

Abstract
To further understand copper-promoted oxidation reactions, the Cu(I) complexes LCuX (L = N,N-di-tert-butyl-N′,N′-dimethylethylenediamine; X = benzoylformate (BF) or p-nitro-benzoylformate) were synthesized, fully characterized by X-ray crystallography and spectroscopy in solution, and their reactivity with O2 at −80 °C examined. Oxidative decarboxylation of the α-ketocarboxylate ligand was observed, but only to a significant extent when cyclohexene, cyclooctene, or acetonitrile was present. Spectroscopic and conductivity data were consistent with mechanistic postulates involving displacement of the α-ketocarboxylate by the additives to a small extent, followed by oxygenation of the LCu(I) moiety to yield copper-oxygen species that subsequently induce decarboxylation. To test these hypotheses, spectroscopic and kinetic studies of the reactions of Bu4NBF with preformed μ-η2:η2-peroxodicopper(II) and/or bis(μ-oxo)dicopper(III) complexes supported by L or N,N,N′,N′-tetramethylpropylenediamine were performed. In an illustration of a new mode of reactivity for such dicopper-oxygen cores, decarboxylation of the added α-ketocarboxylate was observed and the intermediacy of a carboxylate-bridged μ-η2:η2-peroxodicopper(II) complex was implicated.
Introduction
In efforts to understand copper-promoted oxidation reactions important in biology and industry, extensive research has aimed to isolate, characterize, and probe the reactivity of copper-oxygen intermediates.1-6 Particularly well-studied examples of such intermediates include those with bis(μ-oxo)dicopper(III), μ-peroxodicopper(II), and monocopper-superoxo/peroxo cores, which typically are prepared through low temperature reactions of N-donor ligand-supported Cu(I) complexes with O2. Recent interest has focused on the isolation of monocopper-oxo species, [Cu2+-O•- ↔ Cu3+=O], which have been studied by theory,7 postulated as reactive intermediates in enzymes7a-d,8 and some oxidation reactions,9 yet only identified experimentally in the gas phase.7e In one attempt to access such species we decided to explore a route analogous to one that yields related Fe4+=O moieties,10 but where M = Cu instead of Fe (Figure 1).11 In this route that has been postulated to be followed by several known iron-containing metalloenzymes and model complexes, an α-ketocarboxylate co-ligand is oxidatively decarboxylated via O2 activation and nucleophilic attack of the ketocarbonyl carbon. Our previous efforts to extend this chemistry to M = Cu involved reactions of O2 with Cu(I) complexes of α-ketocarboxylates supported by bidentate pyridyl-imine ligands.11 Oxidative decarboxylation ensued, resulting in the formation of the respective carboxylic acid and hydroxylation of a ligand arene substituent. These findings, in conjunction with theoretical calculations, were interpreted to indicate the intermediacy of a highly reactive monocopper-oxo species that was ‘trapped’ in intramolecular fashion by the proximal arene ring of the supporting pyridyl-imine ligand through a mechanism similar to that proposed for Fe/α-ketocarboxylate chemistry (Figure 1).11
Figure 1.
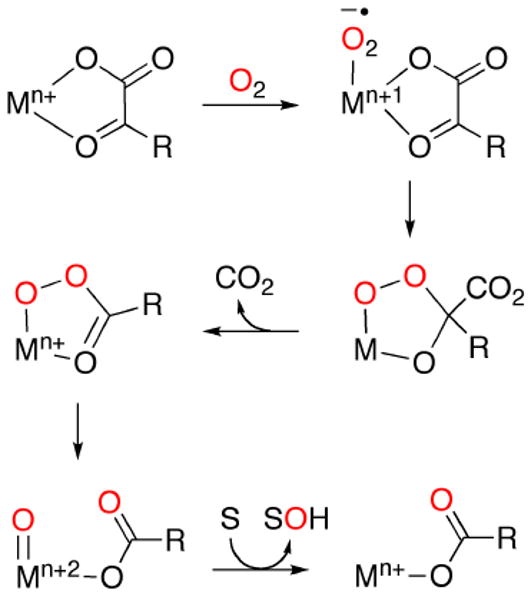
Proposed mechanism for the metal mediated oxidative decarboxylation of an α-ketocarboxylate utilizing O2 (M = Fe, n = 2, or M = Cu, n = 1; S = substrate; R = alkyl or aryl group).
Herein, we report the results of efforts to broaden the scope of the initial studies by examining the effect of using more electron-donating diamine supporting ligands lacking arene substituents (Me4pda and tBu2Me2eda; Figure 2).12,13 We envisioned that such ligands might enable the isolation of a monocopper-oxo species and/or oxidation of an exogenous substrate. Instead, we found that while Cu(I)-α-ketocarboxylates could be isolated using tBu2Me2eda, decarboxylation upon treatment with O2 was insignificant unless alkenes or acetonitrile was present. Subsequent mechanistic studies indicated the involvement of μ-η2:η2-peroxodicopper(II) and/or bis(μ-oxo)dicopper(III) species. These species were independently shown to induce decarboxylation of exogenous α-ketocarboxylate, thus demonstrating a new type of reactivity for such cores.
