

Abstract
The eradication of HIV-1 will likely require novel clinical approaches to purge the reservoir of latently infected cells from a patient. We hypothesize that this therapy should target a wide range of latent integration sites, act effectively against viral variants that have acquired mutations in their promoter regions, and function across multiple HIV-1 subtypes. By using primary CD4+ and Jurkat cell-based in vitro HIV-1 latency models, we observe that single-agent latency reactivation therapy is ineffective against most HIV-1 subtypes. However, we demonstrate that the combination of two clinically promising drugs—namely, prostratin and suberoylanilide hydroxamic acid (SAHA)—overcomes the limitations of single-agent approaches and can act synergistically for many HIV-1 subtypes, including A, B, C, D, and F. Finally, by identifying the proviral integration position of latent Jurkat cell clones, we demonstrate that this drug combination does not significantly enhance the expression of endogenous genes nearest to the proviral integration site, indicating that its effects may be selective.
HIV-1 postintegration latency poses the greatest barrier to complete eradication of the virus from a patient (25). Latent infections have low or no transcriptional activity and fail to generate viral progeny, rendering them untreatable with current antiretroviral treatments that target only actively replicating virus (78). Moreover, latent infections, which persist in resting memory CD4+ T cells with a half-life of up to 44 months (79), provide a permanent reservoir for reactivation and reseeding of the replicating virus (83). Therapeutic reactivation of latent infections, combined with antiretroviral treatments, may accelerate the depletion of latent reservoirs (reviewed in reference 29). However, such latency reactivation strategies have yielded variable results in recent clinical trials (49, 80, 82), underscoring the difficulties associated with purging latent infections. Therefore, the complexities of latency warrant the further development of in vitro analytical and screening assays that can model the conditions of latency and test potential therapies (9).
After viral entry and integration of the viral genome into the host chromosome, the HIV 5′ long terminal repeat (5′ LTR) promoter recruits RNA polymerase II (RNAPII) and other host factors to regulate viral gene expression. Initially, low basal transcription generates primarily abortive transcripts—due to the stalling of RNAPII—and a small fraction of fully elongated viral transcript that is initially spliced to generate mRNA encoding a positive regulator, the transcriptional activator (Tat) (47). Tat protein interacts with cellular positive transcriptional elongation factor B (P-TEFb) (99), and the resulting Tat-P-TEFb complex binds to the trans-activation response (TAR) element at the 5′ end of nascent viral transcripts (3). Here, P-TEFb phosphorylates the C-terminal domain (CTD) of stalled RNAPII to enhance the efficiency of elongation (98). This Tat-mediated transactivation thereby amplifies viral gene expression nearly 100-fold, yielding a strong positive-feedback loop (23).
In addition to Tat and host elongation factors, other cellular activating and repressing mechanisms control transcription and the local chromatin environment of the integrated virus (52, 86). Viral transcription correlates with the recruitment of histone acetyltransferase (HAT) proteins to the LTR and the subsequent acetylation of both histone tails (57) and the Tat protein (41). Alternatively, gene silencing and heterochromatin assembly are driven by the removal of acetyl moieties from histone tails by histone deacetylases (HDACs) (36, 92). Within the U3 enhancer region of viral 5′ LTR, numerous cis-binding elements recruit positive and negative factors that regulate histone modifications and chromatin structure (see Fig. 1B). Among these elements, NFAT and AP-1 sites recruit activating factors (11, 53), while YY1 sites recruit silencing factors, including HDACs (72). Furthermore, κB and Sp1 binding sites promote either transcriptional silencing via HDAC recruitment (58, 92) or activation through recruitment of HATs and transcription factors (2, 26). Collectively, these sites play significant and sometimes synergistic roles in the decision between viral replication versus the establishment of latency (10, 68).
FIG. 1.

Lentiviral latency system and HIV-1 LTR of various subtypes. (A) FACS sorting procedure for polyclonal and clonal populations of LGIT and LG virus infections. Jurkat cells were infected at a low MOI with LGIT (1a) or LG (1b) lentivirus. Six days postinfection, gene expression was strongly stimulated with exogenous Tat protein and HMBA for LGIT virus-infected cells (2a) or Tat protein, HMBA, and TNF-α for LG virus-infected cells (2b). Eighteen hours after stimulation, single GFP-positive (GFP+) cells were sorted from LG virus infections (3b) and cultured for 4 weeks to generate LG clones (4b). Similarly, 18 h after stimulation, polyclonal FACS isolation of GFP+ LGIT virus-infected cells removed uninfected cells (3a). After sorting, GFP+ LGIT-infected cells were cultured under normal conditions for 2 weeks, during which substantial fractions of GFP+ cells relaxed to the off expression mode (4a). FACS was again applied to isolate polyclonal fractions of “infected but off” (GFP−) cells, and these fractions are used as models for latent infections (5b). Likewise, single cells were sorted and expanded to generate LGIT clones, and after 4 weeks of culturing, phenotypic bifurcation (PheB) clones were identified (5a). Off sorts, polyclonal clones that sorted into the off expression mode. (B) Schematic of alignments and DNA-binding elements of U3 regions for subtypes in this study. Binding sites were identified using the Transcription Element Search System (TESS) (http://www.cbil.upenn.edu/cgi-bin/tess/tess). Gray ovals indicate deviations in Sp1 sequence that likely compromise the function of Sp1 site II (for subtypes A2, A, and A/G) and Sp1 site III (for subtypes D, F, and H). Full U3 subtype sequences are supplied in Fig. S6 in the supplemental material. Two distinct isolates of subtype C were analyzed throughout this investigation (C refers to the sequence with GenBank accession no. AF067157, and C′ refers to the sequence with GenBank accession no. AF067154).
In vitro models of HIV latency, often composed of an integrated HIV-1-based vector in CD4+ Jurkat cells, have revealed that nonproductive transcription after viral integration may result from repressed chromatin (37, 92), transcriptional interference from nearby genes (50), the absence of elongation factors (94), and insufficient levels of the viral protein Tat for viral transactivation (89, 90). Using the HIV-1-based LTR-GFP-IRES-Tat (LGIT) (GFP stands for green fluorescent protein and IRES stands for internal ribosome entry site) lentivirus in Jurkat cells, we have previously demonstrated that due to stochastic fluctuations in Tat concentration, clonal populations with single integrations of LGIT lentivirus may phenotypically bifurcate (PheB) into inactive (off) and active (bright) populations (89). Moreover, inactivating point mutations introduced into each of the Sp1 or κB elements in the HIV LTR of the LGIT virus model have revealed that each of these elements uniquely contributes to the recruitment of repressing and activating factors and to the overall stabilities of the off and bright expression modes (10). In particular, mutation of κB site I (mutI NF-κB) decreases recruitment of the NF-κB activating heterodimer p50-RelA, while mutation of Sp1 site III (mutIII Sp1) impedes the recruitment of both p50-RelA and the HAT p300 (10). Since different HIV-1 subtypes and circulating recombinant forms (CRFs) throughout the world have sequence variability within the same Sp1 and κB elements, as well as in other domains throughout U3, these results suggest that subtypes may access distinct latency mechanisms and raise the problematic possibility that each may require a distinct, “tailored” reactivation strategy.
Due to the complex nature of latency, the success of clinical efforts to purge latent reservoirs will depend on the ability to reverse one or more of the possible latency mechanisms (reviewed in reference 29). Resting CD4+ T cells, which maintain low levels of activating factors NF-κB and NFAT (27, 42), provide a cellular environment that favors silenced proviral gene expression and latent infections. In vitro studies suggest that activation of resting CD4+ T cells with proinflammatory cytokines would also reactivate latent infections (63). However, in vivo activation of resting CD4+ T cells with proinflammatory cytokines interleukin 2 (IL-2) and gamma interferon (IFN-γ) or the anti-CD3 monoclonal antibody OKT3 results in long-term depletion of all CD4+ T cells and fails to measurably purge the latent reservoir (46, 87). Moreover, T-cell activation therapies are ineffective against latent infections in actively dividing cells (35) and are unlikely to stimulate latent infections attributed to chromatin silencing or transcriptional interference (reviewed in reference 93).
As a potentially more effective alternative to T-cell activation with cytokines, latency reactivation therapies may utilize pharmacological agents that directly target latency mechanisms. For example, direct activation of the NF-κB pathway with the cytokine tumor necrosis factor alpha (TNF-α) increases the nuclear concentration of the activating p50-RelA heterodimer and induces viral NF-κB-dependent gene expression (19). Similarly, phorbol myristate acetate (PMA) and prostratin, a non-tumor-promoting phorbol ester, stimulate a portion of latent infections by activating the NF-κB and protein kinase C (PKC) pathways to enhance the recruitment of activating factors and P-TEFb to the LTR (84, 91). However, like T-cell activation therapies, these mitogens may fail to target latent infections that arise from transcriptional interference or chromatin silencing (93). To reverse the effects of chromatin silencing, latent infections may require treatment with HDAC inhibitors, such as trichostatin A (TSA) (86), and clinically tested HDAC inhibitors suberoylanilide hydroxamic acid (i.e., SAHA or Vorinostat) (1, 17, 21, 40) and valproic acid (49). Stimulation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by either SAHA or the clinically tested chemotherapeutic hexamethylene bisacetamide (HMBA) (75) may alleviate latency by triggering the localization of P-TEFb to the LTR to enhance viral transcriptional elongation (14, 16, 17). Transcriptional elongation can also be enhanced with okadaic acid, the nonclinical inhibitor of protein phosphatase type 1 (PP1) and type 2A (PP2A) (24), which increases the levels of phosphorylated RNAPII (94). The polyphenol resveratrol, which activates both growth receptor Egr1 (70) and class III HDAC SIRT1 (6), may enhance viral gene expression by upregulating Egr1-dependent growth factors (44) or by promoting the deacetylation of Tat protein via SIRT1 (66), although the precise mechanisms of SIRT1 activation are unclear and contested (65). Finally, the clinically viable DNA methylase inhibitor 5-aza-2′-deoxycytidine (i.e., 5-aza-dC) may have promise, as genomic silencing of latent infections may be regulated by DNA methylation (34, 35). Since each of these drugs targets a distinct mechanism contributing to latency, complete reactivation of the latent reservoir—a heterogeneous population regulated by a variety of distinct mechanisms—may not be feasible with a single agent and instead may require multifaceted combinatorial strategies (69).
In this proof-of-concept study, the following criteria are adopted to evaluate the effectiveness of reactivation strategies that could potentially eradicate latent HIV-1 reservoirs. First, it must be capable of reactivating in a Tat-independent manner, since low or zero levels of Tat exist in latent cells (37, 38). Second, the mechanisms of reactivation must target a wide range of integration sites within latently infected cells, as latency may arise from transcriptional interference from nearby expressing genes or from viral integration in regions of dynamic chromatin or heterochromatin (30, 31, 50, 91). Third, the regimen must stimulate multiple and complementary pathways to maintain effectiveness against potential viral variants and maximize the possible synergy between mechanisms, which could potentially decrease the dosage level and toxicity of each component. Fourth, the therapeutic regimen should optimally target most or all HIV-1 subtypes and CRFs, such that a strategy could be broadly implemented and that individuals with quasispecies infections would be unlikely to develop escape mutants.
Our overall strategy is to explore the ability of single and combinatorial compounds to activate latent HIV-1 in different in vitro latency models. Moreover, to assess the efficacy of such reactivation therapies, we have tested these drugs across a range of conditions, including model virus containing enhancer elements from numerous HIV subtypes, virus with sequence variations in key host transcription factor binding sites, lentiviral vectors that model the Tat feedback loop, and Tat-deficient latent virus. In both peripheral blood mononuclear cell (PBMC)- and Jurkat cell-based systems, our results indicate that certain HIV-1 subtype and promoter mutants that could arise naturally may be resistant to reactivation with any individual antilatency drug. However, we demonstrate that a combination of the NF-κB/PKC activator prostratin with the HDAC inhibitor SAHA—both clinically tested pharmacological agents—synergistically reactivates latent infections across a variety of integration sites, promoter mutants of κB and Sp1 binding sites, and distinct HIV-1 subtypes and CRF isolates. Importantly, our results indicate that the majority of different subtype promoters in either Jurkat or primary CD4+ T-cell latency models are synergistically reactivated by the combination of prostratin and SAHA.
MATERIALS AND METHODS
Lentiviral latency models in Jurkat cells.
