

Abstract
Mouse models of intestinal tumors have advanced our understanding of the role of gene mutations in colorectal malignancy. However, the utility of these systems for studying the role of epigenetic alterations in intestinal neoplasms remains to be defined. Consequently, we assessed the role of aberrant DNA methylation in the azoxymethane (AOM) rodent model of colon cancer. AOM induced tumors display global DNA hypomethylation, which is similar to human colorectal cancer. We next assessed the methylation status of a panel of candidate genes previously shown to be aberrantly methylated in human cancer or in mouse models of malignant neoplasms. This analysis revealed different patterns of DNA methylation that were gene specific. Zik1 and Gja9 demonstrated cancer-specific aberrant DNA methylation, whereas, Cdkn2a/p16, Igfbp3, Mgmt, Id4, and Cxcr4 were methylated in both the AOM tumors and normal colon mucosa. No aberrant methylation of Dapk1 or Mlt1 was detected in the neoplasms, but normal colon mucosa samples displayed methylation of these genes. Finally, p19Arf, Tslc1, Hltf, and Mlh1 were unmethylated in both the AOM tumors and normal colon mucosa. Thus, aberrant DNA methylation does occur in AOM tumors, although the frequency of aberrantly methylated genes appears to be less common than in human colorectal cancer. Additional studies are necessary to further characterize the patterns of aberrantly methylated genes in AOM tumors.
Keywords: DNA methylation, azoxymethane, colorectal cancer, epigenetics
Introduction
Colorectal cancer affects ~148,000 people/year in the United States and is the third most common cancer in men and women in this country [1]. Most colorectal cancers arise from adenomatous polyps, and the risk of developing colon polyps and cancer appears to be a result of the effect of genetic and environmental factors that promote the formation of adenomas and/or the progression of these adenomas to cancer [2]. These environmental and genetic factors contribute to colorectal cancer formation by promoting the accumulation of gene mutations and epigenetic alterations in colon epithelial cells that drive the polyp→cancer formation process.
Many of the genetic events that occur in colon cancer have been identified, and their effects on cancer initiation and progression have been determined through the use of mouse models and human genetics. For instance, the role of APC as an initiating event in the adenoma-cancer progression sequence was determined through studies of the cancer family syndrome, Familial Adenomatous Polyposis, which is caused by germline mutations in APC, and through studies of the ApcMin mouse, which develops intestinal adenomas. Mouse models of cancer have further revealed how gene mutations cooperate in the carcinogenesis process to promote the progression of adenomas initiated by APC mutations [3,4].
Although aberrant DNA methylation has been recently shown to occur commonly in colorectal cancer, the causal role of these epigenetic changes in the process of cancer initiation and promotion is poorly understood at this time. It has been established that the aberrant hypermethylation of tumor suppressor genes can result in their transcriptional silencing, which is the mechanism through which DNA methylation is believed to promote cancer formation. DNA methylation appears to cooperate with concurrent alterations in chromatin structure to repress transcription [5–7]. However, little is known regarding the precise timing of these epigenetic alterations in the transition of normal colon epithelial cells to cancer cells through the polyp→cancer progression sequence. Furthermore, the biological role that these aberrantly methylated genes have on driving the formation of colorectal cancer is also poorly understood [8].
A well-established mouse model of colorectal cancer that has the potential to provide insight into the role of aberrant DNA methylation in the molecular pathogenesis of the polyp→cancer progression sequence is the azoxymethane (AOM) rodent colon cancer model. This model employs the carcinogen AOM to induce neoplasms that recapitulate the adenoma-carcinoma sequence in the mouse colon [9,10]. The AOM model also displays some of the common molecular events seen in human colorectal cancer, including the accumulation of Kras mutations and increased COX2 expression [11–13]
Mouse models have already proven useful in studying the role of DNA methylation in the mouse skin multistage carcinogenesis [14]. Fraga et al assessed the role of DNA methylation in this well-characterized cancer model and found that specific epigenetic events correlated with both initiation steps and with progression steps. They identified several novel genes that were methylated in the mouse model and verified that they are also methylated in primary human cancers [14]. Aberrantly methylated genes have also been identified in mouse models of malignant fibrous histiocytomas, lung cancer, bladder cancer, and leukemia, demonstrating the potential to use mouse models to study the role of epigenetic alterations in cancer initiation and progression [15–19].
Furthermore, with regards to mouse models of intestinal cancer and epigenetic alterations, recently, Hahn et al investigated the glutathione peroxidase Gpx1 and Gpx2 double knockout mouse using the genome wide methylation analytical technique MIRA (Methylated CpG island recovery assay) and identified a group of genes hypermethylated in chronically inflamed, aged, or neoplastic tissue, suggesting that mouse models of intestinal cancer likely display aberrant DNA hypermethylation [20]. It has also already been shown in both the AOM model and mutant Apc mouse models of intestinal cancer that the global DNA hypomethylation observed in human colorectal malignancy is present in tumors arising in these mice suggesting the epigenetic alterations related to DNA methylation will be similar in these models to human colorectal cancer[21–23]. Furthermore, Linhart et al observed in Apcmin/+ mice over expressing DNA methyltransferase 3b1 (Dnmt3b1) that the mice develop larger, more frequent intestinal tumors compared to mice that express normal levels of Dnmt3b1[24]. In aggregate, these studies suggest that alterations in DNA methylation can contribute to tumor pathogenesis in mouse models. Consequently, we assessed the methylation state of tumor suppressor genes in the AOM colon cancer model to determine the role of epigenetic alterations in cancer initiation and progression.
In this study, we have found that AOM induced tumors are globally hypomethylated compared to normal colonic mucosa, an observation frequently seen in human neoplasms [25]. Using methylation specific PCR (MSP), we investigated the methylation status of eleven candidate genes known to be aberrantly methylated in human cancer or mouse tumor models and demonstrate that these genes show gene specific patterns of methylation in the tumors and normal tissues. Of note, two of these genes, Zik1 and Gja9, show aberrant DNA methylation in the tumors that is not present or is present at a low frequency in the normal colon. These results suggest that epigenetic events that recapitulate human colorectal cancer occur in the AOM model.
