

Abstract
Herpes simplex virus type 1 (HSV-1)-induced membrane fusion remains one of the most elusive mechanisms to be deciphered in viral entry. The structure resolution of glycoprotein gB has revealed the presence of fusogenic domains in this protein and pointed out the key role of gB in the entry mechanism of HSV-1. A second putative fusogenic glycoprotein is represented by the heterodimer comprising the membrane-anchored glycoprotein H (gH) and the small secreted glycoprotein L, which remains on the viral envelope in virtue of its non-covalent interaction with gH. Different domains scattered on the ectodomain of HSV-1 gH have been demonstrated to display membranotropic characteristics. The segment from amino acid 626 to 644 represents the most fusogenic region identified by studies with synthetic peptides and model membranes. Herein we have identified the minimal fusogenic sequence present on gH. An enlongation at the N terminus of a single histidine (His) has proved to profoundly increase the fusogenic activity of the original sequence. Nuclear magnetic resonance (NMR) studies have shown that the addition of the N-terminal His contributes to the formation and stabilization of an α-helical domain with high fusion propensity.
Keywords: Glycoprotein, Herpesvirus, Liposomes, Membrane Fusion, NMR, Peptides, Virus Entry
Introduction
Membrane fusion is a fundamental biological process that occurs in physiological conditions as well as in different pathological events. Entry into host cells by enveloped viruses is an example of how this ubiquitous process in biological systems is seized by the pathogen to establish an infection. Viral membrane fusion is mediated and tightly regulated by specific protein machinery whose components are directly encoded by the viral genome. A key role is attributed to surface glycoproteins embedded in the viral envelope. Intact viral fusion proteins as well as synthetic analogues of the fusion domains have been extensively studied in order to understand their mechanistic and structural role during the fusion process occurring between the envelopes of viruses and the membranes of host cells (1–4). Fusion proteins share several common motifs, and one of them is represented by the fusion peptide (FP),3 a short segment rich in hydrophobic residues essential for the fusion process. Fusion peptides are characterized by being active in destabilizing membranes as isolated peptides (5, 6), and mutations within this region in the intact protein often lead to loss in infectivity (7–9). FPs participate in inducing lipid rearrangements giving place to hemifusion and pore formation (1) and are also involved in pore enlargement (10–13). In addition to classical FPs, other domains of the viral envelope glycoproteins are essential for membrane fusion to occur. These membrane-interacting regions are capable of modifying the biophysical properties of phospholipid membranes, suggesting that several segments may have a role in the fusion process (14–16).
The physical event needed to promote membrane fusion induced by enveloped viruses is the insertion of the FP from one of the viral proteins into the cellular membrane to disrupt the normal organization of the lipids in their vicinity. Even though the exact mechanism of insertion of the FP into the target cell membrane is still unknown, several studies suggest that membranotropic regions could potentially cause the membrane distortion necessary for fusion in relation to their high propensity to partition into the membrane interface. Although much information has been obtained in recent years on membrane fusion, the complete mechanisms behind virus-host cell membrane fusion is still unclear; therefore, the elucidation of the nature of the interactions between phospholipids and viral membranotropic domains gains strength in the field of viral-induced membrane fusion.
Herpes simplex virus (HSV), the prototype of the alphaherpesviruses, is a human pathogen that infects epithelial cells before spreading to the peripheral nervous system to establish a life-long latent infection. HSV-1 infection, similarly to other enveloped viruses, is achieved through fusion of the lipid bilayer of the viral envelope with a host cell membrane. The core fusion machinery of herpesviruses is well conserved and consists of the envelope glycoprotein B (gB) and the heterodimeric complex of gH/gL (15–20). Although numerous studies have indicated that both gB and gH/gL exhibit varying degrees of fusogenic properties, the complete fusion is only achieved when the three proteins act together (21–23). Because both glycoproteins (gB and gH) are conserved in all herpesvirus subfamilies, it is likely that they perform a common function in all members of this virus family; they appear to be triggered by the action of glycoprotein gD, which binds cellular receptors, e.g. nectin-1, and either bridges gB and gH/gL into a larger complex (24, 25) or interacts with gB/gH/gL-preformed complexes (26). Notwithstanding how the active complex is assembled, the quartet of envelope glycoproteins promotes fusion of the virion envelope with cellular membranes, and both gB and gH appear to be directly involved in membrane fusion, i.e. in mixing of lipids.
Structural knowledge of the proteins participating in herpesvirus fusion is essential for understanding the molecular mechanism of the entry process. To date only the crystal structure of the HSV-1 gB ectodomain is available and revealed an unexpected structural homology with the fusogenic protein G of vesicular stomatitis virus (27, 28).
The fusogenic properties of gB are now well established (20, 21, 29), and its structural details suggested that it belongs to a third class of fusion proteins where the fusion peptide is represented by a bipartite loop domain. The three classes of viral membrane fusion proteins have been identified largely on the basis of the key structural features of the proteins, and a comprehensive treatment of this subject can be found in recent reviews (1, 30–32). Although no structural data for HSV-1 gH or any of its homologues in other herpesviruses are available, gH has also been considered as a likely fusion effector (15, 16, 19, 33).
HSV-1 gH is an 838-residue type 1 membrane glycoprotein. The ectodomain contains 7 N-glycosylation sites and 8 cysteine residues forming at least 2 disulfide bonds between cysteines 5 and 6 (residue 554 and 589) and cysteines 7 and 8 (residues 652 and 706) (34). Several domains likely to be important for membrane fusion have been identified in the C-terminal region of gH. For example, mutations in the pretransmembrane domain (35), transmembrane region, and cytoplasmic tail (36, 37) affect fusion. Furthermore, peptides matching a number of regions of the gH ectodomain have been shown to interact with membranes and proposed to play a role in the fusion process (15, 16, 33).
gH has been reported to have both features that are characteristic of class I and class II fusion proteins, such as the presence of heptad repeats and a putative fusion peptide region (15, 19, 33, 38). Moreover, peptides spanning the heptad-repeat region of gH inhibit the entry of both human cytomegalovirus (39) and HSV-1 (19, 38), similar to the corresponding entry-inhibitors identified for viruses that have a class I membrane fusion protein.
It has been previously demonstrated that synthetic peptides modeled on HSV-1 gH (gH-(220–262), gH-(381–420), gH-(493–537), gH-(626–644)) are able to induce rapid membrane fusion and act in a synergistic way. These regions in conjunction with the membrane proximal region (16) and the transmembrane anchor of the protein seem to form a continuous tract of hydrophobic membrane-interacting surfaces that could simultaneously destabilize viral and cellular opposing membranes at the point of fusion. gH would seem to play a role in both hemifusion and complete fusion; therefore, the identified membranotropic regions could act in concert or sequentially during either of these steps.
The stretch of gH comprising amino acids 626–644 is particularly interesting; in fact, a peptide modeled on this region is very effective in inducing lipid mixing of model membranes and induce 50% fusion at a low peptide/lipid ratio. Peptide gH-(626–644) has the characteristics of a fusion peptide because of its ability to adopt different conformations when challenged in different environments: TFE, aqueous solution, SDS, and phospholipids vesicles.
Mutations of any residue of the active domain of gH-(626–644) are able to abolish or drastically reduce fusion activity with model membranes (40). A determination of the exact length of this region able to induce fusion might, therefore, shed further light on the function of gH in membrane fusion.
The present study, aimed at minimizing and optimizing the length of HSV-1 gH putative fusion peptide, in fact investigates the relationship between the length of the peptide and membrane fusion. We generated a set of synthetic peptides longer and/or shorter of the known fusogenic sequence (gH-(626–644)) and analyzed them utilizing a combination of biochemical and spectroscopic techniques and nuclear magnetic resonance. The results reported here indicate that this gH membrane-perturbing domain interacts with biological membranes, contributing to the merging of the viral envelope and the cellular membrane, and point out that its activity is largely enhanced by the addition of a single residue of histidine at its N terminus.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
Peptides were synthesized using a standard solid-phase Fmoc (9-fluorenylmethoxycarbonyl) method as previously reported (15). All purified peptides were obtained with good yields (30–40%). Table 1 shows the sequences of all the synthesized peptides. Peptide stock solutions were prepared in 2% dimethyl sulfoxide (DMSO). A scrambled peptide of the native sequence was synthesized, namely SGYARFNLAIAARLLTHTW. Peptide sequences are reported in Table 1.
TABLE 1.
Peptide sequences
| Peptide sequences | ||
|---|---|---|
| gH-(626–644) | NH2-GLASTLTRWAHYNALIRAF-COOH | Starting sequence |
| gH-(621–644) | NH2-AQQTHGLASTLTRWAHYNALIRAF-COOH | Longer and shorter by 5 amino acids |
| gH-(626–649) | NH2-GLASTLTRWAHYNALIRAFVPEAS-COOH | |
| gH-(631–644) | NH2-LTRWAHYNALIRAF-COOH | |
| gH-(626–639) | NH2-GLASTLTRWAHYNA-COOH | |
| gH-(625–644) | NH2-HGLASTLTRWAHYNALIRAF-COOH | N-terminus enlongated by 1 amino acid each |
| gH-(624–644) | NH2-THGLASTLTRWAHYNALIRAF-COOH | |
| gH-(623–644) | NH2-QTHGLASTLTRWAHYNALIRAF-COOH | |
| gH-(622–644) | NH2-QQTHGLASTLTRWAHYNALIRAF-COOH | |
| gH-(625–643) | NH2-HGLASTLTRWAHYNALIRA-COOH | C terminus shortening by 1 amino acid each |
| gH-(625–642) | NH2-HGLASTLTRWAHYNALIR-COOH | |
| gH-(625–641) | NH2-HGLASTLTRWAHYNALI-COOH | |
| gH-(617–644) | NH2-VEVLAQQTHGLASTLTRWAHYNALIRAF-COOH | Longer peptide to be used in NMR experiments |
Liposome Preparation
Large unilamellar vesicles (LUV) consisting of PC/Chol (55/45) and when necessary containing Rho- and NBD phosphatidylethanolamine were prepared as previously reported (15). Lipid concentrations of liposome suspensions were determined by phosphate analysis (41).
Lipid Mixing Assays
Membrane lipid mixing was monitored using the resonance energy transfer assay as previously reported (15). All fluorescence measurements were conducted in PC/Chol (55/45) LUVs. Lipid mixing experiments were repeated at least three times, and results were averaged. Control experiments were performed using the scrambled peptide and DMSO. All the experiments were performed at room temperature and at 37 °C.
Tryptophan Fluorescence Measurements
Tryptophan fluorescence increases with the increase in the environment hydrophobicity, and a blue shift of the emission maxima is observed. Emission spectra of the peptides (4 μm) containing the tryptophan residue in the absence or presence of target vesicles (PC/Chol = 55/45) were recorded between 300 and 400 nm with an excitation wavelength of 295 nm.
The degree of peptide association with lipid vesicles was measured by adding lipid vesicles to 4 μm peptides, and the fluorescence intensity was measured as a function of the lipid/peptide molar ratio in three to four separate experiments. The fluorescence values were corrected by taking into account the dilution factor corresponding to the addition of microliter amounts of liposomes and by subtracting the corresponding blank. The lipid/peptide molar ratio was 200:1.
The binding of hydrophobic peptides to membranes can be described as a partition equilibrium, Xb = KpCf, where Kp is the apparent partition coefficient in units of m−1, Xb is the molar ratio of bound peptide per total lipid, and Cf is the equilibrium concentration of the free peptide in solution. To calculate Xb, we estimated F∞, the fluorescence signal obtained when all the peptide is lipid-bound, either from the plateau region of the titration curve or from a double reciprocal plot of F (total peptide fluorescence) versus CL (total concentration of lipids), as previously suggested by Schwarz et al. (42). (F∞ was obtained by extrapolation of a double reciprocal plot of the total peptide fluorescence versus the total lipid concentration in the outer leaflet, i.e. 1/F versus 1/0.6 CL). Knowing the fluorescence intensities of the free and bound forms of the peptide, the fraction of membrane-bound peptide, fb, could be determined by the formula fb = (F − Fo)/(F∞ − Fo), where F represents the fluorescence of peptide after the addition of the vesicles, and Fo represents the fluorescence of the unbound peptide. Determining the value of fb in turn allows us to calculate the equilibrium concentration of free peptide in the solution, Cf, as well as the extent of peptide binding Xb. It was assumed that the peptides were initially partitioned only over the outer leaflet of the small unilamellar vesicle (60% the total lipid) (43). Therefore, values of Xb were corrected as follows: Xb* = Xb/0.6.
The curve resulting from plotting Xb* versus the concentration of the free peptide, Cf, is referred to as the conventional binding peptide isotherm. Plots of Xb* versus Cf yield straight lines with the slope corresponding to Kp if a simple partition equilibrium is observed. However, when the binding isotherms are not straight lines but deviate to increased binding at higher peptide concentrations, such a deviation is expected for the cooperative binding of peptides that self-associate at the membrane surface. If enough data points of Cf could be collected at very low free peptide concentrations, the surface partition coefficients, Kp, could be estimated from the initial slopes of the curves.
Tryptophan Quenching by Acrylamide
Aliquots of a 4 m solution of the water-soluble quencher were added to the solution containing the peptide (4 μm) in the absence or presence of liposomes at a peptide/lipid molar ratio of 1:200. The maximal concentration of acrylamide is 0.2 mmol/ml. Fluorescence was measured at an excitation wavelength of 295 nm to reduce acrylamide absorbance (and the resulting inner filter effect) and emission at a wavelength of 340 nm to eliminate interference from the Raman band of water (44). The data were analyzed according to the Stern-Volmer equation (45), Fo/F = 1 + Ksv[Q], where Fo and F represent the fluorescence intensities in the absence and the presence of the quencher (Q), respectively, and Ksv is the Stern-Volmer quenching constant, which is a measure of the accessibility of tryptophan to acrylamide. Considering that acrylamide does not significantly partition into the membrane bilayer (44), the value for Ksv can be considered to be a reliable reflection of the bimolecular rate constant for collisional quenching of the tryptophan residue present in the aqueous phase. Accordingly, Ksv is determined by the amount of non-vesicle-associated free peptide as well as the fraction of the peptide residing in the surface of the bilayer.
Binding Analysis by Surface Plasmon Resonance (SPR)
SPR experiments were carried out with a BIAcore 3000 analytical system (Biacore, Uppsala, Sweden) using HPA and L1 sensor chips (BIAcore) as previously reported (46). The HPA sensor chip contains hydrophobic alkanethiol chains that are covalently bound to its gold surface, and a lipid heteromonolayer is created by introducing liposomes to the chip; the complete coverage of the surface with a polar lipid monolayer generates a membrane-like environment where analytes in aqueous buffer interact with a lipid monolayer (47) The L1 sensor chip contains hydrophobic alkanethiol chains with exposed polar headgroups, and a lipid bilayer is being created by introducing liposomes to the chip. The running buffer used for all experiments was phosphate-buffered saline (pH 7.4); the washing solution was 40 mm N-octyl β-d-glucopyranoside. PC/Chol (55/45 w/w) small unilamellar vesicles (80 μl, 0.5 mm) were applied to the chip surface at a flow rate of 2 μl/min. To remove any multilamellar structures from the lipid surface, we used NaOH 10 mm and increased the flow rate to 50 μl/min, which resulted in a stable base line corresponding to the lipid monolayer (or bilayer in the case of L1) linked to the chip surface. The negative control bovine serum albumin was injected (25 μl, 0.1 mg/μl in phosphate-buffered saline) to confirm complete coverage of the nonspecific binding sites. Peptide solutions were injected onto the lipid surface (30 μl at a flow rate of 5 μl/min). Phosphate-buffered saline alone then replaced the peptide solution for 15 min to allow peptide dissociation. SPR detects changes in the reflective index of the surface layer of peptides and lipids in contact with the sensor chip. A sensorgram is obtained by plotting the SPR angle against time. This change in the angle is then translated to response units. Analysis of the peptide-lipid binding event was performed from a series of sensorgrams collected at different peptide concentrations. The sensorgrams for each peptide-lipid bilayer interaction (L1 chip) were analyzed by curve-fitting using numerical integration analysis (48). The BIAevaluation was used to perform complete kinetic analyses of the peptide sensorgrams. Several curve-fitting algorithms were used, but a good fit was obtained only with the two-state reaction model, which was previously used for describing the possible binding mechanisms of antimicrobial peptides (49). The data were fitted globally by simultaneously fitting the sensorgrams obtained at different peptide concentrations, and the two-state reaction model was applied to each data set. This model describes two reaction steps (50) that in terms of peptide-lipid interaction may correspond to (i) peptide (P) binds to lipids (L) to give PL, and (ii) the complex PL changes to PL*, which cannot dissociate directly to P + L and which may correspond to partial insertion of the peptide into the lipid bilayer.
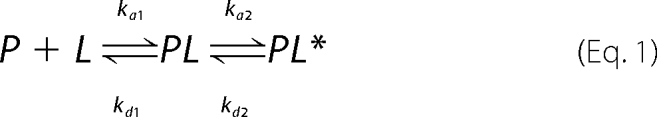 |
The corresponding differential rate equations for this reaction model are represented, where RU1 and RU2 are the response units for the first and second steps, respectively, CA is the peptide concentration, RUmax is the maximum peptide binding capacity (or equilibrium binding response), and ka1, kd1, ka2, and kd2 are the association and dissociation rate constants for the first and second steps, respectively.
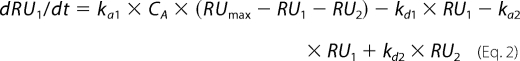 |
Although ka1 has m−1 s−1 units, kd1, ka2, and kd2 have s−1 units; thus, the total affinity constant for the all process, K, has m−1 units. Kinetic data were assessed by using χ2 values, plots of the residuals from the model fitting, and the significance of a parameter assessed by S.D. The quality of the fit to a specific parameter was deemed significant if the S.D. was less than 10%. Except where specifically indicated, all parameter values were significant to the fit.
NMR Measurements
NMR sample solutions (about 1 mm) were prepared by dissolving the crude peptides in TFE-d3/H2O (80/20 v/v). TFE-d3 (99%) was purchased from Cambridge Isotope Laboratories. All the NMR spectra were recorded at 300 K using a Varian UNITY 500 spectrometer operating at 500 MHz. The proton chemical shifts were referenced to the residual TFE methylene resonance (3.88 ppm). Two-dimensional phase-sensitive TOCSY, NOESY and DQFCOSY spectra (51) were collected using the States and Haberkorn method. A spectral width of 6000 Hz was used in both dimensions; typically, 4096 was the number of complex points collected in the ω2 dimension and 512 in the ω1 dimension. The data were zero-filled to 2000 in ω1. Squared-shifted sine-bell functions were applied in both dimensions before Fourier transformation. Two-dimensional total correlation spectroscopy and NOESY experiments were recorded with mixing times of 70 and 150 ms, respectively. Water suppression was achieved utilizing the DPFGSE sequence (52). The data were processed and analyzed using the VNMRJ and XEASY software (53).
Structure Calculation
Experimental distance restraints for structure calculation were derived from the cross-peak volumes in the NOESY spectra, recorded in TFE/H2O (80/20 v/v). The NOESY cross-peaks were manually integrated using the XEASY software and converted to upper distance constraints according to an inverse sixth power peak volume-to-distance relationship for the backbone and to an inverse fourth power function for side chains by using the CALIBA module of the CYANA program (54). Distance constraints for gH-(626–639) together with the obtained scalar coupling constants were then used by the GRIDSEARCH module implemented in CYANA to generate a set of allowable dihedral angles. Structure calculation, which used the torsion angle dynamics protocol of CYANA, was then started from 100 randomized conformers. The 20 conformers with the lowest CYANA target function were further refined in vacuo by means of unrestrained energy minimization using the Discover module of the Insight II software (Accelrys, Inc.). Several cycles of steepest descent were repeated until the energy difference between two successive steps was less than 10−3 kJ mol−1. The structure analysis was performed with the program MOLMOL (55).
Virus Entry Assays
Vero Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. HSV-1-expressing β-galactosidase driven by the cytomegalovirus IE-1 promoter was propagated as previously described (22). Peptides were dissolved in Dulbecco's modified Eagle's medium without serum and used at a range of concentrations. All experiments were conducted in parallel with scrambled peptides and no-peptide controls.
To assess the effect of peptides on inhibition of HSV infectivity, cells were incubated with increasing concentrations of peptide (10, 50, and 100 μm) in the presence of serial dilutions of viral inoculum for 45 min at 37 °C. Nonpenetrated viruses were inactivated by citrate buffer (pH 3.0) after a 45-min incubation at 37 °C. Monolayers were incubated for 48 h at 37 °C in Dulbecco's modified Eagle's medium supplemented with carboxymethylcellulose, fixed, and stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), and plaque numbers were scored. Experiments were performed in triplicate, and the percentage of inhibition was calculated with respect to no-peptide control experiments.
Toxicity
Peptide cytotoxicity was measured by a lactate dehydrogenase assay and carried out according to the manufacturer's instructions using a cytotoxicity detection kit (Roche Diagnostic).
RESULTS
Selection of Peptides
Different membranotropic regions are scattered on the ectodomain of HSV-1 gH envelope glycoprotein (15), and one of the most fusogenic regions along the whole protein is located in the region 626–644. This region could be important in the membrane fusion process; therefore, a set of peptides with longer or shorter sequences compared with the initial sequence was designed and synthesized to be analyzed in the present study. Peptides are reported in Table 1.
Ability of Mutated Peptides to Induce Lipid Mixing
To investigate the fusogenicity of the selected peptides, NBD- and Rho-labeled phosphatidylethanolamine were used as the donor and acceptor of fluorescence energy transfer. A population of LUVs labeled with both was mixed with a population of unlabeled LUVs, and increasing amounts of peptides were added. Dilution of the fluorescent-labeled vesicles via membrane fusion induced by the peptide resulted in a reduction in the fluorescence energy transfer efficiency; hence, dequenching of the donor fluorescence. The dependence of the extent of lipid mixing on the peptide to lipid molar ratio was analyzed; increasing amounts of each peptide were added to a fixed amount of vesicles. To compare the activity of the different peptides, the percentage of lipid mixing as a function of the peptide to lipid molar ratio was calculated. Fig. 1a shows the results of lipid mixing assays in PC/Chol for gH-(626–644) and shorter and longer peptides, namely gH-(621–644), gH-(626–649), gH-(631–644), gH-(626–639), and gH-(617–644) at room temperature. The two shorter peptides (gH-(631–644) and gH-(626–639)) induced lower levels of fusion compared with gH-(626–644), suggesting that shorter peptides were unable to cross or stably position inside the bilayer in accordance with previously reported data (40) where the glycine at the N terminus and the arginine at the C terminus were found to play a vital role in fusion, and their mutation with a serine induced a significant loss of activity. In contrast, the elongated peptides showed a greater fusion activity compared with the starting peptide; in particular, we observed a significant increase of activity with elongation of five residues at the N terminus.
FIGURE 1.

Peptide-promoted membrane fusion of PC/Chol (1:1) LUVs as determined by lipid mixing. Peptide aliquots were added to 0.1 mm LUVs containing 0.6% NBD and 0.6% Rho. The increase in fluorescence was measured after the addition of peptide aliquots. Reduced Triton X-100 (0.05% v/v) was referred to as 100% of fusion. Dose dependence of lipid mixing is reported. Fusion experiments for longer and shorter peptides are reported in panel a, for N-terminal elongated peptides in panel b, and for C-terminal elongated peptides in panel c.
As a consequence of this result, we investigated in deeper details the N terminus of the peptide. Fig. 1b shows results obtained with peptides elongated only at the N terminus, namely, gH625–644 (57), gH624–644, gH623–644, gH622–644, and gH-(621–644). The results clearly indicate that the addition of one histidine residue at the N terminus of the native sequence strongly increases the fusion activity. It is interesting to note that the addition of the glutamines at the N terminus decreases the fusogenicity. We, thus, were able to determine the N-terminal length necessary to obtain the highest level of liposome fusion.
To determine the shortest sequence able to induce fusion, we performed fusion experiments on peptides having a histidine at the N-terminus and being shorter at the C terminus. Our data for peptides gH625–644, gH625–643, gH625–642, and gH625–641 are shown in Fig. 1c. The peptide gH625–641 is significantly impaired in its ability to cause fusion, suggesting that the elimination of four residues at the C terminus causes a drastic reduction of fusogenicity; the two peptides gH625–643 and gH625–642, both, present a reduced activity compared with gH625–644. These data support the idea that a charged residue at the C terminus is important for the localization of the peptide inside the lipid bilayer. No significant levels of fusion were observed with the scrambled version of gH-(626–644). Our data clearly show that the peptide gH625–644 presents a fusion activity greater than the starting sequence gH-(626–644); in particular, at a peptide/lipid ratio of 0.4, the peptide gH-(626–644) presents a percentage of fusion of approximately 40%, whereas gH625–644 is approximately 90%.
Membrane fusion events take place during cell life; thus, a temperature of 37 °C will more closely resemble a natural event. We decided to perform the fusion experiment on the most active peptides at 37 °C (Fig. 2). Our data are particularly interesting and show that the most active peptide is still gH625–644, and the second best peptide is gH625–642, indicating that the histidine and the arginine are fundamental for fusion. It is interesting to compare results obtained for gH-(626–644) and gH625–644 at 37 °C. The peptide gH-(626–644) is able to induce only 20% of fusion at a peptide/lipid ratio of 0.05, whereas gH625–644 was able to induce almost 100% of fusion at the same peptide/lipid ratio, and gH625–642 followed with a percentage of fusion close to 80%. The IC50 of gH625–644 at 37 °C is 2 μm, indicating that this peptide has a strong fusogenic propensity in the analyzed conditions.
FIGURE 2.

Peptide-promoted membrane fusion of PC/Chol (1:1) LUVs as determined by lipid mixing. Peptide aliquots were added to 0. 1 mm LUVs containing 0.6% NBD and 0.6% Rho. The increase in fluorescence was measured after the addition of peptide aliquots. Reduced Triton X-100 (0.05% v/v) was referred to as 100% of fusion. Dose dependence of lipid mixing at 37 °C is reported.
Tryptophan Fluorescence Emission Analysis
We measured the intrinsic fluorescence of gH-(626–644) and gH625–644 (due to the presence of a tryptophan residue in the middle of the sequence) to evaluate the degree of penetration of the peptides into the membrane bilayer. We compared the fluorescence emission spectra in the presence of PC/Chol vesicles with that in buffer (Fig. 3). The fluorescence emission of tryptophan residues increases when the amino acid enters a more hydrophobic environment, and together with an increase in quantum yield, the maximal spectral position may be shifted toward shorter wavelengths (blue shift). For both peptides gH-(626–644) and gH625–644, changes in the spectral properties were observed, suggesting that the single tryptophan residue of each peptide is located in a less polar environment upon interaction with lipids. Emission intensity was enhanced, and the maxima were shifted to lower wavelengths; blue shifts of this magnitude have been observed when amphiphilic tryptophan-containing peptides interact with phospholipid bilayers and are consistent with the indole moiety becoming partially immersed in the membrane, further suggesting that the peptides are capable of penetrating a lipid bilayer (58). A lipid-exposed tryptophan residue located at the center of the hydrocarbon core of a bilayer exhibits a characteristic highly blue shifted emission, with a λ max in the range of 315–318 nm. As a tryptophan residue moves toward the more polar membrane surface, λ max gradually shifts to 335–340 nm. A smaller red shift of the fluorescence for a tryptophan at the bilayer center can also be detected upon helix oligomerization inside the membrane. This shift presumably reflects the change in local environment upon replacement of contacts between tryptophan and lipid with contacts between tryptophan and polypeptide. Our data support the hypothesis that the tryptophan is located inside the membrane and, moreover, that oligomerization processes may have taken place.
FIGURE 3.

Tryptophan fluorescence spectra in buffer and in liposomes for gH-(626–644) and gH625–644.
The increase in fluorescence for tryptophan binding to membrane phospholipids was used for the generation of binding isotherms for gH-(626–644) and gH625–644, from which partition coefficients could be calculated. The concentrations of peptides used were low enough to cause minimal aggregation in the aqueous phase and were assumed not to disrupt the bilayer structure. Small unilamellar vesicles were used in the assay to minimize light-scattering effects. A control experiment was performed by titrating unlabeled peptides with lipids up to the maximal concentration used in the assay. The fluorescence intensity of this solution remained unchanged.
To determine the surface partition coefficient, the fluorescence intensities were converted to moles of bound peptide per moles of lipid and plotted as a function of the free peptide concentration as described under “Experimental Procedures” (Fig. 4). As partition coefficients depend on the concentration of lipid accessible to peptide, the curves obtained by plotting Xb* (the molar ratio of bound peptide per 60% of the total lipid) versus Cf (the equilibrium concentration of free peptide in the solution) are referred to as the conventional binding isotherms. The shape of a binding isotherm of a peptide can provide information on the organization of the peptide within the membrane. A straight line indicates a simple adhesion process. The shape of the binding isotherm of all the peptides tested was not linear, indicating that peptide accumulation at the surface is not a simple phenomenon without cooperative association. In particular, this behavior is the hallmark for peptides that self-associate at membrane surfaces upon partitioning. If aggregation occurred only in the water but not in the bilayer phase, the opposite course of the isotherms should be expected; that is, a steep rise at the origin followed by pronounced flattening. Thus, the shape of the isotherms obtained could be interpreted as reflecting a process whereby peptides first incorporate into the membrane and then aggregate there within. Moreover, there was no evidence of aggregation in water at the concentration used in this experiment (0.1 μm). In the isotherms obtained, the total extent of incorporation (Xb*) slowly increases until a critical concentration is reached where massive internal aggregation apparently starts to develop. The surface partition coefficients Kp were estimated by extrapolating the initial slopes of the curves to Cf values of zero; curves are shown in Fig. 4.
FIGURE 4.

Binding isotherms obtained plotting Xb* versus Cf for gH-(626–644) and gH625–644.
The Kp values for the peptides gH-(626–644) and gH625–644 are shown in Table 2. The Kp value obtained for gH-(626–644) is 1.6 × 104, indicating that the tryptophan in gH-(626–644) is embedded in the bilayer and, thus, that most of the peptide gH-(626–644) is located inside the liposomes. The Kp value for gH625–644 is 2.7 × 104, indicating that also in this peptide the tryptophan residue is located inside the liposomes and is stably inserted.
TABLE 2.
Partition coefficient for the binding of peptides with PC/Chol
| gH-(626–644) | gH-(625–644) | |
|---|---|---|
| Kp | (1.6 ± 0.3) × 104 | (2.7 ± 0.2) × 104 |
Quenching of Tryptophan by Acrylamide
The observed changes in the characteristics of the tryptophan emission upon binding of peptides gH-(626–644) and gH625–644 to lipid vesicles indicate their insertion into the hydrophobic region of the bilayers, with the peptide gH625–644 showing the greatest changes in the spectra and higher Kp values. We also studied the accessibility of the tryptophan residues of membrane-bound peptides toward acrylamide, a neutral, water-soluble, highly efficient quenching molecule that is unable to penetrate into the hydrophobic core of the lipid bilayer. The more deeply a tryptophan residue is buried, the less strongly it is quenched by acrylamide. Stern-Volmer plots for the quenching of tryptophan by acrylamide, recorded in the absence and presence of lipid vesicles, are depicted in Fig. 5. Fluorescence of tryptophan decreased in a concentration-dependent manner by the addition of acrylamide to the peptide solution both in the absence and presence of liposomes without other effects on the spectra. However, in the presence of liposomes, less decrement in fluorescence intensity was evident, thus revealing that tryptophan is less accessible to the quencher in the presence of LUVs. The values for Ksv were lower for gH625–644 (Table 3) in LUVs, suggesting that tryptophan was more buried in the bilayers, becoming more inaccessible for quenching by acrylamide. Comparison of the plots (Fig. 5) and of the values of Ksv (Table 3) for the two peptides confirms that the tryptophan in gH625–644 is more deeply inserted than in the starting peptide. These data further support the hypothesis that the peptide is buried in the membrane, becoming less accessible for quenching by acrylamide.
FIGURE 5.

Stern-Volmer plots of acrylamide quenching of gH-(626-644), L627S, and L631S in buffer (open symbols) and in LUVs (closed symbols).
TABLE 3.
Stern-Volmer (Ksv) quenching constant calculated from the equation Fo/F = 1 + Ksv[Q] (Q, quencher) for gH-(626–644) and gH-(625–644)
| gH-(626–644) | gH-(625–644) | |
|---|---|---|
| Ksv (m−1) in buffer | 14.59 ± 0.4 | 9.66 ± 0.3 |
| Ksv (m−1) in LUVs | 8.68 ± 0.4 | 4.77 ± 0.4 |
Binding Affinity of the Peptides to Lipid Monolayers and Bilayers Measured by SPR
We utilized a BIAcore biosensor method to investigate the mode of action of gH625–644 and compared it with gH-(626–644) (46). In a previous paper (46) we also reported the data for two control peptides: the melittin and the HIV fusion peptide (gp41-FP). In particular, melittin is the major component of the venom of the honeybee Apis mellifera and has been extensively characterized by other groups (59). On the other hand, the fusion peptide of HIV (60) served as a comparison with a well known fusion peptide.
PC/Chol monolayers and bilayers were absorbed onto the HPA and L1 chips, respectively. Sensorgrams of the binding with monolayers and bilayers of PC/Chol are shown in Fig. 6. The sensorgrams revealed that the RU signal intensity increased as a function of the peptide concentration. This indicates that the amount of peptide bound to the lipids is proportional to the peptide concentration. The sensorgrams of the binding of gH625–644 with lipid monolayers showed markedly lower response levels as compared with its binding to bilayers. In contrast, gH-(626–644) showed a similar response to monolayers as compared with bilayers. The inspection of the shape of each sensorgram reveals different binding kinetics with significant differences among the two peptides in the two different lipid surfaces. In particular, the sensorgram obtained for gH625–644 to the L1 chip indicates that it binds to the lipid surfaces in a biphasic manner. The initial association starts as a fast process and then slows down considerably toward the end of the peptide injection. The dissociation follows a similar trend, with the signal falling rapidly at the end of injection as the peptide is no longer present, and the buffer flow removes a large amount of free or weekly bound peptide followed by a much slower step. Moreover, the sensorgrams did not return to zero on the L1 chip, indicating that the peptide remained significantly bound to the surface or inserted into the hybrid bilayer membrane.
FIGURE 6.

Sensorgrams of the binding between various concentrations of gH-(626–644) and gH625–644 with the HPA chip (upper) and the L1 chip (lower).
We employed numerical integration analysis that uses nonlinear analysis to fit an integrated rate equation directly to the sensorgrams (61). When fitting the peptide sensorgrams globally (using different concentrations of the peptides) with the simplest 1:1 Langmuir binding model, a poor fit was obtained (X2 > 100), confirming that this model does not represent the lipid binding mechanism of the peptide. However, a significantly improved fit was obtained using numerical integration of the two-state reaction model of the binding sensorgrams, suggesting that there is likely to be at least two steps involved in the interaction between the peptide and hybrid bilayer membrane surface. In analogy with previous studies of peptide-membrane interactions using SPR (49, 50), the first step may correspond to the actual binding of the peptide to the surface, and the second step is the insertion of the peptide into the hydrophobic core of the membrane. A set of peptide sensorgrams with different peptide concentrations was used to estimate the kinetic parameters. The average values for the rate constants obtained from the two-state model analysis are listed in Table 4 along with the affinity constant values (KA). The data clearly indicate the main influence on the overall binding constant of the fast association rate and slow dissociation rate of the first step. If this step corresponds to the electrostatic interaction, these results clearly indicate that electrostatic forces play an important role in the binding of membrane-active peptides.
TABLE 4.
Association (ka1, ka>2) and dissociation (kd>1, kd>2) rate constants obtained for the HPA and the L1 chip using the two-state model
The affinity constants K1 and K2 are for the first (K1 = ka1/kd1) and for the second (K2 = ka2/kd2) steps respectively, and the affinity constant (KA), determined as (ka1/kd1) × (ka2/kd2), is for the complete binding process. S.D. are reported in parentheses.
| ka1 | kd1 | K1 | ka2 | kd2 | K2 | KA | |
|---|---|---|---|---|---|---|---|
| HPA | |||||||
| gH-(626–644) | (8.14 ± 0.02) × 102 | (4.39 ± 0.05) × 10−2 | 1.8 104 | (6.40 ± 0.07) × 10−4 | (2.13 ± 0.04) × 10−4 | 3.0 | 7.42 × 104 |
| gH-(625–644) | (5.11 ± 0.03) × 101 | (2.51 ± 0.05) × 10−2 | 2.0 103 | (5.22 ± 0.05) × 10−3 | (110 ± 0.06) × 10−4 | 47 | 1.00 × 105 |
| Melittin | (8.60 ± 0.03) × 101 | (7.93 ± 0.01) × 10−3 | 1.1 104 | (1.41 ± 0.05) × 10−3 | (1.83 ± 0.09) × 10−3 | 0.8 | 1.92 × 104 |
| gp41-FP | (1.03 ± 0.02) × 102 | (5.69 ± 0.01) × 10−3 | 1.8 104 | (2.88 ± 0.04) × 10−3 | (1.18 ± 0.05) × 10−4 | 24.4 | 4.62 × 105 |
| L1 | |||||||
| gH-(626–644) | (5.23 ± 0.09) × 102 | (4.38 ± 0.08) × 10−2 | 1.2 104 | (2.41 ± 0.02) × 10−3 | (3.98 ± 0.07) × 10−3 | 0.6 | 6.92 × 104 |
| gH-(625–644) | (7.85 ± 0.03) × 102 | (9.79 ± 0.02) × 10−3 | 8.4 104 | (2.64 ± 0.04) × 10−3 | (2.91 ± 0.07) × 10−3 | 0.9 | 7.21 × 104 |
| Melittin | (4.85 ± 0.02) × 103 | (1.75 ± 0.02) × 10−2 | 2.8 105 | (3.55 ± 0.04) × 10−3 | (2.33 ± 0.02) × 10−3 | 1.5 | 7.01 × 105 |
| gp41-FP | (3.2 ± 0.07) × 101 | (1.92 ± 0.09) × 10−4 | 1.6 105 | (1.11 ± 0.02) × 10−5 | (1.30 ± 0.09) × 10−5 | 0.8 | 3.53 × 105 |
The difference between the affinity of the peptides to monolayers as compared with bilayers should indicate the contribution of the inner leaflet to the binding process (46, 61). In fact, due to structural differences between supported monolayers (HPA chip) and immobilized liposomes (L1 chip), it is possible to differentiate between the surface adsorption and/or partial insertion in the HPA chip and complete insertion into the hydrophobic core of the membrane in the liposomes of the L1 chip. The values of the ratio KA bilayer/KA monolayer for the peptide is 0.7, demonstrating that gH625–644 behaves similarly to the other HSV peptides (all having a KA bilayer/KA monolayer close to 1) and to the HIV fusion peptide, indicating a mechanism of action different from melittin (having a KA bilayer/KA monolayer of 36) (supplemental Table 1S). The values of the ratio KA bilayers/KA monolayer demonstrates that the binding to monolayers is similar to the binding to bilayers, indicating that the peptide is not influenced by the membrane inner layer (40). Thus, fusion peptides do not insert completely into the bilayers but only partially, supporting the idea that the shallow insertion of fusion peptides inside the bilayer perturbs the packing of the lipid polar headgroups and result in local monolayer deformations that can produce an area difference between the membrane leaflets. Consequently, the membrane will develop curvature to compensate for this area asymmetry starting the fusion process.
It is interesting to note that the two peptides gH-(626–644) and gH625–644 have similar K1 values, indicative that the first step corresponding to the electrostatic interaction is similar, whereas they present significantly different K2 values, with gH625–644 more similar to the HIV fusion peptide. This constant corresponds to the second step and in particular to the insertion of the peptide into the hydrophobic core of the membrane, further supporting the hypothesis of the key role played by the insertion of the peptides inside the bilayer for the fusion activity of this viral peptides.
NMR Structure of Synthetic Peptides
There have been numerous structure studies on fusion peptides, but there is not yet clear correlation between structure and fusogenic activity; in particular, fusion peptide can adopt either helical or β-strand conformation. An α-helix conformation has been proposed to be associated with pore formation by peptides in vesicle membranes (62, 63), whereas the extended antiparallel β-structure of the peptides was believed to be involved in the initial steps of the fusion process and is likely to reside primarily at the lipid-water interface; in most studies on viral fusion peptides an α-helical arrangement of the lipid-bound peptide has also been suggested to represent the fusion-active conformation (60, 65). We previously reported (46) the involvement of peptides in a β-conformation in the actual fusion event, although we demonstrated that fusion peptides may adopt an α-helix conformation when more deeply inserted inside the bilayer. A dynamic equilibrium between helix and non-helix forms of the fusion peptide in the membrane environment appears, thus, to be essential for the fusion process. In a previous paper (40), we reported the NMR solution structure of gH-(626–644) fusion peptide and showed that it assumes an α-helix conformation spanning residues 629–638 with 7 hydrogen bonds stabilizing the structure (40). Here, we analyze the solution structure of two peptides derived from HSV-1 gH, both containing the α-helical sequence spanning amino acid 629 to 638, that have been elongated at the N terminus or truncated at the C terminus. The peptide gH-(617–644) is composed of residues 617–644 and is longer than gH-(626–644); the elongation at the N terminus was necessary as the peptide was previously proved to penetrate the bilayer from the N-terminal side (40), and here we found that the addition of one residue at the N terminus was able to significantly improve the fusion activity. Moreover, N-terminal elongation was necessary to determine the conformational (structural) preferences of the histidine residue located at the N terminus. The second peptide, gH-(626–639), is a C-terminal 5-amino acid-truncated gH-(626–644), which could help us in understanding the effective length of the active peptide.
Proton NMR assignment in TFE/H2O (80/20 v/v) was obtained following the standard protocol by means of two-dimensional TOCSY, NOESY, and double quantum-filtered correlation spectroscopy experiments; the 1H chemical shifts for gH-(617–644) and gH-(626–639) are listed in the Biological Magnetic Resonance Bank under deposition numbers 16699 and 16700. The Hα Chemical Shift Index, according to the Wishart method (66), and the NOE connectivity diagrams are reported in Fig. 6. The small differences observed in the chemical shifts of gH-(617–644) with respect to the values reported for gH-(626–644) (40) are indicated by gray bars in Fig. 7.
FIGURE 7.

Chemical shift index secondary structure prediction and NOE effects of gH-(617–644) (upper panel) and gH-(626–639) (lower panel) in TFE/H2O (80/20 v/v) at 300 K. The gray bars reported for gH-(617–644) indicate the Hα chemical shift variations with respect to gH-(626–644) (101).
A total of 235 NOEs (101 interresidues, 134 intraresidues) was obtained for gH-(617–644) from NOESY 150-ms spectrum; these NOEs resulted in 154 meaningful distance constraints and 118 angle constraints used for the structures calculation. 129 NOEs (42 interresidues, 87 intraresidues), 103 JNH-αCH coupling constants (Biological Magnetic Resonance Bank deposition number 16699), 65 meaningful distance constraints, and 68 angle constraints were obtained and used for the structure determination of gH-(626–639). All the relevant NOESY-derived distances are reported in supplemental Tables 2S and 3S. No hydrogen bonding constraints were used in the calculations. All the constraints were used to generate a total of 100 structures, and among them the 20 with the lower target function were selected and energy-minimized. All the obtained structure satisfied the NMR constraint with no NOE violations greater than 0.2 Å. In Fig. 8 the superimposition of the 20 minimized structures is reported for gH-(617–644) and gH-(626–639); the average backbone dihedral angles are reported in supplemental Table 4S, and hydrogen bond distances and angles are reported in Table 5.
FIGURE 8.

Minimized structure of gH-(617–644) (upper structures) and gH-(626–639) (lower structures). Panels a and b, superposition of the 20 lowest energy conformers aligned according to the minimal root mean square deviations of the backbone atoms is shown. Panels c and d, representative structures are shown. Side chains are shown in blue, and side chains assuming α-helical structure in gH-(626–639) are colored in green.
TABLE 5.
Hydrogen bond distances and angles as obtained by Molmol for gH-(617–644) and gH-(626–639)
| Donor | Acceptor | gH-(617–644) |
gH626–639 |
||
|---|---|---|---|---|---|
| H…O distance | N-H…O angle | H…O distance | N-H…O angle | ||
| Å | ° | Å | ° | ||
| Ala628 HN | Thr624 O | 1.6…2.4 | 4…27 | ||
| Ser629 HN | His625 O | 1.8…2.1 | 7…20 | ||
| Thr630 HN | Gly626 O | 1.8…2.3 | 17…30 | ||
| Leu631 HN | Leu627 O | 1.7…1.9 | 1…7 | ||
| Thr632 HN | Ala628 O | 1.9…2.1 | 3…14 | ||
| Arg633 HN | Ser629 O | 1.9…2.2 | 23…31 | 1.6…2.2 | 2…40 |
| Trp634 HN | Thr630 O | 1.8…1.9 | 25…30 | 1.8…2.4 | 19…41 |
| Ala635 HN | Leu631 O | 1.6…1.7 | 4…19 | 2.0…2.4 | 20…28 |
| His636 HN | Thr632 O | 2.3…2.4 | 14…23 | ||
| His636 HN | Arg633 O | 1.8…2.4 | 13…22 | ||
| Asn638 HN | Trp634 O | 1.6…2.0 | 17…39 | ||
| Ala639 HN | Ala635 O | 2.1…2.4 | 9.6…19.5 | ||
| Leu640 HN | Tyr637 O | 2.2…2.4 | 28…38 | ||
| Ile641 HN | Tyr637 O | 1.7…2.2 | 1…15 | ||
| Ala643 HN | Ile641 O | 1.9…2.2 | 16…24 | ||
The NMR structures of both peptides are well defined with root mean square deviations to the mean structure equal to 0.26 Å for the His-625—Leu-640 region of gH-(617–644) and 0.36 Å for the Ala-628—Ala-635 region of gH-(626–639). Both peptides assume an α-helix conformation with disordered N- and C-terminal tails (Fig. 8).
In gH-(617–644) the α-helix begins with histidine 625, spans residues 625–638, and is stabilized by several hydrogen bonds. Hydrogen bonds and dihedral angles obtained for residues Ala-639 and Leu-640 are consistent with the occurrence of the α-helix conformation in ∼50% of the minimized structures. Interestingly, residues 623 and 624 do not show any tendency to assume α-helical conformation as evidenced by their ϕ and ψ angles; this was not the case in gH-(626–644), where Leu-627 and Ala-628 residues preceding the α-helix exhibit average backbone dihedral angles close to the typical α-helix conformation angles (40).
In the case of the peptide gH-(626–639), the α-helix is constituted by residues 630–634 and is stabilized by two hydrogen bonds. A type I β-turn involving residues Arg-633, Trp-634, Ala-635, and His-636 is found in 70% of the final structures.
The three-dimensional structures of gH-(617–644) and of gH-(626–644) (40) underline that the α-helical conformation propensity of the fragment begins with histidine 625 and confirm that the structured region ends with residue 638. In Fig. 9, b and c, the electrostatic surface potentials of gH-(617–644) are reported; polar and charged residues are colored in blue showing the amphipathic profile of the fusion peptide. In particular, the histidine 625 side chain is located on the polar face of the molecule, whereas the tryptophan 634 and tyrosine 637 aromatic rings are still positioned on the same apolar side of the helix. Residue Gly-626, important for the fusogenic activity and highlighted in Fig. 9a in gH 617–644, occupies the second position of the α-helical structure.
FIGURE 9.

α-Helical structure representation of gH-(617–644) (panel a). The helix is depicted in red, His-625 and Gly-626 side chains are reported in gold, and Trp-634 and Tyr-637 side chains are in green. Electrostatic surface potentials are shown in panels a and c. Polar and charged residues are shown in blue, and hydrophobic amino acids are in light gray.
Activity of gH 625–644 Peptide on HSV1 Infectivity
Several reports in the literature (for review, see Ref. 67) describe the potential of fusion peptide analogs of orthomyxoviruses, paramyxoviruses, and HIV as virus entry inhibitors. The accepted view is that the infectivity inhibition may be due to the formation of inactive aggregates between the fusogenic stretches present in the viral protein and the synthetic peptides. These aggregates are formed as a consequence of their intrinsic oligomerization nature and are predicted to stabilize a prefusion intermediate and prevent merging of the bilayers. We, therefore, determined whether longer and shorter peptides compared with gH-(626–644) were efficient at inhibiting HSV infection. To verify that the peptides did not exert toxic effect on Vero cells, monolayers were exposed to different concentrations (10, 50, and 100 μm) of each peptide for 24 h, and cell viability was assayed by an lactate dehydrogenase assay. No statistical difference was observed between the viability of control (untreated) cells and that of cells exposed to the peptides (data not shown).
To test whether modified peptides could affect HSV infectivity, Vero cells were infected with HSV at 37 °C in the presence or absence of each peptide. Plaque formation was measured 48 h post-infection. These results are shown in Fig. 10.
FIGURE 10.

Cells were incubated with increasing concentrations of the peptides (10, 25, 50, and 100 μm) in the presence of the viral inoculum for 45 min at 37 °C. Nonpenetrated virus was inactivated, and cells were incubated for 48 h at 37 °C in Dulbecco's modified Eagle's medium supplemented with carboxymethylcellulose. Plaque numbers were scored, and the percentage of inhibition was calculated with respect to no-peptide control experiments. Data are reported in triplicate, and error bars represent S.D. Inhibition experiments for longer and shorter peptides are reported in panel a, for N-terminal elongated peptides in panel b, and for C-terminal elongated peptides in panel c.
In a dose-dependent inhibition assay of HSV entry, we compared the activity of modified peptides with that of the wild-type gH-(626–644) and found that all peptides exhibited a consistent lower extent of inhibition except gH625–644, which was able to inhibit virus infectivity to a similar extent of gH-(626–644). The inhibition levels are not strong enough to hypothesize their direct development as antivirals but show a direct correlation between the α-helical potential and their inhibitory activity.
DISCUSSION
The mechanism by which herpes simplex virus performs fusion is poorly understood, although evidence has been accumulated in support of models describing the involvement of gB and gH in the fusion event. Furthermore, there are several regions within viral membrane fusion proteins that are involved in the fusion process and are decisive for membrane fusion to take place because destabilization of the lipid bilayer and membrane fusion appear to be the result of the binding and interaction of these segments with the biological membranes. We have shown previously the existence of different membranotropic regions in the HSV gH envelope glycoprotein; one of the most membranotropic regions along the whole glycoprotein is the gH-(626–644), and because this region could be important in the membrane fusion process, we present here the results of the study of the interactions of peptides with longer and shorter sequences derived from this region. It seems, therefore, necessary to elucidate how the capacity of this region to interact and disrupt membranes depends on the amino acid sequence and its length. Several studies have described an effect of the length of the fusion peptide on its fusogenic properties. For example, a peptide corresponding to the 12 first residues of the gp32 SIV protein induces more lipid mixing than a peptide corresponding to the 16 or 24 first residues. However, the minimal HSV-1 fusion peptide, i.e. the shortest peptide capable of inducing significant membrane fusion, has not yet been determined. A helical peptide that inserts obliquely into the membrane induces fusion, and the more tilted the peptide, the more important the fusion. Among the HSV-1 membranotropic domains, gH-(626–644) inserts into the bilayer as an amphiphatic helix; thus, we believe that the minimal fusion peptides of HSV correspond to the shortest helical fragment capable of obliquely inserting into the membrane. In fact, experimental fusion assays on liposomes clearly helped in defining the minimal fusion sequence. In particular, 14 amino acid peptides (gH-(631–644) and gH-(626–639)) induced lower levels of fusion compared with gH-(626–644), suggesting that shorter peptides were unable to cross or stably position inside the bilayer; 24-amino acid peptides showed a greater fusion activity compared with the native peptide, with a significant increase of activity when elongated at the N terminus. Our results further show that the addition of only one amino acid at the N terminus of the native sequence strongly increases the fusion activity. Peptides bearing the histidine at the N terminus but shorter at the C terminus were strongly impaired in their fusion activity, further supporting previous results (40) that pointed out the importance of a charged residue at the C terminus for the localization of the peptide inside the lipid bilayer. In fact, the arginine localized at the C terminus is probably involved in interactions with the polar head groups, and we previously reported that mutation of this arginine severely compromised its fusion activity (40).
Furthermore, we have focused on the possible functional roles of this domain by an in-depth study of the peptide patterned after this domain (gH625–644), and we compared its activity with that of gH-(626–644). The IC50 of gH625–644 at 37 °C is 0.25 μm, indicating that this peptide is a strong fusogenic sequence under this condition and that it is able to induce fusion at a concentration much lower than the native sequence. In particular, we have shown that the gH625–644 peptide interacts with model membranes, penetrates the bilayer with its N terminus, has a tryptophan residue buried inside the bilayer, and adopts a helical conformation with its hydrophobic residues on one face of the helix and polar or charged residues on the opposite face.
The peptide gH625–644 contains particular residues that are crucial for the capacity of the peptide to interact and destabilize target lipid membranes. It is rich in hydrophobic residues, including glycines, leucines, alanines, and aromatic residues such as tryptophan, tyrosines, which are known to preferentially interact at the membrane interface. Both the tryptophan and tyrosine are on the same side of the helix in the three-dimensional structure of gH625–644, forming an amphiphilic helix in which one side is constituted by aromatic and hydrophobic residues, whereas the other side is formed by hydrophilic or small residues. The interaction between the aromatic ring of tryptophan and the side chain of tyrosine is important for maintenance of structural stability during the interaction with the membrane.
Our results strongly support a fundamental role played by the histidine residue at the N terminus of the peptide gH625–644 in its fusion activity. The importance of a single histidine residue as a switch for triggering viral fusion is not novel. It was recently reported (68) that a histidine switch triggers membrane fusion in paramyxoviruses, supporting the importance and specificity of the histidine moiety in activating Paramyxovirus fusion. Considering the pH-independent nature of fusion exhibited by Paramyxovirus, the authors suggest that the pKa of histidine may not influence the observed fusion at neutral pH, in agreement with earlier observations (68). Furthermore, a conserved histidine in one of the fusion loops of Semliki Forest virus E1 protein also plays a fundamental role in membrane fusion (69). There are three possible explanation for the role of histidine in fusion; 1) it could be involved in initial membrane interactions in a process involving multiple protein regions with the membrane during the insertion, 2) it could alter the interaction of the membrane proximal domain with the rest of the protein, or 3) it could be involved in regulating the formation of oligomeric complexes. In our study we can surely eliminate the second explanation, and it may be that the histidine in fusion peptides both helps in initial interactions with the membrane and helps the oligomerization process. This hypothesis is further supported by the fact that the histidine is located at the N terminus, and correct configuration of the N-terminal fusion peptide appears to be crucial for the fusogenic function of several fusion proteins (70, 71) as well as its location in the membrane core after peptide-bilayer interaction (56, 64). We previously demonstrated the importance of a stable penetration of a peptide with fusion activity inside the membrane bilayer (15, 46). In the present work we have demonstrated that the addition of one histidine at the N terminus of the peptide gH-(626–644) is sufficient to make the peptide approximately 8-fold more active.
These findings further support the hypothesis that the activity of fusion peptides, whether they are N-terminal or located inside the protein, is strongly correlated to the following characteristics; (a) the amphipathic character, (b) the high hydrophobicity and high α-helical content, and (c) the high content of aromatic residues and the presence at the C terminus of positive charges.
Recent structural studies have described a new class III of viral fusion proteins (9), among which there is glycoprotein B of HSV-1 and proteins belonging to this class, all shown to possess a bipartite fusion peptide that is composed of two hydrophobic loops at the tip of a four-strand β-sheet region inserted on a pleckstrin domain. The fusion loops are not buried at an oligomeric interface in the putative prefusion conformation but cluster near the viral membrane, and their deep penetration inside the membrane is precluded by the presence of charged and polar residues. Tryptophans and phenylalanines, present in the loops, act as fingers by positioning themselves at the interface between the fatty acid chain and the head group layers of the lipids, but alone they are not sufficient to pull the target membrane toward the viral envelope on the contrary, they may perturb the outer leaflet of the target bilayer. All these data clearly define the existence of fusion peptides bearing different features (helices able to stably insert inside the bilayer (although not completely traversing the bilayer) and loops able to interact with the outer leaflet) and may facilitate the interaction of other membranotropic domains with the bilayer.
Here, a key histidine residue in the most membranotropic domain of gH has been identified. None of the other peptides showed a comparable activity, further confirming the unique role of that amino acid sequence. Our results suggest that studies on membranotropic domains will provide information about virus-target cell fusion and are valuable for further development of intracellular delivery systems with novel mechanism of penetration through the membrane.
Supplementary Material

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1S–4S.
- FP
- fusion peptide
- HSV
- herpes simplex virus
- gB
- glycoprotein B
- gH
- glycoprotein H
- gL
- glycoprotein L
- TFE
- trifluoroethanol
- NBD
- 12-(N-methyl-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl))
- SPR
- surface plasmon resonance
- TOCSY
- two-dimensional total correlation spectroscopy
- NOESY
- two-dimensional nuclear Overhauser effect (NOE) spectroscopy
- HIV
- human immunodeficiency virus
- RU
- response unit
- LUV
- large unilamellar vesicle
- PC/Chol
- phosphatidylcholine/cholesterol.
