

Abstract
Chromogranin A (CHGA/Chga) has been implicated in the genesis of systemic hypertension and consequent cardiac abnormalities. Catestatin (CST) (human CHGA352-372) replacement reduces blood pressure elevation and increases baroreflex sensitivity in Chga knockout (KO) mice. Because of the dampened baroreflex sensitivity, we reasoned that KO mice would display altered heart rate variability (HRV). Thus, we evaluated beat-to-beat measurements in HRV in wild-type (WT) and KO mice, before and after CST replacement. HR dynamics were evaluated by bipolar Einthoven electrocardiogram, with deconvolution into time and frequency domains, as well as Lorenz nonlinear return analyses. At baseline, HR was higher [444 ± 24 beats per minute (bpm)] in KO compared with WT (330 ± 18 bpm) mice. The total power in the HRV spectra was substantially diminished in KO animals. CST increased total power but only in KO mice. Each time-domain parameter was substantially lower in KO compared with WT mice, and the CST in the KO group could reverse the differences. Lorenz analysis revealed reductions in S1 (short axis perpendicular to the line of identity in the ellipse) and S2 (long axis along the line of identity in the ellipse) in KO animals, indicating that regulation of HRV is diminished in the parasympathetic and sympathetic domains. CST replacement caused restoration of both S1 and S2, in the KO group. These data suggest that Chga has a profound effect on autonomic tone to the heart and that its CST fragment is responsible for such actions. The results suggest future strategies for intervention in cardiovascular disorders accompanied by adverse HRV profiles.
The long-term consequences of neonatal inflammation occur in a sex-specific manner, and there is dissociation between physiological and behavioral indicators of inflammation and sickness.
Cardiovascular performance is controlled by the autonomic nervous system (ANS). Beat-to-beat fluctuation in heart rate (HR) is a balanced consequence of the ANS tone to the heart, both sympathetic (increasing HR) and parasympathetic (decreasing HR). The multifunctional protein chromogranin A (CHGA/Chga), the index member of the chromogranin/secretogranin protein family, is involved in several cardio-renal diseases (1,2,3). Its importance in hypertensive end-stage renal disease and as an element in the genesis of hypertension triggered by environmental stress have been described in previous studies (2,4,5,6,7). Along with the release of CHGA on exocytosis by chromaffin cells, there is corelease of its peptide fragment catestatin (CST), with consequent inhibition of its target, the nicotinic acetylcholine receptor (8,9), thereby limiting further catecholamine release. CHGA has further been reported to influence cardiovascular function via the action of its amino terminal (vasostatin) domain on blood vessels (10) and its CST domain on cardiomyocytes (11,12).
Although CHGA is overexpressed in human essential (hereditary) hypertension, plasma CST level is decreased not only in the established cases of hypertension but also in normotensive subjects with a family history of hypertension (13). To get a better insight into the function of Chga, we have generated Chga knockout (KO) mice by targeted ablation of the Chga gene. Consistent with human findings, KO mice displayed high systolic blood pressure (SBP) and diastolic blood pressure and high plasma catecholamines (14). In addition, KO mice showed lack of diurnal variation in SBP (14), echocardiographic evidence of left ventricular hypertrophy, increased ventricular dilation (14), and dampened baroreflex sensitivity (BRS) (15). CST replacement rescued KO mice from high SBP (14) and catecholamines and improved BRS (15). We have recently reported that despite very low insulin level (∼20% of WT) and muscle resistance to insulin, KO mice maintain euglycemic status because of the increased hepatic sensitivity to insulin (16).
A decrease in HR variability (HRV) is longitudinally predictive of increased cardiovascular morbidity and mortality. For example, the earliest sign of cardiac autonomic neuropathy (CAN) is a reduction in HRV, which is detectable at the subclinical stage (17,18). Cardiac autonomic (parasympathetic) function is diminished in diabetic patients before clinical symptoms of neuropathy become evident (19,20,21). As many as 22% of people with type 2 diabetes mellitus suffer from CAN, which leads to impaired regulation of blood pressure (BP), HR, and HRV (22). Silent ischemia is significantly more frequent in patients with than in those without CAN (38 vs. 5%) (23). In addition, HRV measurement is of clinical significance and a prognostic indicator for hypertension (both at the cardiac and adrenal medullary level) (24), postmyocardial infarction (25), and congestive heart failure (26). In all these cases, a significant decrease in the HR fluctuation has been observed. Ceconi et al. (27) reported that serum levels of CHGA are elevated in patients with heart failure. Because KO mice show decreased BRS, we reasoned that KO mice would display altered HRV. Therefore, in the current investigation, we examined both short- and long-term HRV regulation in KO mice compared with wild-type (WT) mice, monitoring temporal and spectral features to identify perturbations of the ANS in HRV via time- and frequency-domain characteristics. In addition, to accomplish our goal, we used return map (Lorenz plot, also known as dynamical systems) analysis of sequential heartbeats (e.g. n + 1 vs. n). We then inspected the data dispersion within Lorenz plots to quantify beat-to-beat variability. Because one of the analytical tools makes use of nonparametric Fast Fourier Transformation (FFT), requiring that biological signals acquired be relatively stable, animals were under sedation to avoid pitfalls encountered in awake animals, as has been demonstrated earlier by many investigators (28,29,30). We thus investigate cardiovascular properties of KO mice to determine whether the catecholamine release-inhibitory CST fragment of Chga may confer benefit onto HRV.
