

Abstract
The aa3-type cytochrome c oxidase from Rhodobacter sphaeroides utilizes two proton-input channels to provide all the protons for chemistry (water formation) and proton pumping. The D-channel is responsible for the uptake of all pumped protons, 4 protons per O2. Several substitutions for either N139 or N207, near the entrance of the D-channel, were previously reported to decouple the proton pump from oxidase activity. In the current work, the characteristics of additional mutations in this region of the protein (N139, N207, N121 and S142) are determined to elucidate the mechanism of decoupling. With the exception of the substitution of a large, hydrophobic residue (N139L), all the mutations of N139 resulted in an enzyme with high oxidase activity but with severely diminished proton pumping stoichiometry. Whereas N207D was previously shown to be decoupled, N207A and N207T exhibit nearly wild type behavior. The new data display a pattern. Small, non-ionizable substitutions for N139 or N121 result in decoupling the proton pump while maintaining high turnover rates. These residues are directly hydrogen bonded to two water molecules (Water6574 and water6584) that are part of the single-file chain of water molecules within the D-channel leading to E286 at the top of the channel. The data suggest that the integrity of this water chain within the D-channel is critical for rapid proton transfer. The mechanism of decoupling is most likely due to slowing the rate of proton delivery below a threshold that is required for protonation of the putative proton loading site. Protons delivered outside this time-window are delivered to the active site where they are consumed in the formation of water. The rate of proton delivery required to protonate the pump site must be significantly faster than the rate of delivery of protons to the catalytic site. For this reason, mutations can result in decoupling the proton pump without slowing the catalytic turnover by the enzyme.
Cytochrome c oxidase couples the redox chemistry of reducing O2 to water to pumping protons across the membrane and generating a proton motive force. During each turnover, eight protons are taken up from N-side of the membrane of which four are used for reduce oxygen to water (chemical protons) and four are pumped out of the membrane (pumped protons) (1). Crystal structures of bacterial cytochrome c oxidase reveal two proton input channels (2-4). The D-channel provides two of the four protons required to reduce O2 to water at the heme-copper active site, and all four protons that are pumped per O2(5, 6). The second channel is designated the K-channel, named for a highly conserved lysine (K362, Rhodobacter sphaeroides numbering). The K-channel delivers the remaining two chemical protons to the active site per O2, and is not directly used for the uptake of pumped protons (7).
The “D-channel” has a highly conserved aspartate residue D132, at the entrance of the channel (8, 9). A string of hydrogen-bonded water molecules connect this aspartate to a highly conserved glutamate (E286). E286 is proposed to be the branch point of both the chemical protons and pumped protons (10). The water string is stabilized by hydrogen bonds to a series of hydrophilic residues: N139, N121, N207, S142, S200, S201 and S197 (2). Asparagine-139 is unique insofar as it interrupts what would otherwise be a continuous hydrogen bonded network of water molecules between D132 and E286 (2).
Mutations which remove the acid group of D132 at the channel entrance by replacing it with either an alanine (D132A) or an asparagine (D132N) severely reduce the oxygen reduction activity (<5%) and abolish proton pumping (8, 9, 11). Similarly, removing the acid group of E286 at the “top” of the D-channel essentially eliminates enzyme turnover (10).
In contrast, several mutations which replace N139 or N207 have been reported to maintain high oxidase activity but with no proton pumping. For example, N139D has twice the oxidase specific activity as does the wild type oxidase, but does not pump protons (12). N207D has equal turnover to the wild type, but no proton pumping (13). N139 and N207, together with N121, form a cluster located about 10 Å “above” D132, the entrance of the D-channel. The X-ray structures show a constriction or “asparagine neck” in the D-channel, disrupting the hydrogen bonded water chain which provides the pathway for rapid proton transfer to E286 and, from there, to either the active site or to the P-side of the membrane (2).
A question of prime interest is how these mutations, near the entrance of the D-channel, decouple the proton pump from oxidase turnover. Previous studies have pointed to a perturbation of E286 caused by the mutations (14). Both the N139D and N207D mutations increase the apparent pKa of E286, as judged by the pH-dependence of rate of proton transfer from E286 to the heme-copper center, measured by the kinetics of the PR→F step in the single-turnover flow-flash reaction of the fully reduced enzyme with O2 (13, 15). Despite the distance of >20Å, an electrostatic effect has been suggested. The higher pKa would lower the rate of proton transfer to the proton acceptor in the pump exit channel, proposed to have a pKa that is slightly higher than that of E286 (about pH 9.4) (12, 14, 15). Proton transfer from E286 to the partially reduced oxygen species at the active site would not be influenced significantly due to the large proton affinity of the proton acceptor. The electrostatic explanation is consistent with the restoration of proton pumping by the double mutant N139D/D132N, which should reduce the negative charge at the entrance of the D-channel, compared to N139D/D132, lowering the pKa towards the value in the wild type oxidase (15).
