

Abstract
An integrated microfluidic device has been developed to perform 1024 in situ click chemistry reactions in parallel using the bovine carbonic anhydrous II (bCAII) click chemistry system as a proof-of-concept study and a rapid hit identification approach using SPE purification and electrospray-ionization mass spectrometry, multiple reaction monitoring (MRM) analysis, all of which improves the sensitivity and throughput of the downstream analysis.
“In situ click chemistry” is a target-guided synthetic (TGS)1–8 method for the fast and efficient production of multiple potential biligand enzymatic inhibitors. One of the best known examples of click chemistry is the assembly of complementary azide and acetylene building blocks inside the binding pockets of the target through a Huisgen cycloaddition reaction.7,9 Over the past six years, the TGS methodology has been successfully applied for the preparation of inhibitors for a variety of targets, such as acetylcholine esterase (AchE),7,10,11 bovine carbonic anhydrase II (bCAII),12,13 protein tyrosine phosphatases,14,15 HIV protease,16 and metalloproteases.17,18 Recently, the application has been extended for in situ templated synthesis of a small molecule at the protein-protein binding site in an important apoptosis pathway – Bcl-XL. 19 However, there have been challenges which hampered the broader application of the approach – namely inadequate flexibility in the platform which restricted the number of possible permutations of a given synthetic route, the high consumption of target proteins and reagents (typically 1–10 nmol range), some of which are scarce and difficult to obtain, and the lack of efficient/sensitive screening technologies for checking reaction products and subsequent hit identification. A solution to these challenges is the development of an operational platform capable of multiplexing in situ click chemistry reactions with simultaneous improvements in the throughput and sensitivity of the hit-identification approach in order to accelerate screening speed and reduce protein/reagent consumption. Most importantly, this new platform must be operated in an automated fashion to avoid human errors, and to enhance operation efficiency and fidelity. With intrinsic advantages including sample/reagent economy,20–29 precise fluidic delivery,30 scalability and automation,31–33 integrated microfluidic devices provide an excellent operational platform for efficiently performing multiplexed in situ click chemistry applications.
Previously, we constructed an integrated microfluidic chip platform for parallel synthesis and screening of 32 in situ click chemistry reactions. 13 In the 1st-generation design, the known bCAII click chemistry system composed of an acetylenic benzenesulfonamide and multiple complementary azides, achieved a 5–12-fold improvement in sample/reagent economy and comparable experimental fidelity over that obtainable with conventional 96-well plates.12 The flexible design provided ample opportunity for further improvement, especially to increase the number of screening reactions, to reduce the consumption of sample/reagent, and to accelerate operational speed for not only the formation of a screening library but also for hit identification.
Herein, we describe a 2nd-generation integrated microfluidic platform in which 1024 click chemistry parallel syntheses were performed with subsequent off-line hit identification. Due to the improved design, the time required for preparing a single click reaction was reduced from 1.0 min (1st-generation) to 17 s (2nd-generation). In parallel, a miniature reverse phase clean-up step (ZipTip®, Fig. S1, ESI†) was developed to remove polar/charged reagents (in this case DMSO and PBS salts) from the reaction mixtures that would otherwise interfere with direct electrospray ionization (ESI) mass spectrometry (MS) used for hit identification. This step eliminated the need for a time-consuming liquid chromatographic (LC) step in the process. Furthermore, by using parent → fragment ion (P → F) transitions, created during collisionally activated decomposition and monitored on a triple quadrupole mass spectrometer, the resulting traces (multiple reaction monitoring (MRM) traces) simplified the hit identification procedure into an easily interpreted, and potentially automatable, format. As a result, the time required for hit identification is significantly reduced (15 s per reaction, 2 min/8 samples) compared to the original LC–MS-based method (40 min per reaction). The combined applications of ZipTip® and ESI-MRM enhanced the sensitivity for hit identification, resulting in reduced enzyme and reactant consumptions. The 2nd-generation platform described here utilized a total reaction volume of approximately 400 nL, containing approximate 12.4 pmol (360 ng) of enzyme, 120 pmol of both the acetylene and azide, compared with the 4 µL reaction mixture containing 0.655 nmol (19 µg) of enzyme, 2.4 nmol of acetylene, and 3.6 nmol of azide, employed in the 1st-generation platform. Overall, a 20–50-fold improvement in sample economy was achieved with the 2nd-generation platform. A side-by-side comparison of sample/reagent consumption and reaction times among the conventional 96-well approach and the two generations of microfluidic platforms is summarized in Table S1(ESI†).