Materials and Methods
Study animals
Generation of KO mice has been described in detail in our previous communication (14). Adult WT and KO mice with mixed genetic background were used in this study. Both WT and KO mice were generated from founders carrying a mixed genotype (50% 129svJ:50% C57BL/6) and then were maintained by brother/sister mating. Mice were housed in cages at 24 C in a facility with 12-h light, 12-h dark cycles, in compliance with the Public Health Service Animal Welfare policy and the American Association for the Accreditation of Laboratory Animal Care, and fed standard chow ad libitum. All experiments were performed under protocols approved by the University of California, San Diego and Veterans Affairs Hospital Animal Care and Use Committee, San Diego, CA.
Protocol
WT (n = 11) and KO (n = 11) mice were used in this study. The mean age and weight for WT were: 120 ± 20 d and 24.3 ± 1.6 g; for KO were: 112 ± 8 d and 25.8 ± 1.2 g, respectively. These differences in age and weight were not statistically significant. Mice were sedated by an ip injection of a 1 μl solution per g body weight, containing Ketamine (100 mg/ml) + Xylazine (20 mg/ml) and Acepromazine (10 mg/ml). Mice were gently strapped onto the surgical table in a fixed supine position with silk tape. The adequacy of sedation was regularly verified by absence of vibrissae twitching and withdrawal reflex to a noxious toe-pinch and was supplemented ip as needed. Body temperature was continuously monitored on an analog thermometer, model 44TA (YSI, Yellow Springs, OH) using a rectal probe and maintained between 36.5 and 38.5 C throughout the experiment using a controlled heating box. The analog output signal fed into the chart recorder (Labchart; AD Instruments, Colorado Springs, CO) and suitably amplified. Animals breathed ambient air spontaneously throughout the experimental run. Investigational agent, a nicotinic cholinergic receptor antagonist (9,31), CST (human CHGA352–372, MW = 2326.8; Imgenex, San Diego, CA) was dissolved in distilled water and injected ip at a volume of 70 μl (6.5 μg/g body weight) from a 1 mm working solution.
Electrophysiological recording
Electrocardiography (EKG) electrodes were attached to all four limbs (including reference electrode) via 30-G stainless steel hypodermic needles (Popper & Sons, Lake Success, NY) so as to record standard bipolar Einthoven derivation (lead I, II, and III) EKG signals. EKG signals were acquired continuously by Gould Universal bio-amplifier (Cleveland, OH) at the frequency range from 0.1 to 3 kHz. The amplified signals were fed into a data acquisition system (PowerLab-8; AD Instruments) and digitally sampled at 2 kHz for eventual off-line measurements and analysis by chart and scope (Labchart 6) software. HR and rhythm were also monitored continuously on a storage oscilloscope (Tektronix 5113, Beaverton, OR) throughout the experiment.
Data editing and evaluation
EKG traces (lead II) over 5-min increments were edited using the Chart software package. High-frequency noise from the EKG was removed using a 45 Hz low-pass filter to enable detection of the R-waves, whenever appropriate. Recording and evaluation of cardiac spectrogram in each (WT and KO) mouse showed that 98% of frequency components (rate and shape information) reside within 0–300 Hz. Digital filter was used in such a way as to preserve these essential frequency components of electrocardiographic discharge. A threshold electrical potential value was set for each trace, and the postthreshold maximum (max) was taken as the R-wave. Each trace was visually inspected for any undetected R-waves, and these were manually inserted, whereas incorrectly detected R-waves or nonnormal beats such as extrasystoles and technically inadequate recordings were excluded.
HR and HRV
HRV quantification was done by the use of generally acceptable methods of 1) frequency-domain analysis, 2) time-domain analysis, and 3) the Lorenz return map analysis.
1) Frequency-domain analysis of HRV was performed using FFT with a Hanning window. Spectral power was calculated on 50% overlapped blocks on sequences of 512 points. The following parameters were calculated: total power (TP), low-frequency (LF) power, high-frequency (HF) power, and the ratio (LF/HF). The different frequency-domain measures of HRV were computed using cut-off frequencies for power in the LF and HF range based on human studies, multiplied by a factor of 10 for HR adjustment (the approximate ratio between murine and human HR) (32). The area under the curve was calculated for the LF (0.4–1.5 Hz), and HF (1.5–4.0 Hz) bands, as previously defined in the mouse species by several investigators (33,34,35,36). TP (variance of normal R-R intervals over temporal segment) was defined as 0–4 Hz. Spectral variability at LF and HF bandwidths was also normalized as parameter ν (normalized units). Normalized power and the LF/HF ratio furnished an assessment of the fractional distribution of power across the frequency axis and emphasized controlled and balanced properties of the arms of the ANS irrespective of TP (37). Five-minute epochs of HR as a function of time were used for power spectrum (power) analysis.
2) Time-domain parameters recorded and calculated included mean R-R interval (in msec), sd of all normal R-R interval (SDNN; in msec), reflecting total autonomic variability, square root mean of squared differences between adjacent normal R-R intervals (RMSSD; in msec), an index for short-term variations in interbeat intervals, primarily of parasympathetic nature, and percentage of normal consecutive interbeat intervals differing by more than 7 msec (NN7%), reflecting cardiac parasympathetic tone, were calculated directly from the sequence of interval times. NN7 is a murine-adjusted value based on pNN50 (one member of a family of time-domain statistics, measuring fraction of consecutive cardiac inter-beat intervals that differ by more than 50 msec for diagnostic and prognostic profiling in patients) determined for humans (32). The mean HR was calculated as the mean sequence of reciprocals of the interval times.