However, the N139T mutation, which also results in decoupling the proton pump, and which has about 40% of the oxidase activity of the wild type, lowers the apparent pKa of E286 (16). Hence, the correlation between the increased pKa of E286 and the lack of proton pumping is not maintained. Recent work on the equivalent of an N139C mutant of the oxidase from Paracoccus denitrificans (N131C) showed that this mutant has high oxidase activity (86%) but only about 10% of the proton pumping stoichiometry as the wild type (17). These data suggest that the electrostatic explanation for the perturbation of E286 is incorrect since cysteine should be neutral under these conditions.
There are additional data to suggest there is a structural perturbation of E286 due to the decoupling mutations (e.g., N139D). FTIR1 difference spectroscopy shows that in decoupled mutants, E286 remains protonated (neutral pH) but is in a slightly altered environment (18). This has also been demonstrated for the equivalent mutation in the oxidase from P. denitrificans (19). The X-ray structure of the N131D mutant of the P. denitrificans oxidase (equivalent to N139D in R. sphaeroides) shows that the mutation results in substantial perturbation of the water chain within the D-channel and also in an alteration of the orientation of the side chain of E278 (E286 in R. sphaeroides) (19).
In the current work, the characteristics of additional mutations in this region of the R. sphaeroides oxidase are reported as part of an effort to determine which features result in the decoupling phenotype. Mutations are reported of N139 (A, C, E, L, Q, S), N121 (A, T, D), S142 (A, D), and N207 (A, T). Double mutants, N207A/S142A and D132N/N139T were also examined. The data show the particular sensitivity of the enzyme to virtually any mutation of N139. The mutations which result in decoupling appear to be those most likely to alter the water chain within the D-channel.
Materials and methods
Site-directed mutagenesis
The mutagenesis/expression protocols have been previously described (12, 13). Briefly, the mutants were constructed using the Quikchange site-directed mutagenesis kit from Stratagene using primers from Integrated DNA Technologies (IDT). The mutations were verified by partially sequencing (UIUC Biotechnology Center) the final expression plasmid. The pRK415 expression plasmid harboring the mutated gene encoding subunit I were transferred into R. sphaeroides JS100 strain by conjugation from E. coli S-17-1 cells by electroporation, then S-17-1 cells were conjugated to R. sphaeroides JS100. The R. sphaeroides JS100 genome lacks the gene for subunit I of the cytochrome aa3 (coxI) and this is complemented by expression of subunit I encoded by the gene on the pRK415 expression plasmid.
Cell growth and enzyme purification
The procedures used to prepare the mutant oxidase are modifications of those previously described (12, 13). The frozen stock of R. sphaeroides cells were first streaked on a Sistrom-agar plate containing 50 μg/mL streptomycin, 50 μg/mL spectinomycin and 1 μg/mL tetracycline (Sistrom-SST plate). The plate was inverted and incubated at 30°C for 3-4 days until colonies were visible. Individual colonies were used to inoculate 120 ml of Sistrom-SST medium and the cells were grown aerobically with shaking at 30°C to early stationary phase. 4 ml aliquots of this culture were used to inoculate 2L flasks containing 1L Sistrom-SST medium, and the cells were grown aerobically at 30°C. When the growth reached early stationary phase, the cells were harvested by centrifugation at 8000 rpm (rcf: 10800g) for 10 min.
Cell pellets suspended in 50 mM potassium phosphate buffer, pH 8.0, containing 1mM EDTA, were homogenized with a glass homogenizer after adding ~20 mg DNase I (Sigma) and 100 μl protease inhibitor (Sigma). The suspended cells were then passed 5 times through a “microfluidizer” (Watts Fluidair, Inc.). Cell debris was removed by spinning the cell lysate at 8000 rpm (rcf: 10800g) for 20 minutes. Cell membranes in the supernatant were collected by ultracentrifugation at 40,000 rpm (rcf: 186000g) for more than 4 hours.
The cell membranes were homogenized in 50 mM potassium phosphate buffer, pH 8.0, containing 40 mM NaCl, and then solublized by adding dodecyl maltoside (DDM), from a 20% stock solution, to a final concentration of 1%, stirred at 4°C for 2 hours and then centrifuged at 40,000 rpm (rcf: 186000g) for 30 min. To the supernatant was added 5-10 ml of Ni-NTA resin plus imidazole (final concentration 5 mM), and the mixture was stirred at 4°C for 2 hours. Then the mixture was loaded into a chromatography column, and washed with 10 column volumes 50 mM potassium phosphate buffer, pH 8.0, containing 40mM NaCl, 15 mM imidazole and 0.1% DDM. The enzyme was then eluted with 50 mM potassium phosphate buffer, pH 8.0, containing 40mM NaCl, 150 mM imidazole and 0.1% DDM. The enzyme was concentrated with a 100-kD cutoff concentrator (Millipore), and the buffer was exchanged to 10 mM Tris-HCl, pH 8.0, containing 0.05% DDM. The protein was aliquoted and flash-frozen with liquid nitrogen and stored at −80°C.
