mmol) was added dropwise. The reaction mixture was allowed to warm to ambient temperature slowly and stirred for an additional 15 hours. At this time the reaction mixture was evaporated to dryness and reconstituted in ethyl acetate (150 mL). The solution was extracted with three 15 mL portions of 1N HCl followed by one 15 mL portion of brine. The organic layer was then dried with MgSO4, filtered through a fritted funnel and dried to an oil. The crude mixture was purified via flash chromatography.
General procedure E: conversion of a primary amide to a nitrile
A primary amide (9.3 mmol) and triethylamine (18.9 mmol) was taken up in 93 mL of anhydrous THF and cooled to 0 °C. Upon cooling, trifluoroacetic anhydride (11.16 mmol) was added drop-wise to the reaction mixture; after 10 minutes of stirring at this low temperature the reaction mixture was allowed to warm to ambient temperature and evaporated to dryness. The crude organic mixture was taken up in 250 mL of EtOAc and extracted with four 15 mL portions of 1N HCl followed by one 10 mL portion of brine. The organic layer was then dried with MgSO4 and evaporated to an oil.
General procedure F: trifluoroacetic acid mediated deprotection of N-Boc protected amines
To a stirring solution of the N-Boc protected amino-nitrile (1.2 mmol) in anhydrous DCM (12 mL) was added TFA, drop-wise (12 mL) at room temperature. After 15 minutes the reaction mixture was evaporated to dryness. Three 5 mL portions of diethyl ether were then added and immediately evaporated to yield the trifluoroacetate salt as an off-white solid. The salts were dried under high vacuum for a minimum of one hour before being carried forth.
(S)-1-(5-(4-octylphenyl)-1,2,4-oxadiazol-3-yl)ethanamine (1)
Compound 5 (0.43 mmol) was dissolved in wet ethanol (20 mL) to this solution was added approximately 200 mg of activated palladium on carbon (30 % w/w). The ambient atmosphere was then replaced with hydrogen and stirred for 15 hours. After this time, the reaction mixture was filtered through celite and concentrated to an oil. After purification by column chromatography (15 – 20 % MeOH in CHCl3) 1 was recovered as an off white solid (0.06 mmol). 1H NMR (500 MHz, DMSO) δ 9.05 (bs, 3H), 8.94 (d, J = 6.2, 1H), 7.91 (d, J = 8.1, 2H), 7.28 (d, J = 7.9, 2H), 4.75 − 4.64 (m, 1H), 2.61 (t, J = 7.6, 2H), 1.56 (dd, J = 6.9, 14.1, 1H),1.50 (d, J = 7.2, 3H), 1.31 − 1.14 (m, 10H), 0.83 (t, J = 6.9, 3H). 13C NMR (126 MHz, DMSO) δ 172.94, 166.97, 146.99, 130.96, 128.53, 128.36, 47.57, 40.45, 40.28, 40.11, 39.95, 39.78, 39.61, 39.45, 35.40, 31.71, 31.18, 29.24, 29.11, 29.04, 22.52, 18.83, 14.41. LCMS and HRMS data was identical to 9.
(S)-Benzyl 1-cyanoethylcarbamate (2)
General procedure E was used to convert 5.7 mmol of N-Cbz-alaninamide to the title product. After flash chromatography, 4.9 mmol of product was recovered. 1H NMR (500 MHz, CDCl3) δ 7.50 − 7.30 (m, 5H), 5.15 (s, 2H), 4.75 − 4.61 (m, 1H), 1.56 (d, J = 7.2, 3H). 13C NMR (126 MHz, CDCl3) δ 154.87, 135.50, 128.66, 128.53, 128.32, 119.14, 67.72, 38.08, 19.55.
(S, Z)-Benzyl 1-amino-1-(hydroxyimino)propan-2-ylcarbamate (3)
The intermediate 2 (4.7 mmol) was dissolved in anhydrous ethanol (7.5 mL). To this solution was added triethylamine (10.81 mmol) and hydroxylamine hydrochloride (10.34 mmol). The reaction mixture was heated to reflux for three hours after which time it was concentrated to an oil and reconstituted in dichloromethane (150 mL). The organic layer was washed with three 10 mL portions of water and one 10 mL portion of brine. The organic solution was dried of MgSO4 and evaporated to dryness. Purification by flash chromatography yielded 2.3 mmol of 3 as an off-white solid. 1H NMR (500 MHz, CD3OD) δ 7.45 − 7.22 (m, 5H), 5.07 (s, 2H), 4.21 (q, J = 7.0, 1H), 1.34 (d, J = 7.1, 3H). 13C NMR (126 MHz, CD3OD) δ 156.56, 156.27, 136.78, 128.04, 127.58, 127.45, 66.13, 18.11.
(R, Z)-Benzyl 1-amino-1-(4-octylbenzoyloxyimino)propan-2-ylcarbamate (4)
General procedure B was used to couple 4 (2.1 mmol) to 4-octylbenzoic acid. After column chromatography, 1.6 mmol of the title product was recovered as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.93 (d, J = 8.2, 2H), 7.35 (s, 5H), 7.25 (d, J = 7.2, 3H), 5.41 − 5.22 (m, 3H), 5.12 (s, 2H), 4.5 − 3.8 (m, 1H), 2.71 − 2.61 (m, 2H), 1.69 − 1.53 (m, 5H), 1.31 − 1.26 (m, 9H), 0.86 (t, J = 6.7, 3H). 13C NMR (126 MHz, CDCl3) δ 164.08, 159.49, 156.77, 148.73, 135.92, 129.45, 128.55, 128.30, 128.07, 126.83, 67.25, 48.22, 36.03, 31.85, 31.15, 29.42, 29.26, 29.22, 22.65, 17.66, 14.11.