Materials and Methods
Generation of AOM induced tumors
The AOM induced colon neoplasms were generated as described previously [26]. At 6 weeks of age, the mice were treated with AOM subcutaneously twice a week for six weeks, and sacrificed at age 22 to 29 weeks.
Tissue Harvesting
Tissue was harvested as described [26] from mice aged 22 to 29 weeks. After the mice were sacrificed, the intestine was flushed with saline, cut longitudinally with scissors and inspected for tumors. Tumors were dissected from normal mucosa with a razor blade and frozen in liquid nitrogen. A sample of each tumor was sectioned and analyzed by standard histological preparation with hematoxylin/eosin staining. Tumor grade was determined by an experienced pathologist (M. Kay Washington) using criteria defined by the Consensus Report of the Pathology of Mouse Models of Intestinal Cancer [9].
To obtain normal tissue for MSP analysis, wild type FVB mice were sacrificed at age 12–24 weeks and normal colonic epithelium scrapings were collected by cutting open the colon longitudinally and scraping the mucosal surface with a glass slide. Scrapings were collected into microcentrifuge tubes and snap frozen in liquid nitrogen. Colonic crypt epithelial cells, used for global methylation analysis, were isolated by first dissecting colonic epithelium (from cecum to rectum) from 12–24 week old mice. After sacrifice, the colon was cut open longitudinally with scissors and soaked in Hanks' Balanced Salt Solution without Calcium or Magnesium (HBSS, Gibco, Carlsbad, CA) for 5 minutes on ice, then incubated for 5 minutes in HBSS + 25% HEPES + 1% Fetal Bovine Serum. The mucosa was then transferred to a 15 ml conical tube with 10 ml HBSS + 10 mM EDTA and vigorously shaken by hand for five minutes, followed by rotation for 20 minutes at 4°C. This process of shaking/rotating was repeated three times. The colon was then removed from the 15 ml conical tube using forceps and the remaining epithelial cell suspension was centrifuged at 2000 rpm for 5 minutes at 4°C. The supernatant was removed and the cell pellet was frozen in liquid nitrogen for future genomic DNA extraction.
DNA extraction and sodium bisulfite treatment
Genomic DNA was isolated either from frozen tissue samples using the Puregene kit (Qiagen, Valencia, CA) or from formalin fixed, paraffin embedded tissue sections using InstaGene Matrix (Bio-Rad, Hercules, CA) as previously published [27]. Enzymatically methylated DNA was generated using SssI methylase (New England Biolabs, Beverly, MA) and used as a positive methylated DNA control for MSP assays. Whole genome amplified (WGA) genomic DNA, which is completely unmethylated, was created using the Repli-g kit (Qiagen, Valencia, CA) following the manufacturer's protocol and was used as a negative control for MSP assays. The resulting DNA was sodium bisulfite modified as previously described [28].
Global methylation analysis
Global DNA methylation was determined using a slot-blot and anti-methyl cytosine antibody method adapted from Tao et al and Vertosick et al[29,30]. Genomic DNA from colonic crypt epithelial cells was isolated from mock treated mice and purified as described. DNA was quantified using Picogreen according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). The DNA (40 ng) was diluted into 50 μl Tris Buffered Saline (TBS) and denatured at 95° C for 10 minutes. Each sample was then slot blotted three times onto an Optitran BA-s Nitrocellulose Transfer membrane (Whatman, Schleicher & Schuell, Germany) and bound to the membrane using a Stratalinker UV Crosslinker (Stratagene, La Jolla, CA) set to 150 mJoules. The cross-linked blot was blocked in 5% fat free milk in TBS for 1 hour at room temperature, then hybridized overnight at 4°C with an anti-5'-methyl cytosine antibody (Eurogentec, San Diego, CA) in 5% milk + TBS at a dilution of 1:500. The blot was washed 3 times in TBS then incubated with an anti-mouse secondary antibody conjugated to an infrared dye (IRD 800) in 5% milk + TBS for 4 hours at room temperature. The blot was washed 3 times with TBS and imaged using a LI-COR infrared scanner (LICOR, Lincoln, NE). Supplementary figure 2 demonstrates linearity of the assay using control calf thymus DNA.
Methylation Specific PCR (MSP)
MSP analysis of bisulfite treated DNA was performed using the methylated and unmethylated primer sets listed in Supplementary Table S1. MSP reactions were performed as previously described [31]. SSSI treated DNA and WGA amplified DNA subjected to bisulfite modification was used as methylated and unmethylated controls for the MSP assays.
Bisulfite Sequencing
Bisulfite treated DNA was PCR amplified with bisulfite sequencing primers generated using MethPrimer, listed in Supplementary Table S1[32]. PCR conditions were as follows: 95° × 15'; (94° × 30”; 60° × 30”; 72° × 30”; 72° × 10') × 35 cycles. PRC amplicons were cloned into a TA vector (Invitrogen, CA), transformed, subjected to DNA extraction (plasmid mini-prep kit, Qiagen) and sequenced on an ABI 3730×l DNA Analyzer in the FHCRC Genomics Shared Resource Core following the manufacturer's suggested protocol.
Quantitative RT-PCR
RNA was isolated from EDTA isolated crypt cells or AOM induced tumors using Trizol (Invitrogen, CA) followed by RNeasy purification (Qiagen, CA) following the manufacturer's instructions. cDNA was synthesized using SSII reverse transcriptase (Invitrogen, CA) per reagent protocol. TaqMan assays (Applied Biosystems, CA) for Zik1 (Assay # Mm00494365) and Gja9 (Assay # Mm00439121) were performed on an Opticon 2 thermocycler (Bio-Rad, CA) in triplicate and normalized using Gusb expression (Assay # Mm01197698). Each assay was repeated three times and error bars were generated by calculation of the standard error of the mean.