Construction of LG and LGIT HIV-1-based plasmids and LGIT virus variants containing two (for κB mutants) or three (for Sp1 mutants) inactivating point mutations have been previously detailed (10, 89). Lentiviral plasmids for LG (pCLG) and LGIT (pCLGIT) were packaged and harvested in HEK 293T cells using 10 μg of vector, 5 μg pMDLg/pRRE, 3.5 μg pVSV-G, and 1.5 μg pRSV-Rev, as previously detailed (89). Culture media were replaced after 12 h, and 24 h later, viral supernatant was passed through a 0.45-μm filter to remove cell debris. The virus was then loaded onto a 20% (wt/wt) sucrose cushion and concentrated by ultracentrifugation in an SW41 rotor (Beckman Coulter, Fullerton, CA) for 1.5 h at 25,000 rpm (107,000 × g) and 4°C. The viral pellet was resuspended in 100 μl of 4°C phosphate-buffered saline (PBS) (pH 7.0) to yield typically between 107 and 108 infectious units/ml. An estimated 103 to 106 infectious units of concentrated virus was used to infect 3 × 105 Jurkat cells. Six days after infection, titer determination curves were constructed by incubating cells with a combination of 5 mM HMBA, 20 ng/ml TNF-α, 400 nM TSA, and 12.5 μg exogenous Tat protein for 18 h and then analyzing GFP expression by flow cytometry to obtain specific titer values. A unique titer determination curve for LG and each LGIT virus variant was used to attain the desired multiplicity of infection (MOI) (0.05 to 0.10).
Primary cell latency model.
Whole-blood samples from three healthy, anonymous donors (9.1, 9.2, and 9.3) were obtained from the City of Hope Donor Apheresis Center (Duarte, CA). Primary blood mononuclear cells (PBMCs) were isolated using Histopaque 1077 (Sigma-Aldrich). Naïve CD4+ T cells were further purified using the naïve CD4+ T-cell biotin antibody cocktail II, antibiotin microbeads, and MACS LS columns (Miltenyi Biotec). The cells were activated with 30 U/ml recombinant IL-2 (rIL-2) (NIH AIDS Reagent Program) and human T-activator CD3/CD28 Dynabeads (Invitrogen). One week after isolation, 105 infectious units of concentrated LGIT virus (subtypes A2, A, B, C, C′, D, and F) were used to infect 1 × 106 CD4+ T cells. Primary cells were cultured in RPMI 1640 medium supplemented with 10% AB human serum (Invitrogen).
Pharmacological treatments.
To determine the theoretical limits of latency reactivation for each Jurkat cell-based lentiviral model, all pharmacological agents were tested at saturating levels for in vitro conditions. In particular, the following drugs were tested at the specified concentrations: 12.5 μg exogenous Tat protein per 3 × 105 cells (NIH AIDS Reagent Program), 20 ng/ml tumor necrosis factor alpha (TNF-α) (Sigma-Aldrich), 10 nM phorbol myristate acetate (PMA) (Sigma-Aldrich), 400 nM trichostatin A (TSA) (Sigma-Aldrich), 5 mM hexamethylene bisacetamide (HMBA) (Sigma-Aldrich), 1 μM prostratin (LC Laboratories), 30 nM okadaic acid (Sigma-Aldrich), 4 μM SAHA (Toronto Research Chemical), 5 mM valproic acid (Sigma-Aldrich), 1 μM 5-aza-deoxycytidine (Sigma-Aldrich), 500 μM (±)-S-nitroso-N-acetylpenicillamine (SNAP) (Calbiochem), 425 μM diethylenetriamine (DETA) 1-substituted diazen-1-ium-1,2-diolates (NONOate) (Cayman Chemical), 20 μg/ml phytohemagglutinin (PHA) (Sigma-Aldrich), 30 μM resveratrol (Sigma-Aldrich), and 500 mM sorbitol (Sigma-Aldrich). After 30-min incubation with 0.5 M sorbitol, Jurkat cells were washed with media and analyzed by flow cytometry 6 h later. Incubation with either resveratrol or 5-aza-deoxycytidine was performed for 48 h prior to flow cytometry analysis. All other drugs were incubated with Jurkat cells or primary CD4+ T cells for 24 h prior to green fluorescent protein (GFP) analysis. For the most efficacious agents (prostratin, SAHA, HMBA, and the combination of prostratin and SAHA), cell viability after drug treatment was analyzed by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] cell viability assay or by propidium iodide (Sigma-Aldrich) and Hoechst 33342 (Invitrogen) staining using flow cytometry (see Fig. S1 in the supplemental material).
Flow cytometry analysis.
To phenotype primary T cells, 5 × 105 cells were stained with the following monoclonal antibodies (Invitrogen): Pacific Blue-labeled anti-CD4, allophycocyanin (APC)-labeled anti-CD45RA, peridinin chlorophyll protein (PerCP)-labeled anti-CD45RO, and anti-CD27 antibody labeled with both APC and Alexa Fluor 750. Stained cells were analyzed by flow cytometry using the CyAn ADP nine-color flow cytometer (Dako) with three laser excitation sources (405 nm, 488 nm, and 635 nm).
Jurkat cells infected with the LGIT or LG lentivirus were sorted with a Dako-Cytomation MoFlo Sorter based on GFP fluorescence. GFP analysis for Jurkat cells was performed using a Beckman-Coulter FC500 flow cytometer. Analysis of flow cytometry was performed with FlowJo (Tree Star, Inc.).
Reagents.
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: p93BR020.1, p90CF056.1, p92UG037.1, p93BR029, p94UG114.1, and p92NG003.1 from Beatrice H. Hahn and Feng Gao and the UNAIDS Network for HIV Isolation and Characterization; p98CN009.8 from Cynthia M. Rodenburg, Beatrice H. Hahn, Feng Gao, and the UNAIDS Network for HIV Isolation and Characterization; p94CY017.41 from Stanley A. Trask, Feng Gao, Beatrice H. Hahn, and the Aaron Diamond AIDS Research Center; p93IN904 and p93IN999 from Kavita Lole, Robert Bollinger, and Stuart Ray; Tat protein from John Brady; and recombinant human IL-2 from Maurice Gately, Hoffmann-La Roche Inc.
Mapping of viral integration sites.
An established method has been used for identifying human immune deficiency virus (HIV-1) integration sites (95). The genomic DNA of infected Jurkat cells was isolated using a DNA minikit (Qiagen) and then restricted by either HpyCH4III or MseI (New England Biolabs [NEB]). The restricted DNA fragments were ligated to preannealed Hpy linker or Mse linker DNA (Hpy linker+, 5′-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGACN-3′; Hpy linker−, 5′-GTCCCTTAAGCGGAG-3′; Mse linker+, 5′-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGAC-3′; Mse linker−, 5′-TAGTCCCTTAAGCGGAG-3′). The ligation products were then used as templates for primary PCR with primers annealing to the HIV LTR and the linkers (HIV-LTR, 5′-AGTGCTTCAAGTAGTGTGTGCCCG-3′; linker primer, 5′-GTAATACGACTCACTATAGGGC-3′) under the following conditions: preincubation at 95°C for 2 min; 30 cycles, with 1 cycle consisting of 30 s at 95°C, 30 s at 55°C, and 2.5 min at 72°C; and the final extension step of 10 min at 72°C. Samples of the initial PCR product were used for nested PCR with primers (HIV LTR nested, 5′-AAAAAGGATCCCCGTCTGTTGTGTGACTCTGGTAACT-3′; linker primer nested, 5′-AAATTAAGCTTAGGGCTCCGCTTAAGGGAC-3′) under the same conditions as primary PCR. The amplified virus-host genome junctions were cloned into pBS SK SP plasmid after restriction with BamHI and HindIII (NEB) and then sequenced. The retroviral integration sites were mapped to the human genome (February 2009 assembly) using the BLAT program on the Ensembl genome browser website (www.ensembl.org). On the basis of the chromosomal locations, various genomic annotations for each retroviral integration site were made via genome browser websites (Ensembl and UCSC [University of California, Santa Cruz] genome browser [www.genome.ucsc.edu]).
mRNA extraction and quantification by RT-PCR.
Reverse transcription-PCR (RT-PCR) was used to determine the LTR-driven gene expression and the expression of nearby genes for three LG clones (BB1, BC5, and DA4) after treatment with antilatency drugs. For each LG clonal population, 2 × 106 Jurkat cells were incubated for 3 h with 5 mM HBMA, 1 μM prostratin, 4 μM SAHA, the combination of 1 μM prostratin and 4 μM SAHA, or dimethyl sulfoxide (DMSO) (vehicle) control. Total mRNA was isolated using RNA STAT-60 reagent (Tel Test), and total cDNA was generated Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). GFP and β-actin primer sets have been described previously (51), and primer sets for endogenous genes were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/) (81). Quantitative PCR (QPCR) primer sequences are provided in Table S1 in the supplemental material. For all samples and primer sets, QPCR conditions included an initial melting step (95°C, 3 min), followed by 35 cycles, with 1 cycle consisting of melting (95°C, 20 s), annealing (55°C, 30 s), and extension (68°C, 20 s) steps. All RT-QPCR measurements were performed in triplicate, and melting curves were generated using the CFX96 real-time PCR detection system (Bio-Rad).
Statistics for reactivation effectiveness.
In the Jurkat experiments shown below in Fig. 2, the polyclonal, infected, off cells were sorted by fluorescence-activated cell sorting (FACS) at 21 days postinfection. On the following day (day 22 postinfection), the same cells were treated with the reactivation drugs. The reported percent reactivation was calculated by subtracting the percentage of off cells after stimulation from the percentage of off cells from an identical, unperturbed, off-sorted sample, and then dividing this amount by the total percentage of off cells in the same unperturbed control. The percent reactivation values for the primary cell experiments in Fig. 4 were calculated by the same method, but these cells were not FACS sorted prior to reactivation. Stimulated and unperturbed (vehicle control) samples were measured by flow cytometry at the same time, and all measurements were performed in triplicate. Reported values are the averages of triplicate measurements, and error bars are standard deviations of these replicates. Statistical significance was determined using both nonparametric (Mann-Whitney-Wilcoxon [MWW]) and parametric (Student's t test) methods with a significance level of α = 0.05 for both. Statistical significance is claimed only when confirmed by both MWW and t test methods.
FIG. 2.

Latency reactivation for LGIT virus mutants and HIV-1 subtypes in the Jurkat cell system. (A) Infection and serial FACS sorting were performed to isolate the infected, off populations for variants of the LGIT virus. These include mutants of subtype B (mutI Sp1 [S1], mutII Sp1 [S2], mutIII Sp1 [S3], mutI NF-κB [N1]), and mutII NF-κB ([N2]) or variants with U3 regions isolated from the following subtypes: B, A2, A, A/G, B/C, C′, C, B/F, D, F, and H (as in Fig. 1B). One day after FACS sorting (day 22 postinfection [Fig. 1A, panels 5b and 6b]), polyclonal off sorts for WT LGIT mutant and HIV-1 subtype variants were treated with the following pharmacological agents to reactivate latent infections: NF-κB/PKC activators TNF-α (white bars), PMA (gray bars), or prostratin (black bars). Data represent the averages of three independent measurements for each drug perturbation, and error bars are standard deviations. For the LGIT mutants of subtype B, upward and downward arrowheads indicate statistically significant deviations from the wild-type subtype B LTR configuration of LGIT (P < 0.05). The broken gray line at 50% reactivation is drawn as a reference marker. (B) Same as in panel A for latency reactivation by TSA (white bars), SAHA (light gray bars), valproic acid (dark gray bars), or HMBA (black bars). (C) Same as in panel A for latency reactivation using the combination of prostratin and SAHA. Asterisks denote statistical synergism by the combination of drugs relative to the reactivation by either individual agent. For details on the quantitative treatment of synergy, see Materials and Methods.
FIG. 4.

Reactivation of latent HIV-1 in primary memory CD4+ T cells. (A) After 14 days of culturing in quiescent conditions (and 28 days after CD4+ T-cell isolation), the cells were treated with antilatency drugs HMBA (H), prostratin (P), SAHA (S), or the combination of prostratin and SAHA (P+S). The data shown include each LGIT subtype for each of three independent donors. Asterisks indicate synergism of the prostratin-SAHA combination with respect to either drug alone. For details on the quantitative treatment of synergy, see Materials and Methods. (B to D) At day 21 postinfection (day 28 postisolation), primary cells were examined for the proviral LTR expression of GFP (x axis) and the surface expression (y axis) of CD4 (B), CD45RO (C), or CD27 (D). (E to G) At days 21 postinfection (same as panels B to D), primary cells were treated with prostratin and SAHA for 24 h and examined for proviral LTR expression of GFP (x axis) and surface expression (y axis) of CD4 (E), CD45RO (F), or CD27 (G).
Statistical analyses for synergism.