For proton pumping assays, the protein was further purified by anion exchange chromatography. 5-10 mg of purified protein were diluted to 4 ml with 10 mM Tris-HCl buffer, pH 8.0, containing 1mM EDTA and 0.05% DDM (buffer A), and loaded onto a DEAE-5PW column (Toso-Haas) attached to an FPLC system (Amersham). The columns were washed with buffer A, and the oxidase was eluted at a flow rate of 0.5 mL/min using a gradient with buffer B (buffer A plus 1 M KCl). The gradient rapidly increased buffer B from 0 to 15% over 0.5 column volume, and then the oxidase was eluted as the gradient was slowly increased from 15% to 45% buffer B over 4 column volumes. The enzyme was collected and concentrated.
UV/Vis spectroscopy
UV/Vis spectra were recorded with a Shimadzu UV/Vis-2101PC spectrophotometer. The concentrated enzyme was diluted with FPLC buffer A. Both oxidized and reduced spectra were recorded. The enzyme was fully reduced by adding a few granules of dithionite. The concentration of the protein was determined using either the reduceD-minus-oxidized spectra: concentration of oxidase (mM) = [A605(red-ox)-A630(red-ox)]/24 mM cm−1, or from the spectrum of the fully reduced enzyme: concentraton of oxidase (mM) = [A604(red)-A640(red)]/40 mM cm−1.
Steady-state activity assay
Steady-state activity was measured polarographically at 30°C in a YSI model 53 oxygen meter, as previously described (12, 13). 1.8 mL of 50 mM potassium phosphate buffer, pH 6.5, containing 0.1% DDM and supplemented with 2.8 mM ascorbate, and 0.55 mM TMPD, was placed in the sample chamber. Horse heart cytochrome c (Sigma) was added to a final concentration of 40 μM. The reaction was initiated by injecting the enzyme into the mixture. The enzyme turnover (number of electrons per second per enzyme) was calculated from the slope of the oxygen-consumption traces.
Reconstitution of the oxidase into proteoliposomes
Cytochrome oxidase vesicles (COVs) were made by the following method (20). Purified asolectin (40 mg/mL) was suspended in 75 mM HEPES-KOH buffer, pH 7.4, containing 2% sodium cholate, and sonicated until the solution was clear. Cytochrome oxidase added to the sonicated asolectin/cholate buffer to a final concentration of 2 μM. Cholate was removed by the addition of Bio-Beads leaving the protein incorporated into the vesicles. Bio-Beads were removed by centrifugation, and the COV solution was dialyzed against 60 mM KCl solution.
Respiratory control ratio
Before a proton pumping measurements, the respiratory control ratio (RCR) was measured as previously described(12, 13) to test the quality of the COVs. The steady state oxidase activity of the reconstituted enzyme was determined using the procedure described above, but without the detergent in the buffer. The oxidase activity was measured in the absence of ionophores, and again after the addition of valinomycin (10 μM) and CCCP (5 μM). The RCR was calculated by dividing the activity in the presence ionophores by the activity in the absence of the ionophores.
Proton pumping measurements
Proton pumping was measured as previously described (13) by determining the pH change in solution after the addition of a limiting amount of oxygen to a solution of COVs with excess reductant. A solution containing 1 mL of 60 mM KCl, 500 μL COVs (about 0.6 μM oxidase) and 40 μM cytochrome c was adjusted to pH 7.4 and placed in the reaction chamber (25°C) fitted with a sensitive pH electrode attached to a computer through an A/D converter and stirred with a magnetic stirrer. The chamber was purged with wet Argon gas for 40 minutes until the solution was anaerobic. Then 50 μM ascorbate was added to reduce the cytochrome c, followed by valinomycin, to a final concentration of 10 μM. Following this, 10 μL of air-saturated water (containing about 256 μM dioxygen), equilibrated at 25°C, was added to the reaction chamber to initiate the reaction, and the pH change recorded. Subsequently, 10 μL of anaerobic 1 mM HCl was added as a calibration. The last step was to repeat the measurement after the addition of the protonophore CCCP (30 μM) to allow rapid equilibration of pH between the inside and outside of the vesicles. The addition of air-saturated water results in alkalinization since 1 H+ is consumed per electron by the chemical reaction.
Results
The four residues in which mutations were made are shown in Figure 1. The UV-vis spectra of all of the mutants appear identical to that of the wild type oxidase, indicating that the hemes are not substantially perturbed by the mutations.