Results
AOM-induced tumors display global DNA hypomethylation compared to normal colonic mucosa
We initially assessed the global DNA methylation status of genomic DNA isolated from AOM induced tumors and compared this to DNA from the colon mucosa. To quantify the amount of total DNA methylation of AOM induced neoplasms, we used a novel anti-methyl cytosine slot-blot method. Single stranded DNA from each normal and tumor sample was spotted in triplicate on a nitrocellulose blot and probed with an antibody that recognizes methylated cytosines. The signals from each spot were quantified using a LI-COR infrared scanner, and the replicates averaged to determine relative methylation levels for each sample. Global methylation levels of five AOM induced colon adenocarcinomas were compared to normal colon crypt epithelial cells by calculating the ratio of methylation signal to total DNA for each sample (Figure 1A). The ratio of signal from the tumor over the normal average represents the fraction of total methylation gained or lost in each sample (Figure 1B). Four out of five tumors were observed to have globally hypomethylated DNA compared to the normal colon epithelium (average decrease of 16.5%, range 4–25%), suggesting that these tumors display decreased DNA methylation across the entire genome, a phenomena observed in human cancer [25]. It is interesting to note that the degree of hypomethylation is specific to an individual tumor as both the T2 and T3 tumors are synchronous tumors from the same mouse. This finding could be due to differing degrees of contamination with normal stromal tissue or may reflect heterogeneity in the underlying molecular pathology of the tumors even in the same individual.
Figure 1. Global methylation analysis of AOM induced tumors.

A.) DNA samples from normal colon (N1 and N2) and colon adenocarcinomas (T1–T5) were heat denatured, spotted on a nitrocellulose membrane in triplicate then immobilized with ultraviolet light. The blot was probed first with an antibody against methylcytosine, then with a secondary antibody conjugated to an infrared dye. Three replicates (Rep 1–3) were performed for each sample. The signal for each band was quantified and the average and standard errors were calculated for each sample. B.) The N1 and N2 samples were averaged to set a normal threshold. Each sample was then graphed as the Log2 ratio of this normal threshold.
The aberrant methylation of tumor suppressor genes occurs in AOM induced colon cancer
To determine if aberrant DNA methylation occurs in tumors in the AOM model, we isolated genomic DNA from both AOM induced colonic adenocarcinomas and age matched normal mouse colon epithelium not exposed to AOM. We identified candidate genes for methylation analysis by surveying the published literature for genes shown to be methylated in mouse models of cancer and/or to be methylated in human cancer. We selected a total of thirteen genes that met these criteria for further analysis and assessed the methylation status of these genes in the AOM induced tumors (Table 1, Figure 2, and Table 2). We identified four patterns of gene methylation which were as follows: 1) genes methylated more frequently in tumor tissue than in normal colon mucosa; 2) genes methylated in both tumor tissue and normal colon mucosa; 3) genes methylated in colon mucosa but not in the AOM induced tumors; 4) no gene methylation in either colon mucosa or tumor tissue. An example of a gene in group 2 is Igfbp3, which is methylated in the majority of tumor samples (70%; N=10), but interestingly, also in a large number of normal epithelium samples (63%; N=8; t=1.0). Methylation was observed in both normal tissue and AOM tumors for Mgmt, Id4, Cdkn2a/p16Ink4a, and Cxcr4. Aberrant methylation of Mgmt was present in 38% of the normal colon samples (N=8) compared to 25% of the tumors (N=12; p=0.64). We observed similar patterns for Id4 (18% methylation in tumors, N=11, versus 43%, N=7, in normal mucosa; p=0.33), Cdkn2a/p16Ink4a (25% of tumor samples, N=12, versus 44%, N=9, in normal colon mucosa; p=0.26), and Cxcr4 (8% of the tumors, N=12, versus 25%, N=8, in normal colon samples; p=0.54). Surprisingly, methylation of Dapk1 and Mlt1 was only detected in normal mucosa suggesting these genes may be hypomethylated in the AOM tumors. Finally, no promoter methylation was identified in p19Arf, Tslc1, Hltf, or Mlh1 in either the AOM induced tumors or normal colon mucosa.
Table I.
Candidate gene list for investigation of methylation in AOM induced tumors
| Gene Name | Symbol | Description | Hypermethylated in human or mouse neoplasms? | Reference |
|---|---|---|---|---|
| Cyclin dependent kinase inhibitor 2a isoforms p16Ink4a and p19ARF | Cdkn2a p16Ink4a Cdkn2a p19ARF | Methylated in many human cancers and mouse models of lung, prostate, and pancreatic cancer | Both | [18,65–67]. |
| Death associated protein kinase | Dapk | Hypermethylated in 81% of colorectal carcinomas and mouse lung tumors | Both | [17,68]. |
| O6-methylguanine-DNA methyltransferase | Mgmt | Frequently methylated in colorectal cancer and in skin tumors mouse models of | Both | [14,69] |
| Inhibitor of DNA Binding 4 | Id4 | Methylated in both human and mouse leukemia | Both | [19] |
| Helicase-like transcription factor | Hltf | Hypermethylated in the progression of normal colonic tissue to adenoma to adenocarcinoma | Human | [35,55,70] |
| MutL homologue 1 | Mlh1 | Frequently methylated in human colon tumors, contributes to microsatellite instability | Human | [40,71,72] |
| Methylated in liver tumor 1 | Mlt1 | Hypermethylated in a SV40 T antigen transgenic mouse model that forms liver tumors. | Mouse | [73] |
| Tumor suppressor in lung cancer 1 | Tslc-1 | Hypermethylated in N-nitrosodiethylamine (DEN) induced murine hepatocelluar carcinomas | Mouse | [74] |
| Insulin-like Growth Factor binding protein 3 | Igfbp3 | Hypermethylated in a mouse carcinogen skin cancer model | Mouse | [14] |
| Zinc finger protein interacting with K protein 1 | Zik1 | Hypermethylated in gastric cancer | Human | [52] |
| Gap junction alpha-10 protein | Gja9 | Hypermethylated in mouse tumors | Mouse | (Conerly, personal communication) |
| C-X-C chemokine receptor type 4 | Cxcr4 | Methylated in a mouse carcinogen skin cancer model | Mouse | [14] |
Figure 2. MSP analysis of representative candidate genes in AOM induced colon adenocarcinomas and normal colon mucosa.