To reliably produce over a thousand reactions in a single operation, the 2nd-generation microfluidic chip (Fig. 1) comprises four components: (i) a pair of microfluidicmultiplexers31 for regulating the 2 × 16 individually addressed reagent inlets; (ii) a 150 nL rotary mixer for mixing reagents for each reaction; (iii) a 250 nL serpentine channel to accommodate additional PBS to give each reaction the volume (400 nL) required for subsequent manipulation, and to complete the mixing of reagents; (iv) a replaceable 20-cm long poly(tetrafluoroethylene) (PTFE) tube (Fig. 1c) connected to the outlet of the chip to serve as a reservoir for accommodating the reaction mixture slugs emerging from the chip. Following our previous protocol,13 a click chemistry library composed of 8 acetylenes (I–VIII, deployed in duplicate) and 16 azides (1–16) was assembled (Table 1). Four different types of reaction conditions were tested in parallel to give 1024 individual reaction mixtures. These four conditions are: (i) 128 duplicated reactions with CuI-catalysis to generate reference products; (ii) 128 duplicated reactions between eight acetylene (I–VIII) and 16 azide (1–16) reactants in the presence of bCAII; (iii) 128 duplicated control reactions performed as in (ii) but in the presence of inhibitor 17 to confirm the active-site specificity of the reactions; and (iv) 128 duplicated blank reactions performed as in (ii), but in the absence of bCAII, to monitor catalysis-independent product formation. Under these conditions, each reaction consumes 20 nL of one acetylene solution (6 mM, 120 pmol), 20 nL of one azide solution (6 mM, 120 pmol), and 110 nL of bCAII (113 µM in PBS) solution. The CuI catalyst solution contained 0.15 mM CuSO4, 3 mM sodium ascorbate and 0.06 mM tris((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)amine in PBS (pH 7.4). The inhibitor 17 was dissolved in DMSO and then diluted with the bCAII solution to give final concentrations of 17 and bCAII of 2 mM and 113 µM, respectively. In the blank reactions, the bCAII solution was replaced with PBS.
Fig 1.

(a) Schematic representation of the 2nd-generation integrated microfluidic platform. The operation of the circuit was computer controlled using color-coded pressure-driven valves: red – positive pressure, off/on; yellow – peristaltic pumping; green – vacuum. (b) Optical image of the actual device. The various channels were loaded with dyes to visualize the different components: red, yellow and green as in part (a) and blue indicated the fluidic channels. (c) The PTFE tubing for off-chip incubation and storage of the reaction mixture slugs. Again, blue and red dyes are used for visualization. Black scale bars are 3 mm.
Table 1.
Structures of the acetylenes I–VIII and the azides 1–16 used for in situ click chemistry screening library in the 2nd-generation microfluidic platform
| Acetylenes I–VIII | Azides 1– 16 |
|---|---|
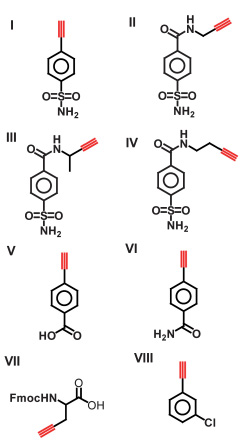 |
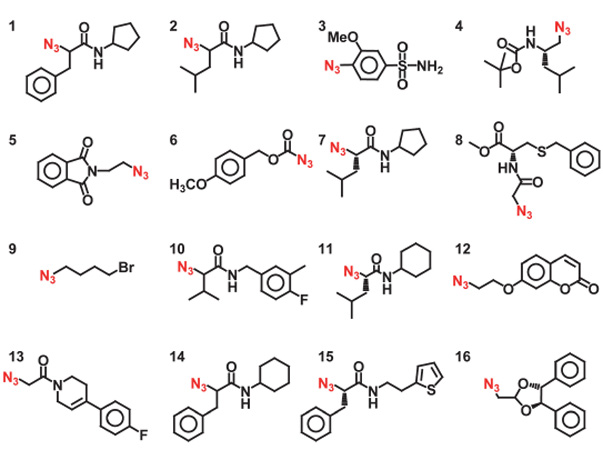 |
By using a computer-controlled interface (LabView, National Instrument Inc.) multiple steps were programmed for automated control of the introduction of all reagents for each reaction. The operation was pneumatically controlled via valve-regulated back-pressure and vacuum suction to control the movement of each reaction slug through the channels (Fig. S1, ESI†). Using the bCAII reaction between acetylene I and azide 1 as an example, the sequence of steps involved in the preparation of each reaction mixture were: firstly, simultaneous introduction of 20 nL of acetylene I and 20 nL of azide 1 solutions into the rotary mixer; secondly, this was followed either by 110 nL of the bCAII solution, or the CuI, the bCAII with inhibitor or PBS solutions to generate the enzymatically catalyzed, CuI-catalyzed, inhibited, and blank reactions, respectively; thirdly, the reaction mixtures were mixed by continual cycling in the rotary mixer; fourthly, additional PBS was introduced to bring the reaction volumes to 400 nL and to flush the mixtures into the serpentine channel used for final mixing; fifthly, the reaction mixture solution was then expelled into the PTFE tube in the form of a slug. To prevent cross contamination between two reaction mixture slugs, air and PBS buffer slugs were introduced in-between each reaction slug.34–36 Eight reaction mixtures were stored in each PTFE tube, which were then manually replaced for the next batch of eight reaction mixtures. Excluding the time required for replacing the PTFE tubes, the entire procedure for preparing the 1024 reaction mixture screening library was completed in 290 minutes (ca. 17 s/cycle). The reaction mixtures were sequentially generated and stored in 128 separate PTFE tubes which were placed in a moisture-regulated incubator at 37 °C for 40 h to complete the reactions. There was no visible volume change of the reaction mixture slugs inside PTFE tubing during the incubation. Once the incubation was completed, the reaction slugs inside the PTFE tubes generated from the reaction conditions (i) and (iv) were pneumatically expelled with water (10 µL) into 200 µL microcentrifuge tubes; those under the conditions (ii) and (iii), were expelled with 10 µL guanidine solution (0.1M) to denature the bCAII and release the triazole products. A miniaturized solid phase extraction procedure (SPE) with ZipTip® (Millipore, C18) pipette tips were then used to pre-purify the products of the reactions (see details in Fig. S2, ESI†). DMSO and PBS salts in the reaction mixtures were not retained, and the retained triazole products were eluted with H2O/acetonitrile/formic acid (50/50/0.1, v/v/v, 20 µL) prior to MS analysis.
The mass spectra and MSMS spectra of each product were first collected from the CuI-catalyzed reactions by direct manual injection of the eluted materials into the ESI source of a triple quadrupole mass spectrometer. Then the remaining elutes were screened for product formation by MRM. The P → F transitions were used to manually identify the presence of eight triazole products from a single injection (Fig. S3, ESI†). The limitation on monitoring only eight reactions simultaneously is imposed by the mass spectrometer used in this work, and could be expanded to one hundred or more with contemporary instrumentation. Continuing the bCAII reaction between acetylene II and azide 1–5, 7, 8, 13, as an example, a positive result among the eight pairs of MRM transitions was recognized by a significant and reproducible peak in the corresponding P → F transition for azides 1 and 13 only, and the absence of peaks in reaction mixtures containing inhibitor 17 and PBS only (Fig. 2). The procedure carries an in-built control in that the intensity of the transitions can be directly compared with the result obtained from CuI-catalysis, so regardless of the efficiency of product ionization and fragment ion yield, the results can be expressed as a percentage of the control reaction. Thus in this example there is a strong hit for II + 1 and II + 13 (about 65 and 60%, respectively) and a modest hit recorded for II + 8, as it is only about 10% of the control (Fig. 2).
Fig 2.

Parallel MRM screening for 8 different products from the 2nd-generation platform between acetylene II and azides 1–5, 7, 8 and 13 under 4 types of reaction conditions in the presence of: i) CuI, (ii) bCAII, (iii) both bCAII and inhibitor 17, (iv) PBS only. These MRM traces were obtained from 5 sequential injections (a–e) of SPE pre-processed samples. The arrows indicate the time of each injection.
All the click chemistry reactions between the acetylenes I–VIII and the azides 1–16 under the 4 reaction conditions (i)-(iv) were screened twice with consistent results which are shown in Fig. S4.† The screening results for the in situ click chemistry reactions between the acetylenes I–VIII and the azides 1–16 (summarized in Table 2) identified 39 hit reactions including 4 modest hit reactions using the 2nd-generation microfluidic platform. We also compared the results of 8 in situ click chemistry reactions between acetylene I and 8 azides 2, 7, 8, 12–16 performed with the 1st- and 2nd-generation platforms (Table 2) but using two different hit-identification methods. The result shows a consistency of 75% (6 out of 8 click reactions showed consistent results), although the current enzyme consumption is only 1/50th of that used by our 1st-generation platform.13
Table 2.
Summary of the screening results from the 256 in situ click chemistry reactions using the nd-generation microfluidic platform including some results from the st-generation one. The results on the first row were obtained via the LC-MS with the 1st-generation platform. For MRM-based hit identification, a threshold of 15% of control is used for a positive result.
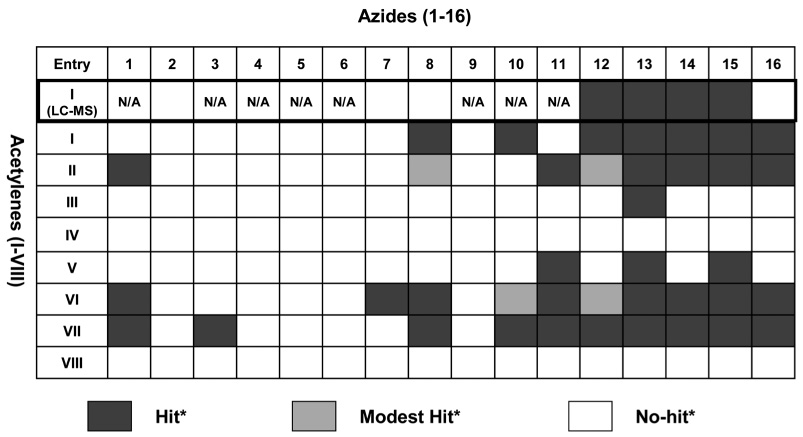 |
Peak heights for the MRM responses were expressed as a percentage of the response obtained with CuI-catalysis. Hit, modest-hit and no-hit reactions were defined as >15%, 5–15% and <5% of the CuI-catalysis response, respectively.
Conclusions
We have developed an improved platform for screening an in situ click chemistry library with 1024 reactions carried out in parallel by combination of integrated microfluidic, SPE purification and ESI-MS MRM technology. The current platform not only accommodates screening of up to 1024 reactions in parallel, but also significantly reduces reagent consumption and screening time. Contemporary instrumentation could be directly interfaced with the next generation of chips to handle on-line screening of large numbers of reactions. Furthermore, the nature of the MRM readout lends itself to computerized interpretation so that the process could be further automated. The construction of a fully automated system has the potential to vastly increase the throughput of enzyme inhibitor discovery.
Supplementary Material
Acknowledgements
We thank Heather Agnew and Prof. J. R. Heath (California Institute of Technology) for kindly providing acetylenes and azides. This research was supported by the DOD-Defense Threat Reduction Agency (W911NF0610243), the NIH-NCI NanoSystems Biology Cancer Center (U54A119347). Y.W and M.S. gratefully acknowledge support by the NSF-PREM program (NSF03564).
Footnotes
Electronic supplementary information (ESI) available: Fig. S1, S2, S3, S4, and Table S1. See DOI: 10.1039/b907430a
Notes and references
- 1.Rideout D. Science. 1986;233:561–563. doi: 10.1126/science.3523757. [DOI] [PubMed] [Google Scholar]
- 2.Huc I, Lehn JM. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2106–2110. doi: 10.1073/pnas.94.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehn JM, Eliseev AV. Science. 2001;291:2331–2332. doi: 10.1126/science.1060066. [DOI] [PubMed] [Google Scholar]
- 4.Ramstrom O, Lehn JM. Nat. Rev. Drug Discovery. 2002;1:26–36. doi: 10.1038/nrd704. [DOI] [PubMed] [Google Scholar]
- 5.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Proc. Natl. Acad. Sci. U. S. A. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demko ZP, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2110–2113. [PubMed] [Google Scholar]
- 7.Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:1053–1101. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Lee LV, Mitchell ML, Huang SJ, Fokin VV, Sharpless KB, Wong CH. J. Am. Chem. Soc. 2003;125:9588–9589. doi: 10.1021/ja0302836. [DOI] [PubMed] [Google Scholar]
- 9.Bock VD, Perciaccante R, Jansen TP, Hiemstra H, van Maarseveen JH. Org. Lett. 2006;8:919–922. doi: 10.1021/ol053095o. [DOI] [PubMed] [Google Scholar]
- 10.Manetsch R, Krasinski A, Radic Z, Raushel J, Taylor P, Sharpless KB, Kolb HC. J. Am. Chem. Soc. 2004;126:12809–12818. doi: 10.1021/ja046382g. [DOI] [PubMed] [Google Scholar]
