

Abstract
Gene expression is regulated both by cis elements, which are DNA segments closely linked to the genes they regulate, and by trans factors, which are usually proteins capable of diffusing to unlinked genes. Understanding the patterns and sources of regulatory variation is crucial for understanding phenotypic and genome evolution. Here, we measure genome-wide allele-specific expression by deep sequencing to investigate the patterns of cis and trans expression variation between two strains of Saccharomyces cerevisiae. We propose a statistical modeling framework based on the binomial distribution that simultaneously addresses normalization of read counts derived from different parents and estimating the cis and trans expression variation parameters. We find that expression polymorphism in yeast is common for both cis and trans, though trans variation is more common. Constraint in expression evolution is correlated with other hallmarks of constraint, including gene essentiality, number of protein interaction partners, and constraint in amino acid substitution, indicating that both cis and trans polymorphism are clearly under purifying selection, though trans variation appears to be more sensitive to selective constraint. Comparing interspecific expression divergence between S. cerevisiae and S. paradoxus to our intraspecific variation suggests a significant departure from a neutral model of molecular evolution. A further examination of correlation between polymorphism and divergence within each category suggests that cis divergence is more frequently mediated by positive Darwinian selection than is trans divergence.
Untangling the genetic basis of phenotypic variation within and between species is a central topic in evolutionary biology. It has long been argued that evolution of protein sequences is insufficient to explain the morphological diversity present in nature (King and Wilson 1975). Consequently, evolution of gene expression has often been invoked as an alternative explanation for phenotypic innovation (Halder et al. 1995; Carroll 2008). As a result, much effort has been devoted to understanding expression evolution in eukaryotes, especially model systems like the budding yeast Saccharomyces cerevisiae (Rockman and Kruglyak 2006). Previous genome-wide studies of expression variation in yeast have taken advantage of full-genome microarrays to determine the linkage relationship between genes with variable expression and the causative mutations. Generally, it is assumed that sets of genes exhibiting local linkage in QTL maps are enriched for cis variation while those with distant linkage are regulated in trans. These QTL studies demonstrated that gene regulation polymorphisms in yeast are common and are dominated by distant linkages (Brem et al. 2002; Yvert et al. 2003). Furthermore, it was shown that many transcripts are linked to only a few “hotspot” regulators. For example, Yvert et al. (2003) reported 1265 variable transcripts regulated by only 13 distant QTLs. Another study of variation in transcript levels has corroborated the ubiquity of transcript variation across many different strains within S. cerevisiae (Kvitek et al. 2008), concluding that expression polymorphism may be under the influence of diversifying selection for adaptation to different environments. It has also been argued that cis expression level polymorphism is under purifying selection, while trans expression polymorphism is under positive selection (Ronald and Akey 2007). Studies in Drosophila took advantage of comparing the allele-specific expression (ASE) patterns of two parental strains to that of their hybrid offspring (i.e., F1 hybrids) to investigate cis and trans expression evolution (Wittkopp et al. 2004, 2008). This experimental design measures the combined expression variation (both cis and trans effects) at a locus through measurement of expression differences between two parental strains. The combined effects of all categories of genetic variation influencing gene regulation can explain expression differences measured between two genetically distinct strains. However, expression differences measured within the F1 hybrids between the same two strains can no longer be attributed to trans-factors, as both genomes share the same cell and the same trans-factors. Consequently, the hybrid experiment measures only cis variation. These experiments in Drosophila on 78 genes showed that cis differences dominate between species more than within species (Wittkopp et al. 2008). ASE polymorphism studies in yeast have found that expression level variation is usually numerically dominated by trans variants (Wang et al. 2007; Sung et al. 2009), even when single-input module genes were chosen to minimize the impact of trans variation (Wang et al. 2007). Additionally, in yeast only 52%–78% of expression QTLs mapped to the same region as the gene they regulate were confirmed to be cis (Ronald et al. 2005), implying that as much as 22%–48% of genes with local linkages might be regulated in trans. Indeed, Ronald et al. (2005) have reported specific instances of trans regulation stemming from local linkage. In these ASE studies, the predominance of trans linkage is generally consistent with the results from QTL mapping (Brem et al. 2002; Yvert et al. 2003), which demonstrated that the majority of linkages are distant and therefore are likely to be driven by a few “hotspot” trans-activating factors that regulate thousands of transcripts spread throughout the genome. In contrast, a recent genome-wide comparison of cis to trans variation between S. cerevisiae and S. paradoxus has shown that cis variation is more common than trans variation, though intraspecific data were not reported (Tirosh et al. 2009).
Counting sequencing reads has long been used to measure the relative copy numbers of those sequences. Bailey et al. (2002) used an increase in read counts to identify increases in copy number due to segmental duplication. More recently, Seoighe et al. (2006) used representation in EST libraries to identify ASE indicative of both genomic imprinting and genetically caused allelic differences. With the advent of high-throughput sequencing technologies like 454 Life Sciences (Roche) sequencing, this perspective has become more quantitative (Springer and Stupar 2007). The application of even higher capacity deep sequencing technologies to quantitative measurement of nucleotide frequencies has obvious benefits for measuring transcriptomes (Nagalakshmi et al. 2008). It has also enabled more accurate measurements of allelic differences in genetically variable nucleotide pools for single nucleotide polymorphism (SNP) discovery (Van Tassell et al. 2008) and ASE (Wang et al. 2008; Bloom et al. 2009).
Here, we investigate the relative contributions of cis and trans regulatory differences to overall expression variation for the entire genome of S. cerevisiae, using Illumina Genome Analyzer (IGA) sequencing of mRNA to measure genome-wide ASE in a co-culture experiment composed of two strains of S. cerevisiae (denoted as BY and RM) and in their F1 hybrid. We report the relative impact of selective constraint on various modes of gene regulation within species across the whole genome for the first time, demonstrating that a large proportion of genes exhibit expression polymorphism, with trans variation dominating over cis, even after removing genes influenced by trans “hotspots” identified by Yvert et al. (2003). Furthermore, to date, a genome-wide comparison of cis and trans variation within and between species has yet to be reported. In this study, we compare our polymorphism data to the divergence data from a recent study (Tirosh et al. 2009), showing not only that cis differences are more common between species than within species, but also that trans variation is much more compatible with a neutral model of selection, whereas many cis variations appear driven to fixation through positive Darwinian selection.