Figure 2.
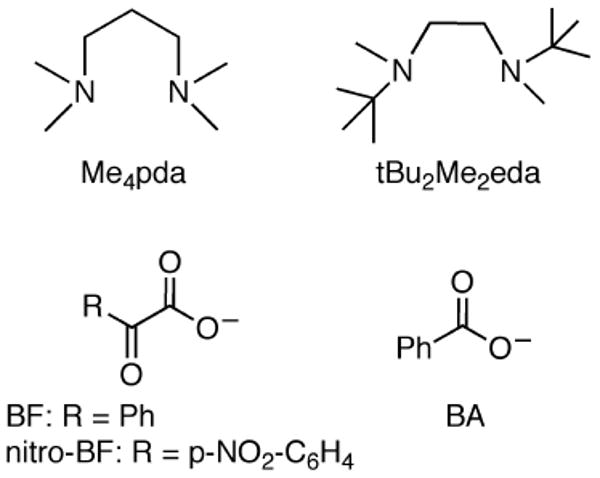
Ligands and abbreviations used in this work.
Experimental Section
General Considerations
All solvents and reagents were obtained from commercial sources unless otherwise noted. All solvents were thoroughly degassed via three cycles of freezing, evacuating, and thawing. THF was dried over Na/benzophenone and distilled under vacuum. CH2Cl2 and CD2Cl2 were dried over CaH2 and distilled under vacuum. Et2O and pentanes were passed through solvent purification columns (Glass Contour, Laguna, CA). All metal complexes were prepared and stored in a Vacuum Atmospheres inert atmosphere glovebox under dry nitrogen or were manipulated under argon or dry nitrogen using standard Schlenk techniques. [LCu(CH3CN)]OTf (L = Me4pda12 or tBu2Me2eda13), Cu4Mes4,14 Bu4NBF (BF = benzoylformate),15 and TlBF16 were prepared by literature methods and Bu4NBA (BA = benzoate) was purchased from Aldrich.
Physical Methods
NMR spectra were recorded on either Varian VI-300, VXR-300, or VI-500 spectrometers at room temperature. Chemical shifts (δ) for 1H and 13C NMR spectra were referenced to residual protium in the deuterated solvent (1H NMR experiments) or the characteristic solvent resonances of the solvent nuclei (13C NMR experiments). Variable temperature 1H NMR spectra were obtained on a Varian VI-300 spectrometer fitted with a liquid nitrogen cryostat. UV-Vis spectra were collected on a HP8453 (190-1000 nm) diode array spectrophotometer. Low temperature UV-Vis experiments were performed using an Unisoku low temperature UV-Vis cell holder. UV-Vis spectra that had drifting baselines due to minor frosting caused by the low-temperature device were corrected when necessary by subtracting the average of a region with no absorbance from the entire spectrum. Resonance Raman spectra were recorded on an Acton 506 spectrometer using a Princeton Instruments LN/CCD-11100-PB/UBAR detector and ST-1385 controller interfaced with Winspec software. The spectra were obtained at -196 °C using backscattering geometry. Excitation at 406.7 nm was provided by a Spectra-Physics BeamLok 2060-KR-V Krypton ion laser. Raman shifts were externally referenced to liquid indene and internally referenced to solvent (CH2Cl2). Elemental analysis was performed by Robertson Microlit Lab (Madison, NJ). Electrospray ionization mass spectrometry (ESI-MS) was performed on a Bruker BioTOF II instrument. All GC-MS experiments were conducted on an Agilent Technologies 7890A GC system and 5975C VL MSD. The GC column was a HP-5ms with dimensions 30 m × 0.250 mm. The standard method used for all runs involved an initial oven temperature of 60 °C (held for 4 min) followed by a 20 °C/min ramp to 230 °C that was held for 15 min. Infrared spectra were collected on a Nicolet Avatar 370 FT-IR equipped with an attenuated total reflectance attachment, using a CaF2 solution cell (International Crystal Laboratories) or a Smart OMNI-Sampler for solid samples.
Electrical conductivity measurements were performed in CH2Cl2 using a Fischer Scientific Accumet Portable AP65 model conductivity bridge with a cell constant of 1.0 cm-1. Anaerobically prepared solutions of (tBu2Me2eda)Cu(BF) in CH2Cl2 (5 ml, 0.1-0.6 mM) were loaded into a 10 mL Schlenk flask, sealed with a glass stopper, and cooled to -78 °C in a dry ice/acetone bath under positive argon pressure. Measurements were taken by removal of the glass stopper under an argon purge and insertion of the probe. Values were acquired after ∼ 5-10 sec to compensate for solution warming from the probe. Samples involving additives were prepared in an identical manner with additives added to stock solutions of the Cu(I) complex followed by serial dilutions. The equivalent conductance Λe was calculated from the conductance measurement and plotted vs. the square root of the concentration. Extrapolation of the linear portion of the plot allowed determination of the equivalent conductance at infinite dilution, Λ0. A plot of (Λo-Λe) vs. the square root of the concentration (Onsager plot) was then constructed.17,18
Global fitting of UV-vis data was performed using Olis GlