

Abstract
Using implicit solvent molecular dynamics and replica exchange simulations, we study the impact of ibuprofen on the growth of wild-type Aβ fibrils. We show that binding of ibuprofen to Aβ destabilizes the interactions between incoming peptides and the fibril. As a result, ibuprofen interference modifies the free energy landscape of fibril growth and reduces the free energy gain of Aβ peptide binding to the fibril by ≃2.5 RT at 360 K. Furthermore, ibuprofen interactions shift the thermodynamic equilibrium from fibril-like locked states to disordered docked states. Ibuprofen's anti-aggregation effect is explained by its competition with incoming Aβ peptides for the same binding site located on the fibril edge. Although ibuprofen impedes fibril growth, it does not significantly change the mechanism of fibril elongation or the structure of Aβ peptides bound to the fibril.
Introduction
A class of diseases, including Alzheimer's, Parkinson's, type II diabetes, and Creutzfeldt-Jakob disease, are linked to aberrant aggregation of polypeptide chains (1). Aggregation pathway proceeds through cascading structural transitions initiated by oligomerization of monomeric chains, which eventually result in the appearance of amyloid fibrils (2). Recent experimental findings suggested that, rather than fibrils, oligomers that are as small as dimers are the primary cytotoxic species (3,4). Irrespective of their cytotoxicity, fibrils are the reservoirs of monomers and, consequently, participate in the equilibrium recycling of polypeptides through different aggregated species (5–7). From the structural perspective, amyloid fibrils display several unique properties:
-
1.
Small sequence homology is observed among amyloidogenic polypeptides;
-
2.
Fibril internal architecture is based on the β-sheet structure (8–12); and
-
3.
Amyloid fibrils are highly resistant to dissociation (13).
Aβ peptides are 39–42 residue amyloidogenic fragments of membrane precursor protein, which are produced in the course of cellular proteolysis (14) (Fig. 1 a). Experimental observations suggest that amyloidogenesis of Aβ peptides is a seminal event in Alzheimer's disease (AD) (15). Consequently, prevention of Aβ aggregation is a viable therapeutic strategy, which could involve the use of small molecular ligands to interfere with amyloid assembly. One such candidate ligand is the nonsteroidal anti-inflammatory drug ibuprofen (16) (Fig. 1 b). Mouse models have shown that it can reduce the extent of Aβ deposition and alleviate memory deficits (17,18). Ibuprofen also decreases the load of Aβ oligomers in mice brains (18). Prophylactic long-term intake of ibuprofen appears to reduce the risk of AD in humans (19), but its clinical use is hampered by side effects.
Figure 1.
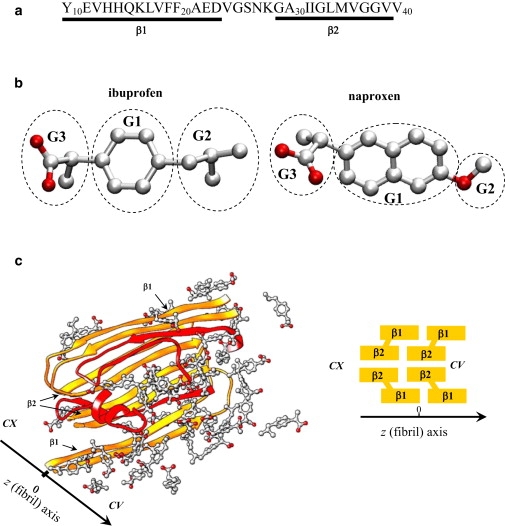
(a) The sequence of Aβ10–40 peptide and the allocation of the β1 and β2 fibril β-strands. (b) The ibuprofen molecule has three structural moieties—the hydrophobic phenyl G1, and isobutyl G2, and hydrophilic carboxylate G3 groups. Naproxen has two polar moieties, the methoxy and carboxylate groups (G2 and G3), linked to the central hydrophobic naphthalene ring (G1). Carbon and oxygen atoms are shown in gray and red. (c) Incoming Aβ10–40 peptides interacting with amyloid fibril in ibuprofen solution. Four Aβ peptides in orange form the fibril fragment. Two incoming peptides in red are bound to the fibril edge. Ibuprofen molecules are in light gray/red. Fibril protofilament consists of four in-registry parallel β-sheets formed by the β1 and β2 strands and has two distinct edges—concave (CV) and convex (CX). Due to indentation of β2, the CV edge has a groove.
Although ibuprofen may play different therapeutic roles in AD, in vitro experiments support direct anti-aggregation effect produced by this ligand. It has been shown that coincubation of ibuprofen with Aβ reduces the accumulation of fibrils (20,21). Ibuprofen also dissociates, at least partially, preformed Aβ fibrils (21). However, little is known about Aβ-ibuprofen interactions on a molecular level. For example:
-
1.
Does ibuprofen decrease the thermodynamic stability of fibrils?
-
2.
Is the anti-aggregation effect due to the competition of ibuprofen and Aβ peptides for the same binding sites in Aβ fibril?
-
3.
Does ibuprofen binding change the fibril growth mechanism and/or the Aβ peptide structure?
These questions can be investigated by molecular dynamics (MD) simulations (22), which have been used to explore the pathways of amyloid assembly (23–26), the conformational ensembles of amyloidogenic peptides (27–29), and the energetics of fibril structures (30,31). More recently, MD simulations probed binding of small ligands to amyloidogenic peptides (32–35).