3) For nonlinear measurements on the return map of R-R intervals (Lorenz plot), the images acquired by the chart software were imported into Image J (National Institutes of Health, Bethesda, MD) for evaluating point dispersion of S1 (short axis perpendicular to the line of identity in the ellipse) and S2 (long axis along the line of identity in the ellipse) (see figure 5). The width and length of the dispersion of data in Lorenz plots reflects the level of short- and long-term beat-to-beat variability, respectively. Such variability is captured by characterizing an ellipse with a long axis S2 (msec), and a short axis S1 (msec), in LabChart 6 (AD Instruments).
Figure 5.

Return maps. Typical representations of Lorenz return maps acquired from WT (top) and KO (bottom) mice. Plotted on abscissa is (R-Rn) against next (R-Rn+1) interval time on the ordinate. KO mice distinctly revealed a compact nature of point dispersion (i.e. narrow HRV) compared with the increased dimension of point's area in the WT mice. Ellipses have two perpendicular axes: long (S2) and short (S1).
Statistical analyses
Data graphs (Kaleidagraph; Synergy software version 4.0; Synergy, Reading, PA) presented as mean ± sem were statistically evaluated by Instat software (GraphPad, San Diego, CA). Comparisons between groups were done by Student's t test. Multiple comparisons were made using two-way ANOVA. Differences were considered significant at P < 0.05.
Results
FFT power spectra of HR dynamics
FFT power spectral analysis (Fig. 1) of HRV in WT and KO mice in the resting state showed periodic components of HRV in two distinct peaks centered around 2.4–3.2 Hz (in the HF band, considered to index cardio-vagal activity), and less than 0.2 Hz (below LF band) in most instances, with occasional smaller peaks at approximately 1 Hz.
Figure 1.
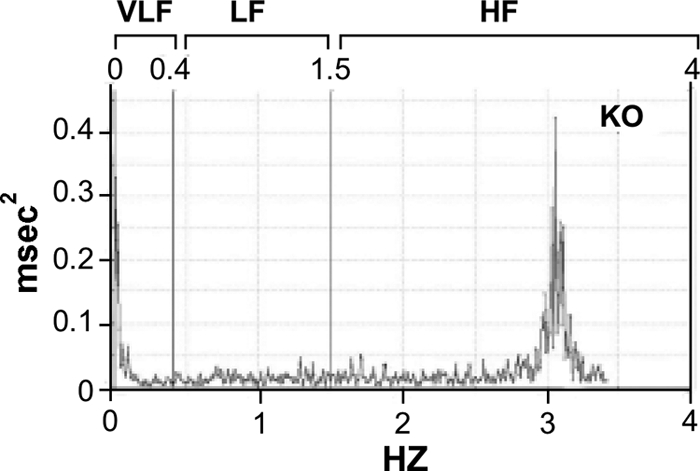
FFT spectrum for HRV obtained from a sedated KO mouse with spontaneous ventilation. The vertical boundaries are VLF < 0.4 Hz, LF = 0.4–1.5 Hz, and HF = 1.5–4 Hz. The results in this animal reveal essentially two spectral peaks; in the HF and in less than 0.4-Hz bands Abscissa (x-axis, in Hz) in the spectrum reflects various frequency bands, whereas the y-axis represents power (msec2) values.
HR
Overall HR is summarized in Fig. 2A. Chga null mice (at 444 ± 24 bpm) showed substantially (P < 0.0003) increased HR over WT mice (at 330 ± 18 bpm). In the presence of CST, tachycardia observed in KO was reduced by approximately 10% (Fig. 2A). On the other hand, in WT mice, an opposite effect (Fig. 2A) to elevate HR was observed. Two-way ANOVA revealed significant difference (P < 0.001) between WT and KO mice. This study also confirmed our earlier observations on overall HR increment in the KO mice (14).
Figure 2.

Bar graphs showing HR and power amplitude derived from FFT analyses of HRV from WT and KO mice at various spectral bands, including: HR (A), TP (B), LF power (C), HF power (D), and LF/HF power (E) ratio and their modification by ip CST. Normalized unit; N.S., not significant (P > 0.05).
HRV frequency domain: Effect on KO and restoration by CST
The TP in the HRV spectra (Fig. 2B) is composed of LF, HF, and frequencies outside of LF and HF power components. TP was substantially diminished in KO animals (P < 0.0009). CST increased TP but only in KO mice (Fig. 2B; P < 0.0001). We found significant difference (P < 0.02) between saline- and CST-treated mice after analysis by two-way ANOVA revealed (Fig. 2B).
At baseline, normalized LF power was comparable in WT and KO mice (Fig. 2C). CST enhanced LF power but only significantly in the KO group (P < 0.007; Fig. 2C). Both the minimum (min) and max LF values shifted to higher levels after CST in each groups (WTmin/max, 3.202/94.727 vs. KOmin/max, 7.838/51.883). Two-way ANOVA resulted in significant difference (P < 0.006) between saline- and CST-treated mice (Fig. 2C).
For normalized HF power, the basal values were comparable in KO vs. WT groups (Fig. 2D), while CST increased HF power in WT (P < 0.03), whereas it reduced HF power in KO group (P < 0.004, Fig. 2D). We noticed significant interaction (P < 0.0007) between strain and peptide when the data were analyzed by two-way ANOVA (Fig. 2D).
The relative strength of the sympatho-vagal balance index LF/HF ratio is shown in Fig. 2E, which shows that the basal sympatho-vagal balance is comparable between WT and KO mice. CST increased the LF/HF ratio only in KO mice (Fig. 2E; P < 0.009). Two-way ANOVA presented significant difference in strain (P < 0.03), peptide (P < 0.02), as well as strain vs. peptide interaction (P < 0.02) (Fig. 2D).
