

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia in the aging population. Prior work has shown that the ε4 allele of apolipoprotein E (apoE4) is a major risk factor for “sporadic” AD, which accounts for >99% of AD cases without a defined underlying mechanism. Recently, we have demonstrated that sulfatides are substantially and specifically depleted at the very early stage of AD. To identify the mechanism(s) of sulfatide loss concurrent with AD onset, we have found that: (1) sulfatides are specifically associated with apoE-associated particles in cerebrospinal fluid (CSF); (2) apoE modulates cellular sulfatide levels; and (3) the modulation of sulfatide content is apoE isoform dependent. These findings not only lead to identification of the potential mechanisms underlying sulfatide depletion at the earliest stages of AD but also serve as mechanistic links to explain the genetic association of apoE4 with AD. Moreover, our recent studies further demonstrated that (1) apoE mediates sulfatide depletion in amyloid-β precursor protein transgenic mice; (2) sulfatides enhance amyloid β (Aβ) peptides binding to apoE-associated particles; (3) Aβ42 content notably correlates with sulfatide content in CSF; (4) sulfatides markedly enhance the uptake of Aβ peptides; and (5) abnormal sulfatide-facilitated Aβ uptake results in the accumulation of Aβ in lysosomes. Collectively, our studies clearly provide a link between apoE, Aβ, and sulfatides in AD and establish a foundation for the development of effective therapeutic interventions for AD.
Keywords: Alzheimer’s disease, Amyloid-beta peptides, Apolipoprotein E, Electrospray ionization mass spectrometry, Lipidomics, Shotgun lipidomics, Sulfatides
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia worldwide. At present, there are no effective therapies which have been shown to delay the onset or progression of AD. Understanding how various factors contribute to AD pathogenesis will likely facilitate the development of effective pharmaceutical treatments. The neurochemical and neuropathological hallmarks of AD include synaptic loss, selective neuronal cell death, decreased levels of certain neurotransmitters, and the presence of abnormal deposits in neurons (neurofibrillary tangles) as well as in the extracellular space (cerebrovascular, diffuse, and neuritic plaques) [1–4]. These extracellular deposits are composed predominantly of amyloid β (Aβ) peptides containing 39–43-amino-acid residues.
Aβ peptides are produced by specific endoproteolytic cleavage of the amyloid-β precursor protein (APP), which is secreted by both neuronal and nonneuronal cells into the extracellular space throughout life. AD patients develop neuritic plaques in brain regions which contain abundant amounts of Aβ filaments and other plaque-associated proteins (e.g., apolipoprotein E (apoE)) [5]. Aβ peptides also accumulate in many nonfilamentous extracellular deposits (i.e., diffuse plaques which are thioflavine-S and Congo red negative). Substantial experimental results indicate the causal relationship of Aβ peptides in the early-onset AD as well as Down syndrome [6, 7], thereby suggesting a central role for Aβ peptides in the pathogenesis of late-onset AD. The available evidence favors a model in which diverse genetic defects lead to a common biochemical abnormality, i.e., initial accumulation of the highly self-aggregating Aβ42 followed by later deposition of Aβ40. One possible mechanism that leads to the buildup of Aβ peptides in sporadic, late-onset AD patients is the dysfunction of one or more of the pathways for Aβ clearance [8]. While the mechanism(s) by which Aβ peptides are normally cleared from the brain is not known, recent evidence suggests that it may involve transport (likely via lipoproteins) through the interstitial space surrounding neuronal cells to the cerebrospinal fluid (CSF) which is in equilibrium with the blood plasma [9]. Alternatively, the clearance of Aβ peptides may involve local uptake and processing by cells (glia and/or neuronal) via receptor-mediated endocytosis and/or phagocytosis (e.g., by astrocytes and microglia) [10]. It has been demonstrated that increases in the cellular uptake of Aβ peptides can be facilitated by the expression of low-density lipoprotein (LDL) receptor-related protein (LRP), thereby supporting an endocytotic process [11]. Indeed, active glial and neuronal processes are found to be in direct contact with Aβ-containing plaques in the AD brain [5]. Furthermore, endosome dysfunction has long been recognized as an early event in the pathogenesis of AD [12], thus strongly supporting the role of endocytosis in Aβ clearance [13, 14].
ApoE is a 34-kDa protein which is associated with most lipoprotein classes in the plasma and high-density lipopro-tein (HDL)-like particles in the central nervous system (CNS) [15, 16]. It plays a key role in systemic transport of cholesterol and other lipids [15, 17]. Therefore, abnormal cholesterol metabolism and homeostasis may be involved in AD pathogenesis [18, 19]. ApoE may have other functions (e.g., in immune regulation [20] and as a molecular chaperone for amyloid proteins under certain disease conditions [21]). In the CNS, apoE is abundantly expressed in astrocytes [22, 23]. There are three common alleles of apoE (ε2, ε3, and ε4) in humans, which are different from each other at two positions: E2 (cys112, cys158), E3 (cys112, arg158), and E4 (arg112, arg158) [15, 17], and among which ε3 is the most common. Cumulative evidence suggests that there are probably several genetic risk factors for “sporadic” AD, which accounts for >99% of all the cases of AD. At present, however, the ε4 allele of apoE is the only single genetic risk factor that has been confirmed by multiple independent studies [24].
Sulfatides are a class of sulfated galactocerebrosides (GalCer; Fig. 1), which mediate diverse biological processes [25–29]. Sulfatides are almost exclusively synthesized by oligodendrocytes in the CNS and are present predominantly in the myelin sheath surrounding axons [25] although a small amount of sulfatides may be synthesized in neurons [29] or astrocyte [30]. Accumulation of sulfatides, due to a deficiency in arylsulfatase A or its coenzymes in lysosomes, is associated with the pathogenesis of metachromatic leukodystrophy [29, 31, 32]. Mice deficient in GalCer and sulfatides resulting from genetic ablation of ceramide galactosyltransferase (Fig. 1) generally die by 3 months of age and demonstrate multiple neurological abnormalities (e.g., abnormal axonal function, dysmyelinosis, and loss of axonal conduction velocity [33–36]). Severe myelin developmental abnormalities, myelin sheath degeneration, and significant increases in the deterioration of nodal/paranodal structures are manifest in sulfatide-deficient mice generated by ablating GalCer sulfotransferase (GST; Fig. 1) [37]. Abnormal sulfatide metabolism and homeostasis have been found in association with multiple other diseases (e.g., cardiovascular diseases [38], colorectal adenocarcinoma [39]). Importantly, we and others have found a substantial loss of sulfatide content in postmortem brain tissues of patients with AD relative to cognitively normal individuals [40–42]. Further analyses of enzymatic levels and/or activities which are involved in sulfatide biosynthesis and degradation indicated that there were no significant differences between AD samples and those from cognitively normal controls [41, 43]. This result is consistent with a genetic study from a Croatian population [44].
Fig. 1.

The chemical structures of sulfatides and their related metabolites and the major enzymes involved in the biosynthesis and degradation of sulfatides. R represents α-hydroxy or nonhydroxy fatty acyl chain
Although sulfatide depletion in AD has been previously described [40], prior insurmountable technical obstacles precluded further investigation of the underlying biochemical mechanisms. Recently, the direct qualitative and quantitative analysis of complex mixtures of lipids by electrospray ionization mass spectrometry (designated shotgun lipidomics; see [45–48] for recent reviews) have made the quantitative analysis of sulfatides as well as over 25 other lipid classes routine in our group [49]. By using shotgun lipidomics, we have demonstrated that sulfatides are depleted up to 90% in cerebral gray matter and up to 50% in white matter in subjects at the very mild stage of AD (mild cognitive impairment, MCI) compared to age-matched cognitively normal controls [41]. Examination of the sulfatide levels in CSF from subjects who have been clinically identified as MCI also displayed substantially lower sulfatide contents relative to controls [50, 51]. Sulfatide contents in postmortem brain samples from subjects with other neurodegenerative diseases (e.g., Parkinson’s disease, dementia with Lewy bodies, and multiple sclerosis) have also been examined [42]. The results suggest that sulfatide deficiency is specific to AD even at the MCI stage and that sulfatides play an important role in AD pathogenesis.
Association of Sulfatides with ApoE Particles in the CNS
We have previously demonstrated that (1) sulfatides are specifically associated with apoE-containing HDL-like particles by both gel filtration column separation and immunoprecipitation/mass spectrometry analysis of human CSF; (2) apoE modulates sulfatide levels in the CNS; and (3) the modulation of CNS sulfatide content is apoE isoform dependent [52]. Specifically, sulfatide content in apoE-associated lipoprotein particles is in the rank order of apoE4 > E3 > E2. This result is consistent with the sulfatide-depleted state of different brain regions of the apoE4 transgenic mice in comparison to the same regions in transgenic mice expressing human apoE3. Although the role of apoE in sulfatide metabolism, trafficking, and homeostasis in peripheral circulatory systems is still unknown, it has been demonstrated that (1) apoE possesses distinct roles in the CNS and in the PNS [53]; (2) apoE is essential to the depletion of sulfatide from human APP mutant transgenic mouse brain in which apoE levels are higher than those in wild-type (WT) littermate controls (see below); and (3) expression of LRP in mouse brain results in a sulfatide-deficient phenotype (unpublished observation). These findings have led us to establishing a working model (Fig. 2) of apoE-mediated sulfatide metabolism, trafficking, and homeostasis [43, 54]. It should be emphasized that the presence of other pathways for sulfatide metabolism and trafficking is not excluded [29].
Fig. 2.
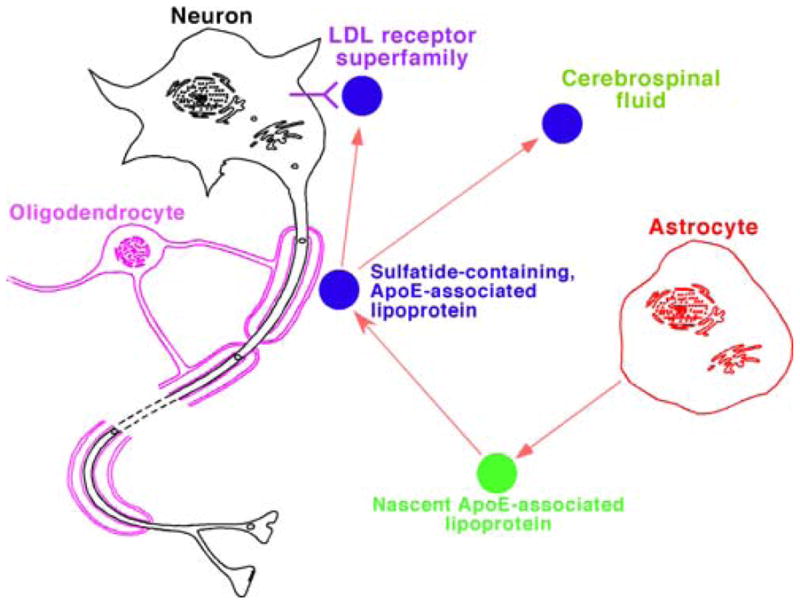
A working model for the metabolism of apolipoprotein E (apoE)-associated lipoproteins which mediate sulfatide homeostasis. In the model, apoE-associated lipoproteins are released from astrocytes, acquire sulfatides from myelin sheath, and then either metabolized through endocytotic pathway through LDL receptors or its family members such as the LDL receptor-related protein or transported to cerebrospinal fluid. (Reprinted from [43] with permission from the International Society for Neurochemistry, Copyright 2007)
According to this working model, apoE-associated particles deficient in sulfatides are released from astrocytes [22, 23, 52] and acquire sulfatides from the myelin sheath (Fig. 2). The mechanism of acquiring sulfatides by apoE particles is still unknown, which may be through a “kiss-and-run” mechanism or through a sulfatide carrier protein including apoE itself. In the working model, these nascent sulfatide-containing apoE-associated lipoprotein particles are then metabolized and degraded through an endocytotic pathway by neuronal cells possessing one or more members of the LDL receptor superfamily (e.g., LRP) [55, 56]. Under normal physiological conditions, the majority of the internalized sulfatides are catabolized to various degradation products (e.g., sulfate, galactose, trans-hexadecenal, sphingosine, etc.) in endosomes and lysosomes [25]. However, a deficiency in sulfatidase or its coenzymes in lysosomes results in the abnormal accumulation of sulfatides in these organelles in metachromatic leukodystrophy [31, 32], which has recently been further demonstrated in a cell model system in our studies [57].
It can be anticipated based upon this working model that (1) sulfatides are present in CSF [50] and are specifically associated with apoE lipoprotein particles [52]; (2) an increased rate of apoE particle uptake and/or an increased expression of lipoprotein receptors can lead to the accelerated degradation of sulfatide-containing, apoE-associated lipoprotein particles, thereby resulting in an accelerated rate of sulfatide depletion from myelin sheath; and (3) sulfatide levels in myelin sheath are dependent on the specific apoE isoforms present since apoE-associated particles transport sulfatides in the rank order of efficiency: apoE4 > E3 > E2 [52]. Collectively, our findings support the hypothesis that apoE is involved in the loss of sulfatide in AD patients, demonstrate that our working model can be utilized to explore the mechanisms by which Apoe4 gene is a major genetic risk factor, and explain why LRP and many proteins involved in endosomal trafficking are associated with AD. Moreover, it should be emphasized that the trafficking of sulfatides by apoE-associated lipoproteins occurs in parallel with the effects of apoE on Aβ clearance and deposition (discussed below).
Association of Aβ Peptides with ApoE Particles
Several previous studies suggest that apoE influences Aβ clearance, deposition, and toxicity in the brain in an isoform-dependent manner [21, 58–62]. In transgenic animal models that develop AD-like amyloidosis and neuritic plaques, the absence of apoE results in little or no Aβ deposition [60, 61, 63]. The expression of human apoE results in an isoform-specific deposition of amyloid within neuritic plaques in decreasing order of potency: apoE4 > E3 > E2 [61, 64]. Our recent studies have demonstrated that the increased production of Aβ in APP transgenic mouse brain requires an increased rate of clearance through the apoE metabolic pathway which occurs through an increase in apoE levels, thereby leading to increased sulfatide trafficking and catabolism (i.e., decreased sulfatide content) in an age-dependent manner (see below) [54]. Together, these data strongly suggest that apoE and its associated lipoproteins (or the component(s) of the apoE-associated particles produced in the brain) are in some manner required for Aβ deposition in these animals. Two nonmutually exclusive possibilities likely explain the influence of apoE on Aβ deposition: (1) the interactions of apoE-associated particles with Aβ peptides facilitate conversion of soluble Aβ to fibrillar Aβ and (2) the interactions of apoE-associated particles with Aβ peptides influence Aβ clearance. Intriguingly, our recent studies provide strong evidence that the sulfatides present in apoE-associated particles facilitate the clearance of extracellular Aβ peptides (see next subsection).
Interaction Between Sulfatides and Aβ Peptides
Prior studies have demonstrated that sulfate ions and the sulfate moieties of proteoglycans are critical for the enhancement of Aβ fibril formation in vitro and in vivo [65, 66]. Other studies have found that heparan sulfate and other proteoglycans accumulate in neurons and in the amyloid deposits of AD and Down syndrome [67, 68]. Recently, we have demonstrated that sulfatides, but not other anionic lipids, present in vesicles robustly facilitate the clearance of extracellular Aβ peptides in an Aβ42-selective manner [69].
Specifically, we determined the effects of sulfatides (complexed to cyclodextrin or guest components in host phosphatidylcholine vesicles) on the clearance of Aβ40 and Aβ42 peptides from the culture media of H4-APPwt cells. It should be noted that H4-APPwt cells normally generate large amounts of Aβ peptides and secrete them into the media [70]. Media sulfatide supplementation in either format markedly lowered the amounts of both Aβ40 and Aβ42 peptides in the media in a dose-dependent manner with selective reduction of Aβ42 [69]. In contrast, similar amounts of anionic lipids including phosphatidylinositol in phosphatidylcholine vesicles only minimally changed the levels of media Aβ peptides under identical experimental conditions. It should be pointed out that phosphatidylinositol species possess a similar chemical structure of head group to sulfatides with an inositol ring and a phosphate, but a very different configuration. Our results indicate that the head group configuration, but not the anionic charge density on the surface of the vesicles, is a determinant factor in mediating the sulfatide-induced decreases in the levels of Aβ peptides present in the media.
Furthermore, through examination of the levels and activities of β-secretase, we have found that the decrease in media Aβ content is due to sulfatide-facilitated clearance through an endocytotic pathway and not lowered Aβ production due to sulfatide supplementation. To further validate this conclusion, we have also performed identical experiments with neuroblastoma (NB) cells which do not produce significant quantities of Aβ peptides. In these experiments, the media of NB cells was replaced with the conditioned media of H4-APPwt cells (i.e., containing high levels of secreted Aβ peptides) prior to addition of sulfatide-containing vesicles to the media. This study revealed that the observed decrease in the concentration of Aβ peptides in the cell culture media was not the result of a sulfatide-dependent reduction in Aβ production, but primarily from increased uptake through an endocytotic pathway.
In addition, determination of the levels of Aβ40, Aβ42, and sulfatides in CSF from AD patients and controls has revealed a highly significant correlation of sulfatide content with Aβ42 content, but not with Aβ40 content (see below). The result strongly suggests a significant interrelationship between Aβ42 and sulfatides. Finally, it should be emphasized that treatment of oligodendrocytes with Aβ peptides did not show any effects on sulfatide mass levels [42].
Interactions Between Sulfatides, Aβ Peptides, and ApoE Isoforms
The collective interactions between sulfatides, apoE isoforms, and Aβ peptides have not been well investigated. However, the results of our recent in vitro and in vivo experiments provide strong evidence that the metabolisms of these components are interwoven and tightly regulated and suggest that any abnormal alteration in one component may result in a pathophysiological condition.