A combination of drugs may act synergistically if their combined activity exceeds the results obtainable by any of the individual components. This investigation employs the fractional product method to quantify whether various drug combinations synergistically reactivate latent infections (88). This simple and classic definition can be derived from the mass-action law principle using the assumptions of first-order behavior and mutually nonexclusive components (13). Both of these assumptions are supported by prior investigations of the coadministration of an NF-κB/PKC activator and an HDAC inhibitor (69) and the cooperative mechanisms of NF-κB and Sp1 factors (68). To evaluate whether synergy exists in a combinatorial drug treatment, consider the antilatency strategies of prostratin (treatment P), SAHA (treatment S), and a combination of the two (treatment PS) at the following drug doses: dose of prostratin [dP] = 1.0 μM prostratin and 0 μM SAHA for treatment P, 0 μM prostratin and dose of SAHA [dS] = 4.0 μM SAHA for treatment S, and dP = 1.0 μM and dS = 4.0 μM for treatment PS. Let μ denote the measured percent reactivation (average of three biological replicate samples) after each drug treatment such that μP = μP(dP, 0), μS = μS(0, dS), and μPS = μPS(dP, dS). For the polyclonal wild-type (WT) LGIT virus (WT.OFF) samples, the measured values are as follows: μP = 57.9%, μS = 38.6%, and μPS = 76.6%, with standard deviations σP = 1.7%, σS = 0.7%, and σPS = 0.9%, respectively. Using the fractional product method, a synergistic effect applies when μPS > 1 − (1 − μP) × (1− μS) and 0.766 > 1 − (1 − 0.579) × (1 − 0.386) = 0.742.
Thus, for the WT.OFF sample, the combination of prostratin and SAHA is synergistic compared to the effects of the individual components. Statistical significance was determined using both nonparametric (Mann-Whitney-Wilcoxon) and parametric (Student's t test) methods with a significance level of α = 0.05 for both. Statistical significance is claimed only when confirmed by both MWW and t test methods.
RESULTS
Establishment of systems to assess reactivation of polyclonal and clonal HIV-1 latency models.
Recent studies have revealed that combinations of drugs may act upon multiple latency mechanisms to provide synergistic reactivation of latent infections (5, 39, 71). Since synergistic reactivation by the combinatorial treatment of NF-κB/PKC activators and HDAC inhibitors is likely mediated by the κB and Sp1 sites within the HIV-1 LTR (68), we first investigated a system in which we could simultaneously explore the regulation of these sites in conjunction with the role of Tat and its feedback loop. In particular, we have previously found that a LTR-GFP-IRES-Tat (LGIT) lentivirus within Jurkat cells can exhibit active (bright) and inactive (off) gene expression modes, and in a process termed phenotypic bifurcation (PheB), clonal populations of LGIT virus-infected Jurkat cells with single viral single integration positions may give rise to both off and bright subpopulations (89). Furthermore, we have previously demonstrated that the Sp1 and κB sites in the viral LTR differentially recruit activating and repressing factors and that mutation of these sites may destabilize both off and bright modes and thereby result in an increased frequency of PheB (10). Since this phenotype is likely driven by stochastic fluctuations in the concentration of Tat (89) and competition between activating and repressing host factors at the promoter (10), such PheB clones can serve as a sensitive means to examine the efficacy of potential antilatency drugs.
As natural variations occur within the Sp1 and κB elements, an individual patient may carry a swarm of LTR variants that can persist for years (62). Sequence variability within the LTR further increases across isolates of HIV-1 subtype B, in which polymorphisms occur throughout each Sp1 and κB element (www.hiv.lanl.gov) (see Fig. S2 in the supplemental material). Since Sp1 and κB sites differentially regulate latency (10), reactivation of latent infections may vary upon the particular configurations of these sites. To determine which Sp1 and κB sites are required for particular reactivating mechanisms, we utilized LTR variants of the LGIT lentivirus containing inactivating mutations in each of the Sp1 (mutI Sp1, mutII Sp1, and mutIII Sp1) and κB (mutI NF-κB and mutII NF-κB) binding sites (10).
Jurkat cells were infected with wild-type LGIT or mutant variant lentivirus at a low MOI (0.05 to 0.10) (Fig. 1A). Six days after infection, gene expression was stimulated with exogenous Tat protein and HMBA to activate the population of “infected but off” cells that remain transcriptionally inactive after infection (Fig. 1A, panel 2a). Eighteen hours after stimulation, FACS was utilized to isolate the polyclonal fraction of GFP-positive (GFP+) cells from uninfected cells (Fig. 1A, panel 3a). After the infected-cell fractions were sorted for wild-type LGIT virus and each Sp1 and κB mutant, the cells were cultured under normal conditions for 2 weeks during which substantial fractions of GFP+ cells relaxed into the off expression mode, generating a bimodal gene expression profile (Fig. 1A, panel 4a). Following this 2-week expansion period, FACS was again applied to isolate the polyclonal off (GFP−) population, which represents the “latent” population of infected cells (Fig. 1A, panel 5b). Similarly, after the same 2-week expansion, single cells were sorted and expanded for 4 weeks until three phenotypic bifurcation (PheB) clones for wild-type LGIT virus and each Sp1 and κB mutant were identified by flow cytometry (Fig. 1A, panel 5a).
Reactivation of polyclonal and clonal LGIT lentivirus populations with mutations in each Sp1 and κB binding element.
After the polyclonal and clonal LGIT virus populations were isolated, each was treated with a variety of pharmacological agents to reactivate latent infections. For both PheB clones and polyclonal clones that sorted into the off expression mode (off sorts), the success of each treatment was evaluated by the percentage change of GFP− (off) cells after stimulation, which corresponds to the percentage of latent infections that were reactivated (referred to as percent reactivation). While the PheB clones in this study represent an important subset of inactive integration sites that exhibit a sensitive Tat-dependent phenotype, the polyclonal off sorts include a larger latent subpopulation that likely encompasses the different phenotypes of PheB and fully silenced clones, such as J-Lats (37). Thus, by examining PheB clones and the polyclonal off sorts for the same Sp1 and κB mutants of LGIT virus, we aim to test the reactivation of each mutant within a system that will be sensitive to stochastic fluctuations in regulating factors but will also access a broad range of silent integration positions and latency mechanisms (Fig. 1A, panels 6a and b).
Throughout this study, we use a variety of pharmacological agents to survey which drugs can effectively reactivate latent infections and whether certain promoter subtypes, mutants, or integration positions might restrict these drugs. In general, the potential clinically viable agents in this study include prostratin, HMBA, SAHA, and valproic acid. However, in many cases, we have also tested the efficacy of potent, nonclinical agents as a benchmark for the clinical alternatives. For example, TNF-α and PMA are tested alongside prostratin, while SAHA and valproic acid are evaluated with the more toxic HDAC inhibitor TSA. Thus, we can evaluate particular drugs, such as prostratin and SAHA, as well as larger classes of drugs (NF-κB/PKC activators and HDAC inhibitors) to evaluate the most effective antilatency agents.
NF-κB and PKC activators require κB site I for latency reactivation.
To determine the contribution of each κB and Sp1 site to reactivation via induction of NF-κB/PKC pathways, each polyclonal off sort was treated with TNF-α, PMA, or prostratin. Treatment with TNF-α reactivated over half of the latent infections for the wild-type LGIT virus off-sorted polyclonal subpopulation (WT.OFF), while treatment with prostratin or PMA achieved approximately 60% reactivation for the same subpopulation (Fig. 2A). Mutation to any Sp1 site did not diminish reactivation via the NF-κB/PKC pathways, as treatment with TNF-α, PMA, or prostratin strongly reactivated the polyclonal off sorts (73% to 93% reactivation) for mutI Sp1 (S1), mutII Sp1 (S2), and mutIII Sp1 (S3) (Fig. 2A). This observation is consistent with our previous findings that Sp1 mutants have a reduced occupancy by histone deacetylase 1 (HDAC1) and, as a result, are more easily reactivated by TNF-α (10). In contrast, mutation of κB site I (N1) dramatically reduced reactivation by TNF-α, PMA, or prostratin for the polyclonal off sort (4% to 14% reactivation) (Fig. 2A), consistent with our previous observations that this mutant fails to sufficiently recruit RelA (10). Although mutII NF-κB (N2) populations were slightly more susceptible to reactivation by TNF-α than mutI NF-κB (20% reactivation), reactivation with either PMA (65%) or prostratin (58%) was statistically indistinguishable from wild-type subtype B LGIT virus (P > 0.05) (Fig. 2A). Reactivation of the PheB clones for each LGIT NF-κB and Sp1 mutant with TNF-α, PMA, or prostratin closely resembled the trends of the polyclonal off sorts (see Fig. S3 in the supplemental material). Collectively, LGIT off sorts and PheB clones revealed that the κB site I (N1)—a relatively well-conserved element with mutations in 2.4% of the 127 subtype B isolates from the LANL database (www.hiv.lanl.gov) (see Fig. S2 in the supplemental material)—plays a critical role in reactivation with NF-κB and PKC activators. Moreover, these results indicate that prostratin is capable of reactivating latent infections comparable to potent, immunogenic (TNF-α) or toxic (PMA) agents.
Latency reactivation with HDAC inhibitors is regulated by Sp1 site III.
Previously, we demonstrated that mutation in any of the three Sp1 sites decreases regulation by HDAC1, which suggests that these mutants may be desensitized to latency reactivation therapies involving HDAC inhibition (10). Treatment with either TSA or SAHA reactivated at least 40% of the WT polyclonal off sorts and outperformed TNF-α for S1, S2, N1, and N2 (P < 0.05) (Fig. 2B). However, S3, which responded with 88% reactivation to TNF-α and 90% reactivation to prostratin, exhibited only 28% reactivation with either TSA or SAHA. These result are consistent with our previous observations that Sp1 site III is particularly important for activation by HDAC inhibitors due to its role in the synergistic and potentially cooperative recruitment of activating factors p300 and RelA (10). Consistent with the polyclonal off sorts, SAHA strongly reactivated all three WT LGIT clones and was similarly effective on PheB clones for both κB mutants, S1, and S2 (see Fig. S3 in the supplemental material). However, like the polyclonal off sorts for mutIII S1, each clone (S3.B3, S3.B6, and S3.C4) exhibited decreased sensitivity to SAHA, and none of the three clones displayed more than 60% reactivation after stimulation (see Fig. S3 in the supplemental material). These results indicate that the efficacy of SAHA is similar to that of the nonclinical HDAC inhibitor TSA but that neither drug is effective in reactivating mutations at site III Sp1.
In addition to SAHA and TSA, we decided to explore the reactivation capabilities of the clinically viable HDAC inhibitor valproic acid, given the recent interest in this particular drug in clinical studies (49, 80). Valproic acid reactivated all LGIT polyclonal off sorts and PheB clones with markedly lower efficacy compared to SAHA (Fig. 2B) (see Fig. S3 in the supplemental material), indicating that SAHA may serve as a more effective clinical alternative to valproic acid. Moreover, these results underscore the importance of Sp1 site III—a moderately conserved element that contains potentially disruptive polymorphisms in 10% of the subtype B isolates from the LANL database (www.hiv.lanl.gov) (see Fig. S2 in the supplemental material)—in reactivating strategies involving HDAC inhibitors.
Elongation agonists and DNA methylase inhibitors are weak activators of latent infections.
In addition to a lack of transcriptional activation, latency may partially result from insufficient transcriptional elongation and Tat transactivation (54). We therefore also utilized the Tat-driven LGIT lentivirus system to examine the effects of the P-TEFb and PI3K/Akt agonist HMBA to determine whether promoting elongation could lead to Tat accumulation and subsequent strong transcriptional activation. HMBA reactivated 13% of WT off sorts and induced statistically equivalent responses from all mutant polyclonal populations except for mutIII Sp1 clones (P > 0.05) (Fig. 2B). Likewise, HMBA was modestly effective in reactivating WT PheB clones but was virtually ineffective for mutIII Sp1 clones (see Fig. S3 in the supplemental material). Therefore, like HDAC inhibitors, HMBA requires an intact site III Sp1 for maximum reactivation but appears to be less effective than SAHA.
We anticipated that okadaic acid, which promotes elongation independently of NF-κB (94), may equally reactivate WT and κB mutant LGIT lentivirus. However, this treatment reactivated merely 4% of WT LGIT off sorts, with statistically indistinguishable results between WT LGIT and all mutants (P > 0.05) (see Fig. S5A in the supplemental material), such that its effects were marginal compared to HDAC inhibitors and NF-κB/PKC activators. Treatment with resveratrol, which may enhance viral transcription by activating Egr1-dependent growth factors or by promoting the deacetylation of Tat protein via SIRT1, was ineffective on all PheB clones (see Fig. S3) and statistically negligible for WT and mutant off sorts (P > 0.05) (see Fig. S5A). Since treatment with either okadaic acid or resveratrol marginally reactivated WT LGIT or any κB or Sp1 mutant, both drugs were not further examined.
Recent investigations have associated latent proviral infections with CpG methylation (34, 39). Thus, the reversal of DNA methylation with the clinically tested methylase inhibitor 5-aza-dC may alleviate gene silencing and help promote reactivation independently of the roles of Sp1 and κB sites. Treatment with 5-aza-dC reactivated merely 4% of WT off sorts and yielded similarly weak reactivation for all other mutants, indicating that DNA methylase inhibition is not sufficient for latency reactivation in this particular model (see Fig. S4 in the supplemental material).
