

Abstract
We have used a new ApoA-I transgenic mouse model to identify by global gene expression profiling, candidate genes that affect lipid and lipoprotein metabolism in response to fenofibrate treatment. Multilevel bioinformatical analysis and stringent selection criteria (2-fold change, 0% false discovery rate) identified 267 significantly changed genes involved in several molecular pathways. The fenofibrate-treated group did not have significantly altered levels of hepatic human APOA-I mRNA and plasma ApoA-I compared with the control group. However, the treatment increased cholesterol levels to 1.95-fold mainly due to the increase in high-density lipoprotein (HDL) cholesterol. The observed changes in HDL are associated with the upregulation of genes involved in phospholipid biosynthesis and lipid hydrolysis, as well as phospholipid transfer protein. Significant upregulation was observed in genes involved in fatty acid transport and β-oxidation, but not in those of fatty acid and cholesterol biosynthesis, Krebs cycle and gluconeogenesis. Fenofibrate changed significantly the expression of seven transcription factors. The estrogen receptor-related gamma gene was upregulated 2.36-fold and had a significant positive correlation with genes of lipid and lipoprotein metabolism and mitochondrial functions, indicating an important role of this orphan receptor in mediating the fenofibrate-induced activation of a specific subset of its target genes.
Keywords: fenofibrate, lipid catabolism, estrogen receptor-related gamma HDL, phospholipid biosynthesis.
Introduction
Fibrates are important lipid-lowering drugs. They have been used in clinical practice for more than three decades for the treatment of primary and secondary dyslipidemias.1, 2 In subjects with low high-density lipoprotein (HDL) levels, fibrates have been shown to reduce triglycerides and increase HDL cholesterol, without significantly affecting low-density lipoprotein (LDL) cholesterol, and to decrease the mortality from coronary artery disease.3 Fibrates in the liver are believed to act, at least in part, through the peroxisome proliferators-activated receptor-alpha (Pparα). It is believed that ligands generated by fibrates bind and activate moderately Pparα. Activated Pparα in turn heterodimerizes with retinoid X receptor alpha (Rxrα), binds to hormone response elements and activates the target genes.4 Previous studies have suggested that fibrates promote lipolysis of triglycerides by increasing the gene expression and the activity of lipoprotein lipase (Lpl),5, 6 increasing the expression of Apoa57, 8 and decreasing the expression of Apoc3.9, 10 Fibrates were shown to promote β-oxidation of fatty acids (FA) by upregulating various genes, including several genes of FA uptake and β-oxidation.11, 12, 13, 14, 15, 16, 17, 18, 19 In rodents, fibrates downregulated the expression of Apoa1,20 hepatic lipase13 and lecithin cholesterol acyltransferase (Lcat)15 genes and upregulated the expression of Apoa221 and phospholipid transfer protein (Pltp).22 Previous studies of human ApoA-I transgenic Mice and rabbits indicated that fenofibrate increase plasma ApoA-I and HDL levels. The animal models used in these studies carry the human transgene under the control of its proximal promoter and also express their endogenous ApoA-I gene.20, 23, 24
To obtain insights on the global molecular changes (34 000 genes) that contribute to lipid lowering and HDL raising effects of fenofibrate, we used a new human ApoA-I mouse model that expresses the human APOA-I gene under the control of the proximal promoter and distal enhancer sequences, in the background of Apoa1-deficient mice.
The fenofibrate treatment did not significantly alter the expression of the human APOA-I gene and plasma ApoA-I levels, but it increased HDL cholesterol to approximately 2-fold and shifted the HDL peak toward lower densities. The expression of other genes involved in the biogenesis of HDL was not affected. The increase in HDL cholesterol was associated with the upregulation of genes involved in phospholipid biosynthesis, phospholipid transfer in plasma and in lipid hydrolysis.
Fenofibrate significantly upregulated genes that are involved in fatty acid transport, β-oxidation and other mitochondrial functions. These changes correlated positively with changes in the estrogen-related receptor gamma (Esrrg) gene and negatively with changes in the T-rich interactive domain 5b (Arid5b) gene, indicating involvement of these transcription factors in the fenofibrate-induced changes in gene expression.
Materials and methods
Animal models
The transgenic mice harboring the human APOA-I gene and its regulatory sequences have been described previously.25 Briefly, the transgenic construct contains 2.1 kb of the 5′ regulatory sequence along with 1.81 kb containing the entire coding sequences of the APOA-I gene, a 1.6-kb segment containing the intragenic sequence and the CAT gene in front of the −890/+24 regulatory sequence of the APOCIII gene, that encompasses the common enhancer of the APOA-I and APOCIII genes. In this construct, the APOCIII gene was replaced by the CAT cDNA sequence.25
Treatment of ApoA-I transgenic mice with fenofibrate
Four-month old transgenic mice harboring the human APOA-I gene and its regulatory sequence25 were separated in four groups of five according to gender. Both the control and the treatment groups were fed for 16 days with a diet containing, as percent of calories, 18.2% proteins, 54.3% carbohydrates, 27.5% fat along with vitamins and minerals as described (Supplementary Table 1).26 The control group continued on the same diet for 16 days, whereas the treatment group received the same diet containing 31 mg fenofibrate/100 g diet. The daily dose of fenofibrate administered in the diet was calculated to be equivalent of 160 mg fenofibrate per 65 kg per day or 2.45 mg per kg body weight per day. This represents the pharmacological dose administered to human subjects. Given that the metabolic rate in mice is 12 × higher than in humans, the equivalent dose for a 25 g mouse was calculated to be 2.46 × 0.025 × 12=0.74 mg day−1. This amount of fenofibrate corresponds to the consumption of ∼2.4 g diet per day that contains ∼10 kcal. Blood was collected from tail vein on days 0 and 16 of the treatment period after 4 h fasting. At the end of the 16th day of the treatment, mice were killed and the livers were collected, frozen in liquid N2 and stored in −80 °C for further analyses. Microarray analysis was carried out in the female mice group and the corresponding controls.
RNA purification and analyses
Total RNA was purified from livers of human ApoA-I transgenic mice that were frozen in liquid nitrogen and stored in −80 °C, using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the instructions of the manufacturer.
The levels of Pparα and glyceraldehyde 3-phosphate dehydrogenase (Gapdh) mRNAs were determined by northern blotting and quantitated by phosphorimaging. The Pparα probe was generated by PCR using as a template for Pparα cDNA clone (cDNA clone MGC:18607 IMAGE: 4222384, Open Biosystems, Huntsville, AL, USA). The primers used to generate the probes were as follows: Sense: 5′-atgccagtactgccgttttc-3′ antisense: 5′-ggccttgaccttgttcatgt-3′. The mGapdh probe was 1.5 kb long and was generated by PstI digestion of a rat Gapdh containing plasmid. The probes were labeled with 32P using the random priming method.27
Real-time quantitative RT-PCR analysis
Hepatic gene expression levels of Esrrg and human APOA-I were determined by reverse transcriptase-PCR (RT-PCR) analysis and normalized against Gapdh expression levels. For these analyses, 1 μg of RNA was initially treated with DNase I (Ambion, Austin, TX, USA, DNA-free and Removal Reagents) according to the manufacturer's instructions. The RNA samples were then reverse-transcribed using the ThermoScript RT-PCR Kit (Invitrogen). Real-time quantitative RT-PCR was carried out using an ABI PRISM 7700 Sequence detector (Applied Biosystems, Foster City, CA, USA). Amplification was performed using SYBR Green PCR Master Mix (Applied Biosystems). The primers used for PCR amplification were as follows: 5′-ggaagaattcgtcaccctca-3′ (Esrrg-forward primer); 5′-ttctgcacagcttccacatc-3′ (Esrrg-reverse primer), 5′-tccatgacaactttggcattg-3′ (Gapdh-forward primer) and 5′-tcacgccacagctttcca-3′ (Gapdh-reverse primer). The primers used for amplification of human APOA-I were purchased from TaqMan gene expression assays (ID HS00163641-mi) that amplify part of exon II and III. The analysis was carried out using the Sequence Detection Software 1.9.1 (Applied Biosystems). Duplicate reactions of each sample were incubated for 2 min at 50 °C, denatured for 10 min at 95 °C and subjected to 40 cycles of annealing at 55 °C for 20 s, with extension at 60 °C for 1 min followed by denaturation at 95 °C for 15 s.