For example, since both Aβ peptides and sulfatides are associated with apoE-containing lipoproteins (see above), the correlation between sulfatides and Aβ42 levels found in CSF would provide in vivo evidence of the interactions between apoE, Aβ, and sulfatides since the apoE levels in CSF from different individuals does not vary significantly [71]. We have determined the levels of sulfatides and Aβ peptides in the CSF of near 100 clinically identified very mild AD patients in conjunction with over 70 age-matched cognitively normal controls (unpublished data). The results demonstrated that the CSF sulfatide levels from AD patients were markedly lower than those obtained from the control samples (p<0.001) as demonstrated previously [41]. We also found that Aβ42 (but not Aβ40) was statistically decreased in very mild AD subjects relative to control subjects (p<0.04). Statistical analyses have demonstrated a significant correlation between the levels of sulfatides and Aβ42 with a linear correlation coefficient of γ=0.593 (p<0.01; unpublished results), indicating a significant interrelationship between Aβ and sulfatides mediated by apoE.
Moreover, we have employed a chemically well-defined vesicular model system containing sulfatides as guest in a host phospholipid matrix to examine the specific interactions between apoE isoforms, sulfatides, and Aβ peptides in a membrane interfacial microenvironment. We have found that sulfatides dramatically enhance the binding of Aβ peptides to apoE-associated vesicles [69]. These results indicate that the difference in the isoform-specific effects of apoE on the clearance of Aβ peptides depend on (1) the differential binding of Aβ peptides to apoE isoform-associated lipoproteins in the absence of sulfatides and (2) the increased affinity of the Aβ/apoE complexes in the presence of sulfatides. Collectively, these results suggest that one role of sulfatides played in the CNS might be to promote the clearance of Aβ peptides through enhancing Aβ binding to membrane surfaces. These results indicate the presence of a selective interaction between sulfatides and Aβ peptides, particularly Aβ42, while the metabolism/clearance of both Aβ peptides and sulfatides is mediated by apoE lipoproteins as carriers and facilitated by interactions of apoE with its receptors. This conclusion has been validated using cell culture model systems in which the role of apoE played in the uptake/clearance of Aβ peptides is manifested through providing a vehicle for transporting sulfatides, thereby Aβ peptides as well as through its binding with the receptors (e.g., LDL receptor and LRP) [69].
Finally, lipidomics analyses have demonstrated that the brain sulfatide content in mouse AD models is specifically depleted in an apoE-dependent manner [54]. Specifically, we have recently tested the interactions between apoE, Aβ, and sulfatides with two well-characterized mouse AD models transgenically expressing the mutants of human APP, i.e., APPsw (also called Tg2576) and APPV717F (also called PDAPP) [63, 72–76]. It is well known that substantial increases in the production of Aβ peptides are manifested in these animal models [73, 75]. Mass spectrometric analyses using a shotgun sphingolipidomics approach [77] have demonstrated that the levels of all sulfatide molecular species (containing various fatty acyl chains with or without a hydroxy moiety) in the cortex of APPsw, Apoe+/+ mice are decreased in an age-dependent manner in comparison to their WT littermate counterparts (Fig. 3a) [54]. Similar findings have also been demonstrated in other cerebral brain regions including hippocampus. In contrast to the depletion of sulfatide, shotgun lipidomics analyses have not shown significant change in the content of other sphingolipid classes including sphingomyelin, GalCer, and ceramide examined in the brain regions of APPsw, Apoe+/+ mice relative to those of their WT littermates. These results indicate that the reduction in sulfatide content is not due to the loss of neuronal myelin sheaths.
Fig. 3.

Temporal changes in the content of total sulfatides in lipid extracts of brain cortices from APP transgenic mice and their wild-type littermates. Lipid extracts of cortices from the wild-type littermate controls (solid squares) and APPsw (a) or APPV717F (b) Tg, Apoe+/+ mice (solid circles) at different ages were prepared using a modified Bligh and Dyer method [53]. The content of each individual sulfatide molecular species after identification was determined in comparison to the selected internal standard after 13C de-isotoping. The data points represent mean ± SD from separate preparations of at least four different animals. *P<0.01 and **P<0.001 compared to the wild-type littermates at the same age. (Modified from [54] with permission)
It is well known that cerebellar Aβ pathology is relatively less severe compared to other brain regions in AD patients in general [78] and in this animal model in particular [74, 75]. To verify that the demonstrated sulfatide depletion in APPsw, Apoe+/+ mouse cortex is related to Aβ pathology (i.e., Aβ production and/or Aβ plaques), but not to mutant APP expression, we have also determined cerebellar sulfatide content in APPsw, Apoe+/+ mice. We have found that the content of sulfatides in the cerebellum of the APPsw, Apoe+/+ mouse was not significantly different from WT littermate controls at <12 months of age, although a relatively lower sulfatide content was present in this region of older APPsw, Apoe+/+ mice (>12 months) in comparison to their WT littermates [54].
In the case of APPV717F, Apoe+/+ which develops AD-type plaques and an Aβ pathology approximately 2 months prior to that observed in the APPsw, Apoe+/+ mouse model [72–75], we have demonstrated three intriguing findings that are related to the depleted sulfatide levels in comparison to those present in APPsw, Apoe+/+ mice (Fig. 3). First, sulfatide deficiency is detected at 6 months of age in APPV717F, Apoe+/+ mouse brain in comparison to 9 months in APPsw, Apoe+/+ mice. Second, the deficiency in sulfatide content in this animal model relative to age-matched WT littermates is much more severe relative to that present in APPsw, Apoe+/+ mice. Third, differences in the deficiencies of sulfatide content increased substantially with age. These observations indicate that sulfatide depletion in APPV717F, Apoe+/+ mouse brain is relatively more severe and occurs at an earlier age in comparison to the APPsw, Apoe+/+ mouse brain.
In contrast to the relatively small changes in cerebellar sulfatide content in APPsw, Apoe+/+ mice over time, a significant decrease in the sulfatide content of cerebellum becomes apparent at 12 months and is significant at 18 months of age in the brain region of APPV717F, Apoe+/+ mice relative to their WT littermate controls (n=4, p<0.01) [54]. These results further support the notion that sulfatide depletion in APPV717F, Apoe+/+ mouse brain is relatively more severe and occurs at an earlier age in comparison to that in APPsw, Apoe+/+ mice. However, similar to APPsw, Apoe+/+ mice, phospholipids and other classes of sphingolipids including sphingomyelin, GalCer, and ceramide did not show significant changes in either the cortex or cerebellum of APPV717F, Apoe+/+ mice at the ages examined relative to age-matched controls.