Results
Orthology and SNP identification
We designed bioinformatics filters based primarily on unambiguous orthology, unique sequence, and presence of SNPs that selected 4442 genes for analysis from an initial pool of 6604 ORFs with untranslated region (UTR) information (Nagalakshmi et al. 2008). (A detailed description of these filters is available in Methods and Supplemental Tables S1–S3.) We identified 893 SNPs caused by putative genome reference sequence errors and corrected them. In total, our analysis incorporated 35,225 SNP sites distributed among 4442 orthologous gene pairs.
Intraspecific genomic DNA sequencing
The cell density ratio between any two strains in a co-culture experiment (dCo) need not be 1, which introduces a systematic bias in mRNA transcript counts between the two strains. To correct for this bias, we sequenced genomic DNA (gDNA) from the same samples that provided the mRNA to estimate both dCo (co-culture) and dHy (F1 hybrid). We estimated that dCo = 1.30 and that, as expected, dHy = 1.00. That dHy is indistinguishable from 1, as expected, indicates that this method is an effective way to estimate d (Supplemental Fig. S1). We used these ratios in the estimation of allelic expression ratios in terms of RM/BY (eCo and eHy), accounting for the effect of cell density (see Methods). We then estimated the means and confidence intervals for cis and trans contributions to expression change in terms of deviations from the null hypotheses: log2(eHy) = log2(eCis) and log2(eCo) = log2(eCis) + log2(eTrans) (see Methods). Our results agree closely with a data set of 227 genes collected in another study using pyrosequencing (Fig. 1; Sung et al. 2009). The correlation between these genes ranges from 0.74 to 0.81, and the regression lines estimated from these comparisons are indistinguishable from the diagonal running through the origin with a slope equal to 1.
Figure 1.

Comparison of two methods of estimating ASE polymorphism. (A–C) Y-axis, log2(eIGA) (IGA transcriptome data), versus x-axis, log2(ePyro) (pyrosequencing). (A,B) comparison of co-culture and hybrid results from the two methods, respectively; (C) comparison between IGA sequencing and pyrosequencing for the value log2(eCo) − log2(eHy), which is also log2(etrans). To test if the regressions differ from equality, we tested the estimated regression coefficients against the null hypothesis H0: m = 1; b = 0. All P-values for these hypothesis tests for all regression parameters are not significant (all P-values > 0.05), indicating that IGA and pyrosequencing give very similar results.
Intraspecific transcriptome sequencing and expression estimation
Among the 4442 genes that passed our bioinformatics filters, 4282 have sequence reads for both alleles in both experiments (Supplemental Tables S1–S3). From 12 channels of IGA sequencing of cDNA for each of the two samples (24 channels in total), we mapped 1.202 and 1.188 million sequence reads from the hybrid sample to BY and RM SNPs, respectively; for the co-culture sample we mapped 1.096 and 1.330 million reads.
In total, 1180 genes (28%) exhibit expression polymorphism at a P-value threshold of 1% (false discovery rate [FDR] < 5%; Supplemental Fig. S2; correlated estimates, see Methods). Next, we classified genes by examining the relationship between cis and trans polymorphisms throughout the genome (Fig. 2D; see Methods section Independent Estimates). The data indicate that trans variation is more frequent, as observed previously (Brem et al. 2002; Yvert et al. 2003), and show greater magnitudes of change 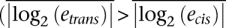 (Wilcoxon rank sum test, all P-values < 0.01) than cis variation. Nearly four times as many genes exhibit only trans change as genes that show only cis change (123 vs. 33). For the 178 genes with significant |log2(ecis)| ≠ |log2(etrans)|, 79% of them (140) are genes where |log2(ecis)| < |log2(etrans)|. Interestingly, 116 genes show unambiguous expression variation for cis and trans variation simultaneously (Fig. 2C,D, “dominant” and “both” categories; our use of “dominant” here should not be confused with genetic dominance; it merely indicates that variation for one type of regulation is greater than the other), while another 589 genes show clear evidence for variation in one category and ambiguous evidence in another (Fig. 2A,C,D, the two “major” categories). Thus, trans differences clearly dominate, influencing 64% (558/863) of differentially expressed genes, though almost half of genes showing expression polymorphism indicate a clear significant cis effect (49% or 421/863). (See Supplemental Fig. S6 for a more detailed description of the classifications above.)
(Wilcoxon rank sum test, all P-values < 0.01) than cis variation. Nearly four times as many genes exhibit only trans change as genes that show only cis change (123 vs. 33). For the 178 genes with significant |log2(ecis)| ≠ |log2(etrans)|, 79% of them (140) are genes where |log2(ecis)| < |log2(etrans)|. Interestingly, 116 genes show unambiguous expression variation for cis and trans variation simultaneously (Fig. 2C,D, “dominant” and “both” categories; our use of “dominant” here should not be confused with genetic dominance; it merely indicates that variation for one type of regulation is greater than the other), while another 589 genes show clear evidence for variation in one category and ambiguous evidence in another (Fig. 2A,C,D, the two “major” categories). Thus, trans differences clearly dominate, influencing 64% (558/863) of differentially expressed genes, though almost half of genes showing expression polymorphism indicate a clear significant cis effect (49% or 421/863). (See Supplemental Fig. S6 for a more detailed description of the classifications above.)
Figure 2.

Genome-wide ASE polymorphism in S. cerevisiae: (A–C) y-axis, log2(ecis), versus x-axis, log2(etrans). (A) Scatter plot of cis and trans estimators with the shading of the points indicating what category individual genes fall into, as determined by their two-dimensional 99% confidence intervals. These classifications are made with respect to the relationship between confidence intervals of each point and the four lines running through the origin, which are the two axes and the two diagonals. Trans-only (t only) and cis-only (c only) indicate that the confidence intervals for the genes, respectively, overlap the log2(ecis) = 0 or the log2(etrans) = 0 axis line only. “Both” refers to the genes that only overlap either the log2(ecis) = log2(etrans) line or the log2(ecis) = −log2(etrans) line (the positive and negative diagonals). Trans-dominant (t dom) and cis-dominant (c dom) genes overlap no lines at all but fall in the quadrants nearest the log2(ecis) = 0 and log2(etrans) = 0 lines, respectively. Trans-major (t maj) overlaps the log2(ecis) = 0 line and at least one of the diagonals, whereas cis-major (c maj) overlaps the log2(etrans) = 0 line and at least one of the diagonals. “ns” indicates nonsignificant genes: each of them overlaps both the log2(ecis) = 0 and the log2(etrans) = 0 lines. A more detailed explanation of how genes are classified can be found in Supplemental Figure S6. (B) A contour plot of the two-dimenstional probability density function of the data (from the two-dimensional kernel density estimator in the MASS library in R) indicating where most genes fall in the cis/trans space using independent estimates of cis and trans. The “elevation” indicated by the contours expresses the probability of a point falling in that region. The total volume beneath the surface sums to unity. (C) A summary of classifications based on the results from Figure 1A. Dark green, trans-only; light green, trans-dominant; dark orange, cis-only; light orange, cis-dominant; hashed regions, the “cis/trans-major” classifications; white, both; gray, nonsignificant genes. (D) The histogram indicates that significant trans changes dominate in comparison to significant cis changes. The left bar of each pair is before discarding the 1265 “hotspot” genes from Yvert et al. (2003), and the right bar is after discarding them. Importantly, trans-dominant and cis-dominant are not to be confused with genetic dominance; instead, they are meant to convey that the magnitude of trans variation is greater than cis variation or the reverse, respectively.
Previous eQTL studies showed a large “hotspot” effect (Brem et al. 2002; Yvert et al. 2003 showed this for 1265/1716 distant eQTLs and 1265/2294 of all eQTLs). To investigate ASE variation for genes less likely to be influenced by these hotspots, we discarded the 1265 genes identified in Yvert et al. (2003) (Fig. 2D). As expected, 76% of the differentially expressed genes discarded fell into either the various trans categories (68%) or the “both” category (8%). In contrast, <2% of the hotspot genes fell into the cis-only category, with the remaining 22% of genes falling into cis categories with a putatively minor trans component. Despite discarding these genes, trans variation remains the prevalent form of variation (Fig. 2D).
Evolutionary constraint in expression polymorphism
To test our ASE data for evolutionary constraint, we compared the magnitudes of our expression ratio estimates between categories of genes predicted to be strongly constrained versus those predicted to be weakly constrained (Fig. 3). For protein–protein interaction networks (Stark et al. 2006; Collins et al. 2007), those genes with the most interactions had significantly lower expression variation than those genes absent from the networks, both for cis and trans polymorphism (Wilcoxon rank sum tests, all P-values < 10−6). Essential genes (genes that cause lethality when deleted; Deutschbauer et al. 2005) also showed significantly less expression variation than nonessential genes for both cis and trans (Wilcoxon rank sum tests, all P-values < 10−15), corroborating a previous study (Ronald and Akey 2007). One important measure of constraint based on sequence evolution is ω (Yang 1997, 2007), the ratio of the rate of nonsynonymous substitution to the rate of synonymous substitution (i.e., Ka/Ks). Our expression estimates show that the value of ω is significantly correlated with both |log2(ecis)| and |log2(etrans)| (all P-values < 0.0001, for Pearson, Kendall, and Spearman correlation coefficients). Comparing the 50th percentile of genes with the lowest ω to those with the highest ω (Fig. 3C) also demonstrates that the magnitude of expression variation in strongly constrained genes is lower than that in less constrained genes.
Figure 3.

Constraint in expression polymorphism. (A) Genes with presently no detected protein–protein interaction (ppi) partners in current data (light bars) versus genes in the upper 50th percentile of those showing interactions (dark bars). (B) Nonessential genes (light bars) are genes whose homozygous knockouts have a fitness of greater than 0.85. Essential genes (light bars) are those genes where homozygous knockouts are lethal. (C) Genes in the lower 50th percentile of ω (dark bars) versus genes in the upper 50th percentile of ω (light bars). Pairs of bars labeled “cis” and “trans” compare the mean expression divergence |log2(e)| between putatively strongly constrained and weakly constrained categories. The category “trans − cis” compares the quantity |log2(etrans)| − |log2(ecis)| between putatively strongly constrained and weakly constrained categories. Significance is indicated as follows: ns, P > 0.05; *, 0.01 < P < 0.05; **, 0.01 < P < 0.001; ***, P < 0.001. Both cis and trans mutations are subject to purifying selection in all comparisons. trans mutations are more sensitive to changes in constraint than are cis changes when constraint is determined by number of protein interactions or essentiality, but not when measured by protein coding sequence (ω).
This correlation between ω and expression is surprising, as it contradicts a previous report that found no such relationship (Ronald and Akey 2007). One possible source of this discrepancy might be because we compared the expression level difference between the set of genes with low ω to those with high ω, while the Ronald and Akey study did the opposite, comparing ω between genes without expression variation to those with expression variation. To reconcile this difference, we performed two additional tests: (1) comparing ω between genes showing a significant P-value (P < 0.01) for the null hypothesis log2(e) = 0 to those that were not significant (P > 0.05) using the Wilcoxon rank sum test; and (2) correlation tests between |log2(e)| and ω, using the Pearson, Kendall, and Spearman methods. For all eight of the tests above (one Wilcoxon test and three correlation tests for both cis and trans), we found that low ω is associated with low expression variation (all P-values < 10−5).
To investigate whether cis or trans variation is more sensitive to purifying selection, we compared the quantity |log2(etrans)| − |log2(ecis)| between constrained and unconstrained categories. For constraint based on both protein–protein interactions and essential/nonessential genes, the quantity |log2(etrans)| − |log2(ecis)| is significantly lower in the strongly constrained categories than in the weakly constrained categories (Fig. 3A,B). Comparing the genes with the lowest ω to those with the highest shows a positive relationship, though the difference is not significant (all correlation P-values > 0.05).
Comparing expression polymorphism to expression divergence
We compared the relative contributions of cis and trans regulatory polymorphism in our data set to cis and trans regulatory divergence between S. cerevisiae and S. paradoxus from a recent publication (Tirosh et al. 2009). We adopted the following perspectives: (1) comparing the relative cis/trans contributions within and between species; and (2) comparing the correlations between polymorphism and divergence between cis and trans. In the first comparison, we test the hypothesis proposed in a recent study in two Drosophila sister species (Fig. 4; Wittkopp et al. 2008). We compared the relationship between the hybrid experiment versus the co-culture/parental experiment within and between species (Fig. 4A,B, respectively), using both major axis (MA) regression and standardized major axis (SMA) regression (Warton et al. 2006). Regression estimates closer to the diagonal indicate that cis predominates, whereas an estimate near the horizontal axis indicates that trans predominates. We show that the interspecific slope is significantly greater than the intraspecific (P < 2.2 × 10−16 for both the MA and the SMA regressions), indicating that the magnitude of cis regulatory variation relative to trans is greater between species than within species.
Figure 4.

Between-species regulatory differences are dominated by cis-regulatory changes more than within-species differences. A and B follow Figure 1 in Wittkopp et al. (2008), by comparing the regression between hybrid and co-culture lines for intraspecific and interspecific variation, respectively. The slope for the interspecific comparison (A) is larger than for the intraspecific comparison (B), regardless of the line-fitting method used (SMA and MA regressions were conducted using the “smatr” package in R (Warton et al. 2006; R Development Core Team 2009). C–E follow Figure 2 in Wittkopp et al. (2008). Measurements of cis between species consistently dominate, regardless of how they are measured. All comparisons are significant, P < 2.2 × 10−16, indicating that cis-regulatory changes dominate between species more than within species.
Next, we investigate three measures of cis variation: the magnitude of cis variation, |log2(ecis)|; the magnitude of cis variation as a proportion of variation measured in the co-culture experiment, |log2(ecis)|/|log2(epar)|; and the magnitude of cis variation as a proportion of the total variation in both cis and trans, |log2(ecis)|/[|log2(ecis)| + |log2(etrans)|]. We plotted each of these values against its quantile (Fig. 4C–E) and found that the interspecific cis values are consistently higher than the intraspecific cis values, regardless of how cis values are scaled (Wilcoxon rank sum test, P-value < 2.2 × 10−16 for all three comparisons). Interestingly, we show that the cis share of total cis + trans variation (Fig. 4E) is significantly higher for divergence than for polymorphism. Next, we conduct a formal test of the neutral mutation hypothesis across the genome by comparing our polymorphism data to the divergence data of Tirosh et al. (2009). We follow the framework of Kreitman and Aguade (1986) by dividing significant expression differences into a 2 × 2 contingency table. The test decisively rejects the predictions of the neutral theory (Table 1A; P < 10−10, Fisher's exact test). Clearly, there are far more cis expression differences between the two species than expected from the within-species polymorphism data, suggesting that natural selection plays an important role in shaping expression variation.
Table 1.
Comparison between polymorphism and divergence in cis and trans mutations

aSignificant nonhomogeneity is evidence of a violation of the neutral theory, as described by Kreitman and Aguade (1986). P-value < 2.2 × 10−16.
bThis enables a test of homogeneity between polymorphism (columns) and divergence (rows), when the categories are divided between significant and nonsignificant genes. The P-values for trans and cis are 2.33 × 10−4 and 0.377, respectively. This result is unchanged if the independent estimates are used (Supplemental Table S4).
cNumbers indicate observations and numbers in parentheses indicate expectations. The numbers in boldface correspond to Figure 5, A and B, respectively.
To trace the source of this pattern, we examined the cis and trans data separately. Interestingly, for significant trans differences, genes showing expression polymorphism tend to show differential expression between species; the correspondence between significantly polymorphic and significantly divergent genes is much greater than expected by chance (Fig. 5A; Table 1B; Fisher's exact test, P < 1 × 10−6). In contrast, no corresponding association is observed for cis variation (Fig. 5B; Table 1B; Fisher's exact test, P > 0.25). Under neutral theory, neutral variation is correlated between polymorphism and divergence, while nonneutral categories will exhibit weaker correlations or even an absence of correlation altogether. This suggests that the number of significant differences for cis-regulatory change may be subject to nonneutral forces. Unlike for qualitative measures discussed above, this pattern does not extend to quantitative measures of expression ratios (Fig. 5C,D; Supplemental Fig. S3C,D).
Figure 5.

Greater correlation between trans polymorphism and divergence than between cis polymorphism and divergence. (A,B) The number of significant genes manifested both between intraspecific measurements and interspecific measurements for trans (A) and cis (B). The histograms indicate the null hypothesis of homogeneous association between significant polymorphisms and significant divergence. Let P be the number of genes with significant expression differences within species and D be the number of differences between species. If P gene names were randomly drawn without replacement from the gene list, the histogram represents the probability that an independent draw of D genes results in x genes common to both lists. Each histogram is the hypergeometric distribution representing the upper lefthand cell in the 2 × 2 table in a Fisher's exact test (Table 1B). For trans genes (A), the cell indicates overlap between significant expression polymorphisms and significant expression divergence and it is significantly higher than expected by chance, while for the cis genes (B), the overlap is well within the range expected simply by randomly shuffling the data. (C,D) The relationship between polymorphism and divergence estimates is shown for genes with significant polymorphism estimates. The regression estimate between trans polymorphism and divergence describes 23% of the variance, while that between cis polymorphism and divergence describes 26%. Each point is composed of genes grouped by bins according to polymorphism estimates. The values are obtained by taking the median of each bin for divergence and polymorphism. Each bin contains 11 or 12 genes.
Discussion
Our results demonstrate that inferring the genetic architecture of expression level evolution can be performed in a straightforward, comprehensive, and rigorous manner. Through a novel application of simple binomial models, we can infer confidence intervals for cis and trans effects while correcting for experimental sampling biases (via the d parameter, Methods; Supplemental Fig. S1). These cis and trans parameter estimates are highly concordant with pyrosequencing experiments (Fig. 1).
Our expression parameter estimates (Fig. 2) demonstrate that, while expression polymorphism is common for both types of regulatory variation, trans differences dominate both in magnitude and in number. Interestingly, even for such a short evolutionary time as the divergence between the BY and the RM strains, many genes exhibit both cis and trans differences simultaneously, indicating that they have sustained at least two mutations affecting the expression phenotype. This prevalence of trans variation persists even when the trans-regulated genes controlled by hotspots (Brem et al. 2002; Yvert et al. 2003) are discarded (Fig. 2D). One central result of molecular evolution is that many if not most nonsynonymous mutations in genes are deleterious. To investigate this perspective in the context of expression polymorphism, we compared expression constraint to constraint in other functional categories. We predict that genes with more interaction partners, essential genes, and genes with a low ω to be more strongly constrained. Comparing the magnitude of expression variation between pairs within each category shows that constraint in expression corresponds to constraint in each of three other measures of constraint (Fig. 3, cis and trans columns). Interestingly, while our results regarding essential genes agree with another report on expression variation (Ronald and Akey 2007), we find conflicting results with regard to ω; we find a correlation between ω and cis variation (Fig. 3; Results), whereas Ronald and Akey do not, perhaps as a result of different means of measuring expression or different subsets comprising our respective comparisons.
To determine which category of gene regulation was more strongly influenced by purifying selection, we compared the difference between trans and cis between various categories (i.e., |log2(etrans)| − |log2(ecis)|, Fig. 3, the trans – cis column). For all functional categories, trans variation was higher in the unconstrained category than in the constrained category, with this difference being statistically significant for protein–protein interactions and essential genes. This stronger filtering of trans polymorphisms in essential genes and in genes with many interaction partners suggests that expression differences between species will be more and more strongly influenced by cis variants compared to trans variants as we move from considering less constrained to more constrained categories. Indeed, it is well established that on very long timescales, important “toolbox” genes can provide much phenotypic innovation despite their highly constrained peptide sequences, but such innovation is strongly influenced by evolution of cis elements (Carroll 2008). However, the opportunity for adaptation through reconfiguring trans-regulators remains an intriguing possibility for genes from less constrained categories. Future investigations of ASE in mutation accumulation lines (MAL) compared to natural lines (NL) (Denver et al. 2005) would enable an examination of the distribution of fitness effects for a wider range of deleterious variants. If our hypothesis above is correct regarding trans variants, the expression ratio of NL versus MAL should be lower in constrained categories and higher in relaxed categories.
In order to extend our inferences to longer timescales, we compared our polymorphism data to a recent data set for expression between S. cerevisiae and S. paradoxus. One key prediction of the neutral theory of molecular evolution is that the degree of polymorphism and the rate of fixation are both increasing functions of the mutation rate (Kimura 1968; Kimura and Ota 1971), leading to the recognition that comparing intraspecific variation (polymorphism) to interspecific variation (divergence) is a powerful strategy for testing hypotheses concerning natural selection (Kreitman and Aguade 1986; Hudson et al. 1987; McDonald and Kreitman 1991; Bustamante et al. 2002). Importantly, such contrasts can distinguish between variation resulting from higher mutation rates (Fisher 1922; Haldane 1927) and variation due to the action of natural selection.
A recent study compared cis and trans expression evolution within and between species for 78 genes in two species of Drosophila (Wittkopp et al. 2008). Plots of hybrid ASE differences versus parental differences showed that interspecific ASE variation fits more closely to the “all cis” line (the diagonal where hybrid = co-culture) than does intraspecific ASE variation (Fig. 1; Wittkopp et al. 2008), though these results were not consistently significant between different partitions of the data (for three-fourths of the partitions, the slopes of one estimate were within the 95% confidence interval [CI] of the other [Wittkopp et al. 2008; Supplemental Table 5], assuming 95% CI = slope, mean ± 1.96 × SE). Similarly, for relative contributions of ASE as measured by cis/(cis + trans), polymorphism and divergence were not significantly different (Fig. 2, column 3; Wittkopp et al. 2008). Interestingly, in one comparison where the authors inferred polymorphism indirectly from divergence data, a cis effect was observed (Fig. 3; Wittkopp et al. 2008).
To test the hypothesis that cis variation is subject to natural selection on a genome-wide scale, we compared our expression polymorphism data to a recent expression divergence data set (Tirosh et al. 2009). Our results strongly suggest that in comparison with intraspecific expression variation, interspecific expression variation is much more strongly shaped by cis evolution. Importantly, regressions between co-culture and hybrid experiments fall significantly closer to the “all cis” (i.e., hybrid = co-culture) line for interspecific comparisons than for intraspecific comparisons (Fig. 4A.B). Moreover, the cumulative cis expression divergence is significantly and consistently above that of cis polymorphism (Fig. 4C), even when cis divergence is scaled by expression variation between the unhybridized parental strains (Fig. 4D). These results strongly suggest that cis variation plays a greater role between species than within species. Most interestingly, when we compare the relative contributions of cis variation to total cis + trans expression variation between polymorphism and divergence, we find that cis divergence plays a larger role than would be predicted from polymorphism data (Fig. 4E), strongly implicating the action of natural selection. We think it possible that a larger sample size in the Drosophila genome might corroborate our results.
We also presented a formal test of the neutral mutation hypothesis across the genome by comparing our significant polymorphic genes to the significant divergent genes from the data of Tirosh et al. (2009). We divide significant expression differences into a 2 × 2 contingency table (Table 1A), as described in Results. If the data follow the predictions of the neutral theory (or alternatively if both categories experience similar selection regimes), then divergence/polymorphism ratios should be similar for both cis and trans categories. A violation of homogeneity within the table is evidence that at least one category violates neutrality, though failure to reject is only a weak indication of neutrality, as it is possible that both categories could be under similar nonneutral selective regimes. The test unequivocally rejects the predictions of the neutral theory (Table 1A; P < 1 × 10−10, Fisher's exact test), strongly suggesting that either there is an excess of divergent cis differences between species as might occur due to positive selection or that there is an excess of polymorphism in trans regulatory variation within species such as might be observed under balancing selection (Kreitman and Aguade 1986; Hudson et al. 1987). Importantly, this test is capable only of determining that the table is highly heterogeneous. It cannot determine the source of the departure from homogeneity.
The test above relies on the correlation between polymorphism and divergence for neutral variants between two different categories. To interrogate this predicted relationship in more detail, we measured the association between significant polymorphism and divergence within cis and trans individually. By doing this, we hope to adduce evidence bearing on which of the two categories is more likely to be neutral. Under neutrality we predict that levels of polymorphism and divergence are mutation driven. If the rate of mutation varies among genes, it should vary in the same manner between polymorphism and divergence for the same gene. For example, genes that are significantly variable within species should be more likely to be variable between species, and genes that are not variable within species should be less likely to be variable between species. Indeed, this pattern is exactly what we observe for trans mutations (Table 1B; Fig. 5A), suggesting that trans differences conform to one prediction of neutral theory. On the other hand, cis differences fit this prediction rather poorly (Table 1B; Fig. 5B). The number of significant cis differences common to both polymorphism and divergence is no greater than expected by chance, indicating that polymorphism and divergence correlate only weakly, arguing against the neutrality of cis variation. Given the relatively low level of cis polymorphism versus the high level of cis divergence combined with the evidence from Table 1B, we suggest that the cis-regulatory differences are under positive selection rather than the alternative that trans polymorphism is under balancing selection. We also examined the correlation between the magnitude of expression divergence and polymorphism (Fig. 5C,D). Unlike the count data described above, for pooled expression variation estimates, polymorphism predicts divergence equally well for both trans variation (r2 = 23%, Fig. 5C) and cis variation (r2 = 26%, Fig. 5D; but see also Supplemental Fig. S3). While for trans variants, whether or not a gene is polymorphic is a good predictor for whether or not it differs between species, the magnitude of expression level polymorphism is only weakly related to the magnitude of expression level divergence. This weak relationship nearly disappears if the genes are not pooled by polymorphism expression level (data not shown), likely because the magnitude of change in expression level can vary greatly between different mutations in the same gene.
These observations indicate that trans variation conforms more closely to the predictions of the neutral theory than cis variation. Thus, our comparisons of polymorphism and divergence data for expression levels strongly suggest that cis evolution strongly shapes differences between species and that such variation is strongly shaped by positive natural selection. Taken together, these analyses paint a comprehensive picture of the selective forces shaping cis and trans evolution and reinforce the idea that cis expression differences play a dominant role in adaptive expression divergence between species.
Methods
Yeast strains and growth conditions
Two culture types were prepared: co-culture and hybrid. The co-culture experiment was prepared from approximately equal amounts of two MATa strains called BY and RM. The hybrid strain was derived by mating BY (MATa) × RM (MATα) and were all grown in standard YPAD medium.
The laboratory strain designated “BY” is officially named BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and is a descendant of S288C. The strain designated “RM” (a gift from Dr. Leland Hartwell, Fred Hutchinson Cancer Research Center) is officially either RM11-1a (MATa lys2Δ0 ura3Δ0 ho∷KAN) or RM11-1α (MATα lys2Δ0 ura3Δ0 ho∷KAN). Both are haploid strains derived from Bb32(3), a natural isolate described previously (Mortimer et al. 1994). We have designated the hybrid of a BY4741 × RM11-1α cross constructed in our lab as WL201. The co-culture sample is simply a mixture of BY4741 and RM11-1a. All yeast strains were grown in YPAD media at 30°C with 250 rpm shaking.
Total RNA extraction and sequencing
To estimate expression ratios for the hybrid and co-culture experiments, total RNA was extracted and purified for mRNA, from which double-stranded cDNA was synthesized, fragmented, and subjected to the Illumina Genome Analyzer (IGA) sequencing protocol.
For expression measurements, overnight subcultures were used to prepare four technical replicates for each of the hybrid (WL201) cultures and BY + RM co-cultures with starting OD600 = 0.1 and harvested when cell density reached OD600 = 1.0. Total RNA was then extracted by the hot acid phenol method (Kohrer and Domdey 1991). Total RNA concentration within each replicate was quantified on a NanoDrop 1000 spectrophotometer (Thermo Scientific). Equal amounts of RNA were pooled within each sample group (i.e., hybrid and co-culture) into a combined RNA sample. The quality of combined RNA was assessed with the BioAnalyzer (Agilent).
The hybrid and co-culture mRNA was purified using oligo(dT) Dynabeads (Invitrogen) according to the manufacturer's protocol. Subsequent reverse transcription was carried out with oligo(dT) primers and the Superscript II kit (Invitrogen) following the manufacturer's instructions. Transcriptome sequencing steps were performed by Fasteris SA, Switzerland. Samples containing cDNAs fragments of 200–400 nucleotides (nt) were then sequenced on 24 lanes (12 for hybrid and 12 for co-culture) of flow cell by an IGA sequencer using Illumina's genomic shotgun protocol, yielding 35-nt-long reads.
Yeast genome sequencing
To estimate relative cell densities in the samples and to confirm SNP assignments, the gDNA from the same hybrid and co-culture samples as used for cDNA sequencing were extracted, fragmented, and subjected to the IGA DNA sequencing protocol. The genomic DNA was extracted using Qiagen Q100 genomic purification kit (Qiagen). Within each sample group, equal amounts of DNA were pooled into a combined DNA sample, analogous to the pooling strategy employed for the transcriptome sequencing above. The combined DNA sample was then fragmented by sonication, and the shotgun libraries were prepared according to Illumina's gDNA protocol. The genomic DNA sequencing was carried out on the Illumina GA-II (IGA-II) sequencer in the High Throughput Sequencing Core Facility of Academia Sinica, yielding reads 40 nt in length.
Mapping IGA reads to the reference genomes
The BY reference genome was downloaded from the SGD project on April 3, 2008 (ftp://ftp.yeastgenome.org/yeast/). The RM reference genome was downloaded from the Saccharomyces cerevisiae RM11-1a Sequencing Project, Broad Institute (http://www.broad.mit.edu). Every cDNA sequence read was used as a query against each reference yeast genome using MEGABLAST with the “wordsize” parameter set to 8 (Zhang et al. 2000), yielding two homology search datasets, one for the BY genome and one for the RM genome. We then recorded all hits with up to two nucleotide mismatches. A mismatch may be due to a sequencing error in the sequence read we obtained, a sequence error in the reference genome(s), or a SNP site between the two reference genomes. For each set of homology search results, we classified each read as uninformative (perfectly matching both genomes); informative only (matching one or two SNPs for one genome); informative and error (matching one SNP and containing one IGA error); error only (containing one or two IGA errors). See Supplemental material for more details for these classifications.
From the 12 channels of cDNA IGA sequencing data for each of the two samples (total 24 channels), we obtained 71,309,740 and 71,549,168 raw reads from the hybrid sample and the co-culture sample, respectively, which were subjected to classification described above (Table S1). Most of the mapped reads matched one place in the genome, uniquely identifying the expression of a single transcript (Table S2).
In order to determine if our read mapping strategy resulted in spurious differential expression (Degner et al. 2009), we examined each combination of experiment (hybrid or co-culture) with strain (BY or RM). By dividing the data for each combination into six channels each, we compared two independent partitions of the same biological material, determining how often the null hypothesis of no differential expression is rejected when differential expression is absent. Our results indicate typical rejection rates (Supplemental Fig. S4).
In order to investigate potential sources of sequencing or amplification bias in our data, we examined the number of times a unique read was represented in each channel of sequencing data. The histograms of expression read-counts compare favorably to a priori estimates based on discrete stochastic models (Supplemental Fig. S5).
Identifying orthologous pairs and polymorphic sites between the BY and RM genomes
Each gene from a refined set of BY gene transcripts with UTR information (Nagalakshmi et al. 2008) was aligned onto the RM genome using BLAT and axtChain (Kent 2002; Kent et al. 2003) to identify its ortholog in the RM genome. These alignments were then used to identify polymorphic sites between the two orthologous genes. We restricted our attention to high scoring pairs (HSPs) derived from the chaining procedure, neglecting reads that mapped to nonhomologous regions.
Exclusion of overlapping gene regions
Transcript sequencing data were derived from double-stranded cDNA; therefore, we were unable to determine which strand reads are mapped to for regions where two transcripts from opposite strands overlap (Nagalakshmi et al. 2008). To avoid misattribution, we first temporarily excluded overlapping regions. Genes whose average read count per base in nonoverlapping regions of the gene were less than 0.025 read/nt were discarded from the data set. Next, we reintegrated reads mapping to regions that no longer overlapped with expressed genes on the opposite strand, yielding 4566 SNP containing gene pairs possessing unique orthologous regions. These genes were subjected to further study (Table S3).
Detecting errors in the genomic sequences of the two strains
If the expression level at a SNP exhibits a particularly strong bias toward one allele, this indicates either a strong pattern of differential expression or an error in the reference genome we aligned the reads to. Our data contained 1490 SNPs exhibiting a very strong bias toward one allele in the cDNA data. By comparing the SNP count data obtained from cDNA to that obtained from gDNA, we classified 893 of such sites as true errors in the reference genomes, 540 as true differential expression, and failed to classify 57 sites. Using this information, we corrected the 893 “true error” SNPs in the reference genomes and discarded from our data set the 57 SNPs which we failed to classify. We then repeated the mapping computation to obtain the final read counts. For more details on this procedure, please consult the Supplemental Materials.
Modeling gene expression as a discrete sampling process
To estimate expression parameters on IGA read data, we formulated our question in terms of the binomial distribution. A normalization parameter is required because differences in total reads between samples occur whenever sampling effort is not evenly distributed between the samples, due to either study design or experimental error. Though some authors recommend employing methods related to standard quantile normalization (Bolstad et al. 2003) for count data (Balwierz et al. 2009), we consider rescaling counts (which contain important information regarding the sampling variance) to be less than ideal. Moreover, we apply no noise correction. There are reasonable physical rationales for noise correction in array studies (Tu et al. 2002). In the case of deep sequencing, however, it is not clear which mapped sequence reads comprise the “signal” and which comprise the “noise.”
Cis and trans parameter estimation
Let a measurement of total informative expression read counts for a gene be N and the read counts for the RM allele be X and for the BY allele be N-X. The data has a binomial distribution with proportion parameter p. Let j represent a single experiment (co-culture or hybrid), e be the expression ratio parameter between RM and BY and d be the normalization ratio parameter between RM and BY. The proportion parameter p can be expressed in terms of d and e:
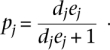 |
The assumptions that the hybrid experiment exhibits only cis variation and the co-culture exhibits a combination of cis and trans variation can be expressed as
Consequently, the binomial proportion parameters can be rewritten as follows:
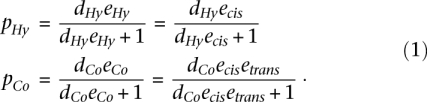 |
Thus, likelihood functions for the hybrid and co-culture experiments can be expressed as:
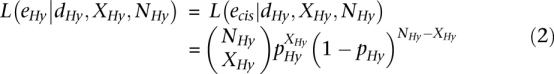 |
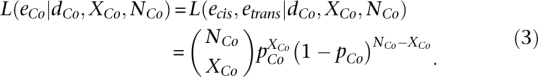 |
Because Equation 3 prevents us from estimating etrans independently of ecis, we can instead examine the product of Equations 3 and 4 to obtain a likelihood function where etrans can be estimated independently from ecis:
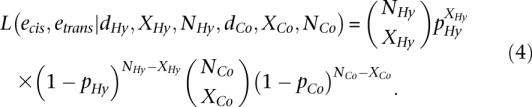 |
We can then obtain expression parameter estimates for ecis from Equations 2 or 4 and for etrans from Equation 4 using standard likelihood maximization methods in R (R Development Core Team 2009).
Independent estimate of cis and trans parameters
When cis and trans are estimated as described above, the estimates are negatively correlated (cf. Fig. 2B and Supplemental Fig. S2B; see Supplemental material for more details). In order to estimate ecis and etrans independently, we divide the hybrid data into two partitions of six channels each. We then estimate ecis from Equation 2 using one partition of the hybrid data and estimate etrans from Equation 4 using the other partition of the hybrid data and all of the co-culture data. For a rationale of when correlated estimates are preferred and when independent estimates are preferred, see the Supplemental material.
Estimation of the normalization parameter
We estimate the normalization parameter d from the gDNA data (which is independent from the cDNA data) as follows:
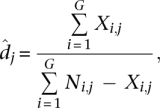 |
where i indexes each gene and j represents the experiment (hybrid or co-culture).
Sequence divergence statistics
Orthologous genes between S. cerevisiae and S. paradoxus were determined from the Fungal Orthogroups Repository (Wapinski et al. 2007). The coding sequences were frame-aligned so that pairwise codon divergence statistics Ka and Ks could be calculated using the PAML package (Yang 1997, 2007). All methods of alignment and statistical calculation follow those used in Emerson et al. (2004), with the exception that MUSCLE (Edgar 2004) was use for alignment.