Combinatorial therapies synergistically reactivate latent κB and Sp1 LTR mutants.
Although individual treatments of NF-κB/PKC activators and HDAC inhibitors were effective in activating substantial fractions of cells in the off expression mode for WT LGIT, the combination of multiple drugs may have synergistic effects. Importantly, costimulation of WT LGIT off sorts with prostratin-SAHA (77% reactivation) (Fig. 2C) or PMA-TSA (87% reactivation) (see Fig. S4 in the supplemental material) synergistically reactivated gene expression, resulting in approximately 30% greater activation than with either prostratin or PMA alone and more than 2-fold greater reactivation than with either SAHA or TSA alone. Although the combinatorial treatment of TSA-HMBA (50% reactivation) is not synergistic, it provides enhancement over individual treatment with either TSA or HMBA by 15% and 37% reactivation, respectively. However, the PMA-TSA-HMBA combination (82% reactivation) provided no further activation relative to PMA-TSA (see Fig. S4), suggesting that HMBA may have redundant effects to the other two agents or that PMA-TSA simply saturate the effects of HMBA. The inclusion of 5-aza-dC also provided no further reactivation when paired with TSA, PMA, or TSA-PMA (see Fig. S4), suggesting that this clinically tested drug is not essential in latency reactivation.
Using both clonal and polyclonal LGIT models, we have demonstrated that mutations in Sp1 site III (S3) severely weaken the effectiveness of HDAC inhibitors, and mutation to κB site I (N1) abrogates the response to NF-κB/PKC activators and HDAC inhibitors (Fig. 2A and B) (see Fig. S3 in the supplemental material). These results indicate that such individual therapy approaches risk the possibility of mutational evasion because relatively minor mutations in noncoding regions, which may readily arise in patients of subtype B infection (see Fig. S2), may severely undermine drug efficacy. We hypothesize that these risks may be greatly tempered by reactivating latent infections with multiple agents, such as the potential synergistic combination of prostratin and SAHA.
For all LGIT mutant polyclonal off sorts, the combinations of prostratin and SAHA or PMA and TSA achieved between 59% (N1) and 99% (S1) reactivation and outperformed every individual component (Fig. 2C) (see Fig. S4 in the supplemental material). Although N1 was resistant to prostratin and PMA and both S3 and N1 were desensitized to SAHA and TSA (Fig. 2C) (see Fig. S4), the prostratin-SAHA or PMA-TSA combination synergistically reactivated both populations and overcame the limitations of mutation to any κB or Sp1 site. Moreover, for N1, either drug combination provided at least 4-fold greater reactivation than prostratin or PMA alone and 2-fold greater reactivation than SAHA or TSA alone. Although the effects were not synergistic for N2, the combination of either prostratin and SAHA or PMA and TSA reactivated almost 30% more latent cells than either SAHA or TSA alone, and approximately 10% more than either prostratin or PMA (Fig. 2C) (see Fig. S4). As observed with WT LGIT, HMBA provided no further reactivation for any mutant when paired with TSA (TSA-HMBA) or when included with PMA and TSA (PMA-TSA-HMBA) (see Fig. S4). Collectively, these results demonstrate that, analogous to highly active antiretroviral therapy (HAART), therapeutic reactivation of latent infections may require a combinatorial approach that performs effectively and often synergistically against different potential promoter architectures and minimizes the likelihood of mutational escape.
Latency reactivation of 11 distinct HIV-1 subtype and recombinant isolates.
Although recent investigations examined the potential reactivation of latent reservoirs using pharmacological agents, these focused exclusively on isolates from subtype B—the subtype most prevalent in the United States and Europe (8, 14, 45, 49). However, due to significant sequence diversity in the LTRs of HIV-1 subtypes (28, 56, 73), non-subtype B isolates may respond distinctly to these agents. In particular, subtypes with variable κB and Sp1 elements may exhibit resistance to drugs, similar to the phenotypes observed for mutIII Sp1 and mutI NF-κB versions of LGIT (Fig. 2A and B) (see Fig. S3 and S4 in the supplemental material). To examine strategies of latency reactivation for various HIV subtypes and to identify potential limitations for divergent promoters, we generated LGIT variants containing U3 regions specific to the following subtypes and recombinants: A, A2, A/G, B, B/C, B/F, D, F, H, and two distinct isolates of C (see Table S2 and Fig. S6 in the supplemental material). As this investigation focuses on the role of the cis-acting elements in reactivation from latency, model LGIT variants were constructed with the U3 regions of each subtype or circulating recombinant forms (CRF) (Fig. 1B), but with the R (including TAR), U5, and Tat regions of subtype B. In addition to having diversity throughout the Sp1 and κB sites, these various U3 enhancer regions also differ in other cis-regulatory elements, including AP-1, YY1, NFAT, COUP-TF, and ILF sites (Fig. 1B). The use of 11 subtype enhancer sequences representing HIV-1 isolates from five continents may help elucidate whether certain subtypes and CRFs would require drug regimens to be “tailored” to their particular promoter architectures.
Although NF-κB/PKC activators stimulated sizable fractions of off sorts for each subtype or CRF (including greater than 50% reactivation of subtype B off sorts), interesting differences emerged among them (Fig. 2A). Activation of NF-κB/PKC with PMA reactivated at least 50% of latent infections for subtypes and CRFs with deviations in Sp1 site II or III (A2, A, A/G, D, F, and H) (Fig. 1B and 2A), consistent with the clonal and polyclonal analyses of LGIT Sp1 mutants. Furthermore, among all subtypes and recombinants tested, the LGIT version for A2, which contains three nucleotide deviations in Sp1 site II (see Fig. S6 in the supplemental material), reactivated most strongly to TNF-α, PMA, and prostratin (68% to 81% reactivation) (Fig. 2A). In contrast, subtype C, which contains an additional κB site, exhibited the weakest response to TNF-α, PMA, and prostratin (38% to 47% reactivation) (Fig. 2A). Interestingly, these results indicate that Sp1 sites are not necessary for effective latency reactivation with PMA or prostratin, but an additional κB site could actually restrict latency reactivation with these NF-κB/PKC activators.
Similarly to NF-κB/PKC stimulation, reactivation with HDAC inhibitors TSA and SAHA effectively reactivated subtypes and CRFs containing nucleotide disparities from subtype B in Sp1 site II (A, A2, and A/G) (Fig. 2B). Subtype recombinant B/F, which has κB and Sp1 sequences identical to those of subtype B, and the three aforementioned subtypes and CRFs (A, A2, and A/G), all displayed over 60% reactivation with TSA and at least 50% reactivation with SAHA. Interestingly, all subtypes that contained deviations in Sp1 site III (D, F, and H) failed to achieve greater than 51% reactivation with TSA or 45% reactivation with SAHA (Fig. 2B), consistent with the observation that mutation of this site (mutIII Sp1) weakens the stimulatory effects of HDAC inhibitors (Fig. 2B). However, stimulation with either TSA or SAHA also failed to reactivate at least 50% of off sorts for CRF B/C and subtypes C and B, indicating that subtypes and recombinants with an intact Sp1 site III may also fail to strongly respond to HDAC inhibition (Fig. 2B). For all subtypes and CRFs, reactivation with valproic acid was dramatically weaker than reactivation with TSA and SAHA, consistent with other LGIT polyclonal and clonal results (Fig. 2B) (see Fig. S3 in the supplemental material). Interestingly, stimulation with HMBA yielded trends similar to those by the HDAC inhibitors and reactivated subtypes and recombinants A, A2, A/G, and B/F by greater than 30% reactivation but failed to reactivate B/C, C, C′, and B beyond 20% (Fig. 2B). Collectively, these results highlight the importance of Sp1 site III for reactivation with HDAC inhibitors, since TSA and SAHA more strongly reactivate subtypes and CRFs with deviations in Sp1 site II (A2, A, and A/G) than those with deviations in Sp1 site III (D, F, and H).
Combinatorial treatment of prostratin and SAHA synergistically reactivate some LGIT subtypes.
Similar to models for subtype B (Fig. 2C) (see Fig. S4 in the supplemental material), costimulation of an NF-κB activator and an HDAC inhibitor produces a synergistic effect for many LGIT subtypes and CRFs compared to the individual components. The combination of prostratin and SAHA reactivated at least 85% of “latent” cells for subtypes A, A/G, B/F, and F. Moreover, this combination exhibited synergistic reactivation on 6 of 11 (55%) subtype isolates, including C′, A, A/G, B, B/F, and F (P < 0.05) (Fig. 2C). These collective results reveal that reactivation of latent infections with individual drugs will likely vary across subtypes and CRFs and that the utilization of only the canonical subtype B as a model for latency may miss the behavior of different subtypes and recombinants.
Direct inhibition of YY1 or activation of AP-1 fails to reactivate most HIV-1 subtypes.
Despite the extensive evidence for the regulation of latency by Sp1 and κB sites (10, 36, 57, 92), the roles of other cis-acting factors, such as YY1 and AP-1, are less defined (33, 96). These unspecified roles are confounded by the distinct genotypic differences in the positions and sequences of YY1 and AP-1 sites across different HIV-1 subtypes and CRFs (Fig. 1B). Since YY1 may promote transcriptional silencing and latency by recruitment of HDAC1 (18), treatment with HDAC inhibitors, including TSA, SAHA, and valproic acid in the LGIT system would likely reverse these effects (Fig. 2B). Likewise, treatment of LGIT system with TNF-α would likely activate AP-1 (85), but any specific regulatory roles of AP-1 might be overshadowed by NF-κB (Fig. 2A). For this study, however, we also aimed to determine whether the broad discrepancies in YY1 and AP-1 binding sites across subtype and CRF isolates (Fig. 1B) may impact latency regulation and specific reactivation strategies.
We thus employed the subtype and CRF variants of LGIT to test whether inhibition of YY1 with either DETA or SNAP may reverse latency for any variant. In contrast to the direct inhibition of HDACs with SAHA, inhibition of the YY1 pathway with either DETA or SNAP marginally reactivated latent infections for all subtypes and CRFs. Although SNAP outperformed DETA for all subtypes and recombinants, it only reactivated at least 5% off sorts from four of the 11 subtypes and CRFs (A2, A, A/G, and B/F) (see Fig. S5B in the supplemental material). Interestingly, higher reactivation occurred specifically for subtypes and CRFs containing a YY1 site at the same position (approximately 229 to 235) within the LTR (see Fig. S5B and S6).
To determine whether AP-1 specifically plays a distinct role in latency reactivation, we have tested each LGIT subtype with sorbitol, which induces hyperosmotic shock and thereby leads to increased binding activity of AP-1 (85). Although activation of both NF-κB and AP-1 with TNF-α moderately activated expression for all subtypes and CRFs (see Fig. S5B in the supplemental material), treatment with sorbitol failed to reactivate any LGIT variant beyond 3% reactivation. Therefore, neither mild activation of AP-1 with sorbitol nor inactivation of YY1 with DETA or SNAP appears sufficient to reactivate latency for any HIV-1 subtype or CRF model in this study.
Generation of in vitro model for HIV-1 latency.
In addition to the Jurkat model for HIV-1 latency described in Fig. 2, we also used human CD4+ primary cells to better model the physiological conditions of latency. Generation of this model begins with naïve CD4+ T cells isolated from whole blood from healthy patients, similar to other systems (7, 39, 59). Consistent with the properties of human naïve CD4+ T cells, isolated cells were positive for CD4, CD45RA, and CD27 surface antigens and largely negative for CD45RO (Fig. 3A to D). After isolation, the cells were activated with CD3/CD28 Dynabeads (Invitrogen) and expanded in activating conditions for 1 week (Fig. 3E). On day 7, the cells were infected with the LGIT lentivirus (MOI of 0.5 to 0.10), including the same subtype LGIT variants A, A2, B, C, C′, D, and F used in the Jurkat cell experiments (Fig. 2). After one more week in activating conditions (day 14 postisolation), the cells were transferred to minimal medium with low levels of interleukins (1 ng/ml IL-7 and 10 U/ml IL-2) to maintain cell viability in resting conditions (Fig. 3E). The cells were cultured for 2 weeks in resting conditions (until postisolation day 28), at which antilatency drugs were used to reactivate latent LGIT virus infections. Latency reactivation was quantified by flow cytometry by measuring the percentage change in GFP+ cells 24 h after drug treatment. Additionally, all procedures for isolation, infection, and reactivation were performed using CD4+ cells from three different healthy donors (9.1, 9.2, and 9.3) to evaluate potential donor variability.
FIG. 3.