Protein analysis
Hepatic homogenates from mice treated with control diet or fenofibrate-containing diet were prepared by extensive shaking of fresh or frozen livers in ice-cold lysis buffer (10 mM NaPO4, pH 7.2, 2 mM EDTA, 10 mM NaN3, 120 mM NaCl and 1% Nonidet P40) supplemented with a mixture of protease inhibitors (Sigma-Aldrich, St Louis, MO, USA). The samples were analyzed by sodium dodecyl sulfate-PAGE and then transferred to polyvinylidene fluoride membranes. The membranes were incubated with one of the following primary antibodies: rabbit polyclonal anti-Pparα (1:250 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or goat polyclonal anti-Gapdh (1:1000 dilution). The membranes were subsequently incubated with a peroxidase-conjugated anti-rabbit or anti-goat (1:500 or 1:2000 dilution, respectively; Sigma-Aldrich) secondary antibody. Protein signals were detected using enhanced chemiluminescence reagents according to the manufacturer's protocol (Amersham Biosciences, Piscataway, NJ, USA).
Plasma lipids, ApoA-I and FPLC profiles
Total cholesterol and triglycerides of the plasma and the Fast Protein Liquid Chromatography (FPLC) fractions were determined as described28 using the WACO (WACO Chemical, Dalton, GA, USA) and INFINITY reagent thermo DNA (Sigma-Aldrich) reagent kits, respectively, according to the manufacturer's instructions. Fractionation of plasma by density gradient ultracentrifugation, electron microscopy of the HDL fractions and non-denaturing two-dimensional gel electrophoresis of plasma was performed as described.28 Plasma ApoA-I was determined by a turbidometric assay using a commercially available kit (AutoKit apoA-I; WACO Chemicals USA) as described.28
Target preparation, microarray hybridization and quality assessment
Total RNA was extracted from the livers of 10 female human ApoA-I transgenic mice. For RNA extraction, each liver specimen was homogenized in Trizol (Life Technologies, Rockville, MD, USA) and choloroform/isoamyl alcohol (49:1). All RNA samples had a 28S/18S rRNA ratio close to 2 on 1.5% agarose gels, and absorbance ratios 260/280 nm between 1.9 and 2.1. A quantity of 6–8 μg of each RNA sample was used for target preparation. Targets were prepared as previously described29 and hybridized to GeneChip Whole Mouse Genome 430 2.0 Arrays (Affymetrix, Santa Clara, CA, USA), which contain 45 000 probe sets representative of over 39 000 transcripts and variants from over 34 000 well-characterized mouse genes. The arrays were washed and stained by the Affymetrix 450 Fluidics station, and scanned using the GeneChip Scanner 3000, which was enabled for high-resolution scanning. The GeneChip Operating Software v1.4 (Affymetrix) was used to determine the expression level and ‘present,' ‘absent' or ‘marginal' call for each probe set.
Approximately 40–50% of the probe sets were called ‘present,' indicating that these transcripts were present at sufficient levels for accurate quantification. Target quality was controlled for by ensuring that the expression of the representative 3′ and 5′ sequences of the control genes β-actin and Gapdh on the microarrays was <3-fold in all 10 data sets. The good hybridization quality was also established through a concentration gradient of spike-in Bacillus subtilis transcripts, which as desired, gave a ‘present' call and increasing expression levels in all data sets.
Analysis of microarray data
The first processing step was performed using the GeneChip Operating Software v1.4. The resulting data were normalized as previously described, to correct for uniform linear aberrations of the reported signals between any two replicate measurements, which may arise from idiosyncrasies during the sample/array processing protocols.30 The data were then filtered to keep only probe sets with more than three ‘present' and one ‘marginal' calls per probe set in the 10 data sets, and therefore a reliable expression measurement. Approximately 22 000 probe sets passed the filtering criteria.
Sample correlation coefficient (CC) analysis was carried out on un-normalized, un-filtered data, to compare the correlation of every pair of data sets/samples. In parallel, hierarchical clustering was performed as described,31 using the Cluster and TreeView Software (Berkeley, CA, USA)32 with centered linear correlation as a measure of similarity using average linkage. For this analysis, only un-normalized data were used, applying a range of standard deviation thresholds.
Significant analysis of microarrays (SAM) was used to identify significant gene expression changes between treated and untreated mice.33 A two-class unpaired data analysis was carried out on normalized and pre-filtered data (as described above) using a Δ threshold of 2.265 and a fold threshold of 2. The ‘Δ' parameter enables the user to examine the effect of the false-positive rate in determining significance, whereas fold is calculated as the ratio of the average expression in treated versus untreated mouse data sets. Only probe sets with 2-fold change and a 0% median false discovery rate, the maximum possible stringency, were selected. Functional annotation of the significantly changed genes based on Gene Ontology categories was performed using the GeneTools software (SCIE-PLAS Ltd, Cambridge, UK).34
Transcription factors identified as significantly changed by SAM in fenofibrate-treated mice were further analyzed by the CC analysis to determine a potential association between transcription factors and changed genes. In particular, we calculated the CC between each of these transcription factors and all other significantly changed genes, taking into consideration the expression pattern of each gene across all samples.
Results
Effects of fenofibrate on hepatic APOA-I mRNA, plasma lipids and ApoA-I and on lipoprotein profiles of the human ApoA-I transgenic mice
The fenofibrate treatment, described in the Experimental Procedures, decreased the levels of hepatic human APOA-I mRNA and plasma ApoA-I of the treated group compared with the control group, although the observed changes were not statistically significant (Figure 1a). However, the plasma cholesterol level was 1.95-fold higher in the treated group than that in the control group (Figure 1b).
Figure 1.

(a) Hepatic human APOA-I mRNA and plasma ApoA-I levels of individual ApoA-I transgenic mice treated with either a control or a fenofibrate-containing diet. (b) FPLC profiles of total cholesterol of ApoA-I transgenic mice fed a diet containing fenofibrate (FF) or without fenofibrate (control). (c and d) Fractionation of the plasma of ApoA-I transgenic mice fed the control (c) or fenofibrate-containing diet (d) by density gradient ultracentrifugation and analysis of the fractions by sodium dodecyl sulfate (SDS)-PAGE. The densities of the fractions are indicated at the top of the figures. (e and f) Electron microscopy pictures of the HDL fractions of ApoA-I transgenic mice fed the control (e) or fenofibrate-containing diets (f). The photomicrographs were taken at × 750 000 magnification and enlarged thrice. (g and h) Two-dimensional gel electrophoresis of plasma of the control (g) or the fenofibrate (h)-containing diets.
FPLC analysis results obtained from the control mice showed that all the cholesterol was distributed in the HDL2 and HDL3 regions. In the fenofibrate-treated mice, the increase in total cholesterol was accompanied by a shift of HDL toward lower densities (HDL2/LDL/IDL) (Figure 1b), which was also confirmed by density gradient ultracentrifugation of plasma. This analysis showed that in the fenofibrate-treated group, the distribution of ApoA-I was shifted toward HDL2/LDL/IDL and the ApoA-I levels appeared reduced (Figures 1c and d). Electron microscopy showed that HDL particles remained spherical (Figures 1e and f). Two-dimensional gel electrophoresis showed that the fenofibrate treatment did not alter the preβ and αHDL subpopulation of HDL (Figures 1g and h).
Fenofibrate induces distinct patterns of gene expression in the human ApoA-I transgenic mice and affects multiple molecular pathways
To determine the effect of fenofibrate treatment on gene expression, we carried out sample CC analysis and hierarchical clustering. Primary evidence confirming the fenofibrate effect on gene expression involved the reduced intergroup (fenofibrate-treated vs untreated control mice) CC values (92–97%) compared with intragroup (treated or untreated mice) values (96–99%). Consistent with this observation of reduced correlation between treated and untreated mice, hierarchical clustering reproducibly distinguished treated from untreated mice based on their global gene expression signatures for all standard deviation thresholds applied and in all iterations (Figure 2a). Therefore, fenofibrate appears to induce several changes in gene expression in the liver.
Figure 2.

(a) Hierarchical clustering of the global gene expression profiles in the liver of control (Ctr) and fenofibrate-treated (Fen). Two distinct branches are evident in the dendrogram, representative of the different patterns of gene expression in Ctr and fenofibrate-treated animals when un-normalized filtered data were used. (b) Major Gene Ontology level-5 categories corresponding to the 244 significantly overexpressed probe sets. An asterisk (*) marks all lipid metabolism-related categories. (c) Major Gene Ontology level-5 categories corresponding to the 69 significantly underexpressed probe sets. An asterisk (*) marks all lipid metabolism-related categories.
SAM was applied on normalized and filtered data to determine high fold and statistically significant gene expression changes induced to the liver of human ApoA-I transgenic mice by fenofibrate. After the application of stringent thresholds (⩾2-fold and 0% false discovery rate), 313 probe sets, representative of 267 different genes and expressed sequence tags (EST), were identified as highly and significantly changed. As some significantly changed genes were represented by multiple significantly changed probe sets, the fold-change values presented for each gene are the average of all its significantly changed probe sets. On the basis of Gene Ontology categorization, the major functional categories of differentially expressed genes were those involved in transport, cell metabolism, including protein, nucleic acid and organic acid, and cellular lipid metabolism (Figures 2b and c). A large number of significantly changed genes encoded for nuclear and mitochondrial proteins, whereas other common categories included genes that encode for endoplasmic reticulum, organelle, membrane and cytoskeleton proteins (Figures 2b and c). The list of the Gene Ontology classifications of all the significantly changed probe sets is presented in the Supplementary Table 2.
Effect of fenofibrate on genes involved in phospholipid biosynthesis and the remodeling of HDL
In an attempt to explain the effects of fenofibrate on plasma lipids and HDL levels, we have analyzed in detail the changes in the expression of hepatic genes involved in lipid and lipoprotein metabolism.
Fenofibrate downregulated 9.75-fold the expression of the Apoa4 gene, and did not significantly alter the expression of human APOA-I and several other genes that are involved in the biogenesis and maturation of HDL such as Abca1, Lcat, Abcg1, as well as the genes encoding proteins such as Apoa2, Apoe and Apoc3, which are components of HDL.35, 36 Fenofibrate upregulated 5.86-fold, the gene Pltp, and caused a moderate 1.51-fold downregulation of Srb1, two genes that are involved in the remodeling of HDL (Table 1).
Table 1.
Fold change of genes implicated in lipid and HDL metabolism in the fenofibrate-treated human apoA-I transgenic mice
| Gene name | Symbol | Fold |
|---|---|---|
| Fatty acid binding protein 3, muscle and heart | Fabp3 | 28.28 |
| Monoacylglycerol-O-acyltransferase 1 | Mogat1 | 19.78 |
| Pancreatic lipase related protein 1 | Pnliprp1 | 19.06 |
| Pyruvate dehydrogenase kinase, isoenzyme 4 | Pdk4 | 8.87 |
| Lipoprotein lipase | Lpl | 8.85 |
| Acyl-CoA thioesterase 1 /// acyl-CoA thioesterase 2 | Acot1 /// Acot2 | 6.59 |
| Solute carrier family 27 (fatty acid transporter), member 1 | Slc27a1 | 6.23 |
| Acyl-CoA thioesterase 2 | Acot2 | 6.13 |
| Phospholipid transfer protein | Pltp | 5.86 |
| Acyl-CoA thioesterase 1 | Acot1 | 5.69 |
| Carnitine palmitoyltransferase 1b, muscle | Cpt1b | 5.67 |
| CD36 antigen | Cd36 | 4.68 |
| Very low density lipoprotein receptor | Vldlr | 4.26 |
| Hydroxysteroid (17-beta) dehydrogenase 11 | Hsd17b11 | 3.96 |
| Phospholipase A2, group VII (platelet-activating factor acetylhydrolase, plasma) | Pla2g7 | 3.91 |
| Malic enzyme, supernatant /// similar to NADP-dependent malic enzyme (NADP-ME) (Malic enzyme 1) | LOC624892 /// LOC677317 /// Mod1 | 3.32 |
| Jun oncogene | Jun | 3.22 |
| Phospholipase A2, group VI | Pla2g6 | 3.20 |
| Enoyl-Coenzyme A, hydratase/3-hydroxyacyl Coenzyme A dehydrogenase | Ehhadh | 3.14 |
| Uncoupling protein 2 (mitochondrial, proton carrier) | Ucp2 | 3.09 |
| Carnitine acetyltransferase | Crat | 3.01 |
| LIM homeobox protein 6 | Lhx6 | 2.93 |
| Acyl-CoA thioesterase 6 | Acot6 | 2.83 |
| Peroxisomal biogenesis factor 11a | Pex11a | 2.76 |
| Phosphatidylinositol membrane-associated 1 | Pitpnm1 | 2.74 |
| Stearoyl-Coenzyme A desaturase 1 | Scd1 | 2.67 |
| ATPase, class II, type 9A | Atp9a | 2.52 |
| Elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 3 | Elovl3 | 2.53 |
| Dodecenoyl-Coenzyme A delta isomerase (3,2 trans-enoyl-Coenzyme A isomerase) | Dci | 2.38 |
| Estrogen-related receptor gamma | Esrrg | 2.36 |
| Uncoupling protein 3 (mitochondrial, proton carrier) | Ucp3 | 2.35 |
| Monoglyceride lipase | Mgll | 2.34 |
| Choline phosphotransferase 1 | Chpt1 | 2.33 |
| Fatty acid binding protein 2, intestinal | Fabp2 | 2.32 |
| Acyl-CoA thioesterase 9 | Acot9 | 2.31 |
| Diacylglycerol kinase, eta | Dgkh | 2.24 |
| Choline kinase alpha | Chka | 2.09 |
| Acyl-CoA thioesterase 4 | Acot4 | 2.08 |
| Acyl-CoA thioesterase 8 | Acot8 | 2.06 |
| E2F transcription factor 8 | E2f8 | 2.06 |
| CD14 antigen | Cd14 | 2.04 |
| 3-hydroxy-3-methylglutaryl-Coenzyme A lyase | Hmgcl | 1.71 |
| 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 | Hmgcs1 | 1.63 |
| Peroxisome proliferator activated receptor gamma | Pparg | 1.59 |
| 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 | Hmgcs2 | 1.30 |
| ATP-binding cassette, sub-family G, member 1 | Abcg1 | 1.29 |
| Catalase | Cat | 1.15 |
| Diacylglycerol O-acyltransferase 1 | Dgat1 | 1.14 |
| Fatty acid synthase | Fasn | 1.13 |
| ATP-binding cassette, subfamily A, member 1 | Abca1 | 1.10 |
| Apolipoprotein A-V | Apoa5 | −1.07 |
| Apolipoprotein A-I-binding protein | Apoa1bp | −1.08 |
| Apolipoprotein A-II | Apoa2 | −1.17 |
| Diacylglycerol O-acyltransferase 2 | Dgat2 | −1.17 |
| Apolipoprotein E | ApoE | −1.19 |
| Acetyl-Coenzyme A carboxylase | Acaca | −1.21 |
| Sterol regulatory element binding transcription factor 1 | Srebf1 | −1.21 |
| Sterol regulatory element binding transcription factor 2 | Srebf2 | −1.22 |
| Lecithin:cholesterol acyltransferase | Lcat | −1.26 |
| Apolipoprotein C-III | Apoc3 | −1.34 |
| 3-hydroxy-3-methylglutaryl-Coenzyme A reductase | Hmgcr | −1.38 |
| Peroxisome proliferator activated receptor alpha | Ppara | −1.39 |
| Scavenger receptor, class B, type I | Srb1 | −1.51 |
| Peroxisome proliferative activated receptor, gamma, coactivator 1 alpha | Ppargc1a (Pgc1a) | −1.54 |
| Peroxisome proliferative activated receptor, gamma, coactivator 1 beta | Ppargc1b (Pgc1b) | −2.03 |
| Kruppel-like factor 13 | Klf13 | −2.04 |
| AT rich interactive domain 5B (Mrf1 like) | Arid5b | −2.08 |
| Hydroxysteroid 11-beta dehydrogenase 1 | Hsd11b1 | −2.20 |
| Ngfi-A binding protein 2 | Nab2 | −2.22 |
| Phospholipase A1 member A | Pla1a | −2.33 |
| Apolipoprotein A-IV | Apoa4 | −9.75 |
Abbreviation: HDL, high-density lipoprotein.
Fold changes reaching statistical significance are marked in bold. Changes in important genes of lipid and HDL metabolism that did not reach statistical significance are also included. For genes represented by more than one probe sets on the 430 2.0 Affymetrix arrays, the average of all the probe sets is presented.
The genes encoding the lipolytic enzymes Lpl and pancreatic lipase-related protein 1 (Pnliprp1) were upregulated 8.85- and 19.06-fold, respectively, whereas Apoa5 that promotes lipolysis37 did not change (Figure 3, Table 1 and Supplementary Table 2).
Figure 3.

Pathways of lipid and lipoprotein metabolism affected by fenofibrate. Upregulation of genes involved in phospholipid biosynthesis, HDL synthesis and remodeling. Upregulation of genes involved in lipolysis, FA transport, β-oxidation, synthesis of unsaturated fatty acids and long-chain fatty acids.
Fenofibrate also affected few genes that are involved in phospholipid biosynthesis. Significant upregulation was observed in monoacylglycerol acyl transferase 1 (Mogat1), diacylglycerol kinase (Dgkh), phosphtidylinositol membrane associated 1 (Pitpnm1), choline phosphotransferase (Chpt1) and choline kinase α (Chkα) genes, which were upregulated 19.78-, 2.24-, 2.74-, 2.33- and 2.09-fold, respectively (Table 1). The corresponding proteins have an important role in phospholipid biosynthesis.38 Some of these changes are expected to have an effect on the synthesis of HDL.39
Effect of fenofibrate on genes affecting triglyceride hydrolysis, FA transport, β-oxidation, other mitochondrial function, fatty acid biosynthesis and carbohydrate metabolism
The monoglycerides generated by the actions of Lpl and Pnliprp, which are upregulated, can be channeled toward phospholipid biosynthesis. Significant upregulation was also observed for the genes encoding the enzyme monoglyceride lipase (Mgll), which catalyzes the hydrolysis of monoglycerides to FA and glycerol.40 Free FA generated by the action of the lipolytic enzymes are channeled toward β-oxidation (Figure 3).
Several genes encoding transporters of FA into the cells including CD36 antigen (CD36), the FA transport protein 1 (Fatp1) or Slc27a1, and FA-binding proteins (Fabp3 and Fabp2), as well as the gene encoding the VLDL receptor (Vldlr) were upregulated 2.3- to 28.3-fold by fenofibrate (Figure 3 and Table 1).
A large number of genes encoding mitochondrial and peroxisomal proteins were upregulated (Supplementary Tables 2 and 3). The majority of these genes are involved in β-oxidation of FA. This includes carnitine O-acetyl transferase (Crat) and carnityl-O-palmitoyl transferase 1b (Cpt1b), which transport activated FA into the mitochondria, as well as several genes involved in the β-oxidation pathway. Among those are genes encoding the mitochondrial enzymes acyl CoA thiol esterases 2 and 9 (Acot2 and Acot9) and the peroxisomal enzymes acyl CoA thiol esterases 4, 6 and 8 (Acot4, Acot6 and Acot8), dodecenoyl CoA delta isomerase (Dci), and enoyl CoA hydratase (Ehhadh). Significant upregulation was also observed for the genes encoding the mitochondrial uncoupling proteins 2 and 3 (Ucp2 and Ucp3) and the cytosolic Acot1 gene. All these genes were upregulated 2.06- to 6.13-fold (Figure 3 and Table 1).
Fenofibrate upregulated the expression of the genes encoding malic enzyme (Mod1), stearoyl CoA desaturase (Scd1) and elongation of very long-chain FAs (Elovl3) 2.67- to 3.32-fold, which may have a role in the biosynthesis of unsaturated FA and long-chain FA (Figure 3 and Table 1).
The expression of genes encoding enzymes of lipogenesis, glycolysis, gluconeogenesis and the Krebs cycle did not change significantly. Few changes pertinent to these pathways include 2.49- to 8.87-fold upregulation of pyruvate dehydrogenase kinase 4 (Pdk4), phosphomannomutase 1 (Pmm1) and galactose kinase-1 (Galk1) genes and a 2.08-fold downregulation of the fructose biphosphatase (Fbp1) gene (Table 1).
Figure 3 depicts the significantly changed genes in response to fenofibrate and their potential contribution in phospholipid biosynthesis, and the remodeling of HDL, as well as in triglyceride hydrolysis, FA transport and lipid catabolism.
Associations between transcription factors and lipid-related genes in response to fenofibrate treatment
Our analysis showed that the transcription factor coding genes Esrrg, Jun oncogene (Jun), Lim homeobox protein 6 (Lhx6) and E2F transcription factor 8 (E2f8) were upregulated 2.36-, 3.22-, 2.93- and 2.06-fold, respectively, whereas the transcription factor coding genes Arid5b, Nigfi A binding protein 2 (Nab2) and Kruppel-like factor 13 (Klf13) were downregulated 2.08-, 2.22- and 2.04-fold, respectively, in response to fenofibrate treatment. To explore the potential transcriptional mechanisms involving these factors, we performed a CC analysis for each of the seven significantly changed transcription factors against the remaining 312 significantly changed probe sets, as identified by SAM.
These 2184 (312 × 7) CC values obtained from this analysis ranged between 0.4189 and 0.9899. The CC value between the multiple probe sets of each significantly changed gene ranged from 0.7561 to 0.9985 and the average CC was 0.9466. Probe sets representing different genes of the same family had significantly lower CC than 0.9400 (for example, Pla2g6 and Pla2g7 had CC=0.6400–0.7500). Therefore, genes with CC ⩾0.9400 are considered to have highly similar expression patterns and could be potentially functionally related. The number of genes that had CC >0.9400 for each of the transcription factors is shown in Figure 4a.
Figure 4.

(a) Significantly changed transcription factor genes and their correlation coefficient (CC >0.94) with other significantly changed genes (>2-fold change, 0% false discovery rate) that are shown in Supplementary Table 2. The gene expression patterns of Esrrg and Arid5b had high CC to the largest numbers of other significantly changed genes. Black bars indicate positive and gray bars indicate negative correlation. (b) Correlation of the expression of Esrrg with several genes of lipid metabolism and few genes of carbohydrate metabolism. All negative correlations are marked with asterisks (*). Genes with the highest (0.97–0.99) CCs are presented in the inner circle. (c) Correlation of the expression of Arid5b with several genes of lipid metabolism and few genes of carbohydrate metabolism. Note that several genes of panels b and c correlate positively with Esrrg and negatively with Arid5b. All negative correlations are marked with asterisks (*). Genes with the highest (0.97–0.99) correlation coefficients are presented in the inner circle. In both panels b and c, genes related to carbohydrate metabolism appear in gray color.
The Esrrg transcription factor gene had the highest CC to the largest number of probe sets (89 probe sets CC >0.9400), and particularly to lipid-related genes (20 probe sets), including lipid metabolism (for example, Mod1, Mogat1, Acot2, Acot8, Pltp, ApoA4, Dci and Cte1) and lipid transport (for example, Slc27a1, Cd36, Vldlr and Atp9a) (Figure 4b and Supplementary Tables 3 and 4). To test the putative role of Esrrg in our data, we analyzed the CC of Esrrg to all 45 000 probe sets on the arrays, and filtered for CC ⩾0.9400. Esrrg correlated strongly with 268 probe sets. Of the 268 probe sets, 71 were related to metabolism, 50 of the genes encode mitochondrial proteins, 34 were lipid related and 21 were specifically involved in electron transport (data not shown). Esrrg may therefore represent a mediator of the fenofibrate effect on lipid metabolism and mitochondrial function.
The gene encoding a Arid5b had a CC >0.9400 with 79 probe sets and showed a highly similar but inverse correlation pattern to that observed for Esrrg (CC=−0.966). For every high positive CC of a probe set to Esrrg, Arid5b showed a high negative CC, and vice versa. Of the 79 probe sets, 16 were lipid-related, 7 were involved in lipid transport, 6 were mitochondrial and 3 were peroxisomal genes (Figure 4c and Supplementary Tables 4 and 5).
Klf13 had negative CC to multiple significantly changed probe sets, several of which were related to lipid metabolism and lipid transport (for example, Vldlr, Acot9, Mogat1 and Slc27a1), cytochrome-related (for example, Cyp4f14 and cytochrome c oxidase subunit Vlb polypeptide 2 (Cox6b2)) and proteolysis-related (for example, Ctse) genes (Supplementary Table 3). Lhx6 correlated positively with six probe sets including Pdk4, whereas Jun, Erf8 and Nab2 correlated positively with four, two and one probe sets, respectively (Supplementary Table 3).
Fenofibrate does not alter hepatic Pparα gene expression, but increase cellular Pparα levels and hepatic Esrrg mRNA levels
The microarray analysis did not detect significant changes in the hepatic expression of the Pparα gene and showed a 1.5- and 2.03-fold reduction in the expression of Pparα coactivators 1a and 1b (Pgc1a and Pgc1b), respectively (Table 1). The lack of increase in PPARα mRNA level was confirmed by northern blot analysis (Figures 5a and c). However, the protein levels of Pparα were increased in the liver of the fenofibrate-treated mice (Figure 5b). In agreement with our microarray results, RT-PCR analysis of hepatic RNA showed that the Esrrg gene was significantly upregulated in fenofibrate-treated ApoA-I transgenic mice (Figure 5d). The combined effect of fenofibrate on the transcription factors Pparα, Esrrg and Arid5b in mitochondrial and cytosolic genes affecting lipid metabolism and HDL synthesis and remodeling is shown in Figure 6 and explained further in the Discussion section.
Figure 5.

Hepatic Pparα and Gapdh, mRNA and protein levels in human ApoA-I transgenic mice that were fed either control diet or fenofibrate-containing diet. (a) Northern blotting analysis of RNA isolated from the livers of control and fenofibrate-treated mice for Pparα and Gapdh expression. (b) Western blotting analysis showing hepatic protein for Pparα and Gapdh protein levels of control and fenofibrate-treated mice. (c) Bar graphs showing the ratio of Pparα/Gapdh levels of Pparα mRNA of mice fed a control or fenofibrate-containing diet as shown in panel a. (d) Hepatic Esrrg mRNA levels of human ApoA-I transgenic mice treated with either control diet or fenofibrate-containing diet.