(S)-Benzyl 1-(5-(4-octylphenyl)-1,2,4-oxadiazol-3-yl)ethylcarbamate (5)
The intermediate 4 (1.5 mmol) was dissolved in DMF (30 mL). 1 g of activated 4 Å molecular sieves were added and the reaction mixture was heated to 110 °C. After 6 hours, the mixture was cooled to ambient temperature and filtered through a medium frit. After diluting with 250 mL of ethyl acetate, the solution was extracted with eight 15 mL portions of water and one 15 mL portion of brine. The organic layer was collected, dried over MgSO4 and evaporated to dryness. Purification by flash chromatography yielded 1.14 mmol of 5. 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 8.3, 2H), 7.41 − 7.28 (m, 7H), 5.42 (d, J = 8.6, 1H), 5.26 − 5.04 (m, 2H), 2.78 − 2.60 (m, 2H), 1.74 − 1.51 (m, 5H), 1.32 − 1.25 (m, 10H), 0.88 (t, J = 6.8, 3H). 13C NMR (126 MHz, CDCl3) δ 176.26, 171.80, 155.46, 148.69, 136.21, 129.17, 128.53, 128.14, 121.37, 67.01, 44.24, 36.08, 31.84, 31.09, 29.41, 29.22, 22.66, 20.72, 14.12.
(S)-N-(1-amino-1-iminopropan-2-yl)-4-octylbenzamide hydrochloride (9)
General procedure A was used to convert 0.24 mmol of 12 to the title product. After previously described recrystallization techniques, 0.078 mmol of a white powdery product was recovered. 1H NMR (500 MHz, DMSO) δ 8.91 (bs, 1H), 8.66 (d, J = 5.2, 1H), 7.86 (d, J = 8.1, 2H), 7.29 (d, J = 8.1, 3H), 7.17 (bs, 1H), 4.65 − 4.47 (m, 1H), 2.61 (t, J = 7.6, 2H), 1.56 (dd, J = 6.9, 14.1, 2H), 1.47 (d, J = 7.2, 3H), 1.32 − 1.14 (m, 10H), 0.83 (t, J = 6.8, 3H). 13C NMR (126 MHz, DMSO) δ 172.75, 167.26, 147.07, 130.91, 128.54, 128.35, 47.91, 35.39, 31.70, 31.21, 29.23, 29.02, 22.52, 18.64, 14.41. LCMS: tR = 8.4 minutes; m/z = 304, 607 (dimer). HRMS m/z calculated for C18H30N3O (M + H): 304.2389. Found: 304.2384
(S)-tert-Butyl 1-cyanoethylcarbamate (10)
Sodium carbonate (1.5 g) was dissolved in 15 mL of water and to this solution was added alaninamide hydrochloride (5.7 mmol). In a separate flask, Di-tert-butyl dicarbonate (11.4 mmol) was dissolved in 1,4-dioxanes (11.4 mL). This organic solution was then added to the aqueous solution and allowed to stir for 15 hours. After this time, the reaction mixture was diluted with 25 mL of 1N HCl and extracted with four 25 mL portions of ethyl acetate. The organic layers were combined, washed with brine, dried over MgSO4, and concentrated to a white solid. This solid was then subjected to general procedure E. After flash chromatography, 3.1 mmol of the title product was recovered. 1H NMR (500 MHz, CDCl3) δ 4.82 (bs, 1H), 4.62 (bs, 1H), 1.54 (d, J = 7.2, 3H), 1.46 (s, 8H). 13C NMR (126 MHz, CDCl3) δ 154.10, 119.53, 37.57, 28.21, 19.62.
(S)-N-(1-Cyanoethyl)-4-octylbenzamide (11)
General procedure F was used to deprotect 10 (1.17 mmol). After standard purification techniques the off white solid was coupled to 4-octylbenzoic acid (1.17 mmol) using general procedure B. Flash chromatography yielded 0.5 mmol of the title product. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 8.1, 2H), 7.25 (dd, J = 4.0, 7.8, 2H), 6.56 (bs, 1H), 5.16 (p, J = 7.2, 1H), 2.64 (t, J = 6.4, 2H), 1.65 (ddd, J = 9.5, 10.2, 19.7, 5H), 1.42 − 1.15 (m, 10H), 0.87 (dd, J = 5.9, 7.0, 3H). 13C NMR (126 MHz, CDCl3) δ 166.58, 148.04, 129.94, 128.80, 127.18, 119.40, 36.24, 35.85, 31.83, 31.13, 29.39, 29.21, 22.63, 19.58, 14.08.
(S)-2-(4-octylbenzamido)propanoic acid (12)
General procedure B was used to couple 0.31 mmol of L-alanine methyl ester to 4-octylbenzoic acid. After standard work up procedures, the recovered material was dissolved in 5 mL of isopropanol. In a separate flask, 0.62 mmol of sodium hydroxide was dissolved in 2 mL of water and poured into isopropanol solution. The mixture was heated to 60 °C for 3 hours at which time the reaction mixture was cooled to ambient temperature and evaporated to dryness. The crude material was taken up in 50 mL of a 4:1 suspension of ethyl acetate to 1N HCl. The aqueous layer was extracted 3 additional times with ethyl acetate. The organic layers were combined, washed with brine and dried over magnesium sulfate. Evaporation of solvent yielded 0.23 mmol of the title product as an off-white solid. 1H NMR (300 MHz, CDCl3) δ 7.71 (d, J = 8.2 Hz, 2H), 7.31 − 7.16 (m, 3H), 6.80 (d, J = 6.9 Hz, 1H), 4.81-4.77 (m, 1H), 2.76 − 2.51 (m, 2H), 1.57 (d, J = 7.1 Hz, 5H), 1.27 (d, J = 7.3 Hz, 11H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 176.41, 168.48, 147.81, 131.10, 129.06, 127.39, 48.95, 36.08, 32.07, 30.81, 29.24, 23.00, 18.37, 14.32.