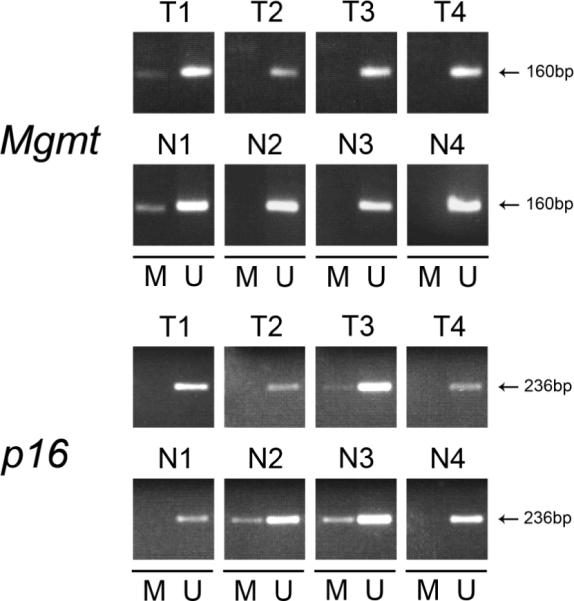
Representative images from MSP assays for Mgmt and Cdkn2A (p16Ink4a) from colon neoplasms (T1–T4) and normal colon (N1–N4) are shown. Methylated genes (M) are present in the normal colon mucosa and colon tumor tissues. Unmethylated (U) genes were identified in both normal and tumor cells, which may represent contamination with heterogeneous tissue elements or a clonal mixture of cells. MSP reactions were performed using the primer sets and annealing parameters listed in Supplementary Table 1. Amplicon size (in bp) is shown. Sssi (methylation positive control) and WGA (methylation negative control) control DNA samples are not shown.
Table 2.
MSP analysis of AOM induced tumors and normal mouse colonic epithelium
| Gene | AOM Tumors: Frequency of Methylated Genes (N, %) | Normal Intestinal Epithelium Frequency of Methylated Genes (N,%) | ||
|---|---|---|---|---|
| Cdkn2a/p16INK4a | 3/12 | (25) | 4/9 | (44) |
| Igfbp3 | 7/10 | (70) | 5/8 | (63) |
| Mgmt | 3/12 | (25) | 3/8 | (38) |
| Id4 | 2/11 | (18) | 3/7 | (43) |
| Cxcr4 | 1/12 | (8) | 2/8 | (25) |
| Dapk1 | 0/8 | (0) | 3/8 | (38) |
| p19Arf | 0/8 | (0) | 0/7 | (0) |
| Mlt1 | 0/6 | (0) | 1/8 | (13) |
| Tslc1 | 0/4 | (0) | 0/1 | (0) |
| Hltf | 0/11 | (0) | - | - |
| Mlh1 | 0/11 | (0) | - | - |
| Gja9 | 14/15 | (93) | 3/6 | (50) |
| Zik1 | 2/15 | (13) | 0/6 | (0) |
Finally, we identified two genes, Gja9 and Zik1, that were aberrantly methylated more commonly in the AOM tumors than in the normal colon mucosa. (Figure 3 and Table 2). Gja9 was methylated in 93% of AOM tumors (N=15) compared to 50% of normal colon (N=6, p=0.06) and Zik1 was methylated in 13% of tumors (N=15) and 0% of normal colonic mucosa (N=6, p=0.37). Furthermore, bisulfite sequencing of 10 clones from an AOM tumor that demonstrated methylation of Gja9 and Zik1 by MSP confirmed the results of the MSP assays. As shown in Figure 3, Gja9 demonstrated dense methylation in several clones, although several were completely unmethylated. Zik1 demonstrated significantly less methylation (present in only 3 of 10 clones), which correlates with the low level of methylation demonstrated by the MSP assay (Figure 2). We propose that the majority of the clones demonstrating no methylation are likely from normal cells that are intermixed in the tumors.
Figure 3. MSP and bisulfite sequencing forGja9 and Zik1.

A1, B1. Schematic diagrams of the promoter region of Gja9 and Zik1, respectively. Location of the transcription start site (Tss) is shown with an arrow and location of MSP and bisulfite sequencing primers are shown with black and grey boxes, respectively. A2, B2. Results of MSP analysis for Gja9 and Zik1 for four AOM tumors (T1–T4) and normal colon samples (N1–N4). Methylated (M) and Unmethylated (U) genes are shown. A3, B3. Results of bisulfite sequencing analysis of the promoter region of Gja9 and Zik1. Ten clones for each gene were isolated and sequenced from a tumor that was shown to carry either methylated Gja9 or Zik1 by MSP. Empty and black circles represent unmethylated and methylated CpG dinucleotides, respectively. A subset of the sequenced clones demonstrates methylation for each gene.
Expression of Zik1 and Gja9 was determined using quantitative RT-PCR. TaqMan assays were performed on AOM tumors and normal colon epithelium. Zik1 demonstrated decreased expression in AOM tumors that carried methylated Zik1 as well as in tumors that carried unmethylated Zik1 when compared to normal colonic epithelium (Figure 4). Interestingly, Gja9 was not expressed in either AOM tumors or normal colon (data not shown).
Figure 4. qRT-PCR analysis of Zik1 expression.