Figure 6.
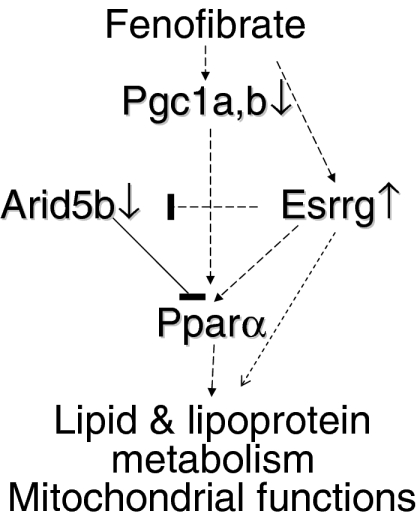
Schematic representation of the effect of fenofibrate on genes affecting lipid and lipoprotein metabolism and mitochondrial functions, as well as the role of the transcription factors Esrrg, Pparα, the coactivators Pgc1a and Pgc1b and Arid5b in this process. Direct effects may involve transcriptional activation of the Pparα gene by Esrrg, stabilization of Pparα by ligands, activation by coactivators and synergistic interactions between Esrrg and Pparα on the promoters of fenofibrate-inducible genes. Indirect effects may involve the Esrrg-mediated negative regulation of Arid5b expression, which may influence Pparα gene expression.
Discussion
This study used a new ApoA-I transgenic mouse model in order to decipher by global gene expression profiling the mechanism through which fenofibrate affect genes of lipid and lipoprotein metabolism.
The multilevel bioinformatical analysis of the microarray data identified significant changes in numerous genes involved in relatively few, yet central biological processes. Although all the observed changes may have important implications, our analysis has focused on the effect of fenofibrate on genes that affect HDL biogenesis and remodeling, as well as lipid metabolism and transcription factors related to the observed changes.
Effect of fenofibrate on the expression of the human ApoA-I gene and on plasma HDL levels
The mouse model used in this study expresses the human APOA-I gene under the control of its regulatory sequences that include the proximal promoter and the APOCIII enhancer in ApoA-I-deficient background. This mouse model has two advantages. It can record reliably the changes in human APOA-I gene transcription in vivo. In addition, due to the inactivation of the endogenous mouse Apoa1 gene, the mouse synthesizes HDL that contains human APOA-I as its major protein.25 The lipid analysis of plasma showed that there was a 1.95-fold increase in the total cholesterol in the fenofibrate-treated mice and a shift of HDL toward lower densities, compared with the control group. The increase in total plasma cholesterol was the result of the increase in HDL cholesterol. The treatment did not change the profile of the HDL subpopulations as determined by two-dimensional gel electrophoresis.
Real-time PCR analysis showed that in the fenofibrate-treated group, there was a small decrease in the expression of the human APOA-I gene that was not statistically significant. A similar small non-statistically significant decrease was also detected in plasma ApoA-I levels of the fenofibrate-treated mice. Two previous studies suggested that fenofibrate caused 2- to 2.6-fold increase in human APOA-I gene expression in ApoA-I transgenic mice, as well as a 3.5- to 7.5-fold increase in plasma ApoA-I levels.20, 23 These increases did not occur in ApoA-I transgenic mice in the PPARα-deficient background.23 In ApoA-I transgenic rabbits, the higher doses of fenofibrate used increased hepatic human ApoA-I mRNA levels and plasma ApoA-I levels approximately 1.6- and 2-fold, respectively.24
It has been reported that fenofibrate downregulates the expression of Apoa1 gene in mice, whereas they upregulate the expression of the human ApoA-I.20 The different responses of the human and mouse Apoa1 genes to the fenofibrate treatment was attributed to differences between the human and rodent Apoa1 promoter.41 This study shows that the in vivo regulation of the human apoA-I gene in mice when it contains its proximal and distal regulatory sequences resembles the regulation of the mouse Apoa1 gene described previously.20
The observed differences between the present and the previous studies,20, 23, 24 can be explained based on the mechanism of transcriptional regulation of the human APOA-I gene that emerged from in vitro and in vivo studies.25 In the transgenic mice used here, the expression of the human APOA-I gene was controlled by the proximal promoter and the distal APOCIII enhancer. In the transgenic mice and rabbits used in previous studies,20, 23, 24 the transgene lacked the APOCIII enhancer and the transcription of the APOA-I gene was driven by the proximal APOA-I promoter alone.42, 43 We have shown previously that in the transgenic mice where the APOCIII enhancer is inactivated, the intestinal transcription of the APOA-I gene is abolished and the hepatic transcription is reduced to 20% compared with mice that carry both the proximal promoter and the APOCIII enhancer.25, 44
The proximal human APOA-I promoter contains two hormone response elements between nucleotides −210 to −197 and −132 to −119, which can bind Rxrα/Pparα heterodimers. In addition, the human APOCIII enhancer contains an hormone response element between nucleotides −736 to −721, which can bind Rxrα/Pparα heterodimers.44 On the basis of our knowledge on how the human APOA-I transgene is regulated in vivo, it is expected that in the fenofibrate-treated mice, the expression of the human APOA-I gene will be controlled by interactions between Rxrα/Pparα heterodimers bound to the proximal promoter and the distal enhancer, as well as the interactions of the distantly bound Rxrα/Pparα heterodimers with SP1.25, 44 These interactions, that may affect positively or negatively gene expression, are not possible in the transgenic mice that lack the APOCIII enhancer used in the previous studies.20, 23, 24
Effect of fenofibrate on the expression of genes that may affect the synthesis of HDL
Current advances in HDL research indicate that the initial step in the biogenesis of HDL is the extracellular lipidation of lipid-poor ApoA-I by the action of ABCA1 lipid transporter.45 Important steps for the maturation of HDL are the esterification of HDL of the nascent particles by Lcat, the transfer of lipids by Pltp, the cholesteryl ester transfer protein and the interaction of the HDL with the HDL receptor (SR-BI) and the ABCG1 lipid transporter.28, 35, 45
The fenofibrate treatment significantly downregulated (9.75-fold) the expression of Apoa4 gene. A 10-fold decrease in the Apoa4 gene expression was previously observed in the rat liver by clofibrate treatment.18 In addition, the fenofibrate treatment caused small but not statistically significant changes in the expression of mouse Apoc3, Apoa2, Apoa5 and Apoe genes, as well as in other genes that are involved in the biogenesis and maturation of HDL such as Abca1, Lcat and Abcg1 (Table 1). Overall, the increase in HDL could not be accounted for by the upregulation of Apoa1 and Apoa2 as suggested previously20, 21 or attributed to the changes in several other genes implicated in the biogenesis of HDL.
Consistent with the previous reports, the fenofibrate treatment upregulated 5.85- and 8.85-fold the expression of the Pltp and Lpl genes, respectively.6, 22 The function of the Pltp protein is to transfer phospholipids from VLDL, IDL and LDL to HDL. It has been shown recently that a 3-fold increase in the Pltp activity in transgenic mice reduces the plasma HDL levels and promotes atherogenesis.46 Thus, the Pltp-mediated phospholipid transfer cannot explain the observed increase and the shift of the HDL peak to the lower density region in response to fenofibrate treatment. On the other hand, increase in Lpl is associated with increased plasma HDL levels.47 Previous studies indicated that fenofibrate promotes the degradation of Srb1.48 Thus, increased degradation of Srb1 combined with the observed 1.51-fold reduction in Srb1 gene expression may have contributed to some extent to the increase in the size of HDL.49
The increase in the HDL levels could be also affected by the increased expression of genes involved in phospholipid biosynthesis. This study showed that the fenofibrate treatment caused significant changes in the genes involved in phospholipid biosynthesis, including Mogat1, Dgkh, Pitpnm1, Chpt1 and Chkα (Table 1). Furthermore, the overexpression of the Mogat1 gene (19.8-fold increase) is expected to promote the generation of diacylglycerol from monoacylglycerol, and the overexpression of Dgkh is expected to further promote the generation of diacylglycerol phosphate, which is used in phospholipid biosynthesis. Finally, the overexpression of Chka will promote phosphocholine biosynthesis and the overexpression of Pitpnm1, which regulates DAG consumption via the CDP–choline pathway, may promote phosphatidylcholine biosynthesis.38 A previous study showed that the biosynthesis of phosphatidylcholine in the liver is important for HDL formation.39 Overall, the upregulation of the genes involved in phospholipid biosynthesis as well as that of Lpl may account for the observed increase in HDL levels, and the modest downregulation of the Srb1 gene may account for the increase in the size of HDL in response to fenofibrate treatment.