(S)-N-(1-amino-1-oxopropan-2-yl)-4-octylbenzamide (13)
General procedure B was used to couple 0.42 mmol of L-alaninamide hydrochloride to 4-octylbenzoic acid. After standard work up and purification procedures, 0.23 mmol of the title product was recovered. 1H NMR (500 MHz, CD3OD) δ 7.79 (d, J = 8.5 Hz 2H), 7.28 (d, J = 8.2 Hz, 2H), 4.56 (q, J = 7.2 Hz, 1H), 2.67 (t, J = 7.5 Hz, 2H), 1.66 − 1.60 (m, 2H), 1.47 (d, J = 7.2 Hz, 3H), 1.37 − 1.19 (m, 10H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ 176.59, 168.54, 147.05, 131.15, 128.13, 127.16, 49.26, 35.32, 31.59, 28.97, 22.29, 16.88, 13.00.
N-(Cyanomethyl)-4-octylbenzamide (14)
General procedure B was used to couple aminoacetonitrile bisulfate (0.42 mmol) to 4-octylbenzoic acid. After flash chromatography, 0.38 mmol of the title product was recovered. 1H NMR (300 MHz, CDCl3) δ 7.70 (d, J = 8.3, 2H), 7.25 (d, J = 7.6, 2H), 6.78 (t, J = 5.5, 1H), 4.37 (d, J = 5.8, 2H), 2.64 (t, J = 9, 2H), 1.62 (dt, J = 7.4, 22.1, 2H), 1.3 – 1.26 (m, 10H), 0.87 (t, J = 6.7, 2H). 13C NMR (75 MHz, CDCl3) δ 174.80, 148.82, 130.03, 129.08, 127.46, 117.30, 77.67, 77.25, 76.83, 36.09, 32.06, 31.35, 29.62, 29.44, 28.19, 22.86, 14.31.
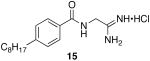
N-(2-amino-2-iminoethyl)-4-octylbenzamide hydrochloride (15)
General procedure A was used to convert 0.38 mmol of 14 to the title product. Upon isolation with standard purification techniques, 0.16 mmol of the title product was recovered as a white solid. 1H NMR (500 MHz, DMSO) δ 8.94 (s, 1H), 7.82 (d, J = 8.0, 4H), 7.29 (d, J = 7.9, 2H), 4.15 (d, J = 5.2, 2H), 2.61 (t, J = 7.4, 2H), 1.68 − 1.47 (m, 2H), 1.25 − 1.21 (m, J 10H), 0.83 (t, J = 6.4, 3H). 13C NMR (126 MHz, DMSO) δ 168.79, 167.52, 147.03, 130.99, 128.61, 128.17, 35.39, 31.70, 31.17, 29.23, 29.10, 29.02, 22.51, 14.40. LCMS: tR = 6.9 minutes; m/z = 290. HRMS m/z calculated for C17H28N3O (M + H): 290.2232. Found: 290.2232
(R)-tert-Butyl 1-cyanoethylcarbamate (16)
To a stirring solution of N-Boc-D-alanine (7.9 mmol) in dichloromethane (16 mL) was added triethylamine (35.55 mmol). This solution was cooled to -25 °C and isobutyl chloroformate (15.8 mmol) was added dropwise. The reaction was stirred at this low temperature for 45 minutes at which time ammonia (23.7 mmol) as a 7N solution in methanol was added and the reaction mixture was allowed to warm to ambient temperature and stirred for an additional 15 hours. After this time, the reaction mixture was concentrated to an oil by rotary evaporation and reconstituted in ethyl acetate (200 mL). The organic layer was washed with four 20 mL portions of 1N HCl and one 15 mL portion of brine and dried over MgSO4. The solvent was evaporated and the intermittent product was dried under vacuum. General procedure E was then used to convert the primary amide to a nitrile. 4 mmol of the title product was recovered as a white powdery solid. 1H and 13C spectral data was identical to that of 10.
(R)-N-(1-Cyanoethyl)-4-octylbenzamide (17)
General procedure F was used to deprotect 16 (1.7 mmol). General procedure B was then used to couple the resulting amine to 4-octylbenzoic acid. After standard purification techniques the title product (0.85 mmol) was recovered. 1H and 13C spectral data were identical to that of 11.
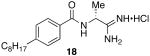
(R)-N-(1-amino-1-iminopropan-2-yl)-4-octylbenzamide hydrochloride (18)
General procedure A was used to convert 17 (0.38 mmol) to the title product. After standard purification techniques, 18 (0.15 mmol) was recovered as a white solid. 1H and 13C spectral data were identical to that of 9. LCMS: tR= 8.4 minutes; m/z = 304. HRMS m/z calculated for C18H30N3O (M + H): 304.2389. Found: 304.2382.
N-(1-Cyanocyclopropyl)-4-octylbenzamide (19)
General procedure C was used to convert 4-octylbenzoic acid (0.53 mmol) to the corresponding acyl chloride. After standard work-up procedures, the acyl-chloride was coupled to 1-amino-1-cyclopropylcarbonitrile hydrochloride (0.41 mmol) using general procedure D. After purification by flash chromatography the title product (0.21 mmol) was recovered. 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 8.3, 2H), 7.21 (d, J = 8.4, 2H), 7.12 (s, 1H), 2.68 − 2.58 (m, 2H), 1.59 (dd, J = 5.8, 8.3, 4H), 1.35 (dd, J = 5.9, 8.3, 2H), 1.32 − 1.20 (m, 10H), 0.87 (t, J = 7.0, 3H). 13C NMR (126 MHz, CDCl3) δ 168.06, 148.02, 129.96, 128.73, 127.27, 120.23, 35.85, 31.83, 31.13, 29.39, 29.21, 22.63, 20.88, 16.91, 14.08.