HRV time domain: Decline in KO and effects of CST
In Fig. 3, A and B, individual time-series profiles of R-R (sequential N-N) intervals timed from WT and KO mice are shown, to evidence a larger pulse interval fluctuation in WT (Fig. 3A) than KO (Fig. 3B) mice, in which the fluctuation width in pulse interval is visibly lower at baseline. Graphical representations of time-domain parameters are shown in Fig. 4. The basal N-N value was different between WT and KO mice (P < 0.01; Fig. 4A). We found significant difference (P < 0.01) between WT and KO mice after analysis by two-way ANOVA (Fig. 4A). SDNN values were significantly lower in KO mice (P < 0.003; Fig. 4B), which was reversed by CST (P < 0.0009; Fig. 4B). Two-way ANOVA showed significant interaction (P < 0.003) between strains vs. peptide (Fig. 4B). Like mean N-N and SDNN, coefficient of variation (in %, as 100 × sd/mean) in N-N intervals (CVNN) was also significantly low (P < 0.009; Fig. 4C) in KO mice, which was inhibited by CST (P < 0.003; Fig. 4C). We found significant interaction (P < 0.003) between strains vs. peptide when the data were analyzed by two-way ANOVA (Fig. 4C). Likewise, NN7% values are lower in KO mice (P < 0.01; Fig. 4D). Two-way ANOVA showed significant difference (P < 0.05) between WT and KO mice (Fig. 4D). Similarly, RMSSD values were substantially lower in KO compared with WT mice (P < 0.006), which was significantly improved by CST (P < 0.03; Fig. 4E). There was a significant interaction (P < 0.05) found between strains vs. peptide (Fig. 4E) by two-way ANOVA analysis.
Figure 3.
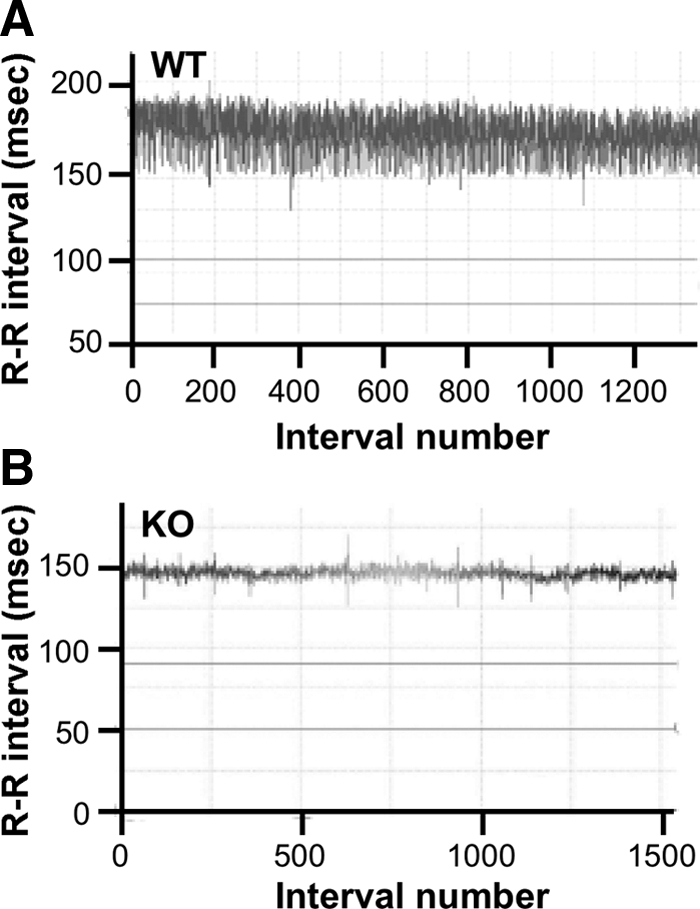
Cardiac rate tachogram in (A) WT and (B) KO mice. The abscissa (x-axis) shows beat number (x, x + 1, x + 2, … , x + n), whereas the ordinate (y-axis) shows the R-R intervals (in msec) of consecutive beats: y1 = (x + 1) − x, y2 = (x + 2) − (x + 1), … , yn = (xn) − (xn − 1).
Figure 4.

Time-domain parameters of HRV in the WT and in the KO mice, including: N-N (difference in R-R between consecutive beats) (A), SDNN (sd of N-N) (B), CVNN (coefficient of variation of N-N) (C), NN7% (% of consecutive beats more than 7 msec different in R-R) (D), and RMSSD (root mean square of sd of N-N) (E). N.S., Not significant (P > 0.05).
Lorenz return map analysis of HRV
Reductions in time-domain parameters in the KO strain were consistent with the results of nonlinear (Lorenz) beat-to-beat measurements of R-R intervals. Examples of Lorenz return maps obtained from WT and KO animals are shown in Fig. 5, where S1 reflects shorter-term (msec-to-msec) predominantly parasympathetic drive, and S2 reflects longer-term (sec-to-sec) variability, primarily of a sympathetic nature, and S1/S2 is an index of vago-sympathetic balance. There were significant reductions in S1 (P < 0.05; Fig. 6A) and S2 (P < 0.005; Fig. 6B) in KO mice, with no change in the ratio of S1/S2. CST intervention improved both the S1 (P < 0.008; Fig. 6A) and S2 (P < 0.004; Fig. 6B) values in KO mice only. In case of S1/S2, CST caused improvement in WT (P < 0.03; Fig. 6C) and decrement in KO mice (P < 0.02; Fig. 6C). Although the two-way ANOVA demonstrated significant difference between saline vs. CST only in S1 parameter (P < 0.04), it shows significant interaction between strain and peptide in S1 (P < 0.04; Fig. 6A), S2 (P < 0.0005; Fig. 6B), as well as S1/S2 (P < 0.001; Fig. 6C) parameters.