Figure 1.

(A) The continuous chain of water molecules from D132 to D286 in the D-channel of the aa3-type oxidase from R. sphaeroides (2). The water hydrogens and orientations were estimated using the software Whatif (24). (B) Structural detail of the region around N139 showing water molecules (red spheres) and other residues discussed in the text. From (PDB code :2GSM).
The steady state activities of all the mutants, as well as wild type oxidases, were measured at pH 6.5. The turnover number (electrons/s) of the wild type enzyme is about 1100 electrons/s), and the activity of each mutant is expressed as the percent of this value in Table 1.
Table 1. Characteristics of the mutations in subunit I of the cytochrome oxidase from R. sphaeroides.
| Activity ( % WT) |
Respiratory control ratio |
Proton pumping ( % WT) |
|
|---|---|---|---|
| Wild type (WT) | 100 | 10 | 100% |
| N139A | 40 | 2.4 | <5% |
| N139C | 110 | 5 | <5% |
| N139D(12, 14) | 200 | 7 | <5% |
| N139E | 110 | 7 | <5% |
| N139L | 7 | 0.7 | <5% |
| N139Q | 60 | 6 | 30% |
| N139S | 90 | 9 | <5% |
| N139T (16) | 40 | 5 | <5% |
| N121A | 110 | 9 | 20% |
| N121T | 100 | 9 | 20% |
| N121D | 70 | 4 | <5% |
| N207D(13) | 100 | 5 | <5% |
| N207A | 90 | 9 | 90% |
| N207T | 90 | 8 | 80% |
| S142A | 80 | 7 | 90% |
| S142D | 2 | 0.45 | <5% |
| N207A/S142A | 80 | 5 | 80% |
| D132N/N139T | 90 | 6 | <5% |
aSteady state oxidase turnover number measured as described in the text. The activity of the wild type is about 1100 e−/sec.
bProton pumping measured as described in the text. The wild type oxidase is assumed to pump 1 H+/e−. The assays often give less than this value for the wild type, so the data are reported relative to the wild type. Due to undetermined systematic errors, the actual pumping of the wild type in these assays was 0.5 to 0.7 H+/e−. Mutations with no detectable proton pumping are indicated as having <5% proton pumping.
Most of the substitutions for N139 result in relatively modest effects on the oxidase activity. As previously reported (12, 14), N139D has twice the turnover of the wild type. N139S, N139C and N139E have about the same activity as the wild type, whereas N139A, N139T (16), N139Q retain about half the specific activity. The only mutation to substantially reduce the oxidase activity is N139L, which has only 7% of the wild type activity.
Remarkably, every mutation of N139 that was examined, besides N139L, has either no detectable proton pumping or substantially reduced proton pumping. Similarly, each of the three mutations of N121 (A, T and D) have substantially reduced proton pumping and close to wild type oxidase activity. Substituting an aspartate for either N121, N207 or N139 results in the elimination of proton pumping. Non-ionizable substitutions for either N139 or N121 (alanine and threonine) also eliminate proton pumping, but this is not observed with N207. Both N207A and N207T appear similar to the wild type oxidase in all respects.
Replacing S142 by an aspartate virtually eliminates oxidase activity, unlike the aspartate substitutions for the nearby asparagines. On the other hand, S142A has oxidase activity and proton pumping comparable to the wild type, similar to N207A. The double mutant, S142A/N207A is also nearly wild type (80% active and 80% proton pumping). The X-ray structure (Figure 2) shows that the side chains of both N207 and S142 are hydrogen bonded to Water6638 (2).
Figure 2.

Structural detail showing the hydrogen bonding of S142 and N207 to Water6638 in the cytochrome oxidase from R. sphaeroides (2). Distances are indicated in Ångstroms. From (PDB code :2GSM).
Previously, it was shown that the loss of oxidase activity and proton pumping caused by the D132N mutation was reversed by N139D (15). The rationale at the time was that N139D restored the charge that had been removed by the D132N mutation. Whereas the D132N mutant is has about 5% oxidase activity, the data in Table 1 shows that N139T restores the activity to the D132N mutant to 90% of the wild type. However, proton pumping is not restored in N139T/D132N.
Only two of the mutations in the set, N139L and S142D, have very low oxidase activity. As a rule, all mutations in the D-channel that have very low oxidase activity (<10%) lack proton pumping, and the same is true for both N139L and S142D. Furthermore, the respiratory control ratio (RCR) of each of these mutants is less than 1 (Table 1), in contrast to all the other mutants, which have RCR values significantly greater than 1. RCR values less than 1 have been previously reported and discussed for both the D132N (11, 15) and G204D (21) mutants. These mutations effectively block proton translocation through the D-channel.