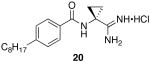
N-(1-carbamimidoylcyclopropyl)-4-octylbenzamide hydrochloride (20)
General procedure A was used to convert 15 (0.17 mmol) to the corresponding amidine. After standard work-up and purification techniques 20 (0.04 mmol) was recovered as a white solid. 1H NMR (500 MHz, CD3OD) δ 7.82 (d, J = 8.3, 2H), 7.30 (d, J = 8.2, 2H), 2.76 − 2.62 (m, 2H), 1.75 (dd, J = 5.9, 8.5, 2H), 1.64 (dd, J = 7.1, 14.3, 2H), 1.56 (dd, J = 6.1, 8.4, 2H), 1.29 (dd, J = 7.1, 15.0, 11H), 0.89 (t, J = 7.0, 3H). 13C NMR (126 MHz, CD3OD) δ 172.52, 169.94, 147.79, 130.21, 128.19, 127.47, 35.32, 32.59, 31.58, 30.98, 29.11, 28.97, 28.82, 22.29, 18.16, 12.99. LCMS: tR = 8.5 minutes; m/z = 316. HRMS m/z calculated for C19H30N3O (M + H): 316.2389. Found: 316.2381.
N-(1-Cyanocyclopropyl)-4-decylbenzamide (21; C10)
General procedure C was used to convert 2.184 mmol of 4-decylbenzoic acid to the corresponding acyl chloride. After work up, this intermediate was immediately subjected to methods described in general procedure D in the presence of 1-amino-1-cyclopropanecarbonitrile hydrochloride (1.68 mmol). After flash chromatography, 1 mmol of the title product was recovered as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (dd, J = 3.2, 8.1, 2H), 7.21 (d, J = 8.0, 2H), 2.66 − 2.58 (m, 2H), 1.59 (d, J = 5.6, 4H), 1.36 (q, J = 5.8, 2H), 1.32 − 1.20 (m, 15H), 0.87 (t, J = 6.9, 3H). 13C NMR (126 MHz, CDCl3) δ 168.16, 148.04, 129.96, 128.74, 127.32, 120.30, 35.86, 31.88, 31.15, 29.59, 29.45, 29.31, 29.23, 22.67, 20.88, 16.91, 14.12.
N-(1-Cyanocyclopropyl)-4-dodecylbenzamide (21; C12)
General procedure C was used to convert 4-dodecylbenzoic acid (0.52 mmol) to the corresponding acyl chloride. After standard work up procedures, the intermediate was immediately subjected to conditions described in general procedure D in the presence of 1-amino-1-cyclopropanecarbonitrile hydrochloride (0.4 mmol). After flash chromatography, 0.15 mmol of the title product was recovered. 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 7.1, 2H), 7.23 (d, J = 7.5, 2H), 6.82 (s, 1H), 2.63 (t, J = 7.7, 2H), 1.65 − 1.57 (m, 4H), 1.35 (t, J = 7.0, 2H), 1.29 − 1.25 (m, 17H), 0.88 (t, J = 6.5, 3H). 13C NMR (126 MHz, CDCl3) δ 167.81, 148.06, 130.01, 128.75, 127.19, 120.07, 35.85, 31.89, 31.11, 29.61, 29.53, 29.42, 29.32, 29.20, 22.66, 20.88, 16.97, 14.08.
N-(1-Cyanocyclopropyl)-4-tetradecylbenzamide (21; C14)
Tetradecanoic acid (0.61 mmol) was subjected to the methods described in general procedure C. After standard work up procedures were used, the intermediate acyl chloride was coupled to 1-amino-1-cyclopropanecarbonitrile hydrochloride (0.47 mmol) using general procedure D. After flash chromatography 0.28 mmol of the title product was isolated. 1H NMR (300 MHz, CDCl3) δ 7.68 (d, J = 8.2, 2H), 7.23 (d, J = 8.2, 2H), 6.85 (s, 1H), 2.63 (t, J = 9, 2H), 1.64 − 1.55 (m, 4H), 1.35 (dd, J = 5.8, 8.5, 2H), 1.25 (s, 20H), 0.87 (t, J = 6.6, 3H). 13C NMR (75 MHz, CDCl3) δ 168.05, 148.30, 130.19, 128.98, 127.43, 120.32, 36.09, 32.15, 31.38, 29.88, 29.68, 29.58, 29.45, 22.92, 21.10, 17.20, 14.36.
N-(1-Cyanocyclopropyl)-4-hexadecylbenzamide (21; C16)
General procedure C was used to convert hexadecanoic acid (0.58 mmol) to the corresponding acyl chloride. After standard work up techniques, the intermediate was coupled to 1-amino-1-cyclopropanecarbonitrile hydrochloride using the methods described in general procedure D. Upon flash chromatography 0.41 mmol of the title product was recovered. 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 8.2, 2H), 7.23 (d, J = 8.1, 2H), 6.81 (s, 1H), 2.63 (t, J = 7.7, 3H), 1.64 − 1.58 (m, 4H), 1.35 (q, J = 4.9, 2H), 1.33 − 1.21 (m, 27H), 0.88 (t, J = 6.9, 3H). 13C NMR (126 MHz, CDCl3) δ 167.79, 148.06, 130.01, 128.75, 127.19, 120.07, 97.26, 35.84, 31.90, 31.11, 29.66, 29.63, 29.54, 29.43, 29.33, 29.20, 22.67, 20.88, 16.96, 14.08.
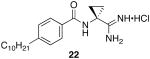
N-(1-carbamimidoylcyclopropyl)-4-decylbenzamide hydrochloride (22)
General procedure A was used to convert 21; C10 (0.12 mmol) to the corresponding amidine. After standard recrystallization techniques, 0.026 mmol of the title product was recovered. 1H NMR (500 MHz, CD3OD) δ 7.81 (d, J = 6.1, 2H), 7.30 (d, J = 6.3, 2H), 2.67 (t, J = 6.7, 2H), 1.75 (s, 2H), 1.63 (s, 2H), 1.56 (s, 2H), 1.29 (d, J = 20.9, 14H), 0.89 (dd, J = 4.5, 7.0, 3H). 13C NMR (126 MHz, CD3OD) δ 172.51, 169.93, 147.78, 130.20, 128.34, 128.04, 127.48, 35.32, 32.59, 31.63, 30.97, 29.27, 28.81, 28.66, 22.30, 18.15, 12.98. LCMS: tR = 10.34; m/z = 344. HRMS m/z calculated for C21H34N3O (M + H): 344.2702. Found: 344.2698.