Isolation and infection of human primary CD4+ T cells. (A to D) Naïve CD4+ T cells were isolated from human whole blood from three donors (9.1, 9.2, and 9.3). Six days after isolation, the cells were analyzed by flow cytometry for expression of surface receptors CD4 (A), CD45RA (B), CD45RO (C), and CD27 (D). Histogram overlays include negative controls (shaded gray) and naïve CD4+ T cells (black outline). Fluorescence channels 6 (FL 6), 8, 4, and 9 are shown in panels A, B, C, and D, respectively. (E) Naïve CD4+ T cells were activated with CD3/CD28 antigen beads for 3 days after isolation to promote T-cell activation and expansion. At 7 days postisolation, cells from each donor were infected by one of seven different LGIT subtype variants at a low MOI. Cells were cultured in activating conditions until day 14, at which cells were transferred to minimal growth medium (1 ng/ml IL-7 and 10 U/ml IL-2).
Latency reactivation in primary CD4+ T cells.
One week after infection (day 14 postisolation), 105 cells from each LGIT lentivirus subtype and donor were strongly stimulated using CD3/CD28 Dynabeads, 1 ng/ml IL-7, and 30 U/ml IL-2 to determine the total percentage of GFP+ cells in T-cell activation conditions. This percentage was set as the baseline to normalize the subsequent latency reactivation following a 2-week period of resting culture conditions (see Fig. S7A and S7B in the supplemental material). After the 2-week resting period (day 28 postisolation), LGIT virus infections were reactivated with HMBA, prostratin, SAHA, or the combination of prostratin and SAHA (Fig. 4A). Consistent with the observations in the Jurkat cell system (Fig. 2B), HMBA was only moderately effective in reactivating latent infections for all LGIT subtypes and reactivated no more than 12% of latent infections for any variant except subtype A (27% reactivation for A9.1 and 19% reactivation for A9.3) (Fig. 4A). In contrast, prostratin reactivated at least 15% of latent infections in cells from at least one donor for all subtypes (Fig. 4A). Interestingly, treatment with SAHA accomplished at least 15% reactivation of all subtypes in at least one donor, except for the two variants of subtype C (3.7% reactivation for C9.1, 4.9% reactivation for C9.2, 1.1% reactivation for C9.3, 6.7% reactivation for C′9.1, 3.8% reactivation for C′9.2, and 2.2% reactivation for C′9.3) (Fig. 4A). These results are strikingly similar to the Jurkat cell-based experiments (Fig. 2), which reveal that subtype C (C and C′) are poorly reactivated by SAHA in comparison to prostratin (Table 1).
TABLE 1.
Summary of latency reactivation with prostratin and SAHA in Jurkat cell and primary CD4+ T-cell models
 |
Importantly, the combination of prostratin and SAHA reactivated latent infections in resting CD4+ primary cells substantially better than either drug alone. In fact, for 17 of 21 total conditions (seven LGIT lentivirus subtypes in three different donors), reactivation with the prostratin-SAHA combination was more effective than either individual component. Furthermore, for 10 of 21 total conditions, including for at least one donor from every LGIT subtype variant except LGIT subtype A, prostratin-SAHA exhibited synergistic reactivation (P < 0.05) (Fig. 4A). Subtype A, though not synergistically reactivated by prostratin-SAHA, was strongly reactivated by prostratin alone (98% reactivation for A9.1, 26% reactivation for A9.2, and 89% reactivation for A9.3) (Fig. 4A). When paired with the Jurkat cell experiments, which also indicate strong reactivation by prostratin for subtype A (Fig. 2A), these results suggest that this particular subtype may not require a combinatorial drug therapy for effective latency reactivation (Table 1). In contrast, subtype variants C and C′ are poorly reactivated by an individual drug and appear to require a combination drug therapy for synergistic reactivation (Table 1).
In addition to the reactivation of latent infections, as measured by GFP expression, cells were stained for specific surface markers to verify that the reactivated LGIT lentivirus infections occurred exclusively in memory CD4+ T cells. After treatment with prostratin-SAHA, antibody staining revealed that all GFP+ cells were also positive for CD4 and CD45RO, a marker for memory T cells (Fig. 4B, C, E, and F). Additionally, we observed significant populations of GFP+ cells either positive or negative for CD27, indicating that this latency reactivation strategy is effective for memory (CD4+ CD45RO+ CD27+) and effector (CD4+ CD45RO+ CD27−) T cells (Fig. 4D and G).
Single-agent and combinatorial reactivation strategies for Tat-deficient LG clones.
To this point, we have examined the reactivation of latency using the LGIT provirus, which enables strong Tat feedback upon transcriptional activation. However, recent studies suggest that some latent infections may arise from HIV-1 variants with impaired Tat transactivation (97). Moreover, since transcriptional silencing during latency precludes the production and accumulation of Tat, we hypothesize that latency reactivation strategies should be effective in Tat-deficient conditions. To model the Tat-deficient conditions of HIV-1 latency, we infected and generated clonal populations of Jurkat cells harboring single integrations of the LTR-GFP (LG) lentivirus, which drives GFP expression from the HIV-1 subtype B LTR (37, 38, 89). Similarly to the infections with the LGIT lentivirus, Jurkat cells were infected at a low multiplicity of infection with the LG lentivirus (MOI of 0.05 to 0.10) prior to FACS sorting (Fig. 1A, panels 1b to 3b). Single GFP+ cells were then sorted by FACS and cultured under normal conditions for 4 weeks to generate clonal populations of LG cells (Fig. 1A, panel 4b). Over this period, a substantial fraction of infected LG cells relaxed into the low GFP− region (off), likely due to the decay of Tat and NF-κB after stimulation.
Five LG clones that exhibited a variety of GFP distributions prior to stimulation (gray-filled histograms in Fig. S8A to S8C in the supplemental material) were selected to examine the reactivation of antilatency drugs in the absence of Tat. However, first, to verify that each LG clone is susceptible to Tat activity, and thus would be capable of viral activation, we have infected each with a Tat-expressing lentivirus. The resulting proviral construct constitutively expresses both Tat and mOrange from the human ubiquitin promoter (UbPr-mOrange-IRES-Tat and OrIT) to enable the identification of Tat-expressing cells by the mOrange fluorescent protein. Each LG clonal population was infected with the OrIT lentivirus (MOI of 0.15 to 0.20) and monitored by flow cytometry for both expression of mOrange (Tat) and GFP (LTR-driven expression). Each of the five LG clones responds to Tat transactivation, as indicated by the GFP+/mOrange+ subpopulations for each two-parameter histogram (Fig. 5A). Thus, each of these five LG clones (without OrIT lentivirus) will be further examined for reactivation by antilatency agents.
FIG. 5.
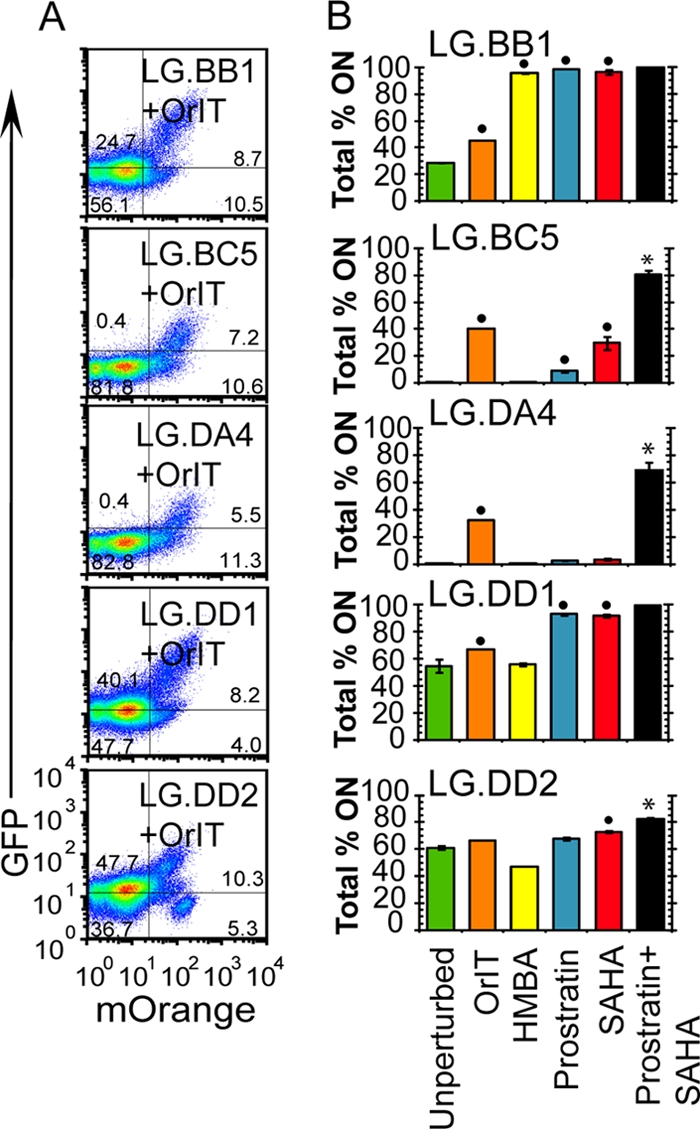
Latency reactivation of LG clones with subtype B LTR. (A) Five HIV-1 subtype B LTR-GFP (LG) clones (BB1, BC5, DA4, DD1, and DD2) were isolated to examine latency reactivation for the Tat-deficient lentivirus (Fig. 1A, panel 4b). Each Jurkat cell clonal population was subsequently infected with another lentivirus (OrIT) that constitutively expresses mOrange and HIV-1 Tat (subtype B) from the human ubiquitin promoter (MOI of 0.15 to 0.20). The cells were analyzed for expression of mOrange (x axis) and GFP (y axis), as shown in two-dimensional (2-D) histograms. Although cells are clonal with respect to the LG (GFP) infection, there are subpopulations that are either responsive (mOrange+/GFP+) or resistant (mOrange+/GFP−) to Tat transactivation. (B) LG clones, as isolated in Fig. 1A, panel 4b, were reactivated with either Tat (OrIT infection) or antilatency agents. As measured by flow cytometry, the percentage of GFP+ cells are indicated for the original LG clone (green bars), the LG population infected with the OrIT lentivirus (orange bars), and each LG clone stimulated with HMBA (yellow bars), prostratin (blue bars), SAHA (red bars), or the combination of prostratin and SAHA (black bars). The position of the off gate is set for uninfected Jurkat cells (GFP−), whereas the on gate indicates GFP+ cells. All data are averages of three biological replicate samples, and error bars are standard deviations. The small black circles indicate at least a 10% increase from the unperturbed samples, and asterisks indicate synergistic reactivation by the drug combination. Drug concentrations are provided in Materials and Methods, and the histograms are provided in Fig. S8 in the supplemental material. For details on the quantitative treatment of synergy, see Materials and Methods.
Since each LG clone exhibits a distinct monomodal gene expression profile in the absence of the OrIT lentivirus, the fractions of GFP+ cells (total percent on) before and after stimulation for each clone are reported (Fig. 5B, 28% on for LG.BB1, 0.1% for LG.BC5, 0.0% for LG.DA4, 54% for LG.DD1, and 61% for LG.DD2). The NF-κB-inducing cytokine TNF-α significantly enhanced gene expression of all clones except LG.DA4 (Fig. 5B) (see Fig. S8A in the supplemental material). Similarly, activation of both NF-κB and PKC pathways with the phorbol ester PMA significantly increased expression for clones LG.BB1, LG.BC5, and LG.DD2. However, treatment with prostratin enhanced expression only in clones LG.BB1 and LG.DD2 (Fig. 5B). Therefore, stimulation of the NF-κB and PKC pathways in Tat-deficient clones failed to activate all five LG clones and thus appeared to preferentially enhance expression only at particular integration sites in the absence of Tat.
Treatment of the LG clones with TSA significantly enhances expression of only three clones (LG.BB1, LG.BC5, and LG.DD1), while SAHA activates all clones except LG.DA4 (Fig. 5B) (see Fig. S8B in the supplemental material). In contrast, treatment with P-TEFb agonist HMBA provides no stimulation for any clones except LG.BB1 (Fig. 5B), suggesting that enhancing elongation is insufficient to reactivate many integration positions. Collectively, these results suggest that HDAC inhibition may provide a more potent reactivation mechanism than enhancing elongation but that neither strategy can reactivate at all integration positions.
Similar to the LGIT virus experiments in Jurkat cells and PBMCs (Fig. 2C and 4A), combinatorial reactivation strongly activated expression of all LG clones, achieving between 68 and 99% GFP+ cells with prostratin-SAHA (Fig. 5B) (see Fig. S8C in the supplemental material), including synergistic reactivation for three clones (LG.BC5, LG.DA4, and LG.DD2). Therefore, the combination of clinically safe drugs prostratin and SAHA effectively activates expression in Tat-deficient systems and dramatically outperforms individual agents.
Integration site analysis for LG clones.