Acknowledgments
W.-H.L. was supported by Academia Sinica, Taiwan; the International E. Balzan Prize Foundation; and NIH grants GM30998 and GM081724. J.J.E. was supported by an Academia Sinica Distinguished Postdoctoral Fellowship. We thank Michael McDonald, Jun-Yi Leu, and Bin He for thoughtful discussion of key issues in early versions of the manuscript. We also thank three anonymous reviewers for helpful suggestions and constructive criticisms.
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE20749.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.101576.109.
References
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007 [DOI] [PubMed] [Google Scholar]
- Balwierz PJ, Carninci P, Daub CO, Kawai J, Hayashizaki Y, Belle WV, Beisel C, van Nimwegen E 2009. Methods for analyzing deep sequencing expression data: Constructing the human and mouse promoterome with deepCAGE data. Genome Biol 10: R79 doi: 10.1186/gb-2009-10-7-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JS, Khan Z, Kruglyak L, Singh M, Caudy AA 2009. Measuring differential gene expression by short read sequencing: Quantitative comparison to 2-channel gene expression microarrays. BMC Genomics 10: 221 doi: 10.1186/1471-2164-10-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755 [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Nielsen R, Sawyer SA, Olsen KM, Purugganan MD, Hartl DL 2002. The cost of inbreeding in Arabidopsis. Nature 416: 531–534 [DOI] [PubMed] [Google Scholar]
- Carroll SB 2008. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell 134: 25–36 [DOI] [PubMed] [Google Scholar]
- Collins SR, Kemmeren P, Zhao X-C, Greenblatt JF, Spencer F, Holstege FCP, Weissman JS, Krogan NJ 2007. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics 6: 439–450 [DOI] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Streelman JT, Kim SK, Lynch M, Thomas WK 2005. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat Genet 37: 544–548 [DOI] [PubMed] [Google Scholar]
- Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G 2005. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 169: 1915–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC 2004. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113 doi: 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Kaessmann H, Betran E, Long M 2004. Extensive gene traffic on the mammalian X chromosome. Science 303: 537–540 [DOI] [PubMed] [Google Scholar]
- Fisher R 1922. On the dominance ratio. Proc R Soc Edinb 42: 321–341 [Google Scholar]
- Haldane J 1927. A mathematical theory of natural and artificial selection, Part V: Selection and mutation. Proc Camb Philol Soc 23: 838–844 [Google Scholar]
- Halder G, Callaerts P, Gehring WJ 1995. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 267: 1788–1792 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci USA 100: 11484–11489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M 1968. Evolutionary rate at the molecular level. Nature 217: 624–626 [DOI] [PubMed] [Google Scholar]
- Kimura M, Ota T 1971. Protein polymorphism as a phase of molecular evolution. Nature 229: 467–469 [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Kohrer K, Domdey H 1991. Preparation of high molecular weight RNA. Methods Enzymol 194: 398–405 [DOI] [PubMed] [Google Scholar]
- Kreitman ME, Aguade M 1986. Excess polymorphism at the Adh locus in Drosophila melanogaster. Genetics 114: 93–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Will JL, Gasch AP 2008. Variations in stress sensitivity and genomic expression in diverse S. cerevisiae isolates. PLoS Genet 4: e1000223 doi: 10.1371/journal.pgen.1000223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654 [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Romano P, Suzzi G, Polsinelli M 1994. Genome renewal: A new phenomenon revealed from a genetic study of 43 strains of Saccharomyces cerevisiae derived from natural fermentation of grape musts. Yeast 10: 1543–1552 [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team 2009. R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- Rockman MV, Kruglyak L 2006. Genetics of global gene expression. Nat Rev Genet 7: 862–872 [DOI] [PubMed] [Google Scholar]
- Ronald J, Akey JM 2007. The evolution of gene expression QTL in Saccharomyces cerevisiae. PLoS One 2: e678 doi: 10.1371/journal.pone.0000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald J, Brem RB, Whittle J, Kruglyak L 2005. Local regulatory variation in Saccharomyces cerevisiae. PLoS Genet 1: e25 doi: 10.1371/journal.pgen.0010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe C, Nembaware V, Scheffler K 2006. Maximum likelihood inference of imprinting and allele-specific expression from EST data. Bioinformatics 22: 3032–3039 [DOI] [PubMed] [Google Scholar]
- Springer NM, Stupar RM 2007. Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 19: 2391–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M 2006. BioGRID: A general repository for interaction datasets. Nucleic Acids Res 34: D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H-M, Wang T-Y, Wang D, Huang Y-S, Wu J-P, Tsai H-K, Tzeng J, Huang C-J, Lee Y-C, Yang P, et al. 2009. Roles of trans and cis variation in yeast intraspecies evolution of gene expression. Mol Biol Evol 26: 2533–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N 2009. A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662 [DOI] [PubMed] [Google Scholar]
- Tu Y, Stolovitzky G, Klein U 2002. Quantitative noise analysis for gene expression microarray experiments. Proc Natl Acad Sci 99: 14031–14036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tassell CP, Smith TPL, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS 2008. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods 5: 247–252 [DOI] [PubMed] [Google Scholar]
- Wang D, Sung H-M, Wang T-Y, Huang C-J, Yang P, Chang T, Wang Y-C, Tseng D-L, Wu J-P, Lee T-C, et al. 2007. Expression evolution in yeast genes of single-input modules is mainly due to changes in trans-acting factors. Genome Res 17: 1161–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sun Q, McGrath SD, Mardis ER, Soloway PD, Clark AG 2008. Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One 3: e3839 doi: 10.1371/journal.pone.0003839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A 2007. Natural history and evolutionary principles of gene duplication in fungi. Nature 449: 54–61 [DOI] [PubMed] [Google Scholar]
- Warton DI, Wright IJ, Falster DS, Westoby M 2006. Bivariate line-fitting methods for allometry. Biol Rev Camb Philos Soc 81: 259–291 [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG 2004. Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88 [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet 40: 346–350 [DOI] [PubMed] [Google Scholar]
- Yang Z 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13: 555–556 [DOI] [PubMed] [Google Scholar]
- Yang Z 2007. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591 [DOI] [PubMed] [Google Scholar]
- Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, Mackelprang R, Kruglyak L 2003. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet 35: 57–64 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W 2000. A greedy algorithm for aligning DNA sequences. J Comput Biol 7: 203–214 [DOI] [PubMed] [Google Scholar]