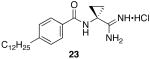
N-(1-carbamimidoylcyclopropyl)-4-dodecylbenzamide hydrochloride (23)
General procedure A was used to convert 21; C12 (0.15 mmol) to the corresponding amidine. Upon previously described recrystallization procedures 0.06 mmol of the title product was recovered. 1H NMR (500 MHz, DMSO) δ 9.08 (s, 1H), 8.66 (bs, 4H), 7.81 (d, J = 6.7, 2H), 7.27 (d, J = 6.9, 2H), 2.60 (s, 2H), 1.65 (s, 2H), 1.54 (s, 2H), 1.37 (s, 2H), 1.21 (s, 19H), 0.83 (s, 3H). 13C NMR (126 MHz, DMSO) δ 172.22, 167.99, 147.01, 131.21, 128.45, 128.32, 35.38, 33.09, 31.74, 31.23, 29.45, 29.27, 29.16, 28.98, 22.54, 18.42, 14.42. LCMS: tR = 10.85 minutes; m/z = 372. HRMS m/z calculated for C23H38N3O (M + H): 372.3015. Found: 302.3005.
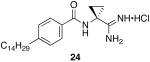
N-(1-carbamimidoylcyclopropyl)-4-tetradecylbenzamide hydrochloride (24)
General procedure A was used to convert 0.26 mmol of 21; C14 to the corresponding amidine. After standard recrystallization techniques, 0.04 mmol of the title product was recovered as a white solid. 1H NMR (500 MHz, DMSO) δ 9.04 (s, 1H), 8.61 (bs, 4H), 7.81 (d, J = 7.8, 2H), 7.27 (d, J = 5.8, 2H), 2.60 (t, J = 7.1, 2H), 1.65 (s, 2H), 1.54 (d, J = 5.5, 2H), 1.37 (s, 2H), 1.24 − 1.21 (m, 21H), 0.83 (t, J = 6.5, 3H). 13C NMR (126 MHz, DMSO) δ 172.23, 168.02, 147.03, 131.24, 128.45, 128.31, 127.93, 35.38, 33.12, 31.72, 31.20, 29.44, 29.25, 29.13, 28.98, 22.52, 18.40, 14.39. LCMS: tR = 12.3 minutes; m/z = 400. HRMS m/z calculated C25H42N3O (M + H): 400.3328. Found: 400.3322.
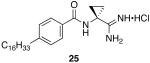
N-(1-carbamimidoylcyclopropyl)-4-hexadecylbenzamide hydrochloride (25)
General procedure A was used to convert 0.18 mmol of 21; C16 to the corresponding amidine. However, instead of methanol, ethanol was used to solubilize the starting material. After standard recrystallization techniques, 0.04 mmol of the title product was recovered as a white solid. 1H NMR (500 MHz, DMSO) δ 9.06 (s, 1H), 8.65 (bs, 4H), 7.81 (d, J = 7.5, 2H), 7.27 (d, J = 7.5, 2H), 2.67 − 2.56 (m, 2H), 1.65 (s, 2H), 1.54 (d, J = 4.0, 2H), 1.37 (s, 2H), 1.21 (bs, 25H), 0.84 (t, J = 5.4, 3H). 13C NMR (126 MHz, DMSO) δ 172.16, 168.01, 147.02, 131.21, 128.45, 128.31, 35.38, 33.11, 31.74, 31.24, 29.46, 29.27, 29.15, 28.99, 22.54, 18.41, 14.42. LCMS: tR = 12.4; m/z = 428. HRMS m/z calculated C27H46N3O (M + H): 428.3641. Found: 428.3642.
1-Cyano-N-(4-decylphenyl)cyclopropanecarboxamide (26; C10)
General procedure B was used to couple 4-decylaniline (1.00 mmol) to 1-cyanocyclopropanecarboxylic acid. After column chromatography, 0.99 mmol of the title product was recovered as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.16 (s, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 2.73 − 2.40 (m, 2H), 1.77 (dd, J = 8.2, 4.4 Hz, 2H), 1.68 − 1.51 (m, 4H), 1.29 (d, J = 10.1 Hz, 14H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.54, 140.27, 134.81, 129.13, 120.87, 120.27, 35.63, 32.16, 31.69, 29.86, 29.75, 29.60, 29.49, 22.95, 18.44, 14.39, 14.32.
1-Cyano-N-(4-dodecylphenyl)cyclopropanecarboxamide (26; C12)
General procedure B was used to couple 4-dodecylaniline (1.00 mmol) to 1-cyanocyclopropanecarboxylic acid. After column chromatography, 0.99 mmol of the title product was recovered as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.18 (s, 1H), 7.41 (d, J = 8.3 Hz, 2H), 7.14 (d, J = 8.3 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 1.83 − 1.67 (m, 2H), 1.67 − 1.45 (m, 4H), 1.28 (s, 18H), 0.90 (t, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.53, 140.24, 134.84, 129.12, 120.88, 120.26, 35.63, 32.19, 31.70, 29.93, 29.77, 29.63, 29.51, 22.96, 18.44, 14.40, 14.31.
1-Cyano-N-(4-tetradecylphenyl)cyclopropanecarboxamide (26; C14)
General procedure B was used to couple 4-tetradecylaniline (1.00 mmol) to 1-cyanocyclopropanecarboxylic acid. After column chromatography, 0.99 mmol of the title product was recovered as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.24 − 8.00 (m, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 2.76 − 2.38 (m, 2H), 1.77 (dd, J = 8.2, 4.4 Hz, 2H), 1.67 − 1.50 (m, 4H), 1.27 (s, 22H), 0.89 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.50, 140.32, 134.74, 129.16, 120.81, 120.28, 35.63, 32.18, 31.70, 29.93, 29.76, 29.63, 29.50, 22.95, 18.45, 14.39.