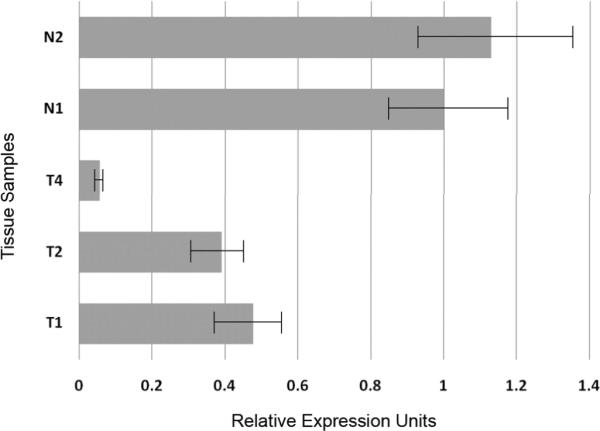
Normalized expression of Zik1 in AOM tumors T1–T4 and normal colonic epithelium N1–N2. The designation of the tumors corresponds to methylation analysis of tumors and normal tissue demonstrated in Figure 3.
We further assessed the methylation status of these genes in the AOM tumors by analyzing the methylation status of all of the genes in each individual tumor to determine if a subset of the tumors displayed an excessive proportion of methylated genes similar to the CpG Island Methylator Phenotype (CIMP) observed in human colon cancer[33,34]. To determine if the tumors could be stratified based on the proportion of aberrantly methylated genes, we compared the tumors based on the number of methylated genes from each individual tumor. For tumors that were analyzed by MSP for greater than 2 genes (N=12), an average 1.4 genes were methylated per tumor (range 0–3). A similar pattern was found in normal colonic epithelium, with a mean of 2.1 genes methylated per sample (range 0–5, p= 0.27, student's t-test). Thus, we did not identify a subset of tumors that displayed a high proportion of methylated genes.
Discussion
The discovery of aberrantly methylated genes in cancer has led to an intense interest in determining the role of these epigenetic events in the molecular pathogenesis of cancer[34–41]. Mouse models of cancer have the potential for determining the causal factors that lead to the hypermethylation of tumor suppressor genes. Consequently, studies of aberrant gene methylation have been carried out in mouse models of lung cancer, skin cancer, leukemia, and prostate cancer[14,19,24,42,43]. These tumor models have included both genetic and carcinogen-induced models and have revealed that aberrant DNA methylation does occur in the tumors arising in these models. For instance, Patel et al have reported that methylene chloride exposure leads to methylation of the p16Ink4a tumor suppressor gene, which is an epigenetic alteration that likely contributes to tumorigenesis in primary lung cancer[44]. Vuillemenot et al showed that lung tumors generated by cigarette smoke or other carcinogens display promoter methylation of the H-cadherin and the progesterone receptor genes [43]. Furthermore, Hinoi et al reported tumor specific hypomethylation in the intestinal neoplasms arising in Apcmin mouse by 5-methylcytosine immunohistochemistry [45]. In the TRAMP prostate cancer model and in the Il15 transgenic mouse model of acute lymphocytic leukemia, restriction landmark genome scanning has been used to identify genes that are aberrantly methylated in the neoplasms in these mice[19,42].
We have now assessed the role of aberrant gene methylation in a model of colon cancer, the AOM induced colon adenocarcinoma model. We first analyzed global DNA methylation using a 5-methyl cytosine antibody method and found that AOM tumors are generally hypomethylated compared to normal mouse colonic epithelium. These results are consistent with previously published studies and confirm the use of our model system as being generalizable to other studies of AOM induced colon cancer [23]. This validation is relevant in light of the considerable variability in susceptibility to AOM induced tumors observed between mouse strains [46–48]
Because one of the most common epigenetic alterations observed in human colorectal cancer is hypomethylation of the tumor genome with selective hypermethylation of the promoter regions of tumor suppressor genes, after identifying global DNA hypomethylation in the AOM tumors, we examined the methylation status of a series of candidate tumor suppressor genes[49–54]. The genes were selected primarily based on published studies showing them to be methylated in cancers occurring in mouse models or in human cancer. We identified different patterns of methylation of the genes when the methylation status of the genes in the tumors was compared to normal colon mucosa. A subset of tumors carried methylated Mgmt, Id4, Cdkn2a (p16Ink4a), and Cxcr4. Mgmt, Id4, and Cxcr4 were methylated in <25% of the tumor samples, with similar frequencies detected in normal colon mucosa. Several of the genes analyzed (Dapk, Mlt1, Tslc1, Hltf, Mlh1, and p19Arf) were not methylated in the AOM induced tumors or in the normal colon mucosa. Two additional genes of note are Dapk and Mlt1, which displayed low levels of methylation in normal colonic mucosa and no methylation in the tumors, suggesting these genes may be hypomethylated in a subset of tumors. These patterns of methylation in normal colon and colon cancer are also observed in human colon and highlight the importance of the reference tissue used to establish the normal methylation state of the gene[55,56].
Finally, we observed aberrant methylation of two genes, Gja9 and Zik1. which represent 15% of the candidate genes assessed. Gja9 (Gap Junction protein alpha-9), also known as Cx36, encodes for a 59 kDa protein involved in gap junction formation and function. It is the main connexin found in the central nervous system that plays an important role in gap junctions responsible for synaptic transmission[57]. Gja9 is also involved in pancreatic beta islet cell function and mice lacking Gja9 demonstrate decreased glucose-induced insulin production[54]. The aberrant DNA methylation of Gja9 has not been reported previously and was identified as a candidate gene based on preliminary results from genome-wide methylation studies that are in progress (Conerly, personal communication). Zik1 (Zinc finger protein-Interacting with K protein 1) was first identified as a transcriptional repressor that binds to the nuclear ribonucleoprotein particle K protein [58]. Zik1 has also been identified by Methylation Sensitive Representational Difference Analysis (MS-RDA) as being hypermethylated in intestinal metaplasia (IM) of the stomach. Mihara et al reported that Zik1 is hypermethylated in 100% of human gastric IM samples (N=16), 80% of gastric cancer cell lines (N=10), and 73% of primary gastric cancer tumor samples (N=15), suggesting that epigenetic silencing of Zik1 may contribute to the pathogenesis of gastric neoplasia [52]. Our discovery that this gene is also hypermethylated in mouse AOM induced tumors suggests that Zik1 may play an important role in the transformation of gastrointestinal mucosa from normal to cancer. We have also demonstrated that Zik1 shows decreased expression in AOM tumors regardless of methylation status, further supporting this hypothesis.