Figure 6.

Return map analysis of R-R intervals showing short axis S1 (A) and long axis S2 (B) magnitudes, as well as S1/S2 ratio (C) as a predominant index of vago-sympathetic balance, in WT and KO mice. N.S., Not significant (P > 0.05).
Discussion
CST effect on cardiovascular system
Although CST was initially identified as a potent endogenous nicotinic-cholinergic antagonist (9), subsequent studies indicate that CST exerts profound effects on cardiovascular system (11,12,13,14,15,38,39; Fung M. M., R. M. Salem, P. Mehtani, B. Thomas, C. F. Lu, B. Perez, F. Rao, M. Stridsberg, M. Ziegler, S. K. Mahata, D. T. O'Connor, submitted for publication). This was apparent after the iv administration of CST to rats that reduced pressor responses to activation of sympathetic outflow by electrical (7.5 V, 20 Hz) stimulation (38). Recent studies indicate that CST caused dose-dependent vasodilation predominantly in female subjects after phenylephrine- induced preconstriction to 69% with an EC50 of approximately 30 nm, which was the same order of magnitude to circulating endogenous CST (4.4 nm) (Fung M. M., R. M. Salem, P. Mehtani, B. Thomas, C. F. Lu, B. Perez, F. Rao, M. Stridsberg, M. Ziegler, S. K. Mahata, D. T. O'Connor, submitted for publication). We also found that despite low CHGA precursor concentrations, female subjects had higher plasma CST levels than males, which reflects increased processing of CHGA-to-CST (Fung M. M., R. M. Salem, P. Mehtani, B. Thomas, C. F. Lu, B. Perez, F. Rao, M. Stridsberg, M. Ziegler, S. K. Mahata, D. T. O'Connor, submitted for publication). These findings indicate that CST may contribute to regulation of endogenous vascular tone and influence the complex predisposition to hypertension.
Consistent with diminished plasma CST not only in established cases of essential (hereditary) hypertension but also in the still-normotensive offspring of patients with hypertension (positive family history: FH+) (13), we detected high BP and higher plasma catecholamines in KO mice (14). Of note, CST replacement rescued KO mice from the high resting BP and plasma catecholamines during normal (14) or after immobilization stress (15), implicating CST as an antihypertensive peptide even in stressful conditions. Because of the potent inhibition of the inotropic and lusitropic properties of the rodent (12) and frog (11) heart, CST is now considered as a novel cardiac modulator, which could protect the heart against excessive sympathetic drive such as that seen in hypertensive cardiomyopathy. CST replacement also restored (elevated) dampened BRS sensitivity in KO mice (15), indicating that CST resets the entire autonomic nervous reflex arc to restore normal cardiovascular function. The present study documents that CST also improves several components of HRV.
HRV in the frequency domain
Mice deficient in the endogenous Chga peptide CST display increased HR, decreased HRV, and altered autonomic HR modulation, when compared with WT mice. Spectral analysis of HR signals in the frequency domain revealed that the HRV of KO mice were decreased overall (TP), but spectral decomposition suggested a most prominent decline in the less than 0.4-Hz band. Controversy surrounding the role of the less than 0.4-Hz band still exists, and no specific guidelines on interpretation of less than 0.4 Hz are consistently observed in the literature (40). It is generally postulated that the primary function of less than 0.4-Hz band lies in the thermogenic and neurohumoral processes; because all experiments in this study were carried out under controlled temperature conditions, any regulatory role in HRV via a thermogenic process can be ruled out, suggesting neurohormonal differences. The observation of significant reductions in the KO group for TP (Fig. 2B) corresponds to similar findings in human subjects with hypertension (41,42), and hyperadrenergic hypertension has been a distinct feature in the KO mouse model (14), whereas human genetic variation at the CHGA locus elevates risk for hypertension, in association with augmented sympathetic tone (13). Reductions in TP are associated with increments in human cardiovascular risk (41,42). The increase in LF power (0.4–1.5 Hz, reflecting a mixture of parasympathetic and sympathetic activity) after CST injection is not likely a result of increasing sympathetic activity, because CST is demonstrated to cause decrease plasma norepinephrine level (9,31,43,44). It is interesting that LF considered to reflect mainly sympathetic activity was negatively correlated with the HR after CST replacement, suggesting that LF changes are most likely driven by baroreflex oscillation and not necessarily by absolute parasympathetic and sympathetic components, and the enhanced LF is now being validated by us to reflect changes in BRS (15).
A decrease in HF power (1.5–4.0 Hz, reflecting predominantly parasympathetic activity) by CST (Fig. 2D), likely reflecting a decline in parasympathetic output, suggests that a decrement in sympathetic tone caused partial restoration (reduction) of overall HR (Fig. 2A). What branch of the ANS responded primarily to CST in increasing TP in the KO animals (overall HRV; Fig. 2B)? LF power (Fig. 2C) increased after CST in the KO, whereas HF power actually decreased (Fig. 2D). Thus, CST likely caused a beneficial change (decline) in sympathetic tone to achieve the overall (TP) favorable change in HRV.