1-Cyano-N-(4-hexadecylphenyl)cyclopropanecarboxamide (26; C16)
General procedure B was used to couple 4-hexadecylaniline (1.00 mmol) to 1-cyanocyclopropanecarboxylic acid. After column chromatography, 0.99 mmol of the title product was recovered as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.29 − 7.98 (m, 1H), 7.43 (t, J = 14.0 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 2.78 − 2.44 (m, 2H), 1.77 (dd, J = 8.2, 4.4 Hz, 2H), 1.67 − 1.50 (m, 4H), 1.27 (s, 26H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.50, 140.29, 134.78, 129.14, 120.83, 120.27, 35.63, 32.19, 31.71, 29.95, 29.77, 29.64, 29.51, 22.96, 18.44, 14.40.
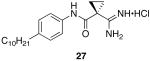
1-carbamimidoyl-N-(4-decylphenyl)cyclopropanecarboxamide hydrochloride (27)
General procedure A was used to convert 0.99 mmol of 26; C10 to the corresponding amidine. After standard recrystallization techniques, 0.51 mmol of the title product was recovered as a white solid. 1H NMR (500 MHz, DMSO) δ 9.69 (s, 1H), 8.96 (s, 4H), 7.63 − 7.31 (m, 2H), 7.17 − 7.04 (m, 2H), 2.66 − 2.30 (m, 2H), 1.66 − 1.34 (m, 6H), 1.19 (dd, J = 30.7, 9.2 Hz, 14H), 0.90 − 0.76 (m, 3H). 13C NMR (126 MHz, DMSO) δ 168.27, 166.44, 138.41, 136.53, 128.64, 128.58, 121.33, 121.28, 34.98, 31.73, 31.44, 29.98, 29.45, 29.30, 29.13, 29.02, 22.54, 15.12, 14.41. LCMS: tR = 9.8 minutes; m/z = 344. HRMS m/z calculated C21H34N3O (M + H): 344.2702. Found: 344.2690.
1-carbamimidoyl-N-(4-dodecylphenyl)cyclopropanecarboxamide hydrochloride (28)
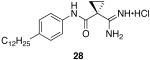
General procedure A was used to convert 0.99 mmol of 26; C12 to the corresponding amidine. After standard recrystallization techniques, 0.45 mmol of the title product was recovered as a white solid. 1H NMR (300 MHz, DMSO) δ 9.57 (s, 1H), 9.16 (s, 2H), 8.91 (s, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 2.48 (m, 2H), 1.52 (m, 6H), 1.21 (s, 18H), 0.83 (s, 3H). 13C NMR (75 MHz, DMSO) δ 168.33, 166.76, 138.72, 136.69, 128.88, 121.61, 35.21, 31.97, 31.67, 30.17, 29.69, 29.53, 29.39, 29.26, 22.77, 15.42, 14.65. LCMS: tR = 11.5 minutes; m/z = 372. HRMS m/z calculated C23H38N3O (M + H): 372.3015. Found: 372.3011.
1-carbamimidoyl-N-(4-tetradecylphenyl)cyclopropanecarboxamide hydrochloride (29)
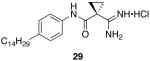
General procedure A was used to convert 0.99 mmol of 26; C14 to the corresponding amidine. After standard recrystallization techniques, 0.37 mmol of the title product was recovered as a white solid 1H NMR (300 MHz, DMSO) δ 9.57 (s, 1H), 9.16 (s, 2H), 8.91 (s, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 2.48 (s, 2H), 1.52 (dd, J = 40.8, 22.8 Hz, 6H), 1.21 (s, 22H), 0.83 (s, 3H). 13C NMR (75 MHz, DMSO) δ 168.33, 166.76, 138.72, 136.69, 128.88, 121.61, 35.21, 31.97, 31.67, 30.17, 29.69, 29.53, 29.39, 29.26, 22.77, 15.42, 14.65. tR = 12.5 minutes; m/z = 400. HRMS m/z calculated C25H42N3O (M + H): 400.3328. Found: 400.3334.
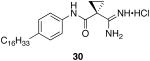
1-carbamimidoyl-N-(4-hexadecylphenyl)cyclopropanecarboxamide hydrochloride (30)
General procedure A was used to convert 0.99 mmol of 26; C16 to the corresponding amidine. However, instead of methanol, ethanol was used to solubilize the starting material. After standard recrystallization techniques, 0.27 mmol of the title product was recovered as a white solid. 1H NMR (500 MHz, DMSO) δ 9.92 (s, 2H), 9.82 − 9.58 (s, 1H), 9.06 (s, 2H), 7.47 (s, 2H), 7.09 (s, 2H), 2.49 (s, 2H), 1.46 (d, J = 62.7 Hz, 6H), 1.21 (s, 26H), 0.83 (s, 3H). 13C NMR (75 MHz, DMSO) δ 168.32, 166.75, 138.71, 136.69, 128.87, 121.59, 35.21, 31.97, 31.69, 30.17, 29.70, 29.54, 29.39, 29.28, 22.78, 15.42, 14.66. tR = 13.2; m/z = 428. HRMS m/z calculated C27H46N3O (M + H): 428.3641. Found: 428.3639.
Sphingosine kinase assay
Human SphK1 and mouse SphK2 cDNAs were used to generate mutant baculoviruses that encoded these proteins. Infection of Sf9 insect cells with the viruses for 72 hours resulted in > 1,000–fold increases in SphK activity in 10,000 × g supernatant fluid from homogenized cell pellets. The enzyme assay conditions were exactly as described16 except infected Sf9 cell extract containing 2-3 μg protein was used as a source of enzyme.