Discussion
Several proposals have been made to explain how some mutations near the entrance of the D-channel result in decoupling proton pumping from oxidase activity while maintaining oxidase activity near wild type levels or, in the case of N139D from R. sphaeroides, even higher. Briefly, these proposals are as follows: 1) An electrostatic perturbation of E286 results in an increase in the apparent pKa of E286, with the consequence of preventing proton transfer to the pump loading site but not to the enzyme active site(22); 2) A structural perturbation of E286, likely transmitted through the alteration of the chain of water molecules in the D-channel, with the same consequence of selectively preventing proton transfer to the pump loading site(19); 3) A local perturbation of the water molecules resulting in slowing proton transfer below a minimum rate needed for proton transfer to the pump loading site(19, 23).
The electrostatic effect was suggested based on the behavior of the N139D (12) and N207D (13) mutations, as well as the N139D/D132N double mutant (15). However, this explanation was made unlikely by the demonstration that N139T also decouples and, in addition, shifts the apparent pKa of E286 in the opposite direction as does N139D (16). Subsequently, the equivalent of the N139C and N139A mutations were shown to be decoupled in the oxidase from P. denitrificans (19). In the current work, N139C, N139A and N139S are also shown to be decoupled while retaining high oxidase activity (Table 1). Even N139Q has substantially lower proton pumping, similar to the behavior of the equivalent mutant in the P. denitrificans oxidase (19). Assuming that the decoupling of the proton pump is due to the same fundamental mechanistic cause, these data rule out a direct electrostatic effect between the residue at position 139 (or 207) and E286 as being the cause of the decoupling phenotype.
In the current work, mutations in N139 as well as in residues in the immediate vicinity of N139 are examined to see if any pattern emerges among those mutations that manifest the decoupling phenotype. The recent 2.0 Å resolution crystal structure of the R. sphaeroides oxidase (PDB code :2GSM) provides structural detail that is particularly useful for this purpose (2). This structure reveals highly structured water molecules in the D-channel. These form a continuous hydrogen-bonded chain from D132 to E286, disrupted only by N139 (2). An optimized orientation of the hydrogen atoms in this chain of water molecules, obtained with the software “Whatif” (24) is shown in Figure 1A. This “proton wire” is essential for rapid proton transfer, which proceeds by the Grotthus mechanism (25). Mutations that disrupt this single-file water chain would be expected to slow down or block proton transfer to E286.
The detailed structure in Figure 1B shows that N139 interrupts the continuous water chain from D132 to E286. Molecular dynamics show that N139 acts as a gate for proton translocation to E286(26). The N139 side chain must move out of the way in order to complete the water chain and provide a pathway for protons to proceed up the D-channel to E286. Large hydrophobic substitutions at this position, e.g., N139L (Table 1), completely block proton flux through the D-channel. The structure makes it clear that N139 plays a unique role, and from the results in the current work, as well as previous studies (19), it appears that only asparagine at this position will allow proper function. This is one important conclusion of the current work.
Figure 3 shows the distances between residues and resolved water molecules, focusing on N139 (Figure 3A), N121 (Figure 3B) and N207 (Figure 3C). The cluster of three asparagines constitutes the “neck” or region of constriction of the water chain. The three asparagines are not, however, equivalent. A schematic is shown in Figure 4. Several points to notice from the structural information are as follows.
Above N139, there is a single-file water chain starting with Water6574, leading to E286. This chain includes Water6574 – Water6584 – Water6563 – Water6613 - Water6575 – Water6558- Water6560 – (E286).
N139 blocks the connection of this water chain to the bulk water phase, and the side chain interacts directly with both Water6574 and Water6584. This conclusion is based on the distances in the reported X-ray structure (2), though the program Whatif (24), which optimizes the water orientations, does not show direct hydrogen bonds. However, the water orientations may be fluctuating rapidly, still allowing for hydrogen bonds to be plausibly formed.
Below N139, Water6614 is hydrogen bonded to both N139 and to D132, which is at the entrance of the D-channel.
The side chains of N139 and N121 are each hydrogen bonded to Water6574 and Water6584, which are components of the water chain leading to E286.
The side chains of N207 and S142 are not directly hydrogen bonded to water molecules within the single-file chain leading to E286. Both N207 and S142 are hydrogen bonded to Water6638 which, in turn, is hydrogen bonded to Water6574.
Oxidase structures (RsCcO 2GSM and 3DTU) (2, 27) indicate that S142 has at least two conformations. One of these conformations is hydrogen bonded to Water6638 but the second conformational state is not. This suggests that the orientation of S142 is not an essential feature of the proton channel. The fact that S142A has near wild type characteristics is consistent with this (Table 1).
Figure 3.