Some of our results differ from those of previously published studies which may reflect differences between the normal methylation states of the primary tissues from which the tumors were derived or differences in the assays used to assess the methylation state in the CpG islands of the genes of interest [14,24,43]. Further studies are needed to reconcile these differences. In addition, we found that the methylation state of Zikl correlates with decresased expression but that Zik1 expression is decreased in AOM tumors regardless of the tumor's methylation status. This finding suggests that Zik1 may play an important role in the formation of AOM tumors and may be downregulated by both genetic and epigenetic mechanisms. Future studies to determine the role of Zik1 in colon cancer carcinogenesis are necessary to better understand these findings. In addition, the identification of Gja9 being a gene that is not expressed in the colon but that shows increased aberrant methylation in AOM tumors compared to normal colon mucosa demonstrates that there are genes similar to methylated VIM, which is observed in human colorectal cancer, in the AOM model. VIM is not expressed in human colon mucosa or colorectal cancer but methylated VIM is commonly detected in colorectal cancers but not in colon mucosa [59].
Importantly, our studies demonstrate that aberrantly methylated genes do arise in the colon cancers arising in the AOM model. In light of our results using a candidate gene approach, we propose that genome wide methods to investigate the DNA methylation status in AOM tumors are likely to identify novel methylated genes that can be used for studying the role of epigenetically altered genes in this model of colorectal cancer. Restriction enzyme based methods, such as Differential Methylation Hybridization (DMH), Methylated CpG Island Amplification (MCA), Restriction Landmark Genomic Scanning (RLGS), or antibody based approaches such as MeDIP, coupled with microarray technology have been effectively used to identify hypermethylated genes in human and mouse tumors[19,42,56,60–64]. Thus, the use of “methylation arrays” to characterize the methylome in AOM induced tumors has the potential to provide a more comprehensive and unbiased assessment of genes methylated in AOM induced tumors.
In summary, we have demonstrated that similar to human colorectal cancer, AOM induced mouse colonic tumors display global DNA hypomethylation and aberrantly hypermethylated genes. Further studies of the epigenetic alterations using genome wide assays should provide a comprehensive assessment of the methylation state of the DNA in these tumors and will demonstrate the potential of this model to be used to further study the molecular pathology of aberrant DNA methylation in colorectal cancer.
Supplementary Material
Supplemental Figure 1: Demonstration of linearity for the global methylation assay. Linearity is shown through the assessment of a concentration curve using known amounts of calf thymus DNA.
Acknowledgments
Grant Support: S.C.B. was supported by pediatric hematology/oncology training grant 2T32CA009351. These studies were also supported by pilot project funding from P30 CA015704 (WMG). We also wish to acknowledge support from the Experimental Histopathology Core and Animal Care Core at the Fred Hutchinson Cancer Research Center and the Mouse Pathology Core Facility at Vanderbilt University Medical Center) for their technical assistance for preparing tissue samples for histological analysis.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annual Reviews Genomics and Human Genetics. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 3.Janssen KP, Abala M, El Marjou F, Louvard D, Robine S. Mouse models of K-ras-initiated carcinogenesis. Biochim Biophys Acta. 2005;1756(2):145–154. doi: 10.1016/j.bbcan.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Munoz NM, Upton M, Rojas A, et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66(20):9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 5.Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11(11):1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 6.Lorincz MC, Schubeler D, Hutchinson SR, Dickerson DR, Groudine M. DNA methylation density influences the stability of an epigenetic imprint and Dnmt3a/b-independent de novo methylation. Mol Cell Biol. 2002;22(21):7572–7580. doi: 10.1128/MCB.22.21.7572-7580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan PS, Shi H, Rahmatpanah F, et al. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res. 2003;63(19):6178–6186. [PubMed] [Google Scholar]
- 8.Rashid A, Shen L, Morris JS, Issa JP, Hamilton SR. CpG island methylation in colorectal adenomas. Am J Pathol. 2001;159(3):1129–1135. doi: 10.1016/S0002-9440(10)61789-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124(3):762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30(2):183–196. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolt AB, Papanikolaou A, Delker DA, Wang QS, Rosenberg DW. Azoxymethane induces KI-ras activation in the tumor resistant AKR/J mouse colon. Mol Carcinog. 2000;27(3):210–218. [PubMed] [Google Scholar]
- 12.DuBois RN, Radhika A, Reddy BS, Entingh AJ. Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors. Gastroenterology. 1996;110(4):1259–1262. doi: 10.1053/gast.1996.v110.pm8613017. [DOI] [PubMed] [Google Scholar]
- 13.Jacoby RF, Llor X, Teng BB, Davidson NO, Brasitus TA. Mutations in the K-ras oncogene induced by 1,2-dimethylhydrazine in preneoplastic and neoplastic rat colonic mucosa. J Clin Invest. 1991;87(2):624–630. doi: 10.1172/JCI115039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraga MF, Herranz M, Espada J, et al. A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res. 2004;64(16):5527–5534. doi: 10.1158/0008-5472.CAN-03-4061. [DOI] [PubMed] [Google Scholar]