- 11.Krasinski A, Radic Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, Kolb HC. J. Am. Chem. Soc. 2005;127:6686–6692. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- 12.Mocharla VP, Colasson B, Lee LV, Roper S, Sharpless KB, Wongand CH, Kolb C. Angew. Chem., Int. Ed. 2005;44:116–120. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- 13.Wang JY, Sui GD, Mocharla VP, Lin RJ, Phelps ME, Kolb HC, Tseng HR. Angew. Chem., Int. Ed. 2006;45:5276–5281. doi: 10.1002/anie.200601677. [DOI] [PubMed] [Google Scholar]
- 14.Srinivasan R, Uttamchandani M, Yao SQ. Org. Lett. 2006;8:713–716. doi: 10.1021/ol052895w. [DOI] [PubMed] [Google Scholar]
- 15.Xie J, Seto CT. Bioorg. Med. Chem. 2007;15:458–473. doi: 10.1016/j.bmc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whiting M, Muldoon J, Lin YC, Silverman SM, Lindstrom W, Olson AJ, Kolb HC, Finn MG, Sharpless KB, Elder JH, Fokin VV. Angew. Chem., Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- 17.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sieber SA, Niessen S, Hoover HS, Cravatt BF. Nat. Chem. Biol. 2006;2:274–281. doi: 10.1038/nchembio781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu XD, Sun JZ, Wang HG, Manetsch R. J. Am. Chem. Soc. 2008;130:13820–13821. doi: 10.1021/ja802683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.deMello AJ. Nature. 2006;442:394–402. doi: 10.1038/nature05062. [DOI] [PubMed] [Google Scholar]
- 21.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 22.Watts P, Haswell SJ. Chem. Soc. Rev. 2005;34:235–246. doi: 10.1039/b313866f. [DOI] [PubMed] [Google Scholar]
- 23.Jahnisch K, Hessel V, Lowe H, Baerns M. Angew. Chem., Int. Ed. 2004;43:406–446. doi: 10.1002/anie.200300577. [DOI] [PubMed] [Google Scholar]
- 24.Geyer K, Codee JDC, Seeberger PH. Chem.–Eur. J. 2006;12:8434–8442. doi: 10.1002/chem.200600596. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi J, Mori Y, Kobayashi S. Chem.–Asian J. 2006;1:22–35. doi: 10.1002/asia.200600058. [DOI] [PubMed] [Google Scholar]
- 26.Mason BP, Price KE, Steinbacher JL, Bogdan AR, McQuade DT. Chem. Rev. 2007;107:2300–2318. doi: 10.1021/cr050944c. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed-Omer B, Brandt JC, Wirth T. Org. Biomol. Chem. 2007;5:733–740. doi: 10.1039/b615072a. [DOI] [PubMed] [Google Scholar]
- 28.Fukuyama T, Rahman T, Sato M, Ryu I. Synlett. 2008:151–163. [Google Scholar]
- 29.Yoshida JI, Nagaki A, Yamada T. Chem.–Eur. J. 2008;14:7450–7459. doi: 10.1002/chem.200800582. [DOI] [PubMed] [Google Scholar]
- 30.Huang B, Wu HK, Bhaya D, Grossman A, Granier S, Kobilka BK, Zare RN. Science. 2007;315:81–84. doi: 10.1126/science.1133992. [DOI] [PubMed] [Google Scholar]
- 31.Thorsen T, Maerkl SJ, Quake SR. Science. 2002;298:580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- 32.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 33.Lee CC, Sui GD, Elizarov A, Shu CYJ, Shin YS, Dooley AN, Huang J, Daridon A, Wyatt P, Stout D, Kolb HC, Witte ON, Satyamurthy N, Heath JR, Phelps ME, Quake SR, Tseng HR. Science. 2005;310:1793–1796. doi: 10.1126/science.1118919. [DOI] [PubMed] [Google Scholar]
- 34.Tice JD, Lyon AD, Ismagilov RF. Anal. Chim. Acta. 2004;507:73–77. [Google Scholar]
- 35.Zheng B, Tice JD, Ismagilov RF. Anal. Chem. 2004;76:4977–4982. doi: 10.1021/ac0495743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garstecki P, Fuerstman MJ, Stone HA, Whitesides GM. Lab Chip. 2006;6:437–446. doi: 10.1039/b510841a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.