

Abstract
Previous studies have shown that small interfering RNA knockdown and pharmacological inhibition of inositol 1,4,5-trisphosphate receptors (IP3Rs) stimulate autophagy. We have investigated autophagy in chicken DT40 cell lines containing targeted deletions of all three IP3R isoforms (triple knock-out (TKO) cells). Using gel shifts of microtubule-associated protein 1 light chain 3 as a marker of autophagy, we find that TKO cells have enhanced basal autophagic flux even under nutrient-replete conditions. Stable DT40 cell lines derived from TKO cells containing the functionally inactive D2550A IP3R mutant did not suppress autophagy in the same manner as wild-type receptors. This suggests that the channel function of the receptor is important in its regulatory role in autophagy. There were no marked differences in the phosphorylation state of AMP-activated protein kinase, Akt, or mammalian target of rapamycin between wild-type and TKO cells. The amount of immunoprecipitated complexes of Bcl-2-Beclin-1 and Beclin-1-Vps34 were also not different between the two cell lines. The major difference noted was a substantially decreased mTORC1 kinase activity in TKO cells based on decreased phosphorylation of S6 kinase and 4E-BP1. The discharge of intracellular stores with thapsigargin stimulated mTORC1 activity (measured as S6 kinase phosphorylation) to a greater extent in wild-type than in TKO cells. We suggest that basal autophagic flux may be negatively regulated by IP3R-dependent Ca2+ signals acting to maintain an elevated mTORC1 activity in wild-type cells and that Ca2+ regulation of this enzyme is defective in TKO cells. The protective effect of a higher autophagic flux in cells lacking IP3Rs may play a role in the delayed apoptotic response observed in these cells.
Keywords: Calcium, Calcium Transport, Inositol Phosphates, mTOR Complex (mTORC), Protein Degradation, Autophagy
Introduction
It is well established that Ca2+ has an important regulatory role in controlling apoptosis (1–3). Inositol 1,4,5-trisphosphate receptors (IP3R)2 participate in this pathway at several levels. First, they provide a conduit for the transfer of Ca2+ between the ER and the mitochondria to sensitize the mechanism that facilitates the release of cytochrome c from the mitochondria (4). Second, IP3Rs interact with, and are regulated by, several proteins that modify apoptotic pathways, including the anti-apoptotic proteins Bcl-2/Bcl-XL (5, 6), cytochrome c (7, 8), and Akt kinase (9–11). Finally, with certain apoptotic stimuli (e.g. staurosporine), IP3Rs support apoptosis independently of the channel function of the receptor via a mechanism that may be linked to a direct role of IP3Rs in activating Ca2+ entry mechanisms across the plasma membrane (12).
Macroautophagy is a proteolytic process in which cytoplasmic constituents (including organelles) are sequestered within double-membraned vesicles (autophagosomes) that ultimately fuse with lysosomes leading to the degradation of their contents (13). A major physiological regulator of this process is nutrient supply, although the process is also regulated by various hormones and can be dysregulated under pathological conditions (14). The complicated steps involved in autophagosome formation and lysosome fusion involve multiple proteins and regulation by many different inputs, including the activities of the mTOR pathway and class III phosphatidylinositol 3-kinase. There have been several reports suggesting that Ca2+ regulates this pathway. Hoyer-Hansen et al. (15) showed that agents that elevated Ca2+ in MCF-7 cells increased the formation of autophagosomes and that this was blocked by treatment with the intracellular Ca2+ chelator BAPTA-AM. However, others have reported that blocking Ca2+ elevations (e.g. with L-type Ca2+ channel antagonists) can enhance autophagy suggesting that Ca2+ has an inhibitory effect on autophagy (16). In addition, the depletion of intracellular stores with thapsigargin has been reported to have both a stimulatory (15, 17) and inhibitory (16, 18, 19) effect on autophagy. Manipulations designed to change the levels of IP3 in cells (e.g. addition of myo-inositol or Li+) alter the rate of starvation-induced autophagy (17). A specific role for IP3Rs in the autophagic process was suggested by the finding that small interfering RNA knockdown or pharmacological blockade of the IP3R with xestospongin B led to an enhancement of autophagy (17). These data suggest that IP3Rs negatively modulate autophagy. However, it is unclear if this involves the channel function of the IP3R, because some effective agents, such as xestospongin B, had no detectable effects on cytosolic or ER luminal Ca2+(17). Disruption of the single IP3R gene in Dictyostelium discoideum impairs an autophagic death pathway (20). The specific autophagic signaling pathway(s) modulated by IP3Rs remains to be identified.
DT40 chicken B-cell lines containing targeted deletion of all three IP3R isoforms (TKO) show a markedly delayed cell death response to various apoptotic stimuli (6, 12, 21). We considered the possibility that adaptive changes in autophagy may have occurred in these cells thereby providing a useful experimental system to investigate the role of IP3Rs in autophagy. In this study, we show that TKO cells have a markedly enhanced rate of autophagy compared with wild-type cells, even under nutrient-replete conditions. The suppression of autophagy required the Ca2+ channel function of the IP3R and was not observed in cell lines transfected with the pore-inactivating D2550A mutant. Several key factors that regulate autophagy were compared in wild-type and TKO cell lines and were not found to be significantly different. These included the activity of AMP and Akt kinase. The differences in basal autophagy could also not be accounted for by altered levels of Beclin-1-Vps34 complexes. Instead, our experiments suggest that altered activity of the mTORC1 complex may be one potential mechanism by which IP3R-mediated Ca2+ fluxes could regulate the autophagic pathway.
MATERIALS AND METHODS
Reagents
RPMI 1640 culture media and G418 sulfate (Geneticin) were obtained from Cellgro-Mediatech (Herndon, VA). Staurosporine, rapamycin, bafilomycin, and okadaic acid were purchased from Sigma. Protogel-stabilized acrylamide solution was from National Diagnostics (Atlanta, GA). Nitrocellulose membrane (0.45 μm) was from Bio-Rad. Polyvinylidene difluoride membrane (Immobilon-P, 0.45 μm) was purchased from Millipore (Bedford, MA).
Antibodies
The following antibodies were obtained from Cell Signaling (MA): LC3B rabbit polyclonal Ab; phospho-mTOR (Ser-2448) rabbit polyclonal Ab; phospho-mTOR (Ser-2481) rabbit polyclonal Ab; total mTOR rabbit polyclonal Ab; phospho-4E-BP1 (Thr-37/46) (236B4) rabbit monoclonal Ab; phospho-Akt (Ser-473) (587F11) mouse monoclonal Ab; total Akt (5G3) mouse monoclonal Ab; phospho-p70 S6 kinase (Thr-389) (108D2) rabbit monoclonal Ab; total p70 S6 kinase rabbit polyclonal Ab; Beclin-1 rabbit polyclonal Ab; phospho-AMPKα (Thr-172) rabbit polyclonal Ab; and total AMPKα rabbit polyclonal Ab. Mouse anti-Bcl-2 monoclonal Ab was purchased from BD Transduction Laboratories. The rabbit polyclonal calnexin Ab has been described previously (22).
DT40 Cell Culture and Incubation Conditions
Wild-type (WT) DT40 cells and IP3R triple knock-out (TKO) cells were the kind gifts of Dr. T. Kurosaki (Kansai Medical University, Moriguchi, Japan). Stable DT40 cells expressing the rat type I IP3R were a gift from Dr. Kevin Foskett (University of Pennsylvania). DT40 cells were grown in RPMI 1640 media supplemented with 10% fetal bovine serum, 1% chicken serum, 100 units/ml penicillin, 100 units/ml streptomycin and maintained at 37 °C in 5% CO2 atmosphere. The stable cell line containing the inactivating D2550A pore mutation was prepared as described previously (12). DT40 cells were seeded at a density of 104 cells/ml and were used for experiments after 24 h. There were no significant differences in the growth rate of the individual cell lines (data not shown).
Measurement of Autophagy
The DT40 cells were treated with staurosporine or rapamycin and then centrifuged at 1,000 × g for 10 min. The cells were washed once in ice-cold phosphate-buffered saline and then lysed in a medium containing 1% Triton X-100 and 50 mm Tris/HCl, pH 7.8, 150 mm NaCl, 2 mm sodium orthovanadate, 10 mm sodium pyrophosphate, 20 mm NaF, 5 nm okadaic acid, and a 1× dilution of a complete protease inhibitor mixture (Roche Diagnostics). The lysates were cleared by centrifugation at 10,000 × g. Protein samples (25 μg) were run on 15% polyacrylamide gels and transferred on polyvinylidene difluoride membranes. Immunoblotting was done using the LC3B antibody. Only the lower band of the LC3 doublet (LC3-II) was quantitated because variable immunodetection and efficiency of transfer of the upper band (LC3-I) has been observed (23). Electron microscopy of DT40 cells was carried out as described by Csordas et al. (24).
RESULTS
Autophagy was measured by immunoblotting for the microtubule-associated light chain 3 (LC3) protein. The covalent modification of the cytosolic LC3-I form with phosphatidylethanolamine generates the LC3-II form, which migrates more rapidly on SDS-PAGE (25). The specific association of LC3-II with autophagosomes has led to this protein being widely used as a marker of autophagy (23). Fig. 1A shows a comparison of LC3-II levels detected by immunoblotting in lysates from wild-type and TKO DT40 cells grown under nutrient-replete conditions. The data show that basal steady-state levels of LC3-II are low in wild-type cells but are ∼4-fold elevated in the TKO cells (Fig. 1B). The induction of apoptosis with staurosporine (STS) over a 6-h period enhanced the levels of LC3-II in wild-type cells and decreased the elevated levels of LC3-II in TKO cells. To determine whether the Ca2+ channel function of IP3Rs was important in mediating its inhibitory effects on autophagy, we utilized a stable DT40 cell line expressing the “pore-dead” D2550A mutant of the rat type I IP3R (26). The expression level of type I IP3R in the mutant has previously been shown to be comparable with wild-type cells (12). The pore-dead cell line also showed elevated levels of LC3-II that were decreased by STS treatment as observed with the TKO cells (Fig. 1, A and B). The wild-type DT40 cells used as a control contain all three chicken IP3R isoforms. Levels of LC3-II in pore-dead cells were also elevated when compared with a stable DT40 cell line expressing the wild-type rat type I IP3R (Fig. 1C). We conclude that the channel function of the IP3R is required for suppressing autophagy.
FIGURE 1.

Lack of functional IP3Rs in DT40 cells is associated with higher levels of basal autophagy. A, WT, IP3R TKO, and the D2550A pore-inactive (pore-dead) DT40 cells lines were incubated in nutrient-replete medium with serum in the presence or absence of staurosporine (1 μm) or bafilomycin A (10 nm) for 6 h. After treatment, the cells were lysed and processed for immunoblotting with LC3 Ab as described under “Materials and Methods.” The same samples were processed on 5% SDS-PAGE to monitor the levels of calnexin used as a loading control. B, levels of the LC3-II band were quantitated densitometrically and normalized to the calnexin levels. The data shown are the mean ± S.E. of 3–5 independent experiments. C, DT40 cell line stably expressing the wild-type rat type I IP3R (type I) and the functionally inactive D2550A mutant in the rat type I background (pore-dead) were directly compared for basal levels of LC3. D, WT, TKO, and pore-dead cell lines were incubated with 10 nm bafilomycin (Baf) A for the indicated times and processed for LC3 immunoblotting as described above.
It has been pointed out that measurements of the steady-state levels of LC3-II may not reflect autophagic flux because the levels are also determined by the degradation of LC3-II upon fusion of autophagosomes with lysosomes (27, 28). Autophagosome/lysosome fusion can be prevented by bafilomycin (29). A 6-h incubation with 10 nm bafilomycin caused a large enhancement in LC3-II levels such that no differences between WT and TKO cells could be distinguished, although the inhibitory effect of STS on LC3-II levels in the TKO cells was still observed (Fig. 1B). Shorter periods of treatment with bafilomycin, which produced submaximal elevations of LC3-II (e.g. after 1 h), still revealed differences between wild-type and TKO cells (Fig. 1D). The basic findings using LC3 immunoblotting were confirmed by analysis of electron microscope images of DT40 cells that showed an enhanced number of debris-containing vacuoles in TKO cells when compared with either wild-type or TKO cells stably expressing the rat type I IP3R (supplemental Fig. S1). These data in DT40 cells are in agreement with previous findings in other cell types that knockdown or inhibition of IP3Rs enhances autophagic flux (17).
Fig. 2 depicts the canonical pathway of autophagy together with known sites that could potentially be regulated by Ca2+ based on information in the literature. Because IP3Rs have an established role in supplying Ca2+ to the mitochondria, which is known to stimulate mitochondrial oxidative metabolism, it is possible that IP3R-deficient DT40 cells might have an increased AMP/ATP ratio. This, in turn, could enhance the activity of AMPK that would stimulate autophagy through the intermediary steps shown in Fig. 2. Calmodulin-dependent kinase kinase-β is an upstream activator of AMPK and has also been proposed to be a site where cytosolic Ca2+ elevations could stimulate autophagy (15). We therefore compared the activation state of AMPK in wild-type, TKO, and pore-dead DT40 cell lines by measuring phosphorylation of the protein at Thr-172 using a phospho-specific Ab. This rabbit polyclonal Ab recognized a doublet of bands in the chicken cells (Fig. 3) of which only the lower band was recognized by the pan-α-subunit AMPK Ab at the appropriate molecular mass of ∼63 kDa. This AMPK band was not significantly different between wild-type, TKO, and pore-dead cells. STS did not affect the phosphorylation state of the AMPK band but substantially decreased the phosphorylation of the upper unknown band in all three cell lines.
FIGURE 2.

Possible sites of regulation of the canonical pathway of autophagy by IP3 receptors and Ca2+. The middle box shows several of the molecular complexes that participate in autophagy. The pathways by which physiological factors such as nutrient availability and growth factors (indicated in red) modulate autophagy are shown schematically. Possible sites at which IP3Rs or elevated cytosolic Ca2+ could also modulate this pathway are indicated in blue. The specific mechanisms are as follows: 1, transfer of Ca2+ from the ER to the mitochondria mediated by IP3Rs could contribute to the maintenance of an elevated “energy state” (high ATP/AMP ratio); 2, Ca2+ acting through calmodulin-dependent kinase kinase-β (CaMKK-β) can activate AMPK (15); 3, Vps34 has also been suggested to be a CaM-dependent enzyme (46); 4, Bcl-2, by binding IP3Rs, can influence the amount bound to the Beclin-1-Vps34 complex (37) ; and 5, activation of calpains has been proposed to inhibit autophagy (16). The only target of calpains in the autophagic pathway that has been described is Atg5 (63). Abbreviations used: FIP200, focal adhesion kinase-interacting protein; Ulk, Unc51-like kinase; TSC, tuberous sclerosis complex protein; Rheb, Ras homology enriched in brain protein; S6K-1, p70 ribosomal protein S6 kinase-1; 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1.
FIGURE 3.
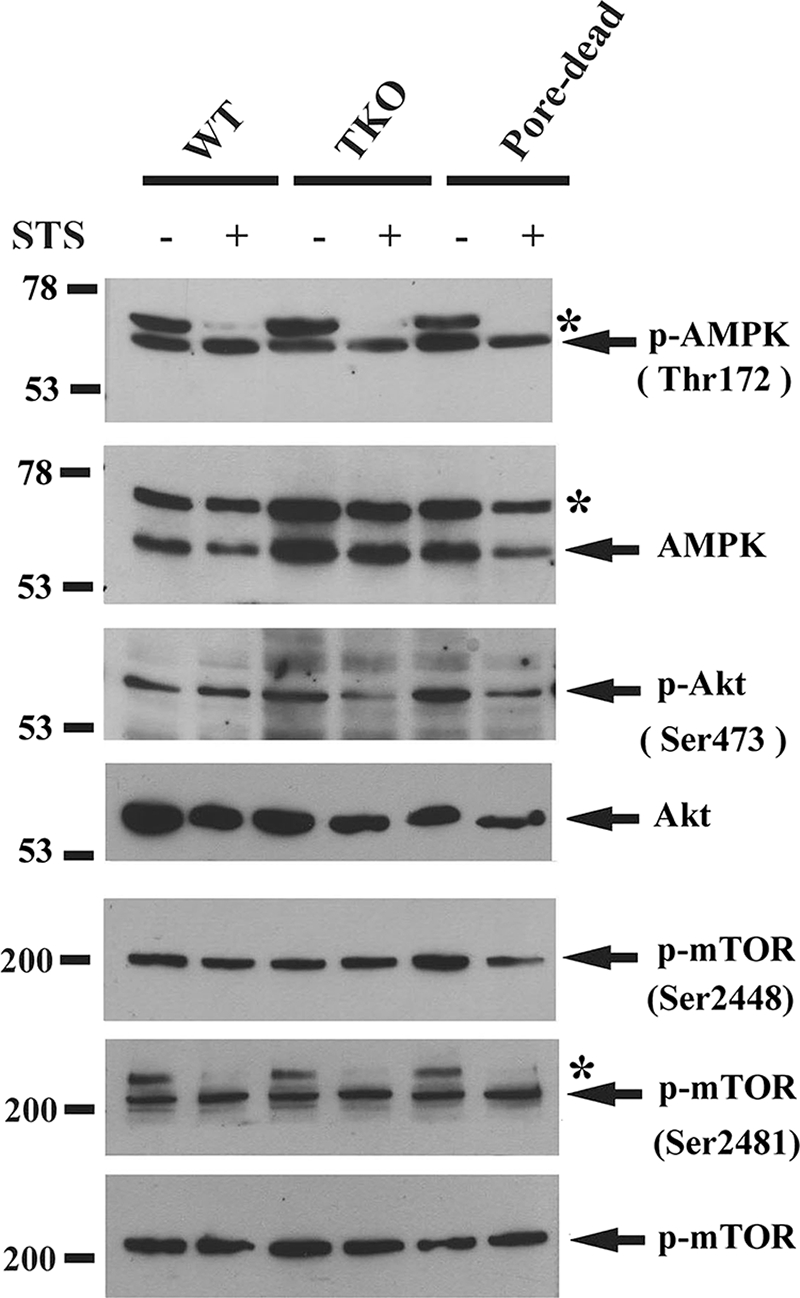
Autophagy is independent of the phosphorylation state of AMPK, Akt, or mTOR. WT, IP3R TKO, and the D2550A pore-inactive (pore-dead) DT40 cells lines were incubated in nutrient-replete medium with serum in the presence or absence of staurosporine (1 μm) for 6 h. After treatment, the cells were lysed in a buffer containing protein phosphatase inhibitors and processed for immunoblotting with the indicated phospho-specific Abs as described under “Materials and Methods.” The data shown is representative of three experiments. * indicates an unknown band.
Activated Akt kinase inhibits autophagy (Fig. 2) (30). Analysis of the phosphorylation state of Akt kinase using phospho-specific Ser-473 Ab indicates a modest increase in activation of Akt in TKO and pore-dead cells. However, these changes are in the opposite direction to account for the stimulated autophagy in these cell lines. STS decreased Ser-437 phosphorylation in TKO and pore-dead cell lines but not in wild-type cells. These changes also do not correlate with the different effects of STS on autophagy in these cell lines. The effects of Akt and AMPK on autophagy are mediated through a pathway that involves TSC and Rheb proteins that regulate the activity of the mTORC1 complex (Fig. 2). High levels of mTOR kinase suppress autophagy by mechanisms that have not been fully characterized but include the phosphorylation of Atg1/Ulk kinases that are involved in the induction of autophagosomes (31). We examined the following two phosphorylation sites on mTOR: Ser-2448 (a site for phosphorylation by several kinases, including Akt (32)) and Ser-2481 (an autophosphorylation site (33)). The basal phosphorylation state of neither site was different in the three different DT40 cell lines (Fig. 3). However, mutation of serines 2448 or 2481 does not alter mTOR kinase activity (32), which is conventionally measured by monitoring the phosphorylation of its substrates. One such substrate is p70 S6 kinase, which is specifically phosphorylated by mTOR on Thr-389 (34). Fig. 4, A and B, shows that there are substantially lower levels of phosphorylated S6 kinase in the TKO and pore-dead cells. We confirmed this finding by examining another mTOR substrate 4E-BP1 using an Ab directed at Ser-65 (35). As with S6 kinase, a lower phosphorylation of 4E-BP1 was observed in TKO and pore-dead cells (Fig. 4A). The phosphorylation state of S6 kinase could be restored by stable transfection of the rat type I IP3R into TKO cells and remained elevated in a DT40 cell line containing only the endogenous chicken type I isoform (DKO-1) (Fig. 4C). We suggest that a lower mTOR kinase activity in the IP3R-deficient cell lines could account for their higher basal autophagy.
FIGURE 4.

Higher autophagic flux in IP3R TKO and pore-dead cell lines is associated with a reduced activity of mTOR. A, WT, IP3R TKO, and the D2550A pore-inactive (pore-dead) DT40 cells lines were incubated in nutrient-replete medium with serum in the presence or absence of staurosporine (1 μm) for 6 h. After treatment, the cells were lysed in a buffer containing protein phosphatase inhibitors and processed for immunoblotting with the indicated Abs against p70 phospho-S6 kinase (S6K) and 4E-BP1, two key substrates of the mTORC1 kinase complex. The data shown are representative of three experiments. B, data from three experiments were quantitated with densitometry and are shown as the means ± S.E. with the wild-type levels set to 100%. C, amount of phospho- and total S6 kinase was measured in lysates from wild-type and TKO DT40 cells as well as cells expressing the chicken (DKO-1) or rat type 1 IP3Rs.
We examined the effects of rapamycin, an inhibitor of mTOR (36), on the levels of LC3-II in wild-type and TKO cells (Fig. 5). In both cell lines, 100 nm rapamycin completely inhibited mTOR activity within 2 h as judged by inhibition of phosphorylation of Thr-389 of S6 kinase (Fig. 5A). Rapamycin produced a smaller and slower decrease in Ser-2448 phosphorylation of mTOR itself, consistent with this site being a relatively insensitive indicator of mTOR kinase activity. Rapamycin elevated the levels of LC3-II in wild-type cells with a peak increase at 4 h, which averaged almost 80% of the elevated levels observed in untreated TKO cells (Fig. 5B). Rapamycin addition to TKO cells caused only a small further increase in LC3-II levels. The results with rapamycin are consistent with the hypothesis that differences in the activity of the mTOR kinase pathway are an important determinant in setting the different levels of basal autophagy observed in wild-type and TKO cells.
FIGURE 5.

Effect of mTORC1 inhibition with rapamycin on autophagy in IP3R wild-type and TKO cells. A, wild-type and TKO DT40 cells were incubated with 100 nm rapamycin for the indicated times. Cell-free lysates were immunoblotted for LC3, phosphorylated mTOR, and S6 kinase and total mTOR, and S6 kinase. The immunoblots shown are representative of three experiments. B, is the quantitation of the levels of LC3-II at the 2-h time point with the levels of the untreated wild-type cells used as the reference control (100%).
Another potential site of autophagy regulation by IP3Rs has been proposed to involve Beclin-1 (37), which stimulates the activity of the class III phosphatidylinositol 3-kinase Vps34 (31, 38). This enzyme has been directly implicated in the formation of autophagosomes from ER membranes (39) and also positively modulates mTOR activity (38). Bcl-2, which inhibits autophagy by sequestering Beclin-1, can also bind to IP3Rs (40). Thus, the presence or absence of IP3Rs could indirectly regulate the amount of Beclin-1-Vps34 complex. This was examined in the experiments shown in Fig. 6. The total levels of Bcl-2 or Beclin-1 were not different in wild-type or TKO cells in the presence or absence of STS (Fig. 6A). Detecting the coimmunoprecipitation of Beclin-1 by Bcl-2 Ab was complicated by the close proximity of the Beclin-1 signal to the heavy chain of IgG. Using a method that minimizes this problem, we found no differences in Bcl-2-Beclin-1 complexes between wild-type and TKO cells (Fig. 6B). The proportion of Beclin-1 complexed with Vps34, determined by immunoprecipitation with Beclin-1 Ab, was also not different in the two cell lines. There was also no evidence that IP3Rs were directly or indirectly complexed with Vps34 (Fig. 6C).
FIGURE 6.
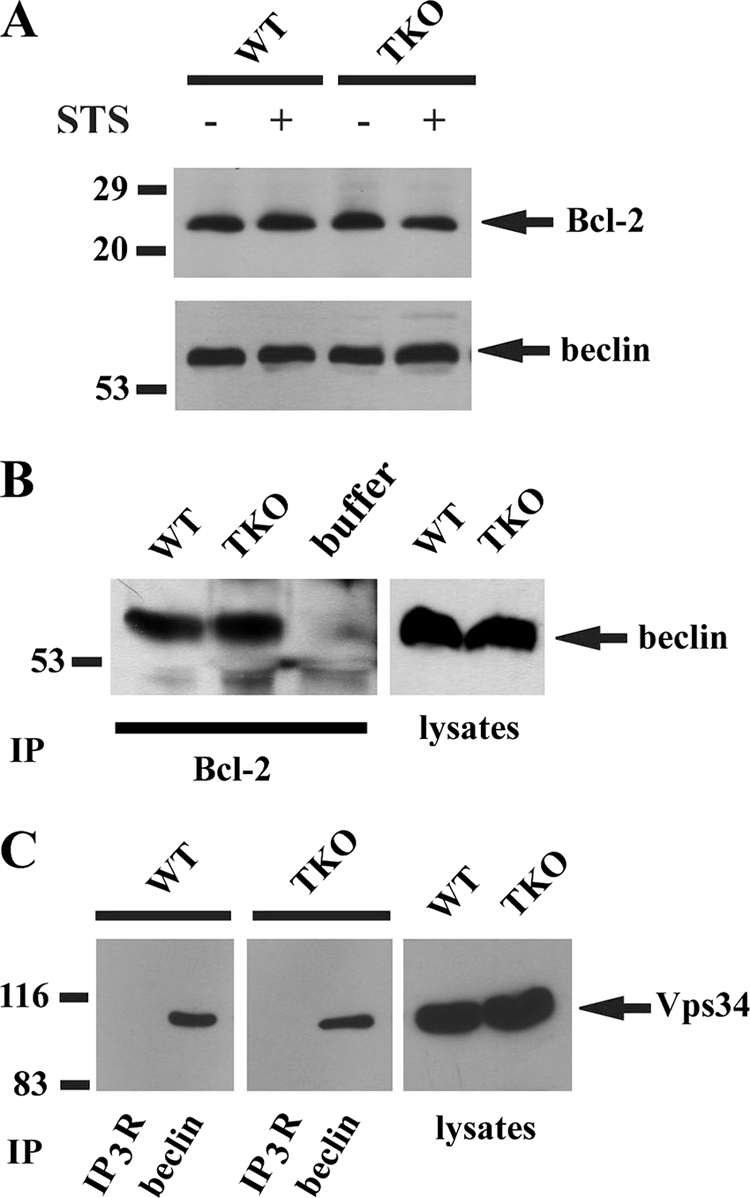
Complex formation of the autophagy regulator Beclin-1 with Bcl-2 and Vps34 in IP3R wild-type and TKO cells. A, levels of Bcl-2 and Beclin-1 in wild-type (WT) and TKO DT40 cell lysates (30 μg) were measured by immunoblotting. B, WT and TKO DT40 cell lysates were immunoprecipitated (IP) with a monoclonal Bcl-2 Ab, and the immunoprecipitates were probed by immunoblotting for the presence of Beclin-1. Immunoprecipitation and immunoblotting were carried out according to the instructions for the TrueBlot kit (eBiosciences, San Diego), which minimizes detection of the IgG band that runs just below the Beclin-1 band. A sample containing buffer alone and Bcl-2 mAb was used to verify the effectiveness of the method. C, Abs to IP3R and Beclin-1 were used to immunoprecipitate 0.5 mg of WT and TKO DT40 cell lysate. The immunoprecipitates were probed by immunoblotting for the presence of Vps34. All data are representative of 2 or 3 experiments.
A mechanism that would be consistent with our experimental observations is that mTORC1 activity in intact cells is dependent on Ca2+ signaling. In unstimulated DT40 cells, the loading of BAPTA-AM is sufficient to enhance LC3-II levels in both wild-type and TKO cells, which is consistent with resting intracellular Ca2+ exerting a tonic negative effect on autophagy (Fig. 7A). Loading with BAPTA-AM also decreased the basal phosphorylation state of S6 kinase in both wild-type and TKO cells (Fig. 7B) confirming observations made in other cell types (41, 42). In wild-type DT40 cells, the addition of thapsigargin to mobilize intracellular Ca2+ stores caused a robust and rapid increase in phospho-S6 kinase levels (>3-fold in 2 min), and levels were entirely suppressed by pretreatment with rapamycin (Fig. 7C). By contrast, thapsigargin did not cause significant changes in pS6K levels in TKO cells over a 10-min period (Fig. 7C). Because the discharge of intracellular Ca2+ stores mediated by thapsigargin is of similar kinetics and magnitude in both cell lines (21), it must be concluded that the mTORC1 complex has a diminished Ca2+ sensitivity in the TKO cells. The possibility that the localization of mTOR protein may be an important factor in these experiments was explored in Fig. 7D. The data show that a large fraction of mTOR was localized in the microsomal membranes, consistent with previous reports that ER localization signals are found in the HEAT domains of mTOR (43). However, localization was not significantly different in WT or TKO cells (Fig. 7D). In addition, localization was not altered by treatment with thapsigargin or A23187 and no evidence for co-immunoprecipitation of mTOR with type I IP3R was obtained (data not shown).
FIGURE 7.

Effect of BAPTA-AM and thapsigargin on mTORC1 activity. A, DT40 cells were incubated with the indicated concentrations of BAPTA-AM for 6 h, and the levels of LC3-II were measured by immunoblotting and quantitated in three separate experiments as shown in the bar graph. B, corresponding changes in the levels of phospho-S6 (p-S6K) kinase and total S6 kinase (S6K) from experiments carried out as in A were measured by immunoblotting. C, changes in the amount of phospho-S6 kinase was measured in DT40 cells after addition of 2 μm thapsigargin (TG). Cell lysates were prepared at the indicated times after thapsigargin treatment. When added, rapamycin (Rap, 50 nm) was preincubated with the cells for 15 min. D, DT40 cells were disrupted by five passes through a 26-gauge needle and centrifuged at 2000 × g for 5 min to remove intact cells. The total homogenate (T) was then centrifuged at 100,000 × g to obtain a membrane (M) and soluble (S) fraction. Equal protein (20 μg) from each fraction was then run on 7% SDS-PAGE and immunoblotted for mTOR. The distribution of ER protein was monitored with calnexin Ab.
DISCUSSION
In this study, we have documented an enhanced autophagic flux in DT40 cell lines in which IP3Rs are absent or are nonfunctional. We hypothesize that the normal responses of wild-type cells to many environmental cues, including growth factors and nutrients, may require periodic elevations of cytosolic Ca2+ that would be severely impaired in cell lines lacking functional IP3Rs. Thus, the TKO/pore-dead cells may exhibit behavior that is characteristic of nutrient insufficiency. In agreement with this, the autophagic response of TKO cells to removal of amino acids from the culture medium was diminished when compared with wild-type cells (data not shown). The dependence of autophagy on IP3Rs is not a unique characteristic of DT40 cells because it has been observed in other cell types in which type I or type III IP3R isoforms have been knocked down by small interfering RNA methods or when channel function was inhibited by xestospongin B (17).
There are several potential sites at which the IP3R protein (or the Ca2+ it translocates) could regulate the canonical autophagic pathway (Fig. 2). The energy status of the cell, sensed by AMPK, is a prime regulator of autophagy. The shuttling of Ca2+ from the ER to the mitochondria is a key function of IP3Rs. The absence of IP3Rs could potentially modify the AMP/ATP ratio of the cell and secondarily influence AMPK activity. In an experimental model where chronic elevation of Ca2+ stimulates autophagy, it has been suggested that Ca2+/calmodulin-dependent kinase kinase-β, an upstream regulator of AMPK phosphorylation, may be involved (15). However, we did not observe any differences in the phosphorylation state of AMPK in wild-type and TKO DT40 cells. Growth factors such as insulin that inhibit autophagy act through Akt kinase. Again, we observed no differences in the phosphorylation state of Akt. The anti-apoptotic protein Bcl-2 inhibits autophagy when specifically targeted to ER membranes (17). The inhibition of autophagy is thought to result from the binding of Bcl-2 to Beclin-1, which is a regulator of Vps34, a class III phosphatidylinositol 3-kinase. Because IP3Rs also bind Bcl-2, it has been suggested that the effects of IP3R knockdown or IP3R inhibition with xestospongin B may be related to an altered availability of Bcl-2 (17, 37). However, we observed no differences in beclin-1-Vps34 complexes in wild-type and TKO cells. In addition, the absence of IP3Rs should make more Bcl-2 available for inhibiting autophagy, whereas a stimulation of autophagy is what is experimentally observed.
The main difference between wild-type and TKO cells that could account for the differences in autophagic flux is the reduced basal activity of the mTORC1 complex in TKO cells, as measured by a reduced phosphorylation of its substrates S6 kinase and 4E-BP1. There is evidence in the literature to suggest that mTORC1 activity in intact cells is regulated by Ca2+. Several studies have reported that enzyme activity is enhanced by thapsigargin or Ca2+-mobilizing stimuli and is inhibited by BAPTA-AM loading of cells (41, 42, 44, 45). The absence of IP3Rs in TKO cells would be expected to diminish Ca2+ signals that act to maintain mTORC1 activity. However, the availability of Ca2+ is not the only factor that is altered because the response to Ca2+ mobilized by thapsigargin is also lost in TKO cells (Fig. 7). This suggests that Ca2+ regulation of the mTORC1 complex is somehow different in TKO cells. Unfortunately, the mechanism(s) by which Ca2+ regulates this enzyme are not well characterized. One possibility is that the components of the mTORC1 complex are assembled or localized differently in the IP3R-deficient cells. Although we noted no differences in the membrane localization of the mTOR protein in TKO cells, it is possible that the other proteins associated with the mTORC1 complex may be targeted differently (e.g. Raptor, mLST8, and Rheb). mTORC1 activity is positively regulated by Vps34, which has been proposed to be activated by Ca2+/CaM (46, 47). Therefore, an alternative possibility is that a decreased activity of mTORC1 in TKO cells is secondary to a decreased activity of the pool of Vps34 involved in regulating autophagy. A more direct role for Vps34 in autophagy has been suggested by recent studies indicating that Vps34 is recruited to specific ER structures that form a platform for the assembly of autophagosomes (39). Thus, it is possible that the localized release of Ca2+ from IP3Rs could directly impede some early ER-dependent step(s) in autophagosome biogenesis.
Previous studies that have examined a role for IP3 or Ca2+ in autophagy have concluded that the regulatory effects are independent of mTOR activation. This was found for lithium (which induces autophagy by a mechanism involving depletion of IP3 levels (48)), for xestospongin-B (37), and for pharmacological compounds that induce autophagy by a mechanism involving inhibition of calpain activity (16). The status of the mTOR pathway was not examined in studies where autophagy was induced by small interfering RNA knockdown of IP3Rs (17). Infection by Toxoplasma gondii stimulates autophagy in the host cell by a mechanism involving Ca2+ elevation without an associated change in mTOR signaling (49). The presence of multiple regulatory mechanisms is not surprising in view of the complexity of the autophagic pathway and the differences in cell types and stimuli.
Recently, Vicencio et al. (37) reported no difference in autophagy between wild-type and TKO DT40 cells. Because knockdown or inhibition of IP3Rs has previously been shown to enhance autophagy (17), the lack of a difference in autophagy between the two cell lines is surprising and was attributed to an adaptive alteration of the TKO DT40 cells or to the continued presence of the ligand binding domain of the type III IP3R in the TKO cells (50). The latter explanation seems unlikely because a functionally inactive IP3R fragment would not be expected to support autophagy based on the experiments with the defective pore mutant. The basis for these discrepant results is not known. We have noted that basal autophagy in wild-type cells is enhanced as the cell density increases with days in culture (data not shown). Thus, differences in autophagy between wild-type and TKO cells may be strongly influenced by the exact growth stage of the cells. In agreement with our results, Cardenas et al. (51) have also reported accelerated autophagy in TKO DT40 cells, although in their study they attributed this to an altered AMPK activity.
Autophagy and apoptosis are interrelated processes (52–54). There are many examples where enhanced autophagy protects cells from apoptotic stimuli (55–57). The enhanced autophagic flux in TKO cells could therefore contribute to the delayed kinetics of cell death observed with several inducers of apoptosis in these cells (12, 21, 58). Activation or inhibition of apoptosis can also influence autophagy (52–54). In wild-type DT40 cells the addition of STS stimulates both apoptosis and autophagy. The enhancement of autophagy by STS has been observed in various cell lines, but the underlying mechanism is unknown (59). Presumably, the action of STS as a protein kinase inhibitor could lead to a decreased phosphorylation of key proteins in the autophagic pathway that are regulated by phosphorylation, e.g. the complex of the mammalian homologues of Atg1 (Ulk1/2)/Atg13/FIP200 (49), Bcl-2 (60), or Beclin-1 (61). In addition, the activation of pro-apoptotic proteins, such as Bad, could diminish the Bcl-2-Bclxl complexed to Beclin-1 (62). In contrast to the stimulation of autophagy seen in wild-type cells, the addition of STS inhibited the elevated levels of autophagy seen in TKO cells. This suggests that regulation by STS is complex, and its stimulatory or inhibitory mode of action must be influenced by the availability of Ca2+ signaling pathways. Ca2+ elevation has been suggested to either stimulate (15) or inhibit autophagy (16). Our results are more consistent with Ca2+ having an inhibitory role. In addition to the calmodulin- and calpain-dependent mechanisms mentioned previously, the possible roles of Ca2+-dependent protein kinases or protein phosphatases have not been investigated. A more complete understanding of the role of Ca2+ will require further studies to assess the relative importance of potential Ca2+ sensors and their sites of action in the autophagic pathway.
Supplementary Material

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- IP3R
- inositol 1,4,5-trisphosphate receptor
- mTOR
- mammalian target of rapamycin
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- ER
- endoplasmic reticulum
- Ab
- antibody
- STS
- staurosporine
- TKO
- triple knock-out
- WT
- wild type
- AMPK
- AMP kinase.
REFERENCES
- 1.Orrenius S., Zhivotovsky B., Nicotera P. (2003) Nat. Rev. Mol. Cell Biol. 4, 552–565 [DOI] [PubMed] [Google Scholar]
- 2.Joseph S. K., Hajnóczky G. (2007) Apoptosis 12, 951–968 [DOI] [PubMed] [Google Scholar]
- 3.Pinton P., Giorgi C., Siviero R., Zecchini E., Rizzuto R. (2008) Oncogene 27, 6407–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szalai G., Krishnamurthy R., Hajnóczky G. (1999) EMBO J. 18, 6349–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rong Y. P., Aromolaran A. S., Bultynck G., Zhong F., Li X., McColl K., Matsuyama S., Herlitze S., Roderick H. L., Bootman M. D., Mignery G. A., Parys J. B., De Smedt H., Distelhorst C. W. (2008) Mol. Cell 31, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C., Wang X., Vais H., Thompson C. B., Foskett J. K., White C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12565–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehning D., Patterson R. L., Sedaghat L., Glebova N. O., Kurosaki T., Snyder S. H. (2003) Nat. Cell Biol. 5, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 8.Boehning D., van Rossum D. B., Patterson R. L., Snyder S. H. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan M. T., Wagner L., 2nd, Yule D. I., Bhanumathy C., Joseph S. K. (2006) J. Biol. Chem. 281, 3731–3737 [DOI] [PubMed] [Google Scholar]
- 10.Szado T., Vanderheyden V., Parys J. B., De Smedt H., Rietdorf K., Kotelevets L., Chastre E., Khan F., Landegren U., Söderberg O., Bootman M. D., Roderick H. L. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchi S., Rimessi A., Giorgi C., Baldini C., Ferroni L., Rizzuto R., Pinton P. (2008) Biochem. Biophys. Res. Commun. 375, 501–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan M. T., Bhanumathy C. D., Schug Z. T., Joseph S. K. (2007) J. Biol. Chem. 282, 32983–32990 [DOI] [PubMed] [Google Scholar]
- 13.Eskelinen E. L. (2008) Int. Rev. Cell Mol. Biol. 266, 207–247 [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N., Klionsky D. J. (2007) Annu. Rev. Nutr. 27, 19–40 [DOI] [PubMed] [Google Scholar]
- 15.Høyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., Bianchi K., Fehrenbacher N., Elling F., Rizzuto R., Mathiasen I. S., Jäättelä M. (2007) Mol. Cell 25, 193–205 [DOI] [PubMed] [Google Scholar]
- 16.Williams A., Sarkar S., Cuddon P., Ttofi E. K., Saiki S., Siddiqi F. H., Jahreiss L., Fleming A., Pask D., Goldsmith P., O'Kane C. J., Floto R. A., Rubinsztein D. C. (2008) Nat. Chem. Biol. 4, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Criollo A., Maiuri M. C., Tasdemir E., Vitale I., Fiebig A. A., Andrews D., Molgó J., Díaz J., Lavandero S., Harper F., Pierron G., di Stefano D., Rizzuto R., Szabadkai G., Kroemer G. (2007) Cell Death Differ. 14, 1029–1039 [DOI] [PubMed] [Google Scholar]
- 18.Gordon P. B., Holen I., Fosse M., Røtnes J. S., Seglen P. O. (1993) J. Biol. Chem. 268, 26107–26112 [PubMed] [Google Scholar]
- 19.Brady N. R., Hamacher-Brady A., Yuan H., Gottlieb R. A. (2007) FEBS J. 274, 3184–3197 [DOI] [PubMed] [Google Scholar]
- 20.Lam D., Kosta A., Luciani M. F., Golstein P. (2008) Mol. Biol. Cell 19, 691–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugawara H., Kurosaki M., Takata M., Kurosaki T. (1997) EMBO J. 16, 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joseph S. K., Boehning D., Bokkala S., Watkins R., Widjaja J. (1999) Biochem. J. 342, 153–161 [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N., Yoshimori T. (2007) Autophagy 3, 542–545 [DOI] [PubMed] [Google Scholar]
- 24.Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., Hajnóczky G. (2006) J. Cell Biol. 174, 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. (2004) J. Cell Sci. 117, 2805–2812 [DOI] [PubMed] [Google Scholar]
- 26.Boehning D., Joseph S. K. (2000) J. Biol. Chem. 275, 21492–21499 [DOI] [PubMed] [Google Scholar]
- 27.Tanida I., Minematsu-Ikeguchi N., Ueno T., Kominami E. (2005) Autophagy 1, 84–91 [DOI] [PubMed] [Google Scholar]
- 28.Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Aliev G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A., Bamber B. A., Bassham D. C., Bergamini E., Bi X., Biard-Piechaczyk M., Blum J. S., Bredesen D. E., Brodsky J. L., Brumell J. H., Brunk U. T., Bursch W., Camougrand N., Cebollero E., Cecconi F., Chen Y., Chin L. S., Choi A., Chu C. T., Chung J., Clarke P. G., Clark R. S., Clarke S. G., Clavé C., Cleveland J. L., Codogno P., Colombo M. I., Coto-Montes A., Cregg J. M., Cuervo A. M., Debnath J., Demarchi F., Dennis P. B., Dennis P. A., Deretic V., Devenish R. J., Di Sano F., Dice J. F., Difiglia M., Dinesh-Kumar S., Distelhorst C. W., Djavaheri-Mergny M., Dorsey F. C., Dröge W., Dron M., Dunn W. A., Jr., Duszenko M., Eissa N. T., Elazar Z., Esclatine A., Eskelinen E. L., Fésüs L., Finley K. D., Fuentes J. M., Fueyo J., Fujisaki K., Galliot B., Gao F. B., Gewirtz D. A., Gibson S. B., Gohla A., Goldberg A. L., Gonzalez R., González-Estévez C., Gorski S., Gottlieb R. A., Häussinger D., He Y. W., Heidenreich K., Hill J. A., Høyer-Hansen M., Hu X., Huang W. P., Iwasaki A., Jäättelä M., Jackson W. T., Jiang X., Jin S., Johansen T., Jung J. U., Kadowaki M., Kang C., Kelekar A., Kessel D. H., Kiel J. A., Kim H. P., Kimchi A., Kinsella T. J., Kiselyov K., Kitamoto K., Knecht E., Komatsu M., Kominami E., Kondo S., Kovács A. L., Kroemer G., Kuan C. Y., Kumar R., Kundu M., Landry J., Laporte M., Le W., Lei H. Y., Lenardo M. J., Levine B., Lieberman A., Lim K. L., Lin F. C., Liou W., Liu L. F., Lopez-Berestein G., López-Otín C., Lu B., Macleod K. F., Malorni W., Martinet W., Matsuoka K., Mautner J., Meijer A. J., Meléndez A., Michels P., Miotto G., Mistiaen W. P., Mizushima N., Mograbi B., Monastyrska I., Moore M. N., Moreira P. I., Moriyasu Y., Motyl T., Münz C., Murphy L. O., Naqvi N. I., Neufeld T. P., Nishino I., Nixon R. A., Noda T., Nürnberg B., Ogawa M., Oleinick N. L., Olsen L. J., Ozpolat B., Paglin S., Palmer G. E., Papassideri I., Parkes M., Perlmutter D. H., Perry G., Piacentini M., Pinkas-Kramarski R., Prescott M., Proikas-Cezanne T., Raben N., Rami