- 15.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36(4):343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 16.Han SY, Iliopoulos D, Druck T, et al. CpG methylation in the Fhit regulatory region: relation to Fhit expression in murine tumors. Oncogene. 2004;23(22):3990–3998. doi: 10.1038/sj.onc.1207526. [DOI] [PubMed] [Google Scholar]
- 17.Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res. 2004;64(11):3844–3848. doi: 10.1158/0008-5472.CAN-03-2119. [DOI] [PubMed] [Google Scholar]
- 18.Schreiner B, Baur DM, Fingerle AA, et al. Pattern of secondary genomic changes in pancreatic tumors of Tgf alpha/Trp53+/− transgenic mice. Genes Chromosomes Cancer. 2003;38(3):240–248. doi: 10.1002/gcc.10285. [DOI] [PubMed] [Google Scholar]
- 19.Yu L, Liu C, Vandeusen J, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet. 2005;37(3):265–274. doi: 10.1038/ng1521. [DOI] [PubMed] [Google Scholar]
- 20.Hahn MA, Hahn T, Lee DH, et al. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res. 2008;68(24):10280–10289. doi: 10.1158/0008-5472.CAN-08-1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaudet F, Hodgson JG, Eden A, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300(5618):489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki K, Suzuki I, Leodolter A, et al. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell. 2006;9(3):199–207. doi: 10.1016/j.ccr.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Tao L, Wang W, Kramer PM, Lubet RA, Steele VE, Pereira MA. Modulation of DNA hypomethylation as a surrogate endpoint biomarker for chemoprevention of colon cancer. Mol Carcinog. 2004;39(2):79–84. doi: 10.1002/mc.20003. [DOI] [PubMed] [Google Scholar]
- 24.Linhart HG, Lin H, Yamada Y, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21(23):3110–3122. doi: 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 26.Biswas S, Chytil A, Washington K, et al. Transforming Growth Factor β Receptor Type II Inactivation Promotes the Establishment and Progression of Colon Cancer. Cancer Res. 2004;64(14):4687–4692. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- 27.Grady WM, Rajput A, Myeroff L, et al. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998;58(14):3101–3104. [PubMed] [Google Scholar]
- 28.Grady WM, Rajput A, Lutterbaugh J, Markowitz S. Detection of aberrantly methylated hMLH1 promoter DNA in the serum of patients with microsatellite unstable colon cancer. Cancer Research. 2001;61:900–902. [PubMed] [Google Scholar]
- 29.Tao L, Li Y, Kramer PM, Wang W, Pereira MA. Hypomethylation of DNA and the insulin-like growth factor-II gene in dichloroacetic and trichloroacetic acid-promoted mouse liver tumors. Toxicology. 2004;196(1–2):127–136. doi: 10.1016/j.tox.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Vertosick FT, Jr., Kelly RH. Antibodies to native and denatured DNA. Quantitation using an immuno-slot-blot technique. J Immunol Methods. 1987;102(1):15–21. doi: 10.1016/s0022-1759(87)80004-2. [DOI] [PubMed] [Google Scholar]
- 31.Petko Z, Ghiassi M, Shuber A, et al. Aberrantly methylated CDKN2A, MGMT, and MLH1 in colon polyps and in fecal DNA from patients with colorectal polyps. Clin Cancer Res. 2005;11:1203–1209. [PubMed] [Google Scholar]
- 32.Liang G, Chan MF, Tomigahara Y, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22(2):480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96(15):8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 35.Bai AHC, Tongg JHM, To K-F, et al. Promoter hypermethylation of tumor-related genes in the progression of colorectal neoplasia. International Journal of Cancer. 2004;112:846–853. doi: 10.1002/ijc.20485. [DOI] [PubMed] [Google Scholar]
- 36.Esteller M. Epigenetic lesions causing genetic lesions in human cancer. promoter hypermethylation of DNA repair genes. Eur J Cancer. 2000;36(18):2294–2300. doi: 10.1016/s0959-8049(00)00303-8. [DOI] [PubMed] [Google Scholar]
- 37.Ferracin M, Gafa R, Miotto E, et al. The methylator phenotype in microsatellite stable colorectal cancers is characterized by a distinct gene expression profile. J Pathol. 2008;214(5):594–602. doi: 10.1002/path.2318. [DOI] [PubMed] [Google Scholar]
- 38.Fleisher AS, Esteller M, Wang S, et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 1999;59(5):1090–1095. [PubMed] [Google Scholar]
- 39.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7(4):536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 40.Kane M, Loda M, Gaida G, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Research. 1997;57:808–811. [PubMed] [Google Scholar]
- 41.Model F, Osborn N, Ahlquist D, et al. Identification and validation of colorectal neoplasia-specific methylation markers for accurate classification of disease. Mol Cancer Res. 2007;5(2):153–163. doi: 10.1158/1541-7786.MCR-06-0034. [DOI] [PubMed] [Google Scholar]
- 42.Camoriano M, Kinney SR, Moser MT, et al. Phenotype-specific CpG island methylation events in a murine model of prostate cancer. Cancer Res. 2008;68(11):4173–4182. doi: 10.1158/0008-5472.CAN-07-6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vuillemenot BR, Hutt JA, Belinsky SA. Gene Promoter Hypermethylation in Mouse Lung Tumors. Mol Cancer Res. 2006 doi: 10.1158/1541-7786.MCR-05-0218. [DOI] [PubMed] [Google Scholar]
- 44.Patel AC, Anna CH, Foley JF, et al. Hypermethylation of the p16 (Ink4a) promoter in B6C3F1 mouse primary lung adenocarcinomas and mouse lung cell lines. Carcinogenesis. 2000;21(9):1691–1700. doi: 10.1093/carcin/21.9.1691. [DOI] [PubMed] [Google Scholar]
- 45.Hinoi T, Akyol A, Theisen BK, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67(20):9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 46.Bissahoyo A, Pearsall RS, Hanlon K, et al. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol Sci. 2005;88(2):340–345. doi: 10.1093/toxsci/kfi313. [DOI] [PubMed] [Google Scholar]
- 47.Nambiar PR, Nakanishi M, Gupta R, et al. Genetic signatures of high- and low-risk aberrant crypt foci in a mouse model of sporadic colon cancer. Cancer Res. 2004;64(18):6394–6401. doi: 10.1158/0008-5472.CAN-04-0933. [DOI] [PubMed] [Google Scholar]