Structural detail of the cytochrome oxidase from R. sphaeroides (2) showing (A) likely hydrogen bonds from N139 to Waters 6614, 6574, 6584 and 6638. (B) Hydrogen bonding of N121 to Water6574 and Water6584, and the hydrogen bonding between these water molecules and Water6638. (C) Hydrogen bonding of N207 to Water6638 and the interactions between waters 6638, 6574, and 6584. The distances from N207 to Water6574, and between Water6638 and Water6584 are too large to predict hydrogen bonds. Distances are indicated in Ångstroms. From (PDB code :2GSM).
Figure 4.
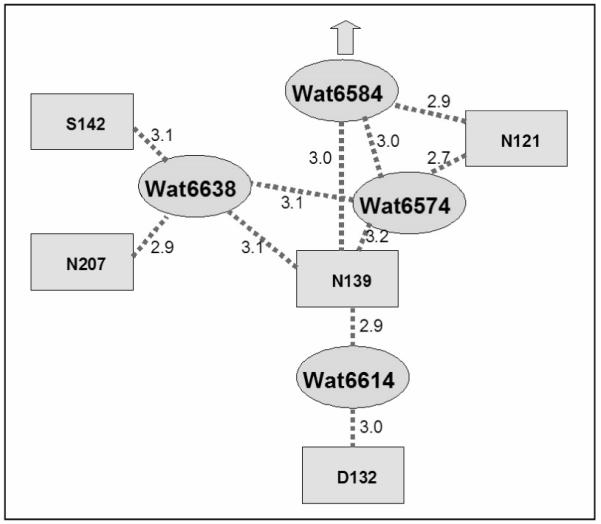
Schematic summarizing the interpretation of the hydrogen bond interactions shown in Figures 2 and 3. Distances are indicated in Ångstroms.
The data (Table 1) show that small, non-ionizable substitutions for N139 or N121 result in decoupling the proton pump. This correlates with the fact that both of these residues directly hydrogen bond to Water6574 and Water6584, which are part of the single-file water chain of the D-channel. In contrast, N207A, N207T and S142A are not significantly different from the wild type oxidase. These two residues are not directly hydrogen bonded to either Water6574 or Water6584 but, are indirectly linked through Water6638. It is concluded that the decoupling due to the non-ionizable substitutions for either N139 or N121 is likely the result of a structural perturbation of the single-file water chain.
Ionizable residues (D or E) have a more potent effect, which depends on position. N139D, N139E and N121D each decouple the proton pump, likely by direct interaction with the single-file water chain. Whereas N207A and N207T pump protons, N207D is decoupled, showing that the perturbation from placing a charged residue at this position is more potent than the perturbation due to either alanine or threonine. S142D, on the other hand, completely blocks oxidase activity, likely due to a structural or electrostatic perturbation that completely disrupts the proton-conducting water chain. An anionic residue at this site could substantially change the orientation of nearby water molecules and the abilities of particular water molecules to act as hydrogen bond donors or acceptors, preventing the Grotthus mechanism from operating. The respiratory control ratio (RCR) of S142D is less than 1, which is similar to other mutations that block all proton flux through the D-channel (11, 13, 21), including N139L (Table 1).
The structure of the N131D mutant of the P. denitrificans oxidase has been reported(19). This is equivalent to N139D in the R. sphaeroides oxidase. Substantial perturbation of the water chain leading to E278 (equivalent to E286 in the R. sphaeroides oxidase) was observed, as well as a change in the orientation of the side chain of E286. In addition, it was shown that the added aspartate (N131D) is deprotonated. Which of these perturbations is the primary cause of the decoupling of the proton pump is unclear. The most likely explanation from the current work is that the alteration of the water chain itself is the immediate cause of the decoupling, since non-ionizable substitutions result in decoupling only when placed at sites in direct contact with Water6574 or Water6584.
Decoupling is most likely caused by the mutations that slow the passage of protons through the region defined by N139, Water6574 and Water6584. This mechanism (19, 23) is consistent with the “kinetic gate” controlling whether protons coming through the D-pathway are directed to the pump loading site or to the catalytic active site (28). There must be a critical time during which protons are directed to the proton loading site. A delay in proton delivery beyond this critical time results in protons being directed to the active site to be consumed in the generation of water. The threshold rate, below which proton pumping is decoupled, must be substantially faster than the rate-limiting step for proton delivery to the active site. Hence, proton pumping can be eliminated without slowing the rate of the oxidase reaction. The nature of the gating, i.e., the opening and closing of proton pathways, and the factors that control gating are not known. The net charge and charge distribution within the active site provide the impetus for the cascade of equilibria that constitute the steps initiated by electron transfer to heme a. Quite possibly, the placement of water molecules in response to the chemistry at the active site provide the critical links in opening/closing of proton pathways.