HRV in the time domain
Strain differences in HRV between WT and KO were perhaps even more apparent in the time domain than in the frequency domain. Time-domain analyses showed fluctuation of width in R-R (N-N) intervals to be substantially reduced in the KO group, along with depression of several associated traits such as mean N-N [arithmetic average of normal (nonectopic) interbeat (R-wave in EKG) interval in msec] (Fig. 4A); SDNN (sd of all normal R-R intervals in msec reflecting global autonomic variability) (Fig. 4B); CVNN (Fig. 4C); NN7% (percentage of normal consecutive interbeat intervals differing by >7 msec reflecting cardiac parasympathetic tone) (Fig. 4D), and RMSSD (square root mean of squared differences between adjacent normal R-R intervals in msec, which is an index for short-term variations and is primarily of parasympathetic in nature) (Fig. 4E) in regulation of HRV. Depression of RMSSD for the KO group at baseline (Fig. 4E) suggests either a deficit of parasympathetic traffic or an increment of sympathetic drive at rest. Such findings in KO mice are reminiscent of findings in human subjects with cardiovascular disease (45), in that the TP is reduced and the largest R-R intervals are lost, such as occurs in CHF (46).
Lorenz return map analysis of HRV
Lorenz analysis also enabled short- and long-term indices of the Lorenz plot, in the form of its indices S1 (short-term parasympathetic regulation) and S2 (longer-term sympathetic regulation). Reductions of S1 and S2 (Fig. 6, A and B) in KO animals indicate that regulation of HRV is diminished both in the parasympathetic and sympathetic domains. Reduction of the S1/S2 quotient in the CST-treated KO group (Fig. 6C) indicates dampened vago-sympathetic balance, consistent with an especially diminished role of parasympathetic modulation of HR.
CST improvement of HRV
Improved HRV by CST in KO mice seem to corroborate clinical findings in modification of human autonomic activity by CST (39). The observed reduction in HR along with improvement in HRV after CST injection and previously reported effect of CST's reversal of SBP and diastolic blood pressure (14) presumably will cause a reduction in myocardial work and oxygen demand, and therefore, could be beneficial to the failing heart and provide additional benefits via attenuation of catecholaminergic drive. Although the anatomical site of action of CST was not directly investigated in this study, the highly cationic nature of CST (9) is reminiscent of the amino acid composition of membrane-transducing peptide domains (47,48), suggesting that CST's autonomic actions might in part occur in the central nervous system, a conclusion in line with the likely sites of CST effects on sympathetic and parasympathetic drive in humans (39).
Conclusion and perspectives
Genetically modified mice lacking Chga and its sympatho-inhibitory fragment CST give rise to outward resemblances to observations in the Framingham study wherein time- and frequency-domain variables of HRV were reduced in untreated hypertensive individuals (49). Based on the animal model presented here, we suggest that CST may have the potential to reverse or minimize such adverse autonomic traits, thereby favorably influence cardiovascular outcomes.
Footnotes
This work was supported by the National Institutes of Health Grant R01 DA011311. Research Career Scientist award from the Department of Veterans Affairs provides the salary support to S.K.M.
Disclosure Summary: The authors have nothing to disclose.
First Published Online April 21, 2010
Abbreviations: ANS, Autonomic nervous system; BP, blood pressure; bpm, beats per minute; BRS, baroreflex sensitivity; CAN, cardiac autonomic neuropathy; CHGA, chromogranin A; CST, catestatin; CVNN, coefficient of variation (in %, as 100 × sd/mean) in N-N intervals; EKG, electrocardiography; FFT, Fast Fourier Transformation; HF, high frequency; HR, heart rate; HRV, HR variability; KO, knockout; LF, low frequency; max, maximum; min, minimum; NN7%, percentage of normal consecutive interbeat intervals differing by more than 7 msec; RMSSD, square root mean of squared differences between adjacent normal R-R intervals (in milliseconds); S1, short axis perpendicular to the line of identity in the ellipse; S2, long axis along the line of identity in the ellipse; SBP, systolic blood pressure; SDNN, sd of all normal R-R intervals (in msec); TP, total power; WT, wild type.