Vascular smooth muscle cell bioassays
Rat aortic vascular smooth muscle cells were cultured in vitro as previously described.28 Total mRNA was extracted using Trizol (Invitrogen), cDNA was synthesized using the iScript cDNA synthesis kit (BioRad), and quantitative real-time polymerase chain reaction (PCR) was used to measure Sphk1, Sphk2 and 18S mRNA expression as previously described.27 Primer sequences: Sphk1 F: GTGCCATCCAGAAACCCCTA, R: AGTCCACGTCAGCAACGAAA; Sphk2 F: TGGGAAGGCATTGTCACTGT, R: AAGAAGCGAGCAGTTGAGCA; 18S F: CGGCTACCACATCCAAGGAA, R: AGCTGGAATTACCGCGGC. Proliferation was assessed by BrdU incorporation (10 μM) based on the Roche BrdU immuno-absorbance assay. Cytotoxic response was determined by the Promega MultiTox-Fluor Assay.
Quantification of S1P by LC/MS
S1P and DHS1P were quantified as their bisacetylated derivatives (BisAcS1P and BisAcDHS1P) using adaptations of previously reported methods.37 Derivatization eliminates sample carryover that is a noted problem with these analytes. In brief, cells were washed in PBS and then scraped into 2 mL of ice cold MeOH which was transferred to glass tubes containing 1 ml CHCl3, 0.5 ml 0.1 M HCl and 50 pmol of C17 S1P. After vortexing, an additional 1 ml of CHCl3 and 1.3 mL 0.1 M HCl were added to the tubes containing the cell suspension. The tubes were vortexed a second time and centrifuged. The lipid-containing organic phase was separated into a second 4 ml glass vial. The organic layer was evaporated to dryness and the material was acetylated with acetic anhydride in pyridine. After a second evaporation, the resulting material was dissolved in MeOH for analysis. Samples were separated on an Aglient Technologies Zorbax Eclipse XDB-C8 column, 4.6×150 mm, 5 mm particle size using methanol/water/HCOOH, 79/20/0.5, v/v, with 5 mM NH4COOH as solvent A and methanol/acetonitrile/HCOOH, 59/40/0.5, v/v, with 5 mM NH4COOH as Solvent B. Analytes were detected by electro-spray ionization tandem mass spectrometry using an ABI 4000 Q-Trap instrument operated in positive ionization mode with optimized declustering potentials, collision energies and exit potentials. The following m/z transitions were monitored: BisAcC17S1P 448/388; BisACS1P, 462/402; BisAcDHS1P, 464/404. Calibration curves were generated using BisAcS1P BisAcDHS1P and BisAcDHS1P that were prepared from S1P, DHS1P and C17S1P obtained from Avanti Polar Lipids and quantified independently by phosphorous analysis.
Liquid chromatography and mass spectrometry for evaluation of chemical purity
All compound subjected to biological evaluation were determined to be >95% pure by LCMS evaluation. High performance liquid chromatography – mass spectrometry (LCMS) was carried out on a Waters 2695 separations module and a Finnigan LCQ series mass spectrometer. All compounds were evaluated for purity using a Thompson Instrument Company Advantage C18 column. Columns were outfitted with 5 μm beads with a 60 Å pore size; columns were 250 mm in length and 4.6 mm in diameter. Mobile phase A consisted of HPLC grade H2O and 0.01% TFA; mobile phase B consisted of MeCN and 0.01% TFA. LCMS identification and purity utilized a binary gradient starting with 90% A and 10% B and linearly increasing to 100% B over the course of 10 minutes, followed by an isocratic flow of 100% B for an additional 10 minutes. A flow rate of 1 mL/minute was maintained throughout the HPLC method. The purity of all products was determined by integration of the total ion count (TIC) spectra and integration of the ultraviolet (UV) spectra at 214 nm; a Waters 486 Tunable Absorbance Detector was used to collect all UV data. Retention times are abbreviated as tR; mass to charge ratios are abbreviated as m/z.
Supplementary Material
Figure 1.

R enantiomer of VPC45129 and its deoxy counterpart, 1.
Scheme 1.

Synthesis of the 5-phenyl-1,2,4-oxadiazole 1.
Scheme 2.

Reductive ring opening and rearrangement of 1.
Scheme 3.

Synthesis of the primary amidinium chloride 9.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 GM067958 to KRL and TLM, T32 GM008715 to PCK; HL081682 to BRW; GM50388 to AJM and American Heart Association SDG to BRW. We also thank Dr. Pam Schoppee-Bortz, PhD, Cardiovascular Division, MSTP-candidate John Hogan, pre-doctoral candidate Monica Lee, ME, Department of Biomedical Engineering, and Amanda M. Gellett, Department of Pharmacology, University of Virginia, for their technical expertise.
Footnotes
Abbreviations: S1P, sphingosine 1-phosphate; SphK1, sphingosine kinase type 1; SphK2, sphingosine kinase type 2; DAG, diacylglycerol; SAR, structure-activity relationship; S1Pn (n = 1 – 5), sphingosine 1-phosphate receptor subtype.
Supplementary Information: Additional 1H and 13C NMR data is available free of charge at http://www.pubs.acs.org.