Although we have demonstrated that treatment with antilatency drugs is capable of activating latent HIV-1 promoters (Fig. 2 to 5), the possibility that the same drugs will nonspecifically and uncontrollably perturb the expression of endogenous genes near the proviral integration site remains. As a final analysis to examine this possibility, we have identified the integration sites of three LG clones (Fig. 6, LG.BB1, LG.BC5, and LG.DA4). The LG integration site of clone BB1 lies outside an area of known transcripts between the Spn (GeneID 6693) and QPRT (GeneID 23475) genes (Fig. 6A). In contrast, the integration site for clone BC5 lies within exon 9 of ATXNL2 (GeneID 11273) in the orientation opposite the orientation of that gene (Fig. 6C). Finally, clone DA4 has an integration site within an intron of the ZMYM2 (GeneID 7750), also in the opposite direction of the gene (Fig. 6E). RT-PCR analyses were then designed to quantify the change of HIV-1 expression as well as the expression of the nearest endogenous genes (see Table S1 in the supplemental material). Each clone was treated with HMBA, prostratin, SAHA, or the combination of prostratin and SAHA and then prepared for RT-PCR 3 h after drug perturbation. For clone BB1, we have quantified the expression of the two nearest genes, while for clones BC5 and DA4, we examined the expression levels of the same genes in positions upstream and downstream of the proviral integration site.
FIG. 6.

Integration site analysis for LG clones after antilatency activation. (A) The integration site of LG clone BB1 was identified at chr16:29684467 (chromosome 16, position 29684467) outside a region of known transcripts and positioned between and in the opposite orientations of the reading frames for SPN (GeneID 6693) and QPRT (GeneID 23475). (B) LG clone BB1 was treated with DMSO (negative control), HMBA, prostratin, SAHA or the combination of prostratin and SAHA. After a 3-h incubation period, RT-PCR was performed on the untreated and treated cells to quantify the expression from the LTR (GFP), the nearest endogenous downstream gene (SPN), and the nearest upstream gene (QPRT). Using the ΔΔCT method, all data were first normalized by the respective expression of β-actin and then by the relative expression of the unperturbed control. All control and drug perturbations were performed in three biological replicate samples, and data represent averages of three independent measurements. Error bars represent standard deviations. Upward and downward arrowheads indicate statistically significant deviations from the DMSO negative control (P < 0.05). Asterisks denote statistically significant synergism of the prostratin-SAHA combination with respect to the individual components. For details on the quantitative treatment of synergy, see Materials and Methods. (C) The integration position of LG clone BC5 was identified at chr16:28841942 inside the reading frame and in the opposite orientation of ATXNL2 (GeneID 11273). (D) Same as in panel B for LG clone BC5. RT-PCR was performed on the untreated and treated cells to quantify the expression from the LTR (GFP), and the mRNA from the ATXNL2 gene upstream (atxUS) and downstream (atxDS) of the LG integration position. (E) The integration position of LG clone DA4 was identified at chr13:20605604 inside the reading frame and in the orientation opposite that of ZMYM2 (GeneID 7750). (F) Same as in panel B for LG clone DA4. RT-PCR was performed on the untreated and treated cells to quantify the expression from the LTR (GFP), and the mRNA from the ZMYM2 gene upstream (zmUS) and downstream (zmDS1 and zmDS2) of the LG integration position. Primers for two different downstream sites of ZMYM2 were used to examine different potential splice variants.
For the LG clone BB1, treatment with HBMA increased the level of GFP mRNA 1.6-fold, with a 25% decrease in expression of Spn and a 33% increase of QPRT (Fig. 6B). Treatment with prostratin enhanced GFP mRNA by nearly 2-fold but resulted in a substantial reduction in the expression of Spn (48%) and a lesser decrease in QPRT (8%). In contrast, SAHA enhanced expression of GFP, Spn, and QPRT by at least 46%. However, treatment with prostratin and SAHA significantly increased GFP mRNA expression 3.1-fold (P < 0.05) without having any significant effect on the expression of either Spn or QPRT (P > 0.05) (Fig. 6A).
Reactivation of LG clone BC5 with either HMBA or prostratin resulted in negligible changes in expression for GFP and the upstream and downstream regions in ATXNL2 (Fig. 6C). Similarly, treatment with SAHA did not enhance the expression of GFP or either ATXNL2 region. However, the prostratin-SAHA combination elevated GFP expression 1.75-fold (P < 0.05), while resulting in statistically insignificant decreases in the expression of the upstream and downstream sites (P > 0.05) (Fig. 6C).
Similarly to BB1, treatment with either HMBA or prostratin on LG clone DA4 resulted in a significant increase in GFP expression (P < 0.05), but no increase in the host gene expression upstream or downstream of the integration site (P > 0.05) (Fig. 6F). However, treatment with SAHA did not statistically enhance the expression of GFP or the endogenous gene ZMYM2 (P > 0.05) (Fig. 6F). Like clones BB1 and BC4, treatment with the prostratin-SAHA combination synergistically activated expression of GFP without increasing the expression of the endogenous gene (Fig. 6F). However, in clones BC5 and DA4, which have integration sites within and in the opposite direction of endogenous genes, the increase of GFP expression appears to loosely correlate with the decrease of expression of the endogenous gene. Though this observation might be the result of transcriptional interference from two opposing promoters, our results would also indicate that the combination of prostratin and SAHA could overcome the effects of transcriptional interference as a mechanism for HIV-1 latency (20, 31, 50).
DISCUSSION
In this proof-of-concept study, we have considered the potential design criteria for effective drug regimens to reactivate latent HIV-1 infections. These results demonstrate that a cocktail therapy, principally composed of clinically viable components SAHA and prostratin, can provide synergistic activation (Fig. 2 and 4 to 6), perform effectively against different HIV-1 subtype enhancers (Fig. 2 and 4), exhibit robustness against viral mutants (Fig. 2), target a wide range of silenced integration sites (Fig. 2 and 4 to 6), and function independently of Tat protein (Fig. 5 and 6).
In this study, we have employed both Jurkat cell and PBMC-based latency models to establish reversible, transcriptionally silent infections in order to test for reactivation upon treatment of various potential drug therapies. The primary cell latency model in this study was adapted from recently reported in vitro systems that allow for long-term culturing of resting memory CD4+ T cells (7, 39, 59). Likewise, we employ LGIT and LG Jurkat cell-based models, similar to other in vitro systems that have proven useful for studying HIV-1 expression and latency (1, 37, 50, 67, 91). Collectively, both in vitro systems enabled the examination of antilatency agents against an assortment of mutant and subtype LTR variants. Interestingly, this approach revealed significant promoter-dependent limitations of each drug that were previously undetected in models that exclusively employed viral isolates from subtype B (17, 45, 91).
Latency reactivation via HDAC inhibitors and NF-κB/PKC activators results from mechanisms that utilize the Sp1 and κB sites (Fig. 1B) and likely involves the removal of repressing HDAC complexes (e.g., HDAC1) and the subsequent recruitment of activating factors (e.g., RelA and p300) (10). These observations suggest that variability in these binding elements, either due to mutations within a subtype or to considerable differences in these elements between subtypes (Fig. 1B), may have a strong impact on emergence from latency. In agreement with our previous findings (10), we have identified the particular importance of each Sp1 and κB element by demonstrating that specific mutants exhibit severely desensitized responses to NF-κB/PKC activators (mutI NF-κB) and HDAC inhibitors (mutIII Sp1) (Fig. 2). Importantly, however, the simultaneous treatment of prostratin and SAHA effectively reactivated “latent” cells for all Sp1 and κB mutants and subtype configurations, suggesting that a combinatorial strategy with these clinically viable drugs may target a broad arrangement of promoter elements and raise the bar for potential viral mutational escape.
Although the mutated versions of LGIT virus were created by direct mutagenesis, rather than isolated from a patient, these models expose limitations in reactivation therapies that target Sp1- and κB-dependent mechanisms. The capacity of Sp1 and κB mutants in LGIT to generate PheB clones that can bifurcate into bright and off populations demonstrates that these mutant promoters could likely establish both active and latent infections. Although natural mutation in any of these sites may weaken the magnitude of transcriptional activation (74) and decrease viral replication rate (12, 32), such mutant viruses would likely remain functionally viable and may ultimately serve as a reservoir for progressive infection after periods of long-term latency (15, 22). Furthermore, even if such weakening polymorphisms arose, it is possible that these unfavorable mutations may be functionally offset by virally advantageous mutations elsewhere in the genome to restore viral expression and fitness (51). Thus, we have employed versions of the LGIT virus with point mutations in the Sp1 and κB sites to highlight the specific roles of these sites and elucidate the potential limitations of individual drug therapies for similar clinical variants.
No individual drug effectively reactivated all latency models; however, combinations of pharmacological agents overcame the limitations of single agents by acting via synergistic mechanisms (see Table S3 in the supplemental material). For example, treatment with the combination of prostratin and SAHA dramatically activated off cells for all five LG clones, with synergistic effects for three of the five clones (Fig. 5B). Importantly, this drug cocktail strongly reactivated each Sp1 and κB mutant version of LGIT, despite our findings that mutation of Sp1 site III diminished SAHA efficacy and mutation of κB site I severely weakened reactivation with prostratin (Fig. 2). Strikingly, in both Jurkat cell- and PBMC-based systems, this potentially therapeutic strategy also synergistically stimulated latent infections across subtype and CRF versions of LGIT virus, despite the diverse arrangements of cis-regulatory binding elements, including Sp1, κB, YY1, NFAT, and AP-1 sites (Fig. 1B).
Although combinatorial treatments with prostratin and SAHA provide synergistic effects for more than half of the subtypes and CRFs in both Jurkat cell and primary CD4+ T-cell systems, the extent of reactivation differs among them (Fig. 2C and 4A). On the basis of our observations for mutant versions of the LGIT virus, subtle yet important deviations in the promoter sequence and architecture, particularly within the Sp1 and κB domains, likely determine the response to either prostratin or SAHA. In particular, HIV-1 genotypes with weakening polymorphisms in Sp1 site II (A, A2, and A/G) are strongly reactivated with individual and combinatorial drug treatments in the Jurkat system (Fig. 2), suggesting that the latent populations of these subtypes and CRFs are destabilized. Similar trends are observed in the primary cell experiments, in which subtype A is strongly reactivated by prostratin alone, while the prostratin-SAHA combination synergistically reactivates latent infections from all three donors infected with LGIT A2 (Fig. 4A). In agreement with the distinct role that Sp1 site III plays in reactivation via HDAC inhibitors (Fig. 2B), subtypes with mutations in Sp1 site III (D, F, and H) responded less favorably to HDAC inhibitors than to NF-κB/PKC activators (Fig. 2B). These trends are consistent with our previous analysis demonstrating that mutation to any Sp1 site in subtype B dramatically destabilized the latent population and that mutIII Sp1 exhibited weakened TSA stimulation (10). Moreover, the dependency on a functional Sp1 site III for effective reactivation by either SAHA or HMBA (Fig. 2B) is interesting, considering that both agents may enhance transcription via stimulation of the PI3K/Akt pathway (16, 17). In general, stimulation of this pathway leads to the phosphorylation of p300 (55), the activation of RelA (61), and the recruitment of P-TEFb to the LTR (17). However, since mutIII Sp1 is deficient in the recruitment of both p300 and RelA (10), the effects of SAHA- or HMBA-induced stimulation of the PI3K/Akt pathway on the LTR are likely minimal for this particular mutant.
Subtype and CRF variants B/C, C, and C′ each contain an additional κΒ or Sp1 site that may strengthen the recruitment of repressive complexes via the p50-p50 homodimer and Sp1 in resting T-cell conditions, thereby stabilizing latent infections at particular integration positions. Interestingly, in the Jurkat system, we observe that each of these subtype promoters is more susceptible to reactivation with prostratin than with SAHA (Fig. 2A and B). Similarly, in the primary cell system, prostratin is more effective than SAHA for all three donors in LGIT C and for two of three donors in LGIT C′. Furthermore, LGIT C′, but not LGIT C, is synergistically reactivated by the combination of prostratin and SAHA in the Jurkat system (Fig. 2C), and the combination of prostratin-SAHA synergistically reactivates C′ for all three donors in the primary cell system (Fig. 4A). These results suggest that mere inhibition of HDACs, without coincident activation of PKC/NF-κB pathways, is insufficient in latency reactivation for subtypes with an additional Sp1 or κB site.
A number of recent studies have revealed that latent infections may frequently arise for integration sites near or within actively expressed genes (20, 30, 31, 50). One particular mechanism to explain this phenotype is transcriptional interference from a nearby gene, which inhibits expression from the viral promoter (48). By identifying the proviral integration sites for three LG clones, we have determined that all lie in the orientation opposite that of the nearest host gene, including two clones with integration sites within the reading frame of the host gene (Fig. 6). These results suggest that transcriptional interference may play a role in the transcriptional silencing of these infections but that the combination of prostratin and SAHA is still sufficient to overcome this possible mechanism. Moreover, we have previously confirmed that LGIT PheB clones are often integrated near actively expressed genes (89), yet that the regulation of active and inactive gene expression states is partially due to differences in the local LTR occupancy of transcription factors and chromatin-modifying factors (10). Therefore, the LG and LGIT PheB clonal latency models both support the notion that latent viruses integrated near actively expressed genes may be significantly reactivated by treatment with the combination of prostratin and SAHA.