References
- Winkler H, Fischer-Colbrie R 1992 The chromogranins A and B: the first 25 years and future perspectives. Neuroscience 49:497–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taupenot L, Harper KL, O'Connor DT 2003 Mechanisms of disease: the chromogranin-secretogranin family. New Engl J Med 348:1134–1149 [DOI] [PubMed] [Google Scholar]
- Montero-Hadjadje M, Vaingankar S, Elias S, Tostivint H, Mahata SK, Anouar Y 2008 Chromogranins A and B and secretogranin II: evolutionary and functional aspects. Acta Physiol 192:309–324 [DOI] [PubMed] [Google Scholar]
- Takiyyuddin MA, Cervenka JH, Hsiao RJ, Barbosa JA, Parmer RJ, O'Connor DT 1990 Chromogranin A. Storage and release in hypertension. Hypertension 15:237–246 [DOI] [PubMed] [Google Scholar]
- Hsiao RJ, Mezger MS, O'Connor DT 1990 Chromogranin A in uremia: progressive retention of immunoreactive fragments. Kidney Int 37:955–964 [DOI] [PubMed] [Google Scholar]
- Chen Y, Rao F, Rodriguez-Flores JL, Mahapatra NR, Mahata M, Wen G, Salem RM, Shih PA, Das M, Schork NJ, Ziegler MG, Hamilton BA, Mahata SK, O'Connor DT 2008 Common genetic variants in the chromogranin A promoter alter autonomic activity and blood pressure. Kidney Int 74:115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DT 1984 Chromogranin A: implications for hypertension. J Hypertens(Suppl 2):S147–S150 [PubMed] [Google Scholar]
- Takiyyuddin MA, Brown MR, Dinh TQ, Cervenka JH, Braun SD, Parmer RJ, Kennedy B, O'Connor DT 1994 Sympatho-adrenal secretion in humans: factors governing catecholamine and storage vesicle peptide co-release. J Auton Pharmacol 14:187–200 [DOI] [PubMed] [Google Scholar]
- Mahata SK, O'Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H, Gill BM, Parmer RJ 1997 Novel autocrine feedback control of catecholamine release. A discrete chromogranin A fragment is a noncompetitive nicotinic cholinergic antagonist. J Clin Invest 100:1623–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aardal S, Helle KB 1992 The vasoinhibitory activity of bovine chromogranin A fragment (vasostatin) and its independence of extracellular calcium in isolated segments of human blood vessels. Regul Pept 41:9–18 [DOI] [PubMed] [Google Scholar]
- Mazza R, Gattuso A, Mannarino C, Brar BK, Barbieri SF, Tota B, Mahata SK 2008 Catestatin (chromogranin A344–364) is a novel cardiosuppressive agent: inhibition of isoproterenol and endothelin signaling in the frog heart. Am J Physiol Heart Circ Physiol 295:H113–H122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelone T, Quintieri AM, Brar BK, Limchaiyawat PT, Tota B, Mahata SK, Cerra MC 2008 The antihypertensive chromogranin a peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology 149:4780–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DT, Kailasam MT, Kennedy BP, Ziegler MG, Yanaihara N, Parmer RJ 2002 Early decline in the catecholamine release-inhibitory peptide catestatin in humans at genetic risk of hypertension. J Hypertens 20:1335–1345 [DOI] [PubMed] [Google Scholar]
- Mahapatra NR, O'Connor DT, Vaingankar SM, Hikim AP, Mahata M, Ray S, Staite E, Wu H, Gu Y, Dalton N, Kennedy BP, Ziegler MG, Ross J, Mahata SK 2005 Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest 115:1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayen JR, Gu Y, O'Connor DT, Mahata SK 2009 Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin A null mouse. Endocrinology 150:5027–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayen JR, Saberi M, Schenk S, Biswas N, Vaingankar SM, Cheung WW, Najjar SM, O'Connor DT, Bandyopadhyay G, Mahata SK 2009 A novel pathway of insulin sensitivity in chromogranin a null mice: a crucial role for pancreastatin in glucose homeostasis. J Biol Chem 284:28498–28509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing DJ, Campbell IW, Clarke BF 1980 The natural history of diabetic autonomic neuropathy. Q J Med 49:95–108 [PubMed] [Google Scholar]
- Watkins PJ, Mackay JD 1980 Cardiac denervation in diabetic neuropathy. Ann Intern Med 92:304–307 [DOI] [PubMed] [Google Scholar]
- Pfeifer MA, Cook D, Brodsky J, Tice D, Reenan A, Swedine S, Halter JB, Porte Jr D 1982 Quantitative evaluation of cardiac parasympathetic activity in normal and diabetic man. Diabetes 31:339–345 [DOI] [PubMed] [Google Scholar]
- Singh JP, Larson MG, O'Donnell CJ, Wilson PF, Tsuji H, Lloyd-Jones DM, Levy D 2000 Association of hyperglycemia with reduced heart rate variability (the Framingham heart study). Am J Cardiol 86:309–312 [DOI] [PubMed] [Google Scholar]
- Villareal RP, Liu BC, Massumi A 2002 Heart rate variability and cardiovascular mortality. Curr Atheroscler Rep 4:120–127 [DOI] [PubMed] [Google Scholar]
- Khandoker AH, Jelinek HF, Palaniswami M 2009 Identifying diabetic patients with cardiac autonomic neuropathy by heart rate complexity analysis. Biomed Eng Online 8:3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SC, Easton JD 2003 Are patients with acutely recovered cerebral ischemia more unstable? Stroke 34:2446–2450 [DOI] [PubMed] [Google Scholar]
- Matveev M, Prokopova R 2002 Diagnostic value of the RR-variability indicators for mild hypertension. Physiol Meas 23:671–682 [DOI] [PubMed] [Google Scholar]
- Kleiger RE, Miller JP, Bigger Jr JT, Moss AJ 1987 Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol 59:256–262 [DOI] [PubMed] [Google Scholar]
- Casolo G, Balli E, Taddei T, Amuhasi J, Gori C 1989 Decreased spontaneous heart rate variability in congestive heart failure. Am J Cardiol 64:1162–1167 [DOI] [PubMed] [Google Scholar]