REFERENCES
- 1.White J. M., Delos S. E., Brecher M., Schornberg K. (2008) Crit. Rev. Biochem. Mol. Biol. 43, 189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison S. C. (2008) Nat. Struct. Mol. Biol. 15, 690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weissenhorn W., Hinz A., Gaudin Y. (2007) FEBS Lett. 581, 2150–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kielian M., Rey F. A. (2006) Nat. Rev. Microbiol. 4, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brasseur R., Pillot T., Lins L., Vandekerckhove J., Rosseneu M. (1997) Trends Biochem. Sci. 22, 167–171 [DOI] [PubMed] [Google Scholar]
- 6.Durell S. R., Martin I., Ruysschaert J. M., Shai Y., Blumenthal R. (1997) Mol. Membr. Biol. 14, 97–112 [DOI] [PubMed] [Google Scholar]
- 7.Zhu N. L., Cannon P. M., Chen D., Anderson W. F. (1998) J. Virol. 72, 1632–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madu I. G., Roth S. L., Belouzard S., Whittaker G. R. (2009) J. Virol. 83, 7411–7421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannah B. P., Heldwein E. E., Bender F. C., Cohen G. H., Eisenberg R. J. (2007) J. Virol. 81, 4858–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shai Y. (2000) Biosci. Rep. 20, 535–555 [DOI] [PubMed] [Google Scholar]
- 11.Tamm L. K., Han X. (2000) Biosci. Rep. 20, 501–518 [DOI] [PubMed] [Google Scholar]
- 12.Skehel J. J., Cross K., Steinhauer D., Wiley D. C. (2001) Biochem. Soc. Trans. 29, 623–626 [DOI] [PubMed] [Google Scholar]
- 13.Martin I., Ruysschaert J. M. (2000) Biosci. Rep. 20, 483–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guillén J., Kinnunen P. K., Villalaín J. (2008) Biochim. Biophys. Acta 1778, 2765–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galdiero S., Falanga A., Vitiello M., Browne H., Pedone C., Galdiero M. (2005) J. Biol. Chem. 280, 28632–28643 [DOI] [PubMed] [Google Scholar]
- 16.Galdiero S., Falanga A., Vitiello M., D'Isanto M., Collins C., Orrei V., Browne H., Pedone C., Galdiero M. (2007) Chembiochem 8, 885–895 [DOI] [PubMed] [Google Scholar]
- 17.Spear P. G., Longnecker R. (2003) J. Virol. 77, 10179–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heldwein E. E., Krummenacher C. (2008) Cell. Mol. Life Sci. 65, 1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galdiero S., Vitiello M., D'Isanto M., Falanga A., Collins C., Raieta K., Pedone C., Browne H., Galdiero M. (2006) J. Gen. Virol. 87, 1085–1097 [DOI] [PubMed] [Google Scholar]
- 20.Galdiero S., Vitiello M., D'Isanto M., Falanga A., Cantisani M., Browne H., Pedone C., Galdiero M. (2008) Chembiochem 9, 758–767 [DOI] [PubMed] [Google Scholar]
- 21.Cai W. H., Gu B., Person S. (1988) J. Virol. 62, 2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forrester A., Farrell H., Wilkinson G., Kaye J., Davis-Poynter N., Minson T. (1992) J. Virol. 66, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian R. P., Geraghty R. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 2903–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atanasiu D., Whitbeck J. C., Cairns T. M., Reilly B., Cohen G. H., Eisenberg R. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avitabile E., Forghieri C., Campadelli-Fiume G. (2007) J. Virol. 81, 11532–11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Avitabile E., Forghieri C., Campadelli-Fiume G. (2009) J. Virol. 83, 10752–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heldwein E. E., Lou H., Bender F. C., Cohen G. H., Eisenberg R. J., Harrison S. C. (2006) Science 313, 217–220 [DOI] [PubMed] [Google Scholar]
- 28.Roche S., Bressanelli S., Rey F. A., Gaudin Y. (2006) Science 313, 187–191 [DOI] [PubMed] [Google Scholar]
- 29.Gage P. J., Levine M., Glorioso J. C. (1993) J. Virol. 67, 2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backovic M., Jardetzky T. S. (2009) Curr. Opin. Struct. Biol. 19, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kielian M. (2006) Virology 344, 38–47 [DOI] [PubMed] [Google Scholar]
- 32.Schibli D. J., Weissenhorn W. (2004) Mol. Membr. Biol. 21, 361–371 [DOI] [PubMed] [Google Scholar]
- 33.Gianni T., Martelli P. L., Casadio R., Campadelli-Fiume G. (2005) J. Virol. 79, 2931–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cairns T. M., Landsburg D. J., Whitbeck J. C., Eisenberg R. J., Cohen G. H. (2005) Virology 332, 550–562 [DOI] [PubMed] [Google Scholar]
- 35.Galdiero M., Whiteley A., Bruun B., Bell S., Minson T., Browne H. (1997) J. Virol. 71, 2163–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harman A., Browne H., Minson T. (2002) J. Virol. 76, 10708–10716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson D. W., Davis-Poynter N., Minson A. C. (1994) J. Virol. 68, 6985–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gianni T., Menotti L., Campadelli-Fiume G. (2005) J. Virol. 79, 7042–7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopper M., Compton T. (2004) J. Virol. 78, 8333–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galdiero S., Falanga A., Vitiello M., Raiola L., Fattorusso R., Browne H., Pedone C., Isernia C., Galdiero M. (2008) J. Biol. Chem. 283, 29993–30009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiske C. H., Subbarow Y. (1925) J. Biol. Chem. 66, 374–389 [Google Scholar]
- 42.Schwarz G., Stankowski S., Rizzo V. (1986) Biochim. Biophys. Acta 861, 141–151 [DOI] [PubMed] [Google Scholar]
- 43.Beschiaschvili G., Seelig J. (1990) Biochemistry 29, 52–58 [DOI] [PubMed] [Google Scholar]
- 44.De Kroon A. I., Soekarjo M. W., De Gier J., De Kruijff B. (1990) Biochemistry 29, 8229–8240 [DOI] [PubMed] [Google Scholar]
- 45.Eftink M. R., Ghiron C. A. (1976) J. Phys. Chem. 80, 486–493 [Google Scholar]
- 46.Galdiero S., Falanga A., Vitiello G., Vitiello M., Pedone C., D'Errico G., Galdiero M. (2010) Biochim. Biophys. Acta 1798, 579–591 [DOI] [PubMed] [Google Scholar]
- 47.Kalb E., Frey S., Tamm L. K. (1992) Biochim. Biophys. Acta 1103, 307–316 [DOI] [PubMed] [Google Scholar]
- 48.Morton T. A., Myszka D. G., Chaiken I. M. (1995) Anal. Biochem. 227, 176–185 [DOI] [PubMed] [Google Scholar]
- 49.Shai Y. (1999) Biochim. Biophys. Acta 1462, 55–70 [DOI] [PubMed] [Google Scholar]
- 50.Mozsolits H., Wirth H. J., Werkmeister J., Aguilar M. I. (2001) Biochim. Biophys. Acta 1512, 64–76 [DOI] [PubMed] [Google Scholar]
- 51.Cavanagh J., Fairbrother W., Palmer A. G., III, Skelton N. J. (1996) Protein NMR Spectroscopy, Principles, and Practice, Academic Press, Inc., San Diego, CA [Google Scholar]
- 52.Hwang T. L., Shaka A. J. (1995) J. Magn. Reson. A 112, 275–279 [Google Scholar]
- 53.Bartels C., Xia T., Billeter M., Guntert P., Wüthrich K. (1995) J. Biomol. NMR 6, 1–10 [DOI] [PubMed] [Google Scholar]
- 54.Herrmann T., Güntert P., Wüthrich K. (2002) J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 55.Koradi R., Billeter M., Wüthrich K. (1996) J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 56.Lüneberg J., Martin I., Nüssler F., Ruysschaert J. M., Herrmann A. (1995) J. Biol. Chem. 270, 27606–27614 [DOI] [PubMed] [Google Scholar]
- 57.Galdiero S., Galdiero M., Pedone C., Falanga A., Vitiello M. (December/11/2009) U. S. Patent Provisional 61/285,619
- 58.Yau W. M., Wimley W. C., Gawrisch K., White S. H. (1998) Biochemistry 37, 14713–14718 [DOI] [PubMed] [Google Scholar]
- 59.Habermann E., Jentsch J. (1967) Hoppe-Seyler's Z. Physiol. Chem. 348, 37–50 [PubMed] [Google Scholar]
- 60.Rafalski M., Lear J. D., DeGrado W. F. (1990) Biochemistry 29, 7917–7922 [DOI] [PubMed] [Google Scholar]
- 61.Papo N., Shai Y. (2003) Biochemistry 42, 458–466 [DOI] [PubMed] [Google Scholar]
- 62.DeGrado W. F., Lear J. D. (1990) Biopolymers 29, 205–213 [DOI] [PubMed] [Google Scholar]
- 63.Parente R. A., Nadasdi L., Subbarao N. K., Szoka F. C., Jr. (1990) Biochemistry 29, 8713–8719 [DOI] [PubMed] [Google Scholar]
- 64.Gordon L. M., Curtain C. C., Zhong Y. C., Kirkpatrick A., Mobley P. W., Waring A. J. (1992) Biochim. Biophys. Acta 1139, 257–274 [DOI] [PubMed] [Google Scholar]
- 65.Lear J. D., DeGrado W. F. (1987) J. Biol. Chem. 262, 6500–6505 [PubMed] [Google Scholar]
- 66.Wishart D. S., Case D. A. (2001) Methods Enzymol. 338, 3–34 [DOI] [PubMed] [Google Scholar]
- 67.Vitiello M., Galdiero M., Galdiero M. (2009) Protein Pept. Lett. 16, 786–793 [DOI] [PubMed] [Google Scholar]
- 68.Krishnan A., Verma S. K., Mani P., Gupta R., Kundu S., Sarkar D. P. (2009) J. Virol. 83, 1727–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chanel-Vos C., Kielian M. (2004) J. Virol. 78, 13543–13552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Freed E. O., Myers D. J. (1992) J. Virol. 66, 5472–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinhauer D. A., Wharton S. A., Skehel J. J., Wiley D. C. (1995) J. Virol. 69, 6643–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.