To further elucidate the mechanisms required for latency reactivation, we have tested several promising compounds, including the natural phorbol ester prostratin and the clinical chemotherapeutic SAHA. However, these drugs have yet to be clinically tested for reactivation of HIV-1 latency, and their individual or combined in vivo effects on global T-cell activation are not fully known. These concerns are strongest with prostratin, which does not induce T-cell proliferation by itself, but can provide a secondary signal in T-cell activation that could lead to inflammation and apoptosis of T cells (8, 43). However, prostratin could inhibit HIV-1 infection in CD4+ T cells at both entry and postentry steps, which may reduce the risk of new infections after latency reactivation (4, 76). Finally, recent studies have identified synthetic PKC activators and HDAC inhibitors that may provide higher efficacy with reduced toxicity and cost compared to prostratin and SAHA (60, 64, 71, 77). Thus, with the development of such new antilatency compounds, the prospects of activating latency across different subtypes and promoter mutants should be explored.
This investigation demonstrates a rigorous in vitro examination of latency reactivation strategies using multiple clonal, polyclonal, and primary cell latency models. We have postulated that the combination of multiple agents may synergistically reactivate latent infections, maintain high efficacy across integration sites, preempt potential mutational escape, and target numerous subtype isolates. Furthermore, in the development of these strategies, we have utilized a number of clinically viable agents, such as prostratin and SAHA, though the simultaneous administration of these agents has not been clinically tested. Therefore, this study provides results that may aid in the design of future in vivo and preclinical studies and further supports the concept of multiagent clinical therapies that aim to reactivate and eradicate the latent reservoir of HIV-infected cells.
Supplementary Material
Acknowledgments
This work was supported by the University of California (UC) Berkeley Chancellor's Opportunity Fellowship (J.C.B.) and the National Institutes of Health (grant R01-GM73058).
We thank Siddharth Dey, Jonathan Foley, and Kathryn Miller-Jensen for helpful discussions and Haitang Li for technical assistance. FACS was performed at the UC Berkeley Flow Cytometry Core Facility with the technical support of Hector Nolla and Alma Valeros and at the Analytical Cytometry Core at the City of Hope under the supervision of Lucy Brown. We also thank Alberto Bosque and Vicente Planelles for detailed protocols and technical discussions regarding primary cell latency models.
Footnotes
Published ahead of print on 31 March 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Archin, N. M., A. Espeseth, D. Parker, M. Cheema, D. Hazuda, and D. M. Margolis. 2009. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retroviruses 25:207-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barboric, M., R. M. Nissen, S. Kanazawa, N. Jabrane-Ferrat, and B. M. Peterlin. 2001. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 8:327-337. [DOI] [PubMed] [Google Scholar]
- 3.Berkhout, B., R. H. Silverman, and K. T. Jeang. 1989. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 59:273-282. [DOI] [PubMed] [Google Scholar]
- 4.Biancotto, A., J. C. Grivel, F. Gondois-Rey, L. Bettendroffer, R. Vigne, S. Brown, L. B. Margolis, and I. Hirsch. 2004. Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J. Virol. 78:10507-10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blazkova, J., K. Trejbalova, F. Gondois-Rey, P. Halfon, P. Philibert, A. Guiguen, E. Verdin, D. Olive, C. Van Lint, J. Hejnar, and I. Hirsch. 2009. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 5:e1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borra, M. T., B. C. Smith, and J. M. Denu. 2005. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 280:17187-17195. [DOI] [PubMed] [Google Scholar]
- 7.Bosque, A., and V. Planelles. 2009. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113:58-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks, D. G., D. H. Hamer, P. A. Arlen, L. Gao, G. Bristol, C. M. Kitchen, E. A. Berger, and J. A. Zack. 2003. Molecular characterization, reactivation, and depletion of latent HIV. Immunity 19:413-423. [DOI] [PubMed] [Google Scholar]
- 9.Burke, B., H. J. Brown, M. D. Marsden, G. Bristol, D. N. Vatakis, and J. A. Zack. 2007. A primary cell model for activation-inducible human immunodeficiency virus. J. Virol. 81:7424-7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burnett, J. C., K. Miller-Jensen, P. S. Shah, A. P. Arkin, and D. V. Schaffer. 2009. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog. 5:e1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canonne-Hergaux, F., D. Aunis, and E. Schaeffer. 1995. Interactions of the transcription factor AP-1 with the long terminal repeat of different human immunodeficiency virus type 1 strains in Jurkat, glial, and neuronal cells. J. Virol. 69:6634-6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, B. K., M. B. Feinberg, and D. Baltimore. 1997. The kappaB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. J. Virol. 71:5495-5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou, T. C., and P. Talalay. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27-55. [DOI] [PubMed] [Google Scholar]
- 14.Choudhary, S. K., N. M. Archin, and D. M. Margolis. 2008. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J. Infect. Dis. 197:1162-1170. [DOI] [PubMed] [Google Scholar]
- 15.Churchill, M. J., D. I. Rhodes, J. C. Learmont, J. S. Sullivan, S. L. Wesselingh, I. R. Cooke, N. J. Deacon, and P. R. Gorry. 2006. Longitudinal analysis of human immunodeficiency virus type 1 nef/long terminal repeat sequences in a cohort of long-term survivors infected from a single source. J. Virol. 80:1047-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras, X., M. Barboric, T. Lenasi, and B. M. Peterlin. 2007. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 3:1459-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contreras, X., M. Schweneker, C. S. Chen, J. M. McCune, S. G. Deeks, J. Martin, and B. M. Peterlin. 2009. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 284:6782-6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coull, J. J., F. Romerio, J. M. Sun, J. L. Volker, K. M. Galvin, J. R. Davie, Y. Shi, U. Hansen, and D. M. Margolis. 2000. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 74:6790-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duh, E. J., W. J. Maury, T. M. Folks, A. S. Fauci, and A. B. Rabson. 1989. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. U. S. A. 86:5974-5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duverger, A., J. Jones, J. May, F. Bibollet-Ruche, F. A. Wagner, R. Q. Cron, and O. Kutsch. 2009. Determinants of the establishment of human immunodeficiency virus type 1 latency establishment. J. Virol. 83:3078-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edelstein, L. C., S. Micheva-Viteva, B. D. Phelan, and J. P. Dougherty. 2009. Activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res. Hum. Retroviruses 25:883-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang, G., H. Burger, C. Chappey, S. Rowland-Jones, A. Visosky, C. H. Chen, T. Moran, L. Townsend, M. Murray, and B. Weiser. 2001. Analysis of transition from long-term nonprogressive to progressive infection identifies sequences that may attenuate HIV type 1. AIDS Res. Hum. Retroviruses 17:1395-1404. [DOI] [PubMed] [Google Scholar]
- 23.Feinberg, M. B., D. Baltimore, and A. D. Frankel. 1991. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. U. S. A. 88:4045-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez, J. J., M. L. Candenas, M. L. Souto, M. M. Trujillo, and M. Norte. 2002. Okadaic acid, useful tool for studying cellular processes. Curr. Med. Chem. 9:229-262. [DOI] [PubMed] [Google Scholar]
- 25.Finzi, D., J. Blankson, J. D. Siliciano, J. B. Margolick, K. Chadwick, T. Pierson, K. Smith, J. Lisziewicz, F. Lori, C. Flexner, T. C. Quinn, R. E. Chaisson, E. Rosenberg, B. Walker, S. Gange, J. Gallant, and R. F. Siliciano. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5:512-517. [DOI] [PubMed] [Google Scholar]
- 26.Furia, B., L. Deng, K. Wu, S. Baylor, K. Kehn, H. Li, R. Donnelly, T. Coleman, and F. Kashanchi. 2002. Enhancement of nuclear factor-kappa B acetylation by coactivator p300 and HIV-1 Tat proteins. J. Biol. Chem. 277:4973-4980. [DOI] [PubMed] [Google Scholar]
- 27.Ganesh, L., E. Burstein, A. Guha-Niyogi, M. K. Louder, J. R. Mascola, L. W. Klomp, C. Wijmenga, C. S. Duckett, and G. J. Nabel. 2003. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature 426:853-857. [DOI] [PubMed] [Google Scholar]
- 28.Gao, F., D. L. Robertson, C. D. Carruthers, S. G. Morrison, B. Jian, Y. Chen, F. Barre-Sinoussi, M. Girard, A. Srinivasan, A. G. Abimiku, G. M. Shaw, P. M. Sharp, and B. H. Hahn. 1998. A comprehensive panel of near-full-length clones and reference sequences for non-subtype B isolates of human immunodeficiency virus type 1. J. Virol. 72:5680-5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geeraert, L., G. Kraus, and R. J. Pomerantz. 2008. Hide-and-seek: the challenge of viral persistence in HIV-1 infection. Annu. Rev. Med. 59:487-501. [DOI] [PubMed] [Google Scholar]
- 30.Han, Y., K. Lassen, D. Monie, A. R. Sedaghat, S. Shimoji, X. Liu, T. C. Pierson, J. B. Margolick, R. F. Siliciano, and J. D. Siliciano. 2004. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 78:6122-6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han, Y., Y. B. Lin, W. An, J. Xu, H. C. Yang, K. O'Connell, D. Dordai, J. D. Boeke, J. D. Siliciano, and R. F. Siliciano. 2008. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 4:134-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrich, D., J. Garcia, F. Wu, R. Mitsuyasu, J. Gonazalez, and R. Gaynor. 1989. Role of SP1-binding domains in in vivo transcriptional regulation of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 63:2585-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He, G., and D. M. Margolis. 2002. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 22:2965-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishida, T., A. Hamano, T. Koiwa, and T. Watanabe. 2006. 5′ long terminal repeat (LTR)-selective methylation of latently infected HIV-1 provirus that is demethylated by reactivation signals. Retrovirology 3:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeeninga, R. E., E. M. Westerhout, M. L. van Gerven, and B. Berkhout. 2008. HIV-1 latency in actively dividing human T cell lines. Retrovirology 5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang, G., A. Espeseth, D. J. Hazuda, and D. M. Margolis. 2007. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 81:10914-10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jordan, A., D. Bisgrove, and E. Verdin. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22:1868-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jordan, A., P. Defechereux, and E. Verdin. 2001. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 20:1726-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kauder, S. E., A. Bosque, A. Lindqvist, V. Planelles, and E. Verdin. 2009. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 5:e1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly, W. K., V. M. Richon, O. O'Connor, T. Curley, B. MacGregor-Curtelli, W. Tong, M. Klang, L. Schwartz, S. Richardson, E. Rosa, M. Drobnjak, C. Cordon-Cordo, J. H. Chiao, R. Rifkind, P. A. Marks, and H. Scher. 2003. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 9:3578-3588. [PubMed] [Google Scholar]
- 41.Kiernan, R. E., C. Vanhulle, L. Schiltz, E. Adam, H. Xiao, F. Maudoux, C. Calomme, A. Burny, Y. Nakatani, K. T. Jeang, M. Benkirane, and C. Van Lint. 1999. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 18:6106-6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinoshita, S., L. Su, M. Amano, L. A. Timmerman, H. Kaneshima, and G. P. Nolan. 1997. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 6:235-244. [DOI] [PubMed] [Google Scholar]
- 43.Korin, Y. D., D. G. Brooks, S. Brown, A. Korotzer, and J. A. Zack. 2002. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J. Virol. 76:8118-8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishnan, V., and S. L. Zeichner. 2004. Host cell gene expression during human immunodeficiency virus type 1 latency and reactivation and effects of targeting genes that are differentially expressed in viral latency. J. Virol. 78:9458-9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulkosky, J., D. M. Culnan, J. Roman, G. Dornadula, M. Schnell, M. R. Boyd, and R. J. Pomerantz. 2001. Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 98:3006-3015. [DOI] [PubMed] [Google Scholar]