A., Reggiori F., Rohrer B., Rubinsztein D. C., Ryan K. M., Sadoshima J., Sakagami H., Sakai Y., Sandri M., Sasakawa C., Sass M., Schneider C., Seglen P. O., Seleverstov O., Settleman J., Shacka J. J., Shapiro I. M., Sibirny A., Silva-Zacarin E. C., Simon H. U., Simone C., Simonsen A., Smith M. A., Spanel-Borowski K., Srinivas V., Steeves M., Stenmark H., Stromhaug P. E., Subauste C. S., Sugimoto S., Sulzer D., Suzuki T., Swanson M. S., Tabas I., Takeshita F., Talbot N. J., Tallóczy Z., Tanaka K., Tanaka K., Tanida I., Taylor G. S., Taylor J. P., Terman A., Tettamanti G., Thompson C. B., Thumm M., Tolkovsky A. M., Tooze S. A., Truant R., Tumanovska L. V., Uchiyama Y., Ueno T., Uzcátegui N. L., van der Klei I., Vaquero E. C., Vellai T., Vogel M. W., Wang H. G., Webster P., Wiley J. W., Xi Z., Xiao G., Yahalom J., Yang J. M., Yap G., Yin X. M., Yoshimori T., Yu L., Yue Z., Yuzaki M., Zabirnyk O., Zheng X., Zhu X., Deter R. L. (2008) Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R., Tashiro Y. (1998) Cell Struct. Funct. 23, 33–42 [DOI] [PubMed] [Google Scholar]
- 30.Degtyarev M., De Mazière A., Orr C., Lin J., Lee B. B., Tien J. Y., Prior W. W., van Dijk S., Wu H., Gray D. C., Davis D. P., Stern H. M., Murray L. J., Hoeflich K. P., Klumperman J., Friedman L. S., Lin K. (2008) J. Cell Biol. 183, 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattingre S., Espert L., Biard-Piechaczyk M., Codogno P. (2008) Biochimie 90, 313–323 [DOI] [PubMed] [Google Scholar]
- 32.Sekuliæ A., Hudson C. C., Homme J. L., Yin P., Otterness D. M., Karnitz L. M., Abraham R. T. (2000) Cancer Res. 60, 3504–3513 [PubMed] [Google Scholar]
- 33.Peterson R. T., Beal P. A., Comb M. J., Schreiber S. L. (2000) J. Biol. Chem. 275, 7416–7423 [DOI] [PubMed] [Google Scholar]
- 34.Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 35.Gingras A. C., Gygi S. P., Raught B., Polakiewicz R. D., Abraham R. T., Hoekstra M. F., Aebersold R., Sonenberg N. (1999) Genes Dev. 13, 1422–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corradetti M. N., Guan K. L. (2006) Oncogene 25, 6347–6360 [DOI] [PubMed] [Google Scholar]
- 37.Vicencio J. M., Ortiz C., Criollo A., Jones A. W., Kepp O., Galluzzi L., Joza N., Vitale I., Morselli E., Tailler M., Castedo M., Maiuri M. C., Molgó J., Szabadkai G., Lavandero S., Kroemer G. (2009) Cell Death Differ. 16, 1006–1017 [DOI] [PubMed] [Google Scholar]
- 38.Backer J. M. (2008) Biochem. J. 410, 1–17 [DOI] [PubMed] [Google Scholar]
- 39.Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., Griffiths G., Ktistakis N. T. (2008) J. Cell Biol. 182, 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rong Y. P., Barr P., Yee V. C., Distelhorst C. W. (2009) Biochim. Biophys. Acta 1793, 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graves L. M., He Y., Lambert J., Hunter D., Li X., Earp H. S. (1997) J. Biol. Chem. 272, 1920–1928 [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov D. D., Sabatini D. M. (2005) J. Biol. Chem. 280, 39505–39509 [DOI] [PubMed] [Google Scholar]
- 43.Liu X., Zheng X. F. (2007) Mol. Biol. Cell 18, 1073–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altamirano F., Oyarce C., Silva P., Toyos M., Wilson C., Lavandero S., Uhlén P., Estrada M. (2009) J. Endocrinol. 202, 299–307 [DOI] [PubMed] [Google Scholar]
- 45.Markova B., Albers C., Breitenbuecher F., Melo J. V., Brümmendorf T. H., Heidel F., Lipka D., Duyster J., Huber C., Fischer T. (2010) Oncogene 29, 739–751 [DOI] [PubMed] [Google Scholar]
- 46.Gulati P., Gaspers L. D., Dann S. G., Joaquin M., Nobukuni T., Natt F., Kozma S. C., Thomas A. P., Thomas G. (2008) Cell Metab. 7, 456–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan Y., Flinn R. J., Wu H., Schnur R. S., Backer J. M. (2009) Biochem. J. 417, 747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarkar S., Floto R. A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L. J., Rubinsztein D. C. (2005) J. Cell Biol. 170, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y., Weiss L. M., Orlofsky A. (2009) J. Biol. Chem. 284, 1694–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guillemette J., Caron A. Z., Regimbald-Dumas Y., Arguin G., Mignery G. A., Boulay G., Guillemette G. (2005) Cell Calcium 37, 97–104 [DOI] [PubMed] [Google Scholar]
- 51.Cardenas C., Cheung K. H., Yang J., Vais H., Foskett J. K. (2008) J. Gen. Physiol. 132, 24a [Google Scholar]
- 52.Maiuri M. C., Zalckvar E., Kimchi A., Kroemer G. (2007) Nat. Rev. Mol. Cell Biol. 8, 741–752 [DOI] [PubMed] [Google Scholar]
- 53.Ferraro E., Cecconi F. (2007) Arch. Biochem. Biophys. 462, 210–219 [DOI] [PubMed] [Google Scholar]
- 54.Thorburn A. (2008) Apoptosis 13, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang C., Kaushal V., Shah S. V., Kaushal G. P. (2008) Am. J. Physiol. Renal Physiol. 294, F777–F787 [DOI] [PubMed] [Google Scholar]
- 56.Wu Y. T., Tan H. L., Huang Q., Kim Y. S., Pan N., Ong W. Y., Liu Z. G., Ong C. N., Shen H. M. (2008) Autophagy 4, 457–466 [DOI] [PubMed] [Google Scholar]
- 57.Abedin M. J., Wang D., McDonnell M. A., Lehmann U., Kelekar A. (2007) Cell Death Differ. 14, 500–510 [DOI] [PubMed] [Google Scholar]
- 58.Assefa Z., Bultynck G., Szlufcik K., Nadif, Kasri N., Vermassen E., Goris J., Missiaen L., Callewaert G., Parys J. B., De Smedt H. (2004) J. Biol. Chem. 279, 43227–43236 [DOI] [PubMed] [Google Scholar]
- 59.Ciechomska I. A., Goemans C. G., Tolkovsky A. M. (2008) Methods Mol. Biol. 445, 175–193 [DOI] [PubMed] [Google Scholar]
- 60.Pattingre S., Bauvy C., Carpentier S., Levade T., Levine B., Codogno P. (2009) J. Biol. Chem. 284, 2719–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zalckvar E., Berissi H., Eisenstein M., Kimchi A. (2009) Autophagy 5, 720–722 [DOI] [PubMed] [Google Scholar]
- 62.Levine B., Sinha S., Kroemer G. (2008) Autophagy 4, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yousefi S., Perozzo R., Schmid I., Ziemiecki A., Schaffner T., Scapozza L., Brunner T., Simon H. U. (2006) Nat. Cell Biol. 8, 1124–1132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.