In this article, to address the questions posed above, we use the atomistic implicit solvent model and replica exchange molecular dynamics (REMD) (36). By using this approach, we have already shown that, consistent with the experiments (37,38), equilibrium fibril growth involves two thermodynamically distinct transitions—docking and locking (26). Docking occurs upon binding disordered Aβ monomers to the fibril without their integration into the fibril structure. During locking, incoming peptides adopt a fibril-like state through activated structural transition. Our preliminary studies have also examined the binding of ibuprofen to Aβ monomers and, separately, to Aβ fibrils (34). Here, through exhaustive REMD simulations, we directly probe the anti-aggregation effect of ibuprofen. Specifically, we compute the free energy landscapes of Aβ fibril growth in the presence of ibuprofen ligands interacting with incoming Aβ peptides and amyloid fibril. The impact of ibuprofen binding on Aβ fibril elongation is revealed by a comparison with a water environment free of ligands (26). In our simulations, we used the twofold symmetry Aβ10–40 fibril structure derived from solid-state NMR experiments (10) (Fig. 1 c). Based on our results, we suggest a rationale for the ibuprofen anti-aggregation effect, and propose a few strategies for its enhancement.
Methods
Molecular dynamics simulations
Simulations of Aβ peptides and ibuprofen (Fig. 1) were performed using the CHARMM MD program (39) and united-atom force-field CHARMM19 coupled with the SASA implicit solvent model (40). Their description, applicability, and testing can be found in our previous studies (41,42). In particular, we have shown that the CHARMM19+SASA force field accurately reproduces the experimental distribution of chemical shifts for Cα and Cβ atoms in Aβ monomers (42,43). Parameterization of ibuprofen (Fig. 1 b) in the CHARMM19 force field has been reported by us earlier (34). According to our tests of the ibuprofen force-field parameters, the in silico distribution of internal dihedral angles in ibuprofen is consistent with the density functional theory calculations and vibrational spectroscopy (34). Arguments for selecting the CHARMM19 force field and the SASA model are presented in the Supporting Material.
The simulation system consists of six Aβ10–40 peptides interacting with Nibu = 60 ibuprofen molecules (Fig. 1). Four peptides are constrained to form a fibril fragment, whereas two unconstrained peptides are free to associate or dissociate from the fibril. The concentration ratio of ibuprofen to Aβ peptides is 10:1, which is only slightly higher than that used in most experiments (20,21). Further description of the simulation system is provided in Supporting Material. In addition, three other Aβ peptide systems were considered:
-
1.
Hexamer consisting of the four-peptide fibril fragment and two incoming peptides in water;
-
2.
Four-peptide fibril fragment without incoming peptides in ibuprofen solution;
-
3.
Monomer in ibuprofen solution.
These systems were studied by us earlier (26,34). Throughout the article, the peptides in orange in Fig. 1 c are referred to as “fibril”, and the red peptides are termed “incoming”.
Replica exchange simulations
Conformational sampling was performed using REMD (36). In total, 24 replicas were distributed linearly in the temperature range from 330 to 560 K with the increment of 10 K. The exchanges were attempted every 20 ps between all neighboring replicas with the average acceptance rate of 24%. Fourteen REMD trajectories were produced, resulting in a cumulative simulation time of 67 μs. Between replica exchanges, the system evolved using NVT underdamped Langevin dynamics with the damping coefficient γ = 0.15 ps−1 and the integration step of 2 fs. Because the initial parts of REMD trajectories are not equilibrated and must be excluded from thermodynamic analysis, the cumulative equilibrium simulation time is reduced to τsim ≈ 56 μs. The REMD trajectories were started with random distributions of incoming peptides and ligands. The convergence of REMD simulations and error analysis are presented in the Supporting Material.
Computation of structural probes
The interactions formed by Aβ peptides and ibuprofen were probed by computing the number of side-chain contacts and hydrogen bonds (HBs). A side-chain contact occurs if the distance between the centers-of-mass of side chains is <6.5 Å. (This cutoff approximately corresponds to the onset of hydration of side chains as the separation distance between them increases.) Computation of contacts formed by ibuprofen is described in the Supporting Material. The backbone HBs between peptide NH and CO groups were assigned according to Kabsch and Sander (44). The same definition was applied to detect HBs between ibuprofen and peptide backbone NH groups. Following our previous studies, we distinguished three classes of HBs. The first includes any peptide-fibril HB, whereas the second and third classes are restricted to the HBs involved in the formation of parallel (antiparallel) β-sheets by incoming peptides on the fibril edge. These HBs are termed parallel (pHB) and anti-parallel (aHB), respectively. Their specific definitions are given in the Supporting Material. An incoming peptide is bound if it forms at least one side-chain contact with the fibril.
Secondary structure in Aβ peptides was computed using the distribution of (ϕ, ψ) backbone dihedral angles. Specific definitions of β-strand and helix states can be found in our previous studies (42). Throughout the article, angular brackets 〈… 〉 imply thermodynamic averages. The distributions of states produced by REMD were analyzed using a multiple histogram method (45).
Results
Ibuprofen suppresses association of Aβ peptides with the fibril
Binding of incoming Aβ peptides to the fibril was probed by computing the thermal averages of the number of hydrophobic contacts 〈Ch(T)〉, the number of HBs 〈Nhb(T)〉, and the number of pHBs 〈Nphb(T)〉 (see Methods). Fig. 2 shows that, in ibuprofen solution, the number of peptide-fibril interactions increases with the decrease in temperature T. Fig. 2, which also displays the peptide-fibril interactions in water (26), reveals that incoming peptides bind with higher affinity to the fibril in the absence of ligands. To compare binding in both environments, we selected the temperature T = 360 K, at which Aβ peptide undergoes locking transition in water (26). In ibuprofen solution, the numbers of peptide-fibril hydrophobic contacts and HBs are 〈Ch〉 ≈ 7.1 and 〈Nhb〉 ≈ 8.0, respectively. Approximately 60% of peptide-fibril HBs are classified as parallel (〈Nphb〉 ≈ 4.7 = 0.6 〈Nhb〉). At the same temperature, the peptide-fibril interactions in water are considerably stronger. For example, 〈Ch〉 is increased to 9.8, whereas the numbers of peptide-fibril HBs, 〈Nhb〉 and 〈Nphb〉, are ≈10.5 and 6.0. Therefore, due to ibuprofen, these peptide-fibril interactions become 20–30% weaker and the ligand appears to destabilize binding of Aβ peptides to the fibril.
Figure 2.
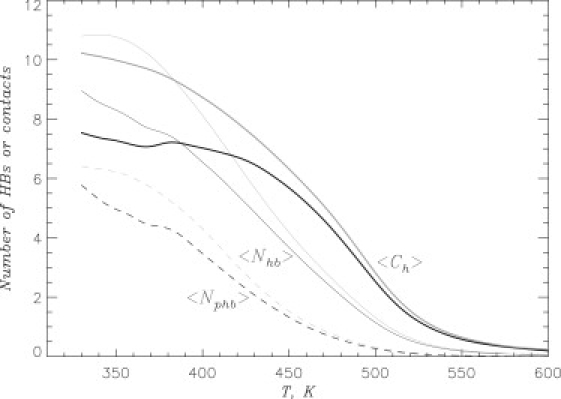
Binding of Aβ10–40 peptides to amyloid fibril is probed by the thermal averages of the number of hydrophobic contacts 〈Ch(T)〉 (thick lines), the number of HBs 〈Nhb(T)〉 (thin lines), and the number of parallel HBs 〈Nphb(T)〉 (dashed lines). The plots show that ibuprofen suppresses Aβ binding to the fibril. The data in solid and shaded representations are obtained in ibuprofen solution and water, respectively.
Ibuprofen binds to Aβ species
To determine the cause of ibuprofen's impact on fibril growth, we studied the interactions of this ligand with Aβ. The inset to Fig. 3 shows the probability Pb(T) of ibuprofen binding to Aβ peptides in hexameric system as a function of temperature. A monotonic increase in Pb(T) as temperature is lowered implicates ibuprofen binding to Aβ peptides with the binding midpoint occurring at Tb ≈ 376 K (Pb(Tb) = 0.5). At 360 K, Pb ≈ 0.63 and the number of bound ligands is 〈L〉 ≈ 38.0. The inset to Fig. 3 also displays the probability Pbi(T) of ibuprofen binding to the aggregation interface (i.e., of interacting simultaneously with the fibril and incoming peptides). We found that Pbi ≈ 0.2, implying that approximately one-third (〈Li〉 ≈ 11.7) of all bound ligands are localized at the interface.
Figure 3.
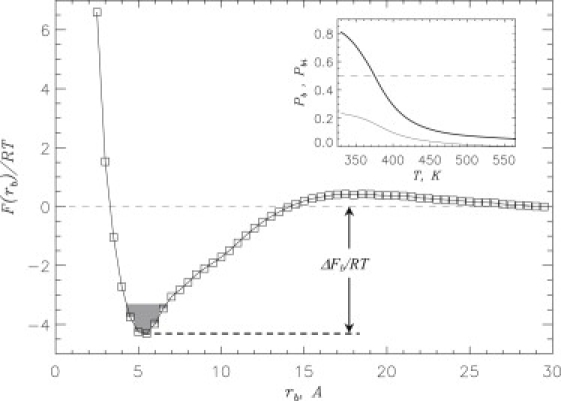
Free energy of ibuprofen ligand F(rb) as a function of the distance rb between ligand and the surface of Aβ hexamer at 360 K. The free energy of binding ΔFb (Table 1) is obtained by integrating over the bound states (shaded) with F(rb) ≤ Fmin + 1.0 RT, where Fmin is the free energy minimum at small rb. The distance rb represents the minimal distance between the ligand and Aβ. The profile F(rb) indicates that binding to Aβ is thermodynamically preferred. The value of F at rb > 29 Å is set to zero. (Inset) Probability Pb(T) of ibuprofen binding to Aβ as a function of temperature T (thick line). The probability Pbi(T) of ibuprofen binding to aggregation interface versus temperature (thin line). The dashed line marks the probability value of 0.5. At T < Tb ≈ 376 K ibuprofen-bound state is thermodynamically preferred (Pb > 0.5). Pb and Pbi are obtained by considering contacts between ibuprofen and Aβ side chains.
Further insight is provided by the binding free energy F(rb) computed as a function of the distance between the ligand and Aβ surface rb. The free energy profile in Fig. 3 reveals a single minimum at rb, 0 ≈ 5 Å, and according to Table 1, the ibuprofen binding free energy is ΔFb ≈ −5.6 RT. The computations of F(rb) separately for the fibril and incoming peptides do not reveal substantial difference in ΔFb (≈ −5.5 RT and ≈ −5.3 RT, respectively). Therefore, there is a strong thermodynamic preference for ibuprofen to bind to Aβ fibril and peptides, and we surmise that this factor destabilizes peptide-fibril interactions.
Table 1.
Binding of ibuprofen to Aβ species at 360 K
Impact of ibuprofen on fibril elongation
The changes in fibril growth induced by ibuprofen can be probed by the free energy landscape of peptide binding. To compare docking transitions in water and ibuprofen environments, we computed the free energy profiles F(C), where C is the number of peptide-fibril side chain contacts. (Because C measures any peptide-fibril interaction without regard to fibril-like content, it is appropriate reaction coordinate for docking.) Fig. 4 a shows that both F(C) plots feature a single minimum, which is consistent with the barrierless nature of binding of Aβ peptides to amyloid fibril (26). More importantly, with respect to water environment, ibuprofen interactions increase the free energy of Aβ bound state by ΔΔFB–U = ΔFB–U(IBU) − ΔFB–U(W) ≈ 2.5 RT (Table 2 and Fig. 4 a) and shift it to smaller C. However, the free energy gain of Aβ binding to the fibril in ibuprofen solution (ΔFB–U ≈ −7.4 RT) is still sufficient to ensure peptide docking to the fibril (Fig. 2).
Figure 4.
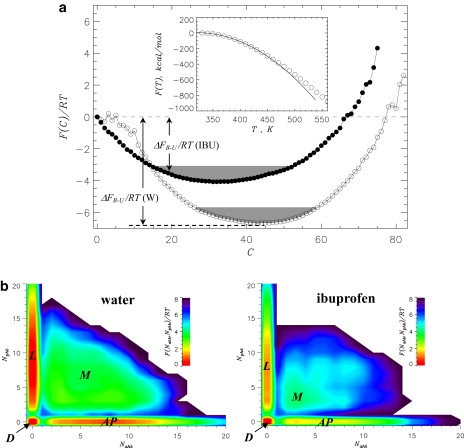
(a) Free energy of incoming Aβ peptide F(C) as a function of the number of peptide-fibril side-chain contacts C in water (open circles, W) and in ibuprofen solution (solid circles, IBU). The free energy of Aβ binding to the fibril is ΔFB–U = FB – FU, where FB and FU = 0 are the free energies of bound (B) and unbound (U, C = 0) states. FB is computed by integrating over the B states (shaded) with F(C) ≤ Fmin + 1.0 RT, where Fmin is the minimum in F(C). The plot shows that ibuprofen destabilizes Aβ binding to the fibril. (Inset) Temperature dependence of the system free energy F(T) calculated self-consistently from the multiple histogram method (45). Quadratic fitting function, from which the docking temperature Td is estimated, is shown by the solid continuous curve. Maximum value of F(T) is set to zero. (b) Free energy surfaces F(Nahb, Nphb) for bound Aβ peptide as a function of the number of antiparallel HBs Nahb and parallel HBs Nphb formed between the peptide and the fibril. The locked (L), antiparallel (AP), docked (D), and mixed (M) states are marked. The free energy landscapes show that, due to ibuprofen, the L state becomes less stable with respect to state D. Panels a and b are computed at 360 K.
Table 2.
Binding of incoming Aβ peptides to amyloid fibril at 360 K
ΔFB–U is the free energy difference between the bound and unbound states (Fig. 4a).
ΔFL–D = FL – FD is the free energy gap between the locked (L) and docked (D) states (Fig. 4b). The free energy of L states FL is obtained by integrating the free energies F(0, Nphb > 0) ≤ FL,min + 1.0 RT, where FL,min is the free energy minimum in the L state.
ΔF′ is the free energy escape barrier for the L state.
The docking temperature Td is estimated from the temperature dependence of the system free energy F(T). If docking is a continuous transition, F(T) can be approximated by the quadratic fitting function −α(T − Td)2, where α is a constant (46). The inset to Fig. 4 a shows that a good fit to F(T) at T ≲ 450 K is obtained when α = 0.019 kcal/(mol K2) and Td = 322 K. For comparison, in water, Td = 380 K (26)—implying that ibuprofen binding decreases the docking temperature by almost 60 K.
To examine the locking transition, we consider the two-dimensional free energy landscape F(Nahb, Nphb), where Nahb and Nphb are the numbers of antiparallel and parallel HBs (see Methods). These structural probes measure the formation of parallel and antiparallel β-sheets by incoming peptides. In Fig. 4 b, four free energy basins can be identified: locked states featuring parallel β-sheets (L:Nphb > 0, Nahb = 0); the states with antiparallel β-sheets (AP:Nphb = 0, Nahb > 0); docked states (D:Nphb = 0, Nahb = 0); and the states with mixed parallel and antiparallel β-sheet structure (M:Nphb > 0, Nahb > 0). There are significant changes in the equilibrium distribution of bound states as L, AP, and M states become less stable with respect to the D state. Indeed, compared to water, the free energy gap ΔFL–D (Table 2) is reduced by ≈1.0 RT in ibuprofen solution. The locking temperature Tl can be obtained by using the probability of occupancy of the L state, PL. Consistent with our previous study (26), we operationally defined the L state as the conformations with Nphb > 3 and Nahb = 0. With this definition, PL = 0.5 at Tl ≈ 330 K in ibuprofen solution. Because in water Tl ≈ 360 K (26), ibuprofen lowers the locking temperature by 30 K. Note that ibuprofen also reduces the escape free energy barriers for the L state (Table 2). Therefore, ibuprofen destabilizes Aβ locked states relative to disordered docked states.
The distribution of peptide-fibril interactions is examined using the thermally averaged map of backbone HBs 〈Nhb(i, j)〉 formed between the fibril residue i and the residue j from incoming peptide. The HB maps 〈Nhb(i, j)〉 shown in Fig. 5 for ibuprofen solution and water display diagonal and off-diagonal traces of peptide-fibrils interactions. Both HB maps are qualitatively similar, but their analysis reveals subtle changes induced by ligand binding. It follows from Fig. 5 that in water, the number of HBs formed by the β1 and β2 regions of incoming peptide, 〈Nhb(β1)〉 and 〈Nhb(β2)〉, are 4.6 and 4.5, respectively (26). In ibuprofen solution, 〈Nhb(β1)〉 and 〈Nhb(β2)〉 are reduced to 3.9 (a 20% change) and 2.7 (a 40% change). Likewise, the values of 〈Nhb(β1)〉 and 〈Nhb(β2)〉 computed for the fibril peptides decrease from 6.7 and 3.2 in water to 5.0 (a 30% change) and 2.6 (a 20% change) in ibuprofen solution. The number of HBs formed between the β1 regions of incoming peptide and the fibril 〈Nhb(β1, β1)〉 (= 2.0) remains unchanged upon ibuprofen binding, although there is a reduction in the number of HBs formed between other Aβ regions. For example, the largest decrease (by 40%) is observed in the number of HBs formed between the fibril β1 and peptide β2 regions (〈Nhb(β1, β2)〉 is reduced from 3.8 to 2.1). As a result, the share of β1−β1 HBs increases from 20% in water to 30% in ibuprofen solution. Because similar observations follow from the analysis of peptide-fibril side-chain contacts, the β1−β1 peptide-fibril interactions appear to be the least affected by ibuprofen.
Figure 5.
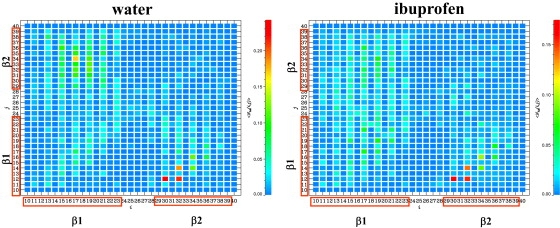
Thermally averaged maps of backbone HBs 〈Nhb(i, j)〉 formed between the fibril residue i and the residue j from incoming peptide. The maps are computed at 360 K. The residues in the β1 and β2 Aβ regions are enclosed in boxes. Similarity in the distribution of 〈Nhb(i, j)〉 in both panels suggests that ibuprofen does not qualitatively change the aggregation interface.
Discussion
Free energy landscape of fibril growth in ibuprofen solution
We showed that ibuprofen binding weakens the interactions between incoming Aβ peptides and the fibril. To analyze fibril elongation we utilized the free energy landscape perspective, which has been useful in protein folding (47). It follows from our results that, due to ibuprofen, the bound state of Aβ peptide becomes less stable (Table 2). Simultaneously, the free energy gap separating the locked and docked states is reduced (Table 2). Consistent with the changes in free energies, the number of peptide-fibril interactions (HBs or side-chain contacts) is reduced by 20–40% (Figs. 2 and 5). Furthermore, in ibuprofen solution, the docking and locking peptide transitions occur at temperatures that are 60 and 30 K lower than in water, respectively.
To rationalize these observations, we compare the binding free energies of Aβ peptides and ibuprofen. In the previous study, we have explored ibuprofen binding to Aβ monomers and fibril fragments (without incoming peptides) (34). Here we recompute the binding free energies ΔFb at 360 K (Table 1) and compare them with the free energy of Aβ binding ΔFB–U (Table 2). Although ibuprofen binding to Aβ fibrils is preferred over interactions with the monomers, both ΔFb values are at least 4.3 RT higher than ΔFB–U. Therefore, in competing for fibril binding sites, Aβ peptides have stronger affinity than ibuprofen. Nevertheless, ibuprofen does reduce the peptide binding free energy gain (ΔFB–U in Table 2) and impedes or even stalls the increase in peptide-fibril interactions at the temperatures below its binding midpoint Tb ≈ 376 K (as seen in Fig. 2 for 〈Nhb(T)〉).
Taken together, our findings suggest that ibuprofen destabilizes, but does not entirely block, Aβ deposition onto the amyloid fibril. Because all bound Aβ states (locked and docked) are destabilized (Fig. 4 a), ibuprofen is expected to slow down fibril elongation.
Molecular basis of ibuprofen anti-aggregation effect
It has been shown in our previous study that ibuprofen preferentially binds to the concave (CV) edge of Aβ fibril (Fig. 1 c and Fig. 5 in (34)). At 330 K the ratio of the numbers of ligands bound to the CV and convex (CX) edges is ∼2:1, whereas at 360 K it becomes 1.4:1. The CV edge is also a primary binding location for incoming Aβ peptides (26). For example, at 360 K the probability of CV binding, PCV, exceeds that of the CX, PCX, as PCV:PCX ≈ 9:1. The binding of Aβ or ibuprofen to the fibril fragment sides is negligible (26,34). These observations suggest that ibuprofen and Aβ compete for the same binding location (the CV edge) on the amyloid fibril. To illustrate this conclusion, we plot in Fig. S2 the probabilities PCV and PCX as a function of temperature in water and ibuprofen solution. At temperatures above the ibuprofen binding midpoint (Tb ≈ 376 K), PCV (and PCX) are similar in both environments. However, at T ≲ Tb in ibuprofen solution, PCV reverses monotonic increase and declines to ≈0.8 at 360 K. Fig. S2 suggests that ibuprofen reduces the difference in the fibril edge affinities for Aβ. Indeed, in water, the free energy of the CV edge Aβ binding is lower than that for the CX by ΔFCV–CX ≈ 2.5RT (26). In ibuprofen solution, ΔFCV–CX is reduced to 1.5 RT. Consequently, ibuprofen anti-aggregation effect can be also explained by the fact that a fraction of Aβ peptides is forced to bind to the low affinity CX edge.
To establish energetic factors controlling ligand binding, we computed the average energy Einter of intermolecular interaction and the average solvation energy Esolv per ibuprofen molecule at 360 K. Upon binding Einter decreases by ΔEinter ≈ 12.5 kcal/mol (from −1.4 to −13.9 kcal/mol), while Esolv increases by 1.0 kcal/mol. The van der Waals (vdW) interactions represent the main contribution to ΔEinter, making up >90% of its value. Therefore, vdW interactions appear to be the main driving factor in ligand binding. This conclusion is supported by the changes in accessible surface area (ASA) occurring upon ibuprofen binding at 360 K. The average ASA values for the three groups G1, G2, and G3 (Fig. 1 b) in unbound ibuprofen are ASAu(G1) = 90 Å2, ASAu(G2) = 161 Å2, and ASAu(G3) = 153 Å2, respectively. For bound ligands we obtained ASAb(G1) = 37 Å2, ASAb(G2) = 83 Å2, and ASAb(G3) = 68 Å2. Therefore, upon binding, the ASAs of these groups are reduced by 53, 78, and 85 Å2, respectively, or by 9, 20, and 17 Å2, respectively, per atom. Assuming that the extent of burial reflects the strength of binding interactions, we conclude that G3 and G2 are the most important for binding. Because the aromatic G1 is sandwiched between G2 and G3, the geometric reasons might limit G1 participation in binding. Note that ibuprofen burial may result from the interactions with the fibril and/or other bound ligands. If one considers the changes in ASA that occur exclusively from interactions with the fibril (i.e., by omitting neighboring ligands), then the ASAs of ibuprofen groups are reduced by 27, 41, and 52 Å2. Therefore, the relative importance of ibuprofen groups for binding does not depend on the details of ASA computations. Consistent with the dominant contribution of vdW interactions, both hydrophilic G3 and hydrophobic G2 groups participate in ligand binding. Similar observations have been made in our previous study that ibuprofen-binding sites are composed of the mixture of hydrophobic and hydrophilic residues (34).
Because polar G3 has somewhat higher binding affinity than the hydrophobic G2 (in terms of ASA changes), ibuprofen binding could be enhanced by G2 modifications, possibly, by functionalizing it with polar atoms and creating a structural motif with two polar atomic groups linked to the central hydrophobic moiety (G1, Fig. 1 b). A similar motif is present in the structure of naproxen (Fig. 1 b), which, consistent with our proposal, binds more tightly to Aβ fibrils than ibuprofen (20). According to competition curves probing the replacement of molecular imaging probe 18FFDDNP, the concentration of ibuprofen must be at least twice larger than that of naproxen to reduce the probe binding by one-half.
To test our proposal in silico, we performed REMD simulations of 40 naproxen molecules coincubated with the fibril fragment formed by four Aβ10–40 peptides. In line with the experiments, we found that naproxen binds with higher affinity to the fibril than ibuprofen. For example, the binding free energy of naproxen ΔFb is ≈ 2.4 RT is lower than for ibuprofen. Furthermore, the midpoint of naproxen binding occurs at Tb = 398 K, which is >30 K higher than Tb = 362 K for ibuprofen (34). The preliminary analysis of naproxen appears to support our proposal concerning the enhancement of ligand binding. However, further studies are needed to evaluate the contribution of the hydrophobic G1 naphthalene group.
An important implication of naproxen binding is that ligand-fibril interactions appear to depend on the chemical structure of the ligands. This conclusion is further supported by the observation that ibuprofen binds with higher affinity to the CV fibril edge by localizing in its groove (Fig. 1 c) (34). The analysis of binding energetics reveals that this binding preference stems from the formation of attractive interligand interactions facilitated by the CV edge geometry. It is possible that ligand-excluded volume also adds to the anti-aggregation effect, but further studies are needed to precisely assess the relative contributions of these factors.
Ibuprofen does not change Aβ aggregation interface
Our data suggest that ibuprofen has no major impact on the Aβ aggregation interface involved in fibril growth. As in water, the interface is polarized, because peptide-fibril interactions preferentially involve the β1 Aβ regions (41,48). Although most peptide-fibril interactions are reduced by 20–40%, the β1-β1 interactions are largely unaffected by ibuprofen. Therefore, ibuprofen further enhances the polarization of aggregation interface. It is also instructive to compute the average registry offsets for peptide-fibril pHBs 〈R(i, j)〉 (see the Supporting Material). We found that in water and in ibuprofen solution, 〈R(i, j)〉 remains almost the same (≈11). Thus, parallel β-sheets formed by incoming peptides on the fibril edge are typically off-registry (〈R〉 >> 1) in both environments.
It is possible that anti-aggregation effect of ibuprofen is due to the changes in the Aβ secondary structure induced by the ligand. To explore this possibility, we computed the fractions of β-strand 〈S〉 and helix 〈H〉 structure in Aβ incoming peptides in ibuprofen solution. Compared to water (26), a small decrease in 〈S〉 is observed from 0.52 to 0.48. The fraction of helix structure 〈H 〉 remains unchanged within the margin of error (0.12 vs. 0.11). Therefore, the impact of ibuprofen binding on secondary structure of bound Aβ peptides is small and the peptides retain mostly β-strand conformations. Consequently, the anti-aggregation effect of ibuprofen is unlikely to be associated with the changes in Aβ secondary structure. This finding suggests that ibuprofen interferes directly with peptide-fibril interactions as described above.
It is also interesting to consider the changes in the thickness D of the layer formed by bound Aβ on the fibril edge. Fig. S3 compares the temperature dependencies D(T) obtained in ibuprofen solution and in water. We showed (26) that the theory of polymer adsorption on attractive walls (49) appears to be applicable to the binding of Aβ peptides to amyloid fibril. As a result similar to polymer adsorption, Aβ binding is represented by the barrierless transition. Exploiting this analogy, D(T) can be fit with the function D(T) ≃ D0/(Tu − T) before unbinding at Tu, where D0 is a constant (49). In water, a single fitting function provides an adequate fit in the entire temperature range. In ibuprofen solution, D(T) requires superposition of two fitting functions with the crossover point at ≈440 K (see the Supporting Material). This suggests that, due to ibuprofen, two Aβ binding regimes are observed. At high temperatures well above the ibuprofen-binding midpoint, Tb ≈ 376 K, peptide-fibril interactions are not affected by ibuprofen and the layer thicknesses D in water and in ibuprofen solution are similar. At lower temperatures (T ≲ 440 K), there is an onset of ibuprofen binding, and deposition of Aβ peptides is affected. As a result, the layer thickness D in ibuprofen solution exceeds that in water by >1 Å. The increase in D signals swelling in the layer of peptides bound to the fibril edge, which is consistent with the free energy analysis above.
Comparison with experiments and simulations
Experimental studies have established an anti-aggregation effect of ibuprofen. For example, ibuprofen reduces the amount of Aβ oligomers in mice brain tissues (18). In vitro studies have found that ibuprofen partly inhibits Aβ1–40 fibril assembly in a concentration-dependent manner when coincubated with fresh (i.e., not-fibrilized) Aβ peptides (20,21). Moreover, if the concentration ratio of ibuprofen to fibril Aβ is ∼22 (which is higher than in our simulations), the ligand completely blocks Aβ1–40 fibril elongation (21). The study of Thomas et al. (50) has used circular dichroism to investigate the changes in secondary structure in preaggregated Aβ25–35 peptides. They demonstrated that, at the ligand/peptide concentration ratio of 8:1, ibuprofen reduces the β-structure content roughly in half due to partial dissociation of the fibrils. These experimental findings support our in silico results suggesting that ibuprofen destabilizes peptide-fibril interactions and the fibril-like locked state. However, we did not observe dissociation of incoming peptides from the fibril induced by ibuprofen as implied by some experimental findings (20,21). The possible reasons for this discrepancy are as follows. Compared to the temperatures used experimentally (∼300 K), our simulation results are obtained at a higher temperature of 360 K, which is also close to the midpoint of ibuprofen binding Tb ≈ 376 K. Weakened ibuprofen-Aβ interactions together with the relatively low ibuprofen/Aβ ratio (compared to (21)) are likely to limit the anti-aggregation action of this ligand in our study.
Experimental data implicates extension of amyloid fibrils via monomer addition to their edges (37,38,51). If ibuprofen affects fibril elongation, it is natural to expect that the ligand binds to the edges of the fibril and directly interferes with peptide-fibril interactions. This is also the conclusion following from our study, which does not support indirect anti-aggregation effect based on secondary structure changes in Aβ. Interestingly, direct interference with fibril formation has been observed for tricyclic planar ligands (33). Using MD simulations, Caflisch et al. (33) have studied 9,10-anthraquinone binding to fibril-forming fragments Aβ14–20. Because this ligand destabilizes the formation of interstrand HBs, it also reduces the accumulation of ordered aggregates.
It is interesting to speculate on the connection between ibuprofen anti-aggregation effect and Aβ mutations. In the previous study, we showed that ibuprofen binding is mostly driven by the geometry of Aβ fibril surface and the ligands tend to concentrate in the groove located on the CV fibril edge (Fig. 1 c) (34). Here we further demonstrated that vdW interactions appear to be the dominant binding factor. If ibuprofen binding is not directly determined by Aβ sequence, then the anti-aggregation affect of ibuprofen should be largely independent on Aβ mutations—provided they do not change the wild-type Aβ fibril structure.
The important question to be addressed in future studies is the impact of ibuprofen on the stability of Aβ oligomers. We have previously showed that, in contrast to fibril binding, ibuprofen does not form large bound clusters when interacting with Aβ monomers (34). When extrapolated to oligomers, these findings suggest that the mechanism of binding to mobile relatively unstructured Aβ species may be quite different from the mechanism observed for the fibrils.
Conclusions
Our study showed that binding of ibuprofen to Aβ destabilizes the interactions between incoming peptides and the amyloid fibril. Ibuprofen binding changes the free energy landscape of fibril growth and reduces the free energy gain of Aβ peptide binding to the fibril by ≃2.5 RT at 360 K. Furthermore, ibuprofen interactions shift the thermodynamic equilibrium from fibril-like locked states to disordered docked states. We explain ibuprofen's anti-aggregation effect by noting that it competes with incoming Aβ peptides for the same binding site on the Aβ fibril surface located on the concave edge. Our simulations also suggest that ibuprofen does not change the mechanism of fibril elongation or the secondary structure of Aβ peptides bound to the fibril. In summary, our simulations offer plausible molecular basis for ibuprofen's anti-aggregation effect known from experimental studies. The analysis of the energetics of ibuprofen binding can be useful in designing new anti-aggregation agents.
Supporting Material
Three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00307-3.
Supporting Material
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health. Fig. 1 c was produced using the UCSF Chimera package (52).
This work was supported by grant No. R01 AG028191 from the National Institute on Aging, of the National Institutes of Health, Bethesda, MD.
References
- 1.Selkoe D.J. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 2.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Shankar G.M., Li S., Selkoe D.J. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy R.M., Pallitto M.M. Probing the kinetics of β-amyloid self-association. J. Struct. Biol. 2000;130:109–122. doi: 10.1006/jsbi.2000.4253. [DOI] [PubMed] [Google Scholar]
- 6.Carulla N., Caddy G.L., Dobson C.M. Molecular recycling within amyloid fibrils. Nature. 2005;436:554–558. doi: 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]
- 7.Martins I.C., Kuperstein I., Rousseau F. Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serpell L.C. Alzheimer's amyloid fibrils: structure and assembly. Biochim. Biophys. Acta. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 9.Burkoth T.S., Benzinger T., Lynn D.G. Structure of the β-amyloid(10–35) fibril. J. Am. Chem. Soc. 2000;122:7883–7889. [Google Scholar]
- 10.Petkova A.T., Yau W.-M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lührs T., Ritter C., Riek R. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson R., Sawaya M.R., Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meersman F., Dobson C.M. Probing the pressure-temperature stability of amyloid fibrils provides new insights into their molecular properties. Biochim. Biophys. Acta. 2006;1764:452–460. doi: 10.1016/j.bbapap.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 14.Shoji M., Golde T.E., Younkin S. Production of the Alzheimer amyloid β-protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 15.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 16.Xia W. Amyloid inhibitors and Alzheimer's disease. Curr. Opin. Investig. Drugs. 2003;4:55–59. [PubMed] [Google Scholar]
- 17.Heneka M.T., Sastre M., Landreth G.E. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ 1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 18.McKee A.C., Carreras I., Dedeoglu A. Ibuprofen reduces Aβ, hyperphosphorylated τ and memory deficits in Alzheimer mice. Brain Res. 2008;1207:225–236. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vlad S.C., Miller D.R., Felson D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agdeppa E.D., Kepe V., Barrio J.R. In vitro detection of (s)-naproxen and ibuprofen binding to plaques in the Alzheimer's brain using the positron emission tomography molecular imaging probe 2-(1-{6-[(2- [18F]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene) malononitrile. Neuroscience. 2003;117:723–730. doi: 10.1016/s0306-4522(02)00907-7. [DOI] [PubMed] [Google Scholar]
- 21.Hirohata M., Ono K., Yamada M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer's β-amyloid fibrils in vitro. Neuropharmacology. 2005;49:1088–1099. doi: 10.1016/j.neuropharm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Ma B., Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Struct. Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 23.Cecchini M., Rao F., Caflisch A. Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J. Chem. Phys. 2004;121:10748–10756. doi: 10.1063/1.1809588. [DOI] [PubMed] [Google Scholar]
- 24.Reddy G., Straub J.E., Thirumalai D. Dynamics of locking of peptides onto growing amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2009;106:11948–11953. doi: 10.1073/pnas.0902473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krone M.G., Hua L., Shea J.E. Role of water in mediating the assembly of Alzheimer amyloid-β Aβ16–22 protofilaments. J. Am. Chem. Soc. 2008;130:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeda T., Klimov D.K. Replica exchange simulations of the thermodynamics of Aβ fibril growth. Biophys. J. 2009;96:442–452. doi: 10.1016/j.bpj.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sgourakis N.G., Yan Y., Garcia A.E. The Alzheimer's peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD / NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang M., Teplow D.B. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008;384:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda T., Klimov D.K. Probing the effect of amino-terminal truncation for Aβ1–40 peptides. J. Phys. Chem. B. 2009;113:6692–6702. doi: 10.1021/jp9016773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchete N.-V., Hummer G. Structure and dynamics of parallel β-sheets, hydrophobic core, and loops in Alzheimer's A β fibrils. Biophys. J. 2007;92:3032–3039. doi: 10.1529/biophysj.106.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng J., Jang H., Nussinov R. Modeling the Alzheimer Aβ 17–42 fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys. J. 2007;93:3046–3057. doi: 10.1529/biophysj.107.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C., Wang Z., Shea J.E. The binding of thioflavin T and its neutral analog BTA-1 to protofibrils of the Alzheimer's disease Aβ(16–22) peptide probed by molecular dynamics simulations. J. Mol. Biol. 2008;384:718–729. doi: 10.1016/j.jmb.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Convertino M., Pellarin R., Caflisch A. 9,10-Anthraquinone hinders β-aggregation: how does a small molecule interfere with Aβ-peptide amyloid fibrillation? Protein Sci. 2009;18:792–800. doi: 10.1002/pro.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raman E.P., Takeda T., Klimov D.K. Molecular dynamics simulations of ibuprofen binding to Aβ peptides. Biophys. J. 2009;97:2070–2079. doi: 10.1016/j.bpj.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chebaro Y., Derreumaux P. Targeting the early steps of Aβ16–22 protofibril disassembly by N-methylated inhibitors: a numerical study. Proteins Struct. Funct. Bioinform. 2009;75:442–452. doi: 10.1002/prot.22254. [DOI] [PubMed] [Google Scholar]
- 36.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;114:141–151. [Google Scholar]
- 37.Esler W.P., Stimson E.R., Maggio J.E. Alzheimer's disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry. 2000;39:6288–6295. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 38.Cannon M.J., Williams A.D., Myszka D.G. Kinetic analysis of β-amyloid fibril elongation. Anal. Biochem. 2004;328:67–75. doi: 10.1016/j.ab.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 39.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1982;4:187–217. [Google Scholar]
- 40.Ferrara P., Apostolakis J., Caflisch A. Evaluation of a fast implicit solvent model for molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2002;46:24–33. doi: 10.1002/prot.10001. [DOI] [PubMed] [Google Scholar]
- 41.Takeda T., Klimov D.K. Probing energetics of Aβ fibril relongation by molecular dynamics simulations. Biophys. J. 2009;96:4428–4437. doi: 10.1016/j.bpj.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda T., Klimov D.K. Interpeptide interactions induce helix to strand structural transition in Aβ peptides. Proteins Struct. Funct. Bioinform. 2009;77:1–13. doi: 10.1002/prot.22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou L., Shao H., Zagorski M.G. Solution NMR studies of the A β(1–40) and A β(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 44.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 45.Ferrenberg A.M., Swendsen R.H. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 1989;63:1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 46.Landau L.D., Lifshitz E.M. Course of Theoretical Physics. Vol. 5. Butterworth-Heinemann; Oxford, UK: 1984. Statistical physics. [Google Scholar]
- 47.Onuchic J.N., Wolynes P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Melquiond A., Dong X., Derreumaux P. Role of the region 23–28 in Aβ fibril formation: insights from simulations of the monomers and dimers of Alzheimer's peptides Aβ40 and Aβ42. Curr. Alzh. Res. 2008;5:244–250. doi: 10.2174/156720508784533330. [DOI] [PubMed] [Google Scholar]
- 49.Grosberg A.Y., Khokhlov A.R. AIP Press; Woodbury, N.Y: 1994. Statistical Physics of Macromolecules. [Google Scholar]
- 50.Thomas T., Nadackal T.G., Thomas K. Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-β aggregation. Neuroreport. 2001;12:3263–3267. doi: 10.1097/00001756-200110290-00024. [DOI] [PubMed] [Google Scholar]
- 51.Ban T., Hoshino M., Goto Y. Direct observation of Aβ amyloid fibril growth and inhibition. J. Mol. Biol. 2004;344:757–767. doi: 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 52.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.