The kinetic model of decoupling described above is an attractive explanation of the mutations described in the current work. However, the model does not readily explain mutations in the D-channel that decouple the proton pump but which have substantially reduced oxidase activity (e.g., D132N) or the double mutant, D132N/N139D, in which the N139D mutation restores proton pumping to D132N but does not fully restore the oxidase activity. There appear to be two separate kinetic ranges in which the rate of proton translocation through the D-channel will result in decoupling from the proton pump. One range is at rates above or near the rate limiting step of oxidase activity, described in this paper, and the second window is below a second threshold corresponding to about 10% of the rate of oxidase activity. Proton pumping is coupled to oxidase activity in between these two time windows.
The perturbation of the water chain by decoupling mutants might also result in a change in conformation of E286, at the top the the D-pathway, and this conformational change could also be the primary cause of decoupling the proton pump. A conformational change this glutamate has been observed in the X-ray structure of the N131D mutation of the P. denitrificans oxidase (19). However, the X-ray structures of two decoupling mutants of the R. sphaeroides oxidase, N139D and N139C, have also been determined, and no change in conformation of E286 is observed in either case (manuscript in preparation). These unpublished structural data, in combination with the current work, strongly support a kinetic mechanism of decoupling caused by local perturbations to the water chain within the D-pathway.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL16101) to R. B. G.
Footnotes
- FTIR
- Fourier transform infrared spectroscopy
- Sistrom-SST plate
- Sistrom-agar plate containing 50 μg/mL streptomycin, 50 μg/mL spectinomycin and 1μg/mL tetracycline
- COV
- cytochrome oxidase vesicles
- RCR
- respiratory control ratio
- TMPD
- N,N,N’,N’-tetramethylphenylenediamine
- CCCP
- Carbonyl cyanide 3-chlorophenylhydrazone
References
- (1).Brzezinski P, Gennis RB. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P, Iwata S. The X-ray Crystal Structures of Wild-type and EQ(I-286) Mutant Cytochrome c Oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- (4).Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 Å Resolution of the Paracoccus denitrificans Two-Subunit Cytochrome c Oxidase Complexed with an Antibody Fv Fragment. Proc. Natl. Acad. Sci. USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Michel H. Cytochrome c Oxidase: Catalytic Cycle and Mechanisms of Proton Pumping - A Discussion. Biochemistry. 1999;38:15129–15140. doi: 10.1021/bi9910934. [DOI] [PubMed] [Google Scholar]
- (6).Wikström M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky MI. The Role of the D- and K-pathways of Proton Transfer in the Function of the Haem-copper Oxidases. Biochem. Biophys. Acta. 2000;1459:514–520. doi: 10.1016/s0005-2728(00)00191-2. [DOI] [PubMed] [Google Scholar]
- (7).Tomson FL, Morgan JE, Gu G, Barquera B, Vygodina TV, Gennis RB. Substitutions for Glutamate 101 in Subunit II of Cytochrome c Oxidase from Rhodobacter sphaeroides Result in Blocking the Proton-conducting K-channel. Biochemistry. 2003;42:1711–1717. doi: 10.1021/bi026750y. [DOI] [PubMed] [Google Scholar]
- (8).Fetter J, Sharpe M, Qian J, Mills D, Ferguson-Miller S, Nicholls P. Fatty Acids Stimulate Activity and Restore Respiratory Control in a Proton Channel Mutant of Cytochrome c Oxidase. FEBS Letters. 1996;393:155–160. doi: 10.1016/0014-5793(96)00874-5. [DOI] [PubMed] [Google Scholar]
- (9).Smirnova IA, Ädelroth P, Gennis RB, Brzezinski P. Aspartate-132 in Cytochrome c Oxidase from Rhodobacter sphaeroides is Involved in a Two-step Proton Transfer during Oxo-Ferryl Formation. Biochemistry. 1999;38:6826–6833. doi: 10.1021/bi982865j. [DOI] [PubMed] [Google Scholar]
- (10).Ädelroth P, Svensson-Ek M, Mitchell DM, Gennis RB, Brzezinski P. Glutamate 286 in Cytochrome aa3 from Rhodobacter sphaeroides is Involved in Proton Uptake During the Reaction of the Fully-reduced Enzyme with Dioxygen. Biochemistry. 1997;36:13824–13829. doi: 10.1021/bi9629079. [DOI] [PubMed] [Google Scholar]