References
- 1.Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006;1758:2016–2026. doi: 10.1016/j.bbamem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Spiegel S, Milstien S. Sphingosine-1-phosphate: and enigmatic signaling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 3.Lynch KR. Lysophospholipid Receptor Nomenclature. Biochim Biophys Acta. 2002;1582:70–71. doi: 10.1016/s1388-1981(02)00138-5. [DOI] [PubMed] [Google Scholar]
- 4.Rosen H, Sanna M, Germana C, Stuart M, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 5.French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 6.Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okada T, Ding G, Sonoda H, Kajimoto T, Haga Y, Khosrowbeygi A, Gao S, Miwa N, Jahangeer S, Nakamura SI. Involvement of N-terminal-extended form of sphingosine kinase 2 in serum-dependent regulation of cell proliferation and apoptosis. J Biol Chem. 2005;280:36318–36325. doi: 10.1074/jbc.M504507200. [DOI] [PubMed] [Google Scholar]
- 8.Johnson KR, Becker KP, Facchinetti MM, Hannun YA, Obeid LM. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA) J Biol Chem. 2002;277:35257–35262. doi: 10.1074/jbc.M203033200. [DOI] [PubMed] [Google Scholar]
- 9.Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- 11.Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, Spiegel S. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weigert A, Schiffmann S, Sekar D, Ley S, Menrad H, Werno C, Grosch S, Geisslinger G, Brüne B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards and anti-inflammatory phenotype. International Journal of Cancer. 2009 doi: 10.1002/ijc.24594. PMID: 19618460. [DOI] [PubMed] [Google Scholar]
- 13.Wong L, Tan SSL, Lam Y, Melendez AJ. Synthesis and evaluation of sphingosine analogues as inhibitors of sphingosine kinases. J Med Chem. 2009;52:3618–3626. doi: 10.1021/jm900121d. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Disc. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- 15.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 16.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 17.Kharel Y, Lee S, Snyder AH, Sheasley-O'neill SL, Morris MA, Setiady Y, Zhu R, Zigler MA, Burcin TL, Ley K, Tung KSK, Engelhard VH, Macdonald TL, Pearson-White S, Lynch KR. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005;280:36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 18.Dev KK, Mullershausen F, Mattes H, Kuhn RR, Bilbe G, Hoyer D, Mir A. Brain sphingosine-1-phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol Ther. 2008;117:77–93. doi: 10.1016/j.pharmthera.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of Para-Alkyl Aryl Amide Analogues of Sphingosine-1-Phosphate: Discovery of Potent S1P Receptor Agonists. Bioorg Med Chem Lett. 2003;13:3401–3404. doi: 10.1016/s0960-894x(03)00812-6. [DOI] [PubMed] [Google Scholar]
- 20.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of 4(5)-phenylimidazole-based analogues of Sphingosine-1-Phosphate and FTY720: Discovery of Potent S1P1 Receptor Agonists. Bioorg Med Chem Lett. 2005;15:3568–3572. doi: 10.1016/j.bmcl.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 21.Foss FW, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of α-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg Med Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu R, Snyder AH, Kharel K, Schaffter L, Sun Q, Kennedy PC, Lynch KR, Macdonald TL. Asymmetric Synthesis of Conformationally Constrained Fingolimod Analogues-Discovery of an Orally Active Sphingosine 1-Phosphate Receptor Type-1 Agonist and Receptor Type-3 Antagonist. J Med Chem. 2007;50:6428–6435. doi: 10.1021/jm7010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foss FW, Mathews TP, Kharel Y, Kennedy PC, Snyder AH, Davis MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of sphingosine kinase substrates as sphingosine 1-phosphate receptor prodrugs. Bio Org Med Chem. 2009;17:6123–6136. doi: 10.1016/j.bmc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyrkov A, Usova A. Reduction of Substituted 5-(Nitromethyl)-3-phenyl-1, 2, 4-oxadiazoles. Russ Jour Org Chem. 2004;40:286–287. [Google Scholar]
- 25.Kitagawa H, Ozawa T, Takahata S, Iida M, Saito J, Yamada M. Phenylimidazole derivatives of 4-pyridone as dual inhibitors of bacterial enoyl-acyl carrier protein reductases FabI and FabK. J Med Chem. 2007;50:4710–4720. doi: 10.1021/jm0705354. [DOI] [PubMed] [Google Scholar]
- 26.Schaefer FC, Peters GA. Base-Catalyzed Reaction of Nitriles with Alcohols. A Convenient Route to Imidates and Amidine Salts. J Org Chem. 1961;26:412–418. [Google Scholar]
- 27.Tang FY, Qu LQ, Xu Y, Ma RJ, Chen SH, Li G. Practical Synthesis of Structurally Important Spirodiamine Templates. Synth Comm. 2007;37:3793–3799. [Google Scholar]
- 28.Wamhoff BR, Lynch KR, Macdonald TL, Owens GK. Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle cell phenotype. ATVB. 2008;28:1454–1461. doi: 10.1161/ATVBAHA.107.159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 30.Yokota S, Taniguchi Y, Kihara A, Mitsutake S, Igarashi Y. Asp177 in C4 domain of mouse sphingosine kinase 1a is important for the sphingosine recognition. FEBS Lett. 2004;578:106–110. doi: 10.1016/j.febslet.2004.10.081. [DOI] [PubMed] [Google Scholar]
- 31.Koppel I, Koppel J, Leito I, Grehn L. Basicity of 3-aminopropionamidine derivatives in water and dimethyl sulphoxide. J Phys Org Chem. 1996;9:265–268. [Google Scholar]
- 32.Buehrer BM, Bell RM. Inhibition of sphingosine kinase in vitro and in platelets. J Biol Chem. 1992;267:3154–3159. [PubMed] [Google Scholar]
- 33.Lynch KR, Macdonald TM. Sphingosine 1-phosphate chemical biology. Biochim Biophys Acta. 2008;1781:508–512. doi: 10.1016/j.bbalip.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 35.Olivera A, Kohama T, Tu Z, Milstien S, Spiegel S. Purification and characterization of rat kidney sphingosine kinase. J Biol Chem. 1998;273:12576–12583. doi: 10.1074/jbc.273.20.12576. [DOI] [PubMed] [Google Scholar]
- 36.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 37.Berdyshev EV, Gorshkova IA, Garcia JGN, Natarajan V, Hubbard WC. Quantitative analysis of sphingoid base-1-phosphates as bisacetylated derivatives by liquid chromatography-tandem mass spectrometry. Anal Biochem. 2005;339:129–136. doi: 10.1016/j.ab.2004.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.