- Ceconi C, Ferrari R, Bachetti T, Opasich C, Volterrani M, Colombo B, Parrinello G, Corti A 2002 Chromogranin A in heart failure; a novel neurohumoral factor and a predictor for mortality. Eur Heart J 23:967–974 [DOI] [PubMed] [Google Scholar]
- Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE 2001 Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol 37:2136–2143 [DOI] [PubMed] [Google Scholar]
- Madeddu P, Salis MB, Emanueli C 1999 Altered baroreflex control of heart rate in bradykinin B2-receptor knockout mice. Immunopharmacology 45:21–27 [DOI] [PubMed] [Google Scholar]
- Touma F, Chew VS, Chua WC, Jelinek H, Wong PT, Spence I, McLachlan CS 2009 Chronic high dose captopril decreases total heart rate variability and increases heart rate in C57BL/6J mice. Int J Cardiol 136:211–213 [DOI] [PubMed] [Google Scholar]
- Mahata SK, Mahata M, Wakade AR, O'Connor DT 2000 Primary structure and function of the catecholamine release inhibitory peptide catestatin (chromogranin A344-364): identification of amino acid residues crucial for activity. Mol Endocrinol 14:1525–1535 [DOI] [PubMed] [Google Scholar]
- Bigger Jr JT, Fleiss JL, Steinman RC, Rolnitzky LM, Schneider WJ, Stein PK 1995 RR variability in healthy, middle-aged persons compared with patients with chronic coronary heart disease or recent acute myocardial infarction. Circulation 91:1936–1943 [DOI] [PubMed] [Google Scholar]
- Uechi M, Asai K, Osaka M, Smith A, Sato N, Wagner TE, Ishikawa Y, Hayakawa H, Vatner DE, Shannon RP, Homcy CJ, Vatner SF 1998 Depressed heart rate variability and arterial baroreflex in conscious transgenic mice with overexpression of cardiac Gsα. Circ Res 82:416–423 [DOI] [PubMed] [Google Scholar]
- Rinne P, Harjunpää J, Scheinin M, Savontaus E 2008 Blood pressure regulation and cardiac autonomic control in mice overexpressing α- and γ-melanocyte stimulating hormone. Peptides 29:1943–1952 [DOI] [PubMed] [Google Scholar]
- Adachi Y, Nakajima Y, Satomoto M, Morita K, Doi M, Sato S 2006 The heart rate variability in mice: telemetric evaluation of endotoxin shock. Masui 55:436–440 [PubMed] [Google Scholar]
- Desjardins F, Lobysheva I, Pelat M, Gallez B, Feron O, Dessy C, Balligand JL 2008 Control of blood pressure variability in caveolin-1-deficient mice: role of nitric oxide identified in vivo through spectral analysis. Cardiovasc Res 79:527–536 [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI 2000 Phenotypic screening for heart rate variability in the mouse. Am J Physiol Heart Circ Physiol 279:H733–H740 [DOI] [PubMed] [Google Scholar]
- Kennedy BP, Mahata SK, O'Connor DT, Ziegler MG 1998 Mechanism of cardiovascular actions of the chromogranin A fragment catestatin in vivo. Peptides 19:1241–1248 [DOI] [PubMed] [Google Scholar]
- Rao F, Wen G, Gayen JR, Das M, Vaingankar SM, Rana BK, Mahata M, Kennedy BP, Salem RM, Stridsberg M, Abel K, Smith DW, Eskin E, Schork NJ, Hamilton BA, Ziegler MG, Mahata SK, O'Connor DT 2007 Catecholamine release-inhibitory peptide catestatin (chromogranin A(352-372)): naturally occurring amino acid variant Gly364Ser causes profound changes in human autonomic activity and alters risk for hypertension. Circulation 115:2271–2281 [DOI] [PubMed] [Google Scholar]
- Task force of the European Society of Cardiology and the North American Society of pacing and electrophysiology 1996 Heart rate variability: standards of measurements, physiological interpretation and clinical use. Circulation 93:1043–1065 [PubMed] [Google Scholar]
- Piccirillo G, Munizzi MR, Fimognari FL, Marigliano V 1996 Heart rate variability in hypertensive subjects. Int J Cardiol 53:291–298 [DOI] [PubMed] [Google Scholar]
- Gibelin P, Spillner E, Bonnan S, Chevallier T 2003 Non-invasive blood pressure variability in chronic heart failure: characteristics and prognostic value. Arch Mal Coeur Vaiss 96:955–962 [PubMed] [Google Scholar]
- Wen G, Mahata SK, Cadman P, Mahata M, Ghosh S, Mahapatra NR, Rao F, Stridsberg M, Smith DW, Mahboubi P, Schork NJ, O'Connor DT, Hamilton BA 2004 Both rare and common polymorphisms contribute functional variation at CHGA, a regulator of catecholamine physiology. Am J Hum Genet 74:197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahata SK, Mahata M, Wen G, Wong WB, Mahapatra NR, Hamilton BA, O'Connor DT 2004 The catecholamine release-inhibitory “catestatin” fragment of chromogranin a: naturally occurring human variants with different potencies for multiple chromaffin cell nicotinic cholinergic responses. Mol Pharmacol 66:1180–1191 [DOI] [PubMed] [Google Scholar]
- Evrengul H, Tanriverdi H, Kose S, Amasyali B, Kilic A, Celik T, Turhan H 2006 The relationship between heart rate recovery and heart rate variability in coronary artery disease. Ann Noninvasive Electrocardiol 11:154–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponikowski P, Anker SD, Chua TP, Szelemej R, Piepoli M, Adamopoulos S, Webb-Peploe K, Harrington D, Banasiak W, Wrabec K, Coats AJ 1997 Depressed heart rate variability as an independent predictor of death in chronic congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 79:1645–1650 [DOI] [PubMed] [Google Scholar]
- Wadia JS, Dowdy SF 2003 Modulation of cellular function by TAT mediated transduction of full length proteins. Curr Protein Pept Sci 4:97–104 [DOI] [PubMed] [Google Scholar]
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, Dowdy SF 2009 Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotechnol 27:567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh JP, Larson MG, Tsuji H, Evans JC, O'Donnell CJ, Levy D 1998 Reduced heart rate variability and new-onset hypertension: insights into pathogenesis of hypertension: the Framingham heart study. Hypertension 32:293–297 [DOI] [PubMed] [Google Scholar]