The lipid lowering and other actions of fenofibrate
As depicted in Figure 3, fenofibrate significantly upregulated the genes involved in lipolysis of Lpl, Pnliprp1 and Mgll, as well as several genes involved in FA transport and β-oxidation. The combined action of Lpl and Pnliprp1 enzymes converts triglycerides to monoglycerides, which can be converted to free FAs and glycerol by the action of Mgl1.40 The hydrolysis of triglycerides of chylomicrons and VLDL by the Lpl reduces plasma triglyceride levels and promotes the clearance of the resulting lipoprotein remnants by the LDL receptor, which recognizes and endocytoses ApoB- and ApoE-containing lipoproteins.50, 51 The monoglycerides that are produced by the action of Lpl and Pnliprp1 can be used for the synthesis of phospholipids that are incorporated into the HDL and other lipoproteins. The free FA produced by the action of the lipolytic enzymes are transferred by the lipid transporters into the mitochondria and undergo β-oxidation.
Acetyl CoA generated by the β-oxidation enters the Krebs cycle for energy production. We observe significant upregulation of the gene encoding Pdk4, which inactivates the pyruvate dehydrogenase complex, by phosphorylating the pyruvate decarboxylase activity of the complex. This ensures the use of fat rather than carbohydrate as a source of energy production for the cell.52 Thee genes related to glycolysis, gluconeogenesis and the Krebs cycle did not show significant changes in expression (Table 1). The observed downregulation of Fbp1 in response to fenofibrate treatment is also consistent with inhibition of gluconeogenesis. Finally, the upregulation of Galk1 will favor the entry of galactose into the glycolytic pathway.
In this study, we did not observe changes in the expression of other genes such as Apoc3 and Apoa5 that have been implicated in plasma triglyceride clearance.7, 8, 9 Thus, it appears that the increased expression of Lpl and Pnliprp1 genes that facilitates lipolysis and lipoprotein remnant clearance, combined with increased uptake and β-oxidation of FA, may account for the lowering of plasma triglycerides in response to fenofibrate treatment. The upregulation of Up2 and Up3 by fenofibrate may represent a complementary mechanism to reduce the concentration of the reactive oxygen species.53 Previous studies have also described upregulation of some but not all of the genes of lipid metabolism shown in Table 1 and Figure 3, including triglyceride lipolysis,5, 6 FA transport54, 55 and β-oxidation.11, 12, 56, 57, 58
In agreement with previous studies,58 we did not find significant changes in the lipogenic genes in response to fenofibrate treatment. The upregulation of both Scd-1, which has been observed previously,59 and Elovl3 suggest that fibrates may promote synthesis of unsaturated FA and long-chain FA. Free FA, that are the substrates of Scd1 and Elovl3, may be produced from the hydrolysis of VLDL by the action of Lpl, by de novo synthesis of FA in the cytoplasm, or by both processes. Acetyl CoA required for FA synthesis can be generated in the cytosol from citrate by the action of citrate lyase. Mitochondrial citrate is exchanged for the cytosolic malate, which can be produced by Mod1. The upregulation of the gene encoding Mod1 by fenofibrate is consistent with previous findings60 and will favor increased production of acetyl CoA in the cytoplasm.
The role of transcription factors in the fenofibrate-induced activation of the target genes
Genes with high CC to each other are likely to participate in common molecular pathways and/or to be co-regulated at the gene transcriptional level. The microarray analysis showed that the transcription factors Esrrg, Lhx6 and Jun and E2f8 were upregulated approximately 2- to 4-fold, whereas the transcription factors Arid56, Nab2 and Klf3 were downregulated approximately 2-fold in response to fenofibrate treatment (Table 1). The 2.36-fold upregulation of Essrg was confirmed by RT-PCR analysis.
The Esrrg gene had the highest CC to the other significantly changed genes, particularly those involved in lipid metabolism and transport (Supplementary Table 3). Esrrg, together with Esrra and Esrrb, belong to the family of orphan nuclear hormone receptors identified initially based on their homology to the estrogen receptor ERα.61 The estrogen receptor-related proteins encoded by the corresponding Esrra, b and g genes, have been implicated in the regulation of genes involved in energy production, cellular FA uptake, FA oxidation, mitochondrial electron transport and oxidative phosphorylation.62, 63 Esrra- and Esrrg-mediated regulation of target gene occurs in part through the direct activation of Pparα gene transcription, using Ppargc 1α or 1b (Pgc1a and Pgc1b) as coactivators.64, 65, 66, 67
In this study, fenofibrate did not cause significant changes in the hepatic expression of the Pparα gene (1.39-fold reduction) as assessed by the microarray analysis and confirmed by northern blotting. However, the protein levels of Pparα were increased in the liver of the fenofibrate-treated mice. This observation is consistent with previous findings showing that fibrates decrease Pparα mRNA levels68, 69 but increased protein levels.68 This effect may be due to the binding of fenofibrate to Pparα that may also increase its stability in addition to increasing its transcriptional activity. Small upregulation (1.59-fold) was observed in the Pparγ gene (Table 1). In addition, the fenofibrate treatment caused 1.5- and 2.03-fold reduction in the expression of Pgc1a and Pgc1b genes, respectively, which are key coactivators of genes involved in mitochondrial oxidative metabolism as well as other cellular energy metabolic pathways.67, 70, 71, 72, 73, 74 It is possible that Esrrg may bind to the promoters of fenofibrate-inducible genes and in synergy with Pparα may activate their transcription. It has been reported that in mouse fibroblast double-deficient for Pparα and Esrra, the addition of Esrrα or Pparα alone fails to activate several Pparα target genes, and supports the concept of synergy between these two transcription factors in the activation of Pparα-inducible genes.75 It is interesting that the increase in Esrrg gene expression is sufficient to activate Pparα and its target genes despite the decrease in expression of the coactivators Pgc1a and Pgc1b genes.
The Arid5b gene expression showed a strong negative correlation to Esrrg, and to most of the lipid metabolism genes for which Esrrg had a positive correlation. Arid5b is considered as a negative regulator of transcription, although its precise functions have not been clearly established.76 In our case, it is possible that the reduced expression of Arid5b may contribute to the increased expression of Esrrg or it may enhance its transcriptional activation functions. It is also possible that Arid5b acts as a repressor of the Pparα promoter activity, and when it is downregulated, either directly by fenofibrate or indirectly by Esrrg, it may promote activation of the Pparα gene and the fenofibrate-inducible genes, even when the Pgc1 levels are reduced.
On the basis of the CC analysis, it appears that downregulation of Klf13 may favor the transcriptional activation of other genes involved in lipid metabolism.
The combined effect of fenofibrate on the transcription factors Pparα, Esrrg, Arid5b and other transcription factors affecting lipid and lipoprotein metabolism is shown in Figure 6. In this scheme Pparα, in synergy with Esrrg, controls directly the expression of specific genes of lipid and lipoprotein metabolism and mitochondrial functions. It is also possible that Esrrg may contribute by inhibiting Arid5b gene expression, thus preventing further downregulation of Pparα gene expression.
Currently, there is an intense interest to increase HDL levels in humans by affecting either ApoA-I synthesis or any other protein involved directly or indirectly in the biogenesis of HDL. This study, in addition to its contribution to the molecular mechanism underlying the pharmacological action of the fibrates, establishes that the new human ApoA-I transgenic mouse model described here is an optimal model to study molecules that affect the synthesis of HDL.
Supplementary Material
Acknowledgments
We thank Iordanes Karagiannides and Andrzej Prohczuk for technical assistance, and Anne Plunkett for preparing the paper. This work was supported by grants from the National Institutes of Health (HL48739 and HL68216), two 6th Framework Programmes of the European Union (No. LSHM-CT-2006-0376331 and LSHG-CT-2006-037277 (Valapodyn)), the Biomedical Research Foundation of the Academy of Athens, the Hellenic Cardiological Society and the John F Kostopoulos Foundation.