- (11).Fetter JR, Qian J, Shapleigh J, Thomas JW, Garcia-Horsman A, Schmidt E, Hosler J, Babcock GT, Gennis RB, Ferguson-Miller S. Possible Proton Relay Pathways in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. USA. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pawate AS, J. M, Namslauer A, Mills DA, Brzezinski P, Ferguson-Miller S, Gennis RB. A Mutation in Subunit I of Cytochrome Oxidase from Rhodobacter sphaeroides Results in an Increase in Steady-State Activity But Completely Eliminates Proton Pumping. Biochemistry. 2002;45:13417–13423. doi: 10.1021/bi026582+. [DOI] [PubMed] [Google Scholar]
- (13).Han D, Namslauer A, Pawate A, Morgan JE, Nagy S, Vakkasoglu AS, Brzezinski P, Gennis RB. Replacing Asn207 by aspartate at the neck of the D channel in the aa3-type cytochrome c oxidase from Rhodobacter sphaeroides results in decoupling the proton pump. Biochemistry. 2006;45:14064–14074. doi: 10.1021/bi061465q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Namslauer A, Pawate A, Gennis RB, Brzezinski P. Redox-Coupled Proton Translocation in Biological Systems: Proton Shuttling in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. USA. 2003;100:15543–15547. doi: 10.1073/pnas.2432106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brändén G, Pawate AS, Gennis RB, Brzezinski P. Controlled Uncoupling and Recoupling of Proton Pumping in Cytochrome c Oxidase. Proc Natl Acad Sci U S A. 2006;103:317–322. doi: 10.1073/pnas.0507734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lepp H, Salomonsson L, Zhu JP, Gennis RB, Brzezinski P. Impaired proton pumping in cytochrome c oxidase upon structural alteration of the D pathway. Biochim Biophys Acta. 2008;1777:897–903. doi: 10.1016/j.bbabio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Olkhova E, Helms V, Michel H. Titration Behavior of Residues at the entrance of the D-Pathway of Cytochrome c Oxidase from Paracoccus denitrificans Investigated by Continuum Electrostatic Calculations. Biophysical Journal. 2005;89:2324–2331. doi: 10.1529/biophysj.105.062091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vakkasoglu AS, Morgan JE, Han D, Pawate AS, Gennis RB. Mutations which decouple the proton pump of the cytochrome c oxidase from Rhodobacter sphaeroides perturb the environment of glutamate 286. FEBS Lett. 2006;580:4613–4617. doi: 10.1016/j.febslet.2006.07.036. [DOI] [PubMed] [Google Scholar]
- (19).Durr KL, Koepke J, Hellwig P, Muller H, Angerer H, Peng G, Olkhova E, Richter OM, Ludwig B, Michel H. A D-pathway mutation decouples the Paracoccus denitrificans cytochrome c oxidase by altering the side-chain orientation of a distant conserved glutamate. J Mol Biol. 2008;384:865–877. doi: 10.1016/j.jmb.2008.09.074. [DOI] [PubMed] [Google Scholar]
- (20).Jasaitis A, Verkhovsky MI, Morgan JE, Verkhovskaya ML, Wikström M. Assignment and Charge Translocation Stoichiometries of the Major Electrogenic Phases in the Reaction of Cytochrome c Oxidase with Dioxygen. Biochemistry. 1999;38:2697–2706. doi: 10.1021/bi982275l. [DOI] [PubMed] [Google Scholar]
- (21).Han D, Morgan JE, Gennis RB. G204D, a Mutation that Blocks the Proton-conducting D-channel of the aa3-type Cytochrome c Oxidase from Rhodobacter sphaeroides. Biochemistry. 2005;44:12767–12774. doi: 10.1021/bi051141m. [DOI] [PubMed] [Google Scholar]
- (22).Namslauer A, Aagaard A, Katsonouri A, Brzezinski P. Intramolecular Proton-Transfer Reactions in a Membrane-Bound Proton Pump: The Effect of pH on the Peroxy to Ferryl Transition in Cytochrome c Oxidase. Bichemistry. 2003;42:1488–1498. doi: 10.1021/bi026524o. [DOI] [PubMed] [Google Scholar]
- (23).Wikstrom M, Verkhovsky MI. Mechanism and energetics of proton translocation by the respiratory heme-copper oxidases. Biochim Biophys Acta. 2007;1767:1200–1214. doi: 10.1016/j.bbabio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- (24).Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- (25).Agmon N. The Grötthuss Mechanism. Chem. Phys. Lett. 1995;244:456–462. [Google Scholar]
- (26).Henry RM, Yu CH, Rodinger T, Pomes R. Functional hydration and conformational gating of proton uptake in cytochrome c oxidase. J Mol Biol. 2009;387:1165–1185. doi: 10.1016/j.jmb.2009.02.042. [DOI] [PubMed] [Google Scholar]
- (27).Qin L, Mills DA, Buhrow L, Hiser C, Ferguson-Miller S. A Conserved Steroid Binding Site in Cytochrome c Oxidase. Biochemistry. 2008;47:9931–9933. doi: 10.1021/bi8013483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Popovic DM, Stuchebrukhov AA. Electrostatic Study of the Proton Pumping Mechanism in Bovine Heart Cytochrome c Oxidase. JACS. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]