To identify the cause(s) underlying the demonstrated sulfatide depletion in brain tissues of the investigated AD animal models, we have performed shotgun sphingolipidomics analyses of lipid extracts from the brain tissues of APP mutant transgenic mice on an apoE-deficient background (i.e., APPsw, Apoe−/− and APPV717F, Apoe−/−) mice and have compared the results to those from the corresponding Apoe+/+ mice. As anticipated, shotgun sphingolipidomics analysis has extended our previous observation [52] that the sulfatide content in the brain tissues of apoE-null mice dramatically accumulated in comparison to WT mice due to a deficiency in sulfatide transport and/or degradative pathways. Intriguingly, the sulfatide levels in the cortices of both APPsw, Apoe−/− and APPV717F, Apoe−/− mice are essentially identical to that of Apoe−/− mice [54]. These results indicate that apoE plays a key role in the sulfatide depletion which occurs in both APPsw, Apoe+/+ and APPV717F, Apoe+/+ mice.
To interpret these observations, we have hypothesized that the excessive production of Aβ peptides in APP transgenic mice likely requires a compensatory process resulting in an increased rate of clearance of Aβ peptides. It is well known that apoE-associated lipoproteins play an important role in the process of Aβ clearance in the brain either through transport of the Aβ-containing apoE-associated lipoproteins to the peripheral circulatory system or by a local endocytotic process through the LDL superfamily of receptors (see above) [79, 80]. Following this line of reasoning, the elevated demand for the clearance of excessive Aβ peptides through either transport to the circulatory system or through localized degradation in endocytotic vesicular organelles in the brain would lead to an upregulation of apoE expression. Indeed, a higher level of apoE in APP transgenic mice relative to the respective controls has been previously reported [81] and has been confirmed in our studies (Fig. 4). The increased apoE level in APP transgenic mice strongly suggests a state of increased apoE production and metabolism, thereby leading to the loss of sulfatide. Since sulfatide levels in the CNS are mediated by apoE in an apoE-isoform-dependent manner, we have believed that changes in the metabolism of the apoE-associated lipoproteins, apoE expression levels, and/or apoE isoforms likely contribute to alterations in the sulfatide content in the CNS (Fig. 2) [43]. The increased chronic endocytotic uptake and endosomal/lysosomal degradation of sulfatides associated with Aβ-containing, apoE-associated lipoprotein particles inevitably result in a deficient content of sulfatides in the APP transgenic mice as shown. However, it should be specifically pointed out that the increased apoE levels might be due to a reduced uptake of apoE via endocytosis. If this was the case, the sulfatide content should be higher as similarly demonstrated in Apoe−/− mouse brain. This is in contrast to our experimental observation that sulfatide is reduced accompanying an increased level of apoE.
Fig. 4.
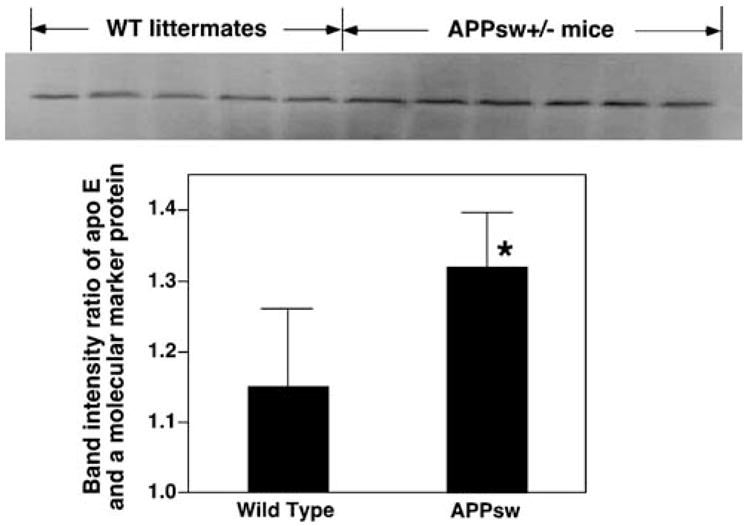
The increased mass levels of apoE in APPsw mouse cortex. The mass levels of cortical apoE in APPsw mice at 12 months of age were determined by Western blot analysis as described previously [54]. *P<0.01 compared to the wild-type littermates at the same age
Sequelae of the Abnormal Interactions of Sulfatides, ApoE Isoforms, and Aβ Peptides
Multiple lines of evidence as discussed above, in combination with the profound depletion of sulfatides present even at the MCI stage of AD [41, 42, 50], are consistent with the central roles of sulfatides in facilitating the apoE-mediated clearance of Aβ peptides through the interactions of these components under normal physiological conditions. Obviously, the consequence of this process under pathophysiological conditions is the resultant myelin dysfunction following sulfatide depletion of the myelin sheath. Specifically, when the clearance pathway for Aβ peptides becomes overwhelmed (e.g., in the cases of APP transgenic mice), sulfatides are depleted by two possible mechanisms: (1) through accelerated apoE-mediated trafficking and metabolism, thereby indicative of increased processing and turnover of sulfatides and (2) through consumption of sulfatides in the process of Aβ clearance.
Such sequelae were also manifested in other cases. For example, when cholesterol biosynthesis is inhibited through microinjection of high doses of cholesterol-lowering drugs (i.e., statins) into mouse brain, we have found the depletion of sulfatides in the cortex from the drug-treated mice (unpublished observation). Specifically, we have found a significant increase in apoE levels (p<0.01, n=4), a substantial reduction in cholesterol content, and a significant decrease in sulfatide content (12.8±0.5 vs. 10.3±0.6, p<0.001) in the statin-treated mouse cortex in comparison to controls. In the CNS, cholesterol is primarily synthesized in astrocytes and transported to neurons through apolipoproteins and other transport proteins in the CSF. Acute inhibition of cholesterol synthesis with a high dose of statin-type drug would perturb cholesterol homeostasis (i.e., distribution and transport) and subsequently alter apoE levels. The experimental results indicate that the increased neuronal demand for cholesterol accompanying the reduction de novo cholesterol synthesis in cortex after statin treatment leads to (1) decreased apolipoprotein cholesterol content; (2) putative increased trafficking of apoE-associated lipoprotein particles and subsequent increased metabolic flux of sulfatides; and (3) decreased sulfatide content. These results strongly support our working model of apoE-mediated sulfatide metabolism (Fig. 2). It should be emphasized that apoE-mediated cholesterol metabolism may independently play roles in AD pathogenesis as well as other pathological conditions [82, 83], which will not be further discussed herein.
Accordingly, these as well as other observations of the interactions between apoE, Aβ peptides, and sulfatides have led us to having a model which illustrates the position of sulfatide depletion in AD pathogenesis (Fig. 5). This model indicates that multiple factors, each of which causes increased apoE metabolism and/or expression, result in an accelerated sulfatide metabolism and a deficient sulfatide content in the CNS (Fig. 5). Sulfatide deficiency itself, in turn, could lead to the axonal dysfunction and cause dementia although other pathways causing these sequelae definitely exist. This model is consistent with the etiological recognition that AD is multifactorial with apoE4 as a major genetic risk factor.
Fig. 5.

Proposed mechanisms and roles of sulfatide depletion in AD pathogenesis
As demonstrated with transgenic animal models, sulfatide content in brain tissues of apoE4 carriers is significantly lower than that in apoE3 ones [52]. This reduced pool of sulfatides is likely at the edge of the amount of sulfatides which are required to maintain the normal neuronal functions, thereby being sensitive to be depleted with upregulation of apoE metabolism or expression. Therefore, our model provides insights into the biochemical mechanism(s) underlying apoE4 as a major genetic risk of AD. In addition, LRP and many other proteins which are involved in the endocytotic process are also the risk factors for AD to a certain degree. According to the proposed model (Fig. 5), this can be interpreted as their association with apoE metabolism. Their upregulation and/or increased turnover may lead to the depletion of sulfatides, but they are only secondary to apoE metabolism.
Finally, the current model may provide a foundation for the development of effective therapeutic interventions for AD. Following this line of reasoning, any approaches that lead to downregulation of apoE metabolism but balance the clearance of Aβ peptides might be useful for increases in life span and delaying the AD onset. To this end, dietary restriction or modest exercise, both of which can modulate apoE levels to a certain degree, may serve on this purpose [84]. Development of sulfatide-based therapeutic treatment has its potential. Finally, it is also possible to develop drugs to enhance the biosynthesis of sulfatides for the treatment of AD. Collectively, our studies not only have provided the insights into the biochemical mechanisms responsible for sulfatide depletion as well as revealed the relationship of sulfatide metabolism with apoE-mediated Aβ clearance but also might lead to a new direction for therapeutic treatment of AD.
Acknowledgments
This work was supported by the National Institutes of Health/National Institute on Aging Grants R01 AG23168 and R01 AG31675. XH serves as a consultant for the LipoSpectrum LLC.
References
- 1.Yankner BA. New clues to Alzheimer’s disease: unraveling the roles of amyloid and tau. Nat Med. 1996;2:850–852. doi: 10.1038/nm0896-850. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 3.Holtzman DM. Abeta conformational change is central to Alzheimer’s disease. Neurobiol Aging. 2002;23:1085–1088. doi: 10.1016/s0197-4580(02)00040-4. [DOI] [PubMed] [Google Scholar]
- 4.Morishima-Kawashima M, Ihara Y. Alzheimer’s disease: beta-amyloid protein and tau. J Neurosci Res. 2002;70:392–401. doi: 10.1002/jnr.10355. [DOI] [PubMed] [Google Scholar]
- 5.McGeer EG, McGeer PL. Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. The ups and downs of Abeta. Nat Med. 2006;12:758–759. doi: 10.1038/nm0706-758. discussion 759. [DOI] [PubMed] [Google Scholar]
- 7.Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol. 1985;17:278–282. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 8.Fan J, Donkin J, Wellington C. Greasing the wheels of Abeta clearance in Alzheimer’s disease: the role of lipids and apolipoprotein E. Biofactors. 2009;35:239–248. doi: 10.1002/biof.37. [DOI] [PubMed] [Google Scholar]
- 9.Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]
- 12.Nixon RA. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 13.Knauer MF, Soreghan B, Burdick D, Kosmoski J, Glabe CG. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc Natl Acad Sci U S A. 1992;89:7437–7441. doi: 10.1073/pnas.89.16.7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burdick D, Kosmoski J, Knauer MF, Glabe CG. Preferential adsorption, internalization and resistance to degradation of the major isoform of the Alzheimer’s amyloid peptide, A beta 1-42, in differentiated PC12 cells. Brain Res. 1997;746:275–284. doi: 10.1016/s0006-8993(96)01262-0. [DOI] [PubMed] [Google Scholar]
- 15.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 16.DeMattos RB, Brendza RP, Heuser JE, Kierson M, Cirrito JR, Fryer J, Sullivan PM, Fagan AM, Han X, Holtzman DM. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem Int. 2001;39:415–425. doi: 10.1016/s0197-0186(01)00049-3. [DOI] [PubMed] [Google Scholar]
- 17.Plump AS, Breslow JL. Apolipoprotein E and the apolipoprotein E-deficient mouse. Annu Rev Nutr. 1995;15:495–518. doi: 10.1146/annurev.nu.15.070195.002431. [DOI] [PubMed] [Google Scholar]
- 18.Shobab LA, Hsiung G-YR, Feldman HH. Cholesterol in Alzheimer’s disease. Lancet Neurology. 2005;4:841–852. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- 19.Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem. 2008;283:11302–11311. doi: 10.1074/jbc.M801520200. [DOI] [PubMed] [Google Scholar]
- 20.Curtiss LK, Edgington TS. Identification of a lymphocyte surface receptor for low density lipoprotein inhibitor, an immunoregulatory species of normal human serum low density lipoprotein. J Clin Invest. 1978;61:1298–1308. doi: 10.1172/JCI109047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 22.Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci U S A. 1985;82:203–207. doi: 10.1073/pnas.82.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 24.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77. doi: 10.1146/annurev.ne.19.030196.000413. [DOI] [PubMed] [Google Scholar]
- 25.Vos JP, Lopes-Cardozo M, Gadella BM. Metabolic and functional aspects of sulfogalactolipids. Biochim Biophys Acta. 1994;1211:125–149. doi: 10.1016/0005-2760(94)90262-3. [DOI] [PubMed] [Google Scholar]
- 26.Ishizuka I. Chemistry and functional distribution of sulfoglycolipids. Prog Lipid Res. 1997;36:245–319. doi: 10.1016/s0163-7827(97)00011-8. [DOI] [PubMed] [Google Scholar]
- 27.Ikami T, Ishida H, Kiso M. Synthesis and biological activity of glycolipids, with a focus on gangliosides and sulfatide analogs. Methods Enzymol. 2000;311:547–568. doi: 10.1016/s0076-6879(00)11105-x. [DOI] [PubMed] [Google Scholar]
- 28.Marcus J, Popko B. Galactolipids are molecular determinants of myelin development and axo-glial organization. Biochim Biophys Acta. 2002;1573:406–413. doi: 10.1016/s0304-4165(02)00410-5. [DOI] [PubMed] [Google Scholar]
- 29.Eckhardt M, Hedayati KK, Pitsch J, Lullmann-Rauch R, Beck H, Fewou SN, Gieselmann V. Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy. J Neurosci. 2007;27:9009–9021. doi: 10.1523/JNEUROSCI.2329-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isaac G, Pernber Z, Gieselmann V, Hansson E, Bergquist J, Mansson JE. Sulfatide with short fatty acid dominates in astrocytes and neurons. Febs J. 2006;273:1782–1790. doi: 10.1111/j.1742-4658.2006.05195.x. [DOI] [PubMed] [Google Scholar]
- 31.von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy: lysosomal disorders. In: Sachdev HS, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited diseases. McGraw-Hill; New York: 2001. pp. 3695–3724. [Google Scholar]
- 32.Molander-Melin M, Pernber Z, Franken S, Gieselmann V, Mansson JE, Fredman P. Accumulation of sulfatide in neuronal and glial cells of arylsulfatase A deficient mice. J Neurocytol. 2004;33:417–427. doi: 10.1023/B:NEUR.0000046572.53905.2c. [DOI] [PubMed] [Google Scholar]
- 33.Bosio A, Binczek E, Stoffel W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc Natl Acad Sci U S A. 1996;93:13280–13285. doi: 10.1073/pnas.93.23.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- 35.Coetzee T, Dupree JL, Popko B. Demyelination and altered expression of myelin-associated glycoprotein isoforms in the central nervous system of galactolipid-deficient mice. J Neurosci Res. 1998;54:613–622. doi: 10.1002/(SICI)1097-4547(19981201)54:5<613::AID-JNR6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 36.Bosio A, Bussow H, Adam J, Stoffel W. Galactosphingolipids and axono-glial interaction in myelin of the central nervous system. Cell Tissue Res. 1998;292:199–210. doi: 10.1007/s004410051051. [DOI] [PubMed] [Google Scholar]
- 37.Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372–381. doi: 10.1002/glia.20292. [DOI] [PubMed] [Google Scholar]
- 38.Hu R, Li G, Kamijo Y, Aoyama T, Nakajima T, Inoue T, Node K, Kannagi R, Kyogashima M, Hara A. Serum sulfatides as a novel biomarker for cardiovascular disease in patients with end-stage renal failure. Glycoconj J. 2007;24:565–571. doi: 10.1007/s10719-007-9053-0. [DOI] [PubMed] [Google Scholar]
- 39.Morichika H, Hamanaka Y, Tai T, Ishizuka I. Sulfatides as a predictive factor of lymph node metastasis in patients with colorectal adenocarcinoma. Cancer. 1996;78:43–47. doi: 10.1002/(SICI)1097-0142(19960701)78:1<43::AID-CNCR8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 40.Svennerholm L, Gottfries CG. Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type I) and demyelination in late-onset form (type II) J Neurochem. 1994;62:1039–1047. doi: 10.1046/j.1471-4159.1994.62031039.x. [DOI] [PubMed] [Google Scholar]
- 41.Han X, Holtzman DM, McKeel DW, Jr, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- 42.Cheng H, Xu J, McKeel DW, Jr, Han X. Specificity and potential mechanism of sulfatide deficiency in Alzheimer’s disease: an electrospray ionization mass spectrometric study. Cell Mol Biol. 2003;49:809–818. [PubMed] [Google Scholar]
- 43.Han X. Potential mechanisms contributing to sulfatide depletion at the earliest clinically recognizable stages of Alzheimer’s disease: a tale of shotgun lipidomics. J Neurochem. 2007;103 (1):171–179. doi: 10.1111/j.1471-4159.2007.04708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bognar SK, Furac I, Kubat M, Cosovic C, Demarin V. Croatian population data for arylsulfatase a pseudodeficiency-associated mutations in healthy subjects, and in patients with Alzheimer-type dementia and Down syndrome. Arch Med Res. 2002;33:473–477. doi: 10.1016/s0188-4409(02)00392-2. [DOI] [PubMed] [Google Scholar]
- 45.Han X, Gross RW. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: a bridge to lipidomics. J Lipid Res. 2003;44:1071–1079. doi: 10.1194/jlr.R300004-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Han X, Gross RW. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of the cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 47.Han X, Gross RW. Shotgun lipidomics: multi-dimensional mass spectrometric analysis of cellular lipidomes. Expert Rev Proteomics. 2005;2:253–264. doi: 10.1586/14789450.2.2.253. [DOI] [PubMed] [Google Scholar]
- 48.Han X. Neurolipidomics: challenges and developments. Front Biosci. 2007;12:2601–2615. doi: 10.2741/2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang K, Cheng H, Gross RW, Han X. Automated lipid identification and quantification by multi-dimensional mass spectrometry-based shotgun lipidomics. Anal Chem. 2009;81:4356–4368. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han X, Fagan AM, Cheng H, Morris JC, Xiong C, Holtzman DM. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann Neurol. 2003;54:115–119. doi: 10.1002/ana.10618. [DOI] [PubMed] [Google Scholar]
- 51.Irizarry MC. A turn of the sulfatide in Alzheimer’s disease. Ann Neurol. 2003;54:7–8. doi: 10.1002/ana.10642. [DOI] [PubMed] [Google Scholar]
- 52.Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system: modulation of sulfatide content. J Biol Chem. 2003;278:8043–8051. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- 53.Cheng H, Jiang X, Han X. Alterations in lipid homeostasis of mouse dorsal root ganglia induced by apolipoprotein E deficiency: a shotgun lipidomics study. J Neurochem. 2007;101:57–76. doi: 10.1111/j.1471-4159.2006.04342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng H, Zhou Y, Holtzman DM, Han X. Apolipoprotein E mediates sulfatide depletion in amyloid precursor protein transgenic animal models of Alzheimer’s disease. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2008.1007.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Uden E, Kang DE, Koo EH, Masliah E. LDL receptor-related protein (LRP) in Alzheimer’s disease: towards a unified theory of pathogenesis. Microsc Res Tech. 2000;50:268–272. doi: 10.1002/1097-0029(20000815)50:4<268::AID-JEMT3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 56.Arelin K, Kinoshita A, Whelan CM, Irizarry MC, Rebeck GW, Strickland DK, Hyman BT. LRP and senile plaques in Alzheimer’s disease: colocalization with apolipoprotein E and with activated astrocytes. Brain Res Mol Brain Res. 2002;104:38–46. doi: 10.1016/s0169-328x(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 57.Zeng Y, Cheng H, Jiang X, Han X. Endosomes and lysosomes play distinct roles in sulfatide-induced neuroblastoma apoptosis: potential mechanisms contributing to abnormal sulfatide metabolism in related neuronal diseases. Biochem J. 2008;410:81–92. doi: 10.1042/BJ20070976. [DOI] [PubMed] [Google Scholar]
- 58.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 60.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 61.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 63.Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT. Apolipo-protein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol. 2000;47:739–747. [PubMed] [Google Scholar]
- 64.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 65.Fraser PE, Nguyen JT, Chin DT, Kirschner DA. Effects of sulfate ions on Alzheimer beta/A4 peptide assemblies: implications for amyloid fibril-proteoglycan interactions. J Neurochem. 1992;59:1531–1540. doi: 10.1111/j.1471-4159.1992.tb08470.x. [DOI] [PubMed] [Google Scholar]
- 66.Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem. 1999;72:1681–1687. doi: 10.1046/j.1471-4159.1999.721681.x. [DOI] [PubMed] [Google Scholar]
- 67.Snow AD, Mar H, Nochlin D, Sekiguchi RT, Kimata K, Koike Y, Wight TN. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am J Pathol. 1990;137:1253–1270. [PMC free article] [PubMed] [Google Scholar]
- 68.van Horssen J, Wilhelmus MM, Heljasvaara R, Pihlajaniemi T, Wesseling P, de Waal RM, Verbeek MM. Collagen XVIII: a novel heparan sulfate proteoglycan associated with vascular amyloid depositions and senile plaques in Alzheimer’s disease brains. Brain Pathol. 2002;12:456–462. doi: 10.1111/j.1750-3639.2002.tb00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeng Y, Han X. Sulfatides facilitate apolipoprotein E-mediated amyloid-b peptide clearance through an endocytotic pathway. J Neurochem. 2008;106:1275–1286. doi: 10.1111/j.1471-4159.2008.05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murphy MP, Das P, Nyborg AC, Rochette MJ, Dodson MW, Loosbrock NM, Souder TM, McLendon C, Merit SL, Piper SC, Jansen KR, Golde TE. Overexpression of nicastrin increases Abeta production. FASEB J. 2003;17:1138–1140. doi: 10.1096/fj.02-1050fje. [DOI] [PubMed] [Google Scholar]
- 71.Fagan AM, Younkin LH, Morris JC, Fryer JD, Cole TG, Younkin SG, Holtzman DM. Differences in the Abeta40/Abeta42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Ann Neurol. 2000;48:201–210. [PubMed] [Google Scholar]
- 72.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano P, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 73.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 75.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 76.Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement. 2009;5:340–347. doi: 10.1016/j.jalz.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang X, Cheng H, Yang K, Gross RW, Han X. Alkaline methanolysis of lipid extracts extends shotgun lipidomics analyses to the low abundance regime of cellular sphingolipids. Anal Biochem. 2007;371:135–145. doi: 10.1016/j.ab.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gearing M, Schneider JA, Robbins RS, Hollister RD, Mori H, Games D, Hyman BT, Mirra SS. Regional variation in the distribution of apolipoprotein E and A beta in Alzheimer’s disease. J Neuropathol Exp Neurol. 1995;54:833–841. doi: 10.1097/00005072-199511000-00010. [DOI] [PubMed] [Google Scholar]
- 79.Bales KR, Dodart JC, DeMattos RB, Holtzman DM, Paul SM. Apolipoprotein E, amyloid, and Alzheimer disease. Mol Interv. 2002;2:363–375. doi: 10.1124/mi.2.6.363. [DOI] [PubMed] [Google Scholar]
- 80.Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuo YM, Crawford F, Mullan M, Kokjohn TA, Emmerling MR, Weller RO, Roher AE. Elevated A beta and apolipoprotein E in A betaPP transgenic mice and its relationship to amyloid accumulation in Alzheimer’s disease. Mol Med. 2000;6:430–439. [PMC free article] [PubMed] [Google Scholar]
- 82.Michikawa M. Cholesterol paradox: is high total or low HDL cholesterol level a risk for Alzheimer’s disease? J Neurosci Res. 2003;72:141–146. doi: 10.1002/jnr.10585. [DOI] [PubMed] [Google Scholar]
- 83.Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, Nolan D, Gandy SE, Martins RN. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–736. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- 84.Nichol K, Deeny SP, Seif J, Camaclang K, Cotman CW. Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 2009;5:287–294. doi: 10.1016/j.jalz.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]