Abbreviations
- Acot1, Acot2, Acot4, Acot6, Acot8 and Acot9
acyl CoA thiol esterases 1, 2, 4, 6, 8 and 9
- Atp9a
ATPase class II type 9a
- Arid5b
ATP-rich interactive domain 5b
- Crat
carnitine O-acetyl transferase
- Cpt1b
carnityl-O-palmitoyl transferase 1b
- Chkα
choline kinase α
- Chpt1
choline phosphotransferase
- CC
correlation coefficient
- Cox6b2
cytochrome c oxidase subunit Vlb polypeptide 2
- Dci
dodecenoyl CoA delta isomerase
- Dgkh
diacylglycerol kinase
- E2f8
E2F transcription factor 8
- Elovl3
elongation of very long-chain fatty acids
- Ehhadh
enoyl CoA hydratase
- Esrrg
estrogen receptor-related g
- Esrrs
estrogen receptor-related proteins
- ∣FA
fatty acid
- GAPDH
glyceraldehydes 3-phosphate dehydrogenase
- Jun
Jun oncogene
- Klf13
Kruppel-like factor 13
- Lhx6
Lim homeobox protein 6
- Lpl
lipoprotein lipase
- Mod1
malic enzyme
- Mgll
monoglyceride lipase
- Mogat1
monoglycerol acyltransferase
- Nab2
Nigfi A binding protein 2
- Pnliprp1
pancreatic lipase-related protein 1
- Pparα
peroxisome proliferator-activated receptor-alpha
- Pitpnm1
phosphatidyl inositol membrane associated 1
- Pltp
phospholipid transfer protein
- Pmm1
phosphomannomutase 1
- Pgc1a and Pgc1b
PPARα coactivators 1a and 1b
- Pdk4
pyruvate dehydrogenase kinase 4
- Rxrα
retinoid X receptor alpha
- SAM
significant analysis of microarray
- Scd1
stearoyl CoA desaturase
- Ucp2 and Ucp3
uncoupling protein 2 and 3
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website (http://www.nature.com/tpj)
References
- Summary of the second report of the National Cholesterol Education Program (NCEP). Expert Panel on Detection Evolution Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II). Scott MG, David Bilheimer, Alan Chait, Luther TC, Margo Denke, Richard JH JAMA 19932693015–3023.et al [PubMed] [Google Scholar]
- Roberts WC. Safety of fenofibrate—US and worldwide experience. Cardiology. 1989;76:169–179. doi: 10.1159/000174488. [DOI] [PubMed] [Google Scholar]
- Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- Fruchart JC, Duriez P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today (Barc) 2006;42:39–64. doi: 10.1358/dot.2006.42.1.963528. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, et al. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996;15:5336–5348. [PMC free article] [PubMed] [Google Scholar]
- Desager JP, Horsmans Y, Vandenplas C, Harvengt C. Pharmacodynamic activity of lipoprotein lipase and hepatic lipase, and pharmacokinetic parameters measured in normolipidaemic subjects receiving ciprofibrate (100 or 200 mg/day) or micronised fenofibrate (200 mg/day) therapy for 23 days. Atherosclerosis. 1996;124:S65–S73. doi: 10.1016/0021-9150(96)05859-5. [DOI] [PubMed] [Google Scholar]
- Vu-Dac N, Gervois P, Jakel H, Nowak M, Bauge E, Dehondt H, et al. Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J Biol Chem. 2003;278:17982–17985. doi: 10.1074/jbc.M212191200. [DOI] [PubMed] [Google Scholar]
- Prieur X, Coste H, Rodriguez JC. The human apolipoprotein AV gene is regulated by peroxisome proliferator-activated receptor-alpha and contains a novel farnesoid X-activated receptor response element. J Biol Chem. 2003;278:25468–25480. doi: 10.1074/jbc.M301302200. [DOI] [PubMed] [Google Scholar]
- Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, et al. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95:705–712. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu-Dac N, Schoonjans K, Laine B, Fruchart JC, Auwerx J, Staels B. Negative regulation of the human apolipoprotein A-I promoter by fibrates can be attenuated by the interaction of the peroxisome proliferator-activated receptor with its response element. J Biol Chem. 1994;269:31012–31018. [PubMed] [Google Scholar]
- Yadetie F, Laegreid A, Bakke I, Kusnierczyk W, Komorowski J, Waldum HL, et al. Liver gene expression in rats in response to the peroxisome proliferator-activated receptor-alpha agonist ciprofibrate. Physiol Genomics. 2003;15:9–19. doi: 10.1152/physiolgenomics.00064.2003. [DOI] [PubMed] [Google Scholar]
- Cariello NF, Romach EH, Colton HM, Ni H, Yoon L, Falls JG, et al. Gene expression profiling of the PPAR-alpha agonist ciprofibrate in the cynomolgus monkey liver. Toxicol Sci. 2005;88:250–264. doi: 10.1093/toxsci/kfi273. [DOI] [PubMed] [Google Scholar]
- Staels B, Peinado-Onsurbe J, Auwerx J. Down-regulation of hepatic lipase gene expression and activity by fenofibrate. Biochim Biophys Acta. 1992;1123:227–230. doi: 10.1016/0005-2760(92)90115-c. [DOI] [PubMed] [Google Scholar]
- Staels B, van Tol A, Andreu T, Auwerx J. Fibrates influence the expression of genes involved in lipoprotein metabolism in a tissue-selective manner in the rat. Arterioscler Thromb. 1992;12:286–294. doi: 10.1161/01.atv.12.3.286. [DOI] [PubMed] [Google Scholar]
- Staels B, van Tol A, Skretting G, Auwerx J. Lecithin:cholesterol acyltransferase gene expression is regulated in a tissue-selective manner by fibrates. J Lipid Res. 1992;33:727–735. [PubMed] [Google Scholar]
- Staels B, Auwerx J. Perturbation of developmental gene expression in rat liver by fibric acid derivatives: lipoprotein lipase and alpha-fetoprotein as models. Development. 1992;115:1035–1043. doi: 10.1242/dev.115.4.1035. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Goel SK, Nemali MR, Carrino JJ, Laffler TG, Reddy MK, et al. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci USA. 1986;83:1747–1751. doi: 10.1073/pnas.83.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B, van Tol A, Verhoeven G, Auwerx J. Apolipoprotein A-IV messenger ribonucleic acid abundance is regulated in a tissue-specific manner. Endocrinology. 1990;126:2153–2163. doi: 10.1210/endo-126-4-2153. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Staels B, Grimaldi P, Auwerx J. Acyl-CoA synthetase mRNA expression is controlled by fibric-acid derivatives, feeding and liver proliferation. Eur J Biochem. 1993;216:615–622. doi: 10.1111/j.1432-1033.1993.tb18181.x. [DOI] [PubMed] [Google Scholar]
- Berthou L, Duverger N, Emmanuel F, Langouet S, Auwerx J, Guillouzo A, et al. Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. J Clin Invest. 1996;97:2408–2416. doi: 10.1172/JCI118687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu-Dac N, Schoonjans K, Kosykh V, Dallongeville J, Fruchart JC, Staels B, et al. Fibrates increase human apolipoprotein A-II expression through activation of the peroxisome proliferator-activated receptor. J Clin Invest. 1995;96:741–750. doi: 10.1172/JCI118118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouly M, Masson D, Gross B, Jiang XC, Fievet C, Castro G, et al. Induction of the phospholipid transfer protein gene accounts for the high density lipoprotein enlargement in mice treated with fenofibrate. J Biol Chem. 2001;276:25841–25847. doi: 10.1074/jbc.M101160200. [DOI] [PubMed] [Google Scholar]
- Duez H, Lefebvre B, Poulain P, Torra IP, Percevault F, Luc G, et al. Regulation of human apoA-I by gemfibrozil and fenofibrate through selective peroxisome proliferator-activated receptor alpha modulation. Arterioscler Thromb Vasc Biol. 2005;25:585–591. doi: 10.1161/01.ATV.0000154140.73570.00. [DOI] [PubMed] [Google Scholar]
- Hennuyer N, Poulain P, Madsen L, Berge RK, Houdebine LM, Branellec D, et al. Beneficial effects of fibrates on apolipoprotein A-I metabolism occur independently of any peroxisome proliferative response. Circulation. 1999;99:2445–2451. doi: 10.1161/01.cir.99.18.2445. [DOI] [PubMed] [Google Scholar]
- Kan HY, Georgopoulos S, Zannis V. A hormone response element in the human apolipoprotein CIII (ApoCIII) enhancer is essential for intestinal expression of the ApoA-I and ApoCIII genes and contributes to the hepatic expression of the two linked genes in transgenic mice. J Biol Chem. 2000;275:30423–30431. doi: 10.1074/jbc.M005641200. [DOI] [PubMed] [Google Scholar]
- Cathcart ES, Gonnerman WA, Elliot-Bryant R, Hajri T, Hayes KC. Dietary modulation of apolipoprotein serum amyloid A (apoSAA) metabolism and prevention of amyloidosis in aging C57BL/6J and SJL/J mice. Nutr Biochem. 1997;8:328–333. [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Chroni A, Duka A, Kan HY, Liu T, Zannis VI. Point mutations in apolipoprotein a-I mimic the phenotype observed in patients with classical lecithin:cholesterol acyltransferase deficiency. Biochemistry. 2005;44:14353–14366. doi: 10.1021/bi050962o. [DOI] [PubMed] [Google Scholar]