- 46.Lafeuillade, A., C. Poggi, S. Chadapaud, G. Hittinger, M. Chouraqui, M. Pisapia, and E. Delbeke. 2001. Pilot study of a combination of highly active antiretroviral therapy and cytokines to induce HIV-1 remission. J. Acquir. Immune Defic. Syndr. 26:44-55. [DOI] [PubMed] [Google Scholar]
- 47.Laspia, M. F., A. P. Rice, and M. B. Mathews. 1989. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 59:283-292. [DOI] [PubMed] [Google Scholar]
- 48.Lassen, K., Y. Han, Y. Zhou, J. Siliciano, and R. F. Siliciano. 2004. The multifactorial nature of HIV-1 latency. Trends Mol. Med. 10:525-531. [DOI] [PubMed] [Google Scholar]
- 49.Lehrman, G., I. B. Hogue, S. Palmer, C. Jennings, C. A. Spina, A. Wiegand, A. L. Landay, R. W. Coombs, D. D. Richman, J. W. Mellors, J. M. Coffin, R. J. Bosch, and D. M. Margolis. 2005. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lenasi, T., X. Contreras, and B. M. Peterlin. 2008. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 4:123-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leonard, J. N., P. S. Shah, J. C. Burnett, and D. V. Schaffer. 2008. HIV evades RNA interference directed at TAR by an indirect compensatory mechanism. Cell Host Microbe 4:484-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewinski, M. K., D. Bisgrove, P. Shinn, H. Chen, C. Hoffmann, S. Hannenhalli, E. Verdin, C. C. Berry, J. R. Ecker, and F. D. Bushman. 2005. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 79:6610-6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li, C., C. F. Lai, D. S. Sigman, and R. B. Gaynor. 1991. Cloning of a cellular factor, interleukin binding factor, that binds to NFAT-like motifs in the human immunodeficiency virus long terminal repeat. Proc. Natl. Acad. Sci. U. S. A. 88:7739-7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin, X., D. Irwin, S. Kanazawa, L. Huang, J. Romeo, T. S. Yen, and B. M. Peterlin. 2003. Transcriptional profiles of latent human immunodeficiency virus in infected individuals: effects of Tat on the host and reservoir. J. Virol. 77:8227-8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu, Y., C. E. Denlinger, B. K. Rundall, P. W. Smith, and D. R. Jones. 2006. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J. Biol. Chem. 281:31359-31368. [DOI] [PubMed] [Google Scholar]
- 56.Lole, K. S., R. C. Bollinger, R. S. Paranjape, D. Gadkari, S. S. Kulkarni, N. G. Novak, R. Ingersoll, H. W. Sheppard, and S. C. Ray. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lusic, M., A. Marcello, A. Cereseto, and M. Giacca. 2003. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 22:6550-6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marban, C., L. Redel, S. Suzanne, C. Van Lint, D. Lecestre, S. Chasserot-Golaz, M. Leid, D. Aunis, E. Schaeffer, and O. Rohr. 2005. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res. 33:2318-2331. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Marini, A., J. M. Harper, and F. Romerio. 2008. An in vitro system to model the establishment and reactivation of HIV-1 latency. J. Immunol. 181:7713-7720. [DOI] [PubMed] [Google Scholar]
- 60.Marquez, N., M. A. Calzado, G. Sanchez-Duffhues, M. Perez, A. Minassi, A. Pagani, G. Appendino, L. Diaz, M. A. Munoz-Fernandez, and E. Munoz. 2008. Differential effects of phorbol-13-monoesters on human immunodeficiency virus reactivation. Biochem. Pharmacol. 75:1370-1380. [DOI] [PubMed] [Google Scholar]
- 61.Mayo, M. W., C. E. Denlinger, R. M. Broad, F. Yeung, E. T. Reilly, Y. Shi, and D. R. Jones. 2003. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-kappa B through the Akt pathway. J. Biol. Chem. 278:18980-18989. [DOI] [PubMed] [Google Scholar]
- 62.Michael, N. L., L. D'Arcy, P. K. Ehrenberg, and R. R. Redfield. 1994. Naturally occurring genotypes of the human immunodeficiency virus type 1 long terminal repeat display a wide range of basal and Tat-induced transcriptional activities. J. Virol. 68:3163-3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moriuchi, H., M. Moriuchi, S. B. Mizell, L. A. Ehler, and A. S. Fauci. 2000. In vitro reactivation of human immunodeficiency virus 1 from latently infected, resting CD4+ T cells after bacterial stimulation. J. Infect. Dis. 181:2041-2044. [DOI] [PubMed] [Google Scholar]
- 64.Nakagawa, Y., R. C. Yanagita, N. Hamada, A. Murakami, H. Takahashi, N. Saito, H. Nagai, and K. Irie. 2009. A simple analogue of tumor-promoting aplysiatoxin is an antineoplastic agent rather than a tumor promoter: development of a synthetically accessible protein kinase C activator with bryostatin-like activity. J. Am. Chem. Soc. 131:7573-7579. [DOI] [PubMed] [Google Scholar]
- 65.Pacholec, M., J. E. Bleasdale, B. Chrunyk, D. Cunningham, D. Flynn, R. S. Garofalo, D. Griffith, M. Griffor, P. Loulakis, B. Pabst, X. Qiu, B. Stockman, V. Thanabal, A. Varghese, J. Ward, J. Withka, and K. Ahn. 2010. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285:8340-8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pagans, S., A. Pedal, B. J. North, K. Kaehlcke, B. L. Marshall, A. Dorr, C. Hetzer-Egger, P. Henklein, R. Frye, M. W. McBurney, H. Hruby, M. Jung, E. Verdin, and M. Ott. 2005. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 3:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pearson, R., Y. K. Kim, J. Hokello, K. Lassen, J. Friedman, M. Tyagi, and J. Karn. 2008. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 82:12291-12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perkins, N. D., N. L. Edwards, C. S. Duckett, A. B. Agranoff, R. M. Schmid, and G. J. Nabel. 1993. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 12:3551-3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quivy, V., E. Adam, Y. Collette, D. Demonte, A. Chariot, C. Vanhulle, B. Berkhout, R. Castellano, Y. de Launoit, A. Burny, J. Piette, V. Bours, and C. Van Lint. 2002. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J. Virol. 76:11091-11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ragione, F. D., V. Cucciolla, V. Criniti, S. Indaco, A. Borriello, and V. Zappia. 2003. p21Cip1 gene expression is modulated by Egr1: a novel regulatory mechanism involved in the resveratrol antiproliferative effect. J. Biol. Chem. 278:23360-23368. [DOI] [PubMed] [Google Scholar]
- 71.Reuse, S., M. Calao, K. Kabeya, A. Guiguen, J. S. Gatot, V. Quivy, C. Vanhulle, A. Lamine, D. Vaira, D. Demonte, V. Martinelli, E. Veithen, T. Cherrier, V. Avettand, S. Poutrel, J. Piette, Y. de Launoit, M. Moutschen, A. Burny, C. Rouzioux, S. De Wit, G. Herbein, O. Rohr, Y. Collette, O. Lambotte, N. Clumeck, and C. Van Lint. 2009. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS One 4:e6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Romerio, F., M. N. Gabriel, and D. M. Margolis. 1997. Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. J. Virol. 71:9375-9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roof, P., M. Ricci, P. Genin, M. A. Montano, M. Essex, M. A. Wainberg, A. Gatignol, and J. Hiscott. 2002. Differential regulation of HIV-1 clade-specific B, C, and E long terminal repeats by NF-kappaB and the Tat transactivator. Virology 296:77-83. [DOI] [PubMed] [Google Scholar]
- 74.Ross, E. K., A. J. Buckler-White, A. B. Rabson, G. Englund, and M. A. Martin. 1991. Contribution of NF-kappa B and Sp1 binding motifs to the replicative capacity of human immunodeficiency virus type 1: distinct patterns of viral growth are determined by T-cell types. J. Virol. 65:4350-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rowinsky, E. K., D. S. Ettinger, L. B. Grochow, R. B. Brundrett, A. E. Cates, and R. C. Donehower. 1986. Phase I and pharmacologic study of hexamethylene bisacetamide in patients with advanced cancer. J. Clin. Oncol. 4:1835-1844. [DOI] [PubMed] [Google Scholar]
- 76.Rullas, J., M. Bermejo, J. Garcia-Perez, M. Beltan, N. Gonzalez, M. Hezareh, S. J. Brown, and J. Alcami. 2004. Prostratin induces HIV activation and downregulates HIV receptors in peripheral blood lymphocytes. Antivir. Ther. 9:545-554. [PubMed] [Google Scholar]
- 77.Savarino, A., A. Mai, S. Norelli, S. El Daker, S. Valente, D. Rotili, L. Altucci, A. T. Palamara, and E. Garaci. 2009. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology 6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sedaghat, A. R., J. D. Siliciano, T. P. Brennan, C. O. Wilke, and R. F. Siliciano. 2007. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog. 3:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Siliciano, J. D., J. Kajdas, D. Finzi, T. C. Quinn, K. Chadwick, J. B. Margolick, C. Kovacs, S. J. Gange, and R. F. Siliciano. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9:727-728. [DOI] [PubMed] [Google Scholar]
- 80.Siliciano, J. D., J. Lai, M. Callender, E. Pitt, H. Zhang, J. B. Margolick, J. E. Gallant, J. Cofrancesco, Jr., R. D. Moore, S. J. Gange, and R. F. Siliciano. 2007. Stability of the latent reservoir for HIV-1 in patients receiving valproic acid. J. Infect. Dis. 195:833-836. [DOI] [PubMed] [Google Scholar]
- 81.Spandidos, A., X. Wang, H. Wang, and B. Seed. 2010. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 38:D792-D799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steel, A., S. Clark, I. Teo, S. Shaunak, M. Nelson, B. Gazzard, and P. Kelleher. 2006. No change to HIV-1 latency with valproate therapy. AIDS 20:1681-1682. [DOI] [PubMed] [Google Scholar]
- 83.Strain, M. C., H. F. Gunthard, D. V. Havlir, C. C. Ignacio, D. M. Smith, A. J. Leigh-Brown, T. R. Macaranas, R. Y. Lam, O. A. Daly, M. Fischer, M. Opravil, H. Levine, L. Bacheler, C. A. Spina, D. D. Richman, and J. K. Wong. 2003. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc. Natl. Acad. Sci. U. S. A. 100:4819-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sung, T. L., and A. P. Rice. 2006. Effects of prostratin on cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 3:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taher, M. M., J. D. Oakley, C. Hershey, and K. Valerie. 2000. Activation of NF-kappa B and p38 MAP kinase is not sufficient for triggering efficient HIV gene expression in response to stress. Biochemistry 39:1709-1715. [DOI] [PubMed] [Google Scholar]
- 86.Van Lint, C., S. Emiliani, M. Ott, and E. Verdin. 1996. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 15:1112-1120. [PMC free article] [PubMed] [Google Scholar]
- 87.van Praag, R. M., J. M. Prins, M. T. Roos, P. T. Schellekens, I. J. Ten Berge, S. L. Yong, H. Schuitemaker, A. J. Eerenberg, S. Jurriaans, F. de Wolf, C. H. Fox, J. Goudsmit, F. Miedema, and J. M. Lange. 2001. OKT3 and IL-2 treatment for purging of the latent HIV-1 reservoir in vivo results in selective long-lasting CD4+ T cell depletion. J. Clin. Immunol. 21:218-226. [DOI] [PubMed] [Google Scholar]
- 88.Webb, J. L. 1963. Effect of more than one inhibitor, vol. 1. Academic Press, New York, NY.
- 89.Weinberger, L. S., J. C. Burnett, J. E. Toettcher, A. P. Arkin, and D. V. Schaffer. 2005. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 122:169-182. [DOI] [PubMed] [Google Scholar]
- 90.Weinberger, L. S., R. D. Dar, and M. L. Simpson. 2008. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat. Genet. 40:466-470. [DOI] [PubMed] [Google Scholar]
- 91.Williams, S. A., L. F. Chen, H. Kwon, D. Fenard, D. Bisgrove, E. Verdin, and W. C. Greene. 2004. Prostratin antagonizes HIV latency by activating NF-{kappa}B. J. Biol. Chem. 279:42008-42017. [DOI] [PubMed] [Google Scholar]
- 92.Williams, S. A., L. F. Chen, H. Kwon, C. M. Ruiz-Jarabo, E. Verdin, and W. C. Greene. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 25:139-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams, S. A., and W. C. Greene. 2007. Regulation of HIV-1 latency by T-cell activation. Cytokine 39:63-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Williams, S. A., H. Kwon, L. F. Chen, and W. C. Greene. 2007. Sustained induction of NF-{kappa} B is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 81:6043-6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu, X., Y. Li, B. Crise, and S. M. Burgess. 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science 300:1749-1751. [DOI] [PubMed] [Google Scholar]
- 96.Yang, X., Y. Chen, and D. Gabuzda. 1999. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-kappaB. J. Biol. Chem. 274:27981-27988. [DOI] [PubMed] [Google Scholar]
- 97.Yukl, S., S. Pillai, P. Li, K. Chang, W. Pasutti, C. Ahlgren, D. Havlir, M. Strain, H. Gunthard, D. Richman, A. P. Rice, E. Daar, S. Little, and J. K. Wong. 2009. Latently-infected CD4+ T cells are enriched for HIV-1 Tat variants with impaired transactivation activity. Virology 387:98-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou, M., M. A. Halanski, M. F. Radonovich, F. Kashanchi, J. Peng, D. H. Price, and J. N. Brady. 2000. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol. Cell. Biol. 20:5077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu, Y., T. Pe'ery, J. Peng, Y. Ramanathan, N. Marshall, T. Marshall, B. Amendt, M. B. Mathews, and D. H. Price. 1997. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 11:2622-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.