- 48.Guda K, Upender MB, Belinsky G, et al. Carcinogen-induced colon tumors in mice are chromosomally stable and are characterized by low-level microsatellite instability. Oncogene. 2004;23(21):3813–3821. doi: 10.1038/sj.onc.1207489. [DOI] [PubMed] [Google Scholar]
- 49.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 50.Feinberg AP. Alterations in DNA methylation in colorectal polyps and cancer. Prog Clin Biol Res. 1988;279:309–317. [PubMed] [Google Scholar]
- 51.Goelz SEVB, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228(4696):187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- 52.Mihara M, Yoshida Y, Tsukamoto T, et al. Methylation of multiple genes in gastric glands with intestinal metaplasia: A disorder with polyclonal origins. Am J Pathol. 2006;169(5):1643–1651. doi: 10.2353/ajpath.2006.060552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tran R, Kashmiri SV, Kantor J, et al. Correlation of DNA hypomethylation with expression of carcinoembryonic antigen in human colon carcinoma cells. Cancer Res. 1988;48(20):5674–5679. [PubMed] [Google Scholar]
- 54.Wellershaus K, Degen J, Deuchars J, et al. A new conditional mouse mutant reveals specific expression and functions of connexin36 in neurons and pancreatic beta-cells. Exp Cell Res. 2008;314(5):997–1012. doi: 10.1016/j.yexcr.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 55.Kim YH, Petko Z, Dzieciatkowski S, et al. CpG island methylation of genes accumulates during the adenoma progression step of the multistep pathogenesis of colorectal cancer. Genes Chromosomes Cancer. 2006;45(8):781–789. doi: 10.1002/gcc.20341. [DOI] [PubMed] [Google Scholar]
- 56.Shen L, Kondo Y, Guo Y, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3(10):e181. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cummings DM, Yamazaki I, Cepeda C, Paul DL, Levine MS. Neuronal coupling via connexin36 contributes to spontaneous synaptic currents of striatal medium-sized spiny neurons. J Neurosci Res. 2008;86(10):2147–2158. doi: 10.1002/jnr.21674. [DOI] [PubMed] [Google Scholar]
- 58.Denisenko ON, O'Neill B, Ostrowski J, Van Seuningen I, Bomsztyk K. Zik1, a transcriptional repressor that interacts with the heterogeneous nuclear ribonucleoprotein particle K protein. J Biol Chem. 1996;271(44):27701–27706. doi: 10.1074/jbc.271.44.27701. [DOI] [PubMed] [Google Scholar]
- 59.Chen WD, Han ZJ, Skoletsky J, et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J Natl Cancer Inst. 2005;97(15):1124–1132. doi: 10.1093/jnci/dji204. [DOI] [PubMed] [Google Scholar]
- 60.Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet. 1999;8(3):459–470. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- 61.Jacinto FV, Ballestar E, Ropero S, Esteller M. Discovery of epigenetically silenced genes by methylated DNA immunoprecipitation in colon cancer cells. Cancer Res. 2007;67(24):11481–11486. doi: 10.1158/0008-5472.CAN-07-2687. [DOI] [PubMed] [Google Scholar]
- 62.Lewin J, Plum A, Hildmann T, et al. Comparative DNA methylation analysis in normal and tumour tissues and in cancer cell lines using differential methylation hybridisation. Int J Biochem Cell Biol. 2007;39(7–8):1539–1550. doi: 10.1016/j.biocel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 63.Weber M, Davies JJ, Wittig D, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37(8):853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 64.Yan PS, Efferth T, Chen HL, et al. Use of CpG island microarrays to identify colorectal tumors with a high degree of concurrent methylation. Methods. 2002;27(2):162–169. doi: 10.1016/s1046-2023(02)00070-1. [DOI] [PubMed] [Google Scholar]
- 65.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2(10):731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 66.Blanco D, Vicent S, Fraga MF, et al. Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway. Neoplasia. 2007;9(10):840–852. doi: 10.1593/neo.07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morey SR, Smiraglia DJ, James SR, et al. DNA methylation pathway alterations in an autochthonous murine model of prostate cancer. Cancer Res. 2006;66(24):11659–11667. doi: 10.1158/0008-5472.CAN-06-1937. [DOI] [PubMed] [Google Scholar]
- 68.Mittag F, Kuester D, Vieth M, et al. DAPK promotor methylation is an early event in colorectal carcinogenesis. Cancer Lett. 2006;240(1):69–75. doi: 10.1016/j.canlet.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 69.Abdel-Fattah R, Glick A, Rehman I, Maiberger P, Hennings H. Methylation of the O6-methylguanine-DNA methyltransferase promoter suppresses expression in mouse skin tumors and varies with the tumor induction protocol. Int J Cancer. 2006;118(3):527–531. doi: 10.1002/ijc.21316. [DOI] [PubMed] [Google Scholar]
- 70.Moinova HR, Chen WD, Shen L, et al. HLTF gene silencing in human colon cancer. Proc Natl Acad Sci U S A. 2002;99(7):4562–4567. doi: 10.1073/pnas.062459899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. PNAS. 1998;95(12):6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuismanen SA, Holmberg MT, Salovaara R, et al. Epigenetic phenotypes distinguish microsatellite-stable and -unstable colorectal cancers. Proc Natl Acad Sci U S A. 1999;96(22):12661–12666. doi: 10.1073/pnas.96.22.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tateno M, Fukunishi Y, Komatsu S, et al. Identification of a novel member of the snail/Gfi-1 repressor family, mlt 1, which is methylated and silenced in liver tumors of SV40 T antigen transgenic mice. Cancer Res. 2001;61(3):1144–1153. [PubMed] [Google Scholar]
- 74.Tsujiuchi T, Sugata E, Masaoka T, et al. Expression and DNA methylation patterns of Tslc1 and Dal-1 genes in hepatocellular carcinomas induced by N-nitrosodiethylamine in rats. Cancer Sci. 2007;98(7):943–948. doi: 10.1111/j.1349-7006.2007.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Demonstration of linearity for the global methylation assay. Linearity is shown through the assessment of a concentration curve using known amounts of calf thymus DNA.