- Sanoudou D, Haslett JN, Kho AT, Guo S, Gazda HT, Greenberg SA, et al. Expression profiling reveals altered satellite cell numbers and glycolytic enzyme transcription in nemaline myopathy muscle. Proc Natl Acad Sci USA. 2003;100:4666–4671. doi: 10.1073/pnas.0330960100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanoudou D, Kang PB, Haslett JN, Han M, Kunkel LM, Beggs AH. Transcriptional profile of postmortem skeletal muscle. Physiol Genomics. 2004;16:222–228. doi: 10.1152/physiolgenomics.00137.2003. [DOI] [PubMed] [Google Scholar]
- Greenberg SA, Sanoudou D, Haslett JN, Kohane IS, Kunkel LM, Beggs AH, et al. Molecular profiles of inflammatory myopathies. Neurology. 2002;59:1170–1182. doi: 10.1212/wnl.59.8.1170. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisvag V, Junge FK, Bergum H, Jolsum L, Lydersen S, Gunther CC, et al. GeneTools—application for functional annotation and statistical hypothesis testing. BMC Bioinformatics. 2006;7:470. doi: 10.1186/1471-2105-7-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannis VI, Zanni EE, Papapanagiotou A, Kardassis D, Chroni A. High-Density Lipoproteins: From Basic Biology to Clinical Aspects. Wiley-VCH: Weinheim; 2006. ApoA-I functions and synthesis of HDL: insights from mouse models of human HDL metabolism; pp. 237–265. [Google Scholar]
- Kypreos KE, Zannis VI. Pathway of biogenesis of apolipoprotein E-containing HDL in vivo with the participation of ABCA1 and LCAT. Biochem J. 2007;403:359–367. doi: 10.1042/BJ20061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- Litvak V, Dahan N, Ramachandran S, Sabanay H, Lev S. Maintenance of the diacylglycerol level in the Golgi apparatus by the Nir2 protein is critical for Golgi secretory function. Nat Cell Biol. 2005;7:225–234. doi: 10.1038/ncb1221. [DOI] [PubMed] [Google Scholar]
- Jacobs RL, Devlin C, Tabas I, Vance DE. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem. 2004;279:47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- Fredrikson G, Tornqvist H, Belfrage P. Hormone-sensitive lipase and monoacylglycerol lipase are both required for complete degradation of adipocyte triacylglycerol. Biochim Biophys Acta. 1986;876:288–293. doi: 10.1016/0005-2760(86)90286-9. [DOI] [PubMed] [Google Scholar]
- Vu-Dac N, Chopin-Delannoy S, Gervois P, Bonnelye E, Martin G, Fruchart JC, et al. The nuclear receptors peroxisome proliferator-activated receptor alpha and Rev-erbalpha mediate the species-specific regulation of apolipoprotein A-I expression by fibrates. J Biol Chem. 1998;273:25713–25720. doi: 10.1074/jbc.273.40.25713. [DOI] [PubMed] [Google Scholar]
- Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci USA. 1991;88:434–438. doi: 10.1073/pnas.88.2.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh A, Ito Y, Breslow JL. High levels of human apolipoprotein A-I in transgenic mice result in increased plasma levels of small high density lipoprotein (HDL) particles comparable to human HDL3. J Biol Chem. 1989;264:6488–6494. [PubMed] [Google Scholar]
- Zannis VI, Kan HY, Kritis A, Zanni EE, Kardassis D. Transcriptional regulatory mechanisms of the human apolipoprotein genes in vitro and in vivo. Curr Opin Lipidol. 2001;12:181–207. doi: 10.1097/00041433-200104000-00012. [DOI] [PubMed] [Google Scholar]
- Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- Samyn H, Moerland M, Van Gent T, van HaperenR, Metso J, Grosveld F, et al. Plasma phospholipid transfer activity is essential for increased atherogenesis in PLTP transgenic mice: a mutation-inactivation study. J Lipid Res. 2008;49:2504–2512. doi: 10.1194/jlr.M800080-JLR200. [DOI] [PubMed] [Google Scholar]
- Brunzell JD, Deeb SS.Familial lipoprotein lipase deficiency, apoC-II deficiency, and hepatic lipase deficiency The Metabolic and Molecular Bases of Inherited Disease 2001McGraw-Hill: New York; 2789–2816.In: Scriver CR, Beaudet AL, Valle D, Sly WS, (eds). [Google Scholar]
- Lan D, Silver DL. Fenofibrate induces a novel degradation pathway for scavenger receptor B-I independent of PDZK1. J Biol Chem. 2005;280:23390–23396. doi: 10.1074/jbc.M502777200. [DOI] [PubMed] [Google Scholar]
- Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kypreos KE, Zannis VI. LDL receptor deficiency or apoE mutations prevent remnant clearance and induce hypertriglyceridemia in mice. J Lipid Res. 2006;47:521–529. doi: 10.1194/jlr.M500322-JLR200. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Hobbs HH, Brown MS.Familial hypercholesterolemia The Metabolic and Molecular Bases of Inherited Disease 2001McGraw-Hill: New York; 2863–2913.In: Scriver CR, Beaudet AL, Valle D, Sly WS, (eds). [Google Scholar]
- Holness MJ, Bulmer K, Gibbons GF, Sugden MC. Up-regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) protein expression in oxidative skeletal muscle does not require the obligatory participation of peroxisome-proliferator-activated receptor alpha (PPARalpha) Biochem J. 2002;366 (Part 3:839–846. doi: 10.1042/BJ20020754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARalpha and PPARgamma activators. J Biol Chem. 1997;272:28210–28217. doi: 10.1074/jbc.272.45.28210. [DOI] [PubMed] [Google Scholar]
- Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16710–16714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- Kelly LJ, Vicario PP, Thompson GM, Candelore MR, Doebber TW, Ventre J, et al. Peroxisome proliferator-activated receptors gamma and alpha mediate in vivo regulation of uncoupling protein (UCP-1, UCP-2, UCP-3) gene expression. Endocrinology. 1998;139:4920–4927. doi: 10.1210/endo.139.12.6384. [DOI] [PubMed] [Google Scholar]
- Hunt MC, Nousiainen SE, Huttunen MK, Orii KE, Svensson LT, Alexson SE. Peroxisome proliferator-induced long chain acyl-CoA thioesterases comprise a highly conserved novel multi-gene family involved in lipid metabolism. J Biol Chem. 1999;274:34317–34326. doi: 10.1074/jbc.274.48.34317. [DOI] [PubMed] [Google Scholar]
- Guo Q, Wang PR, Milot DP, Ippolito MC, Hernandez M, Burton CA, et al. Regulation of lipid metabolism and gene expression by fenofibrate in hamsters. Biochim Biophys Acta. 2001;1533:220–232. doi: 10.1016/s1388-1981(01)00156-1. [DOI] [PubMed] [Google Scholar]
- Miller CW, Ntambi JM. Peroxisome proliferators induce mouse liver stearoyl-CoA desaturase 1 gene expression. Proc Natl Acad Sci USA. 1996;93:9443–9448. doi: 10.1073/pnas.93.18.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowska J, Kochan Z, Zelewski L, Swierczynski J. Tissue-specific effect of clofibrate on rat lipogenic enzyme gene expression. Eur J Pharmacol. 1999;370:329–336. doi: 10.1016/s0014-2999(99)00129-6. [DOI] [PubMed] [Google Scholar]
- Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- Sladek R, Bader JA, Giguere V. The orphan nuclear receptor estrogen-related receptor alpha is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol. 1997;17:5400–5409. doi: 10.1128/mcb.17.9.5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Kelly DP. A role for estrogen-related receptor alpha in the control of mitochondrial fatty acid beta-oxidation during brown adipocyte differentiation. J Biol Chem. 1997;272:31693–31699. doi: 10.1074/jbc.272.50.31693. [DOI] [PubMed] [Google Scholar]
- Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha) J Biol Chem. 2003;278:9013–9018. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Ohizumi H, Fujitani Y, Nemoto T, Tanaka T, Takahashi N, et al. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc Natl Acad Sci USA. 2003;100:12378–12383. doi: 10.1073/pnas.2135217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A. Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab. 2002;282:E1180–E1190. doi: 10.1152/ajpendo.00471.2001. [DOI] [PubMed] [Google Scholar]
- Frederiksen KS, Wulf EM, Wassermann K, Sauerberg P, Fleckner J. Identification of hepatic transcriptional changes in insulin-resistant rats treated with peroxisome proliferator activated receptor-alpha agonists. J Mol Endocrinol. 2003;30:317–329. doi: 10.1677/jme.0.0300317. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, et al. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- St Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A, Wilsker D, Moran E. DNA-binding properties of ARID family proteins. Nucleic Acids Res. 2005;33:66–80. doi: 10.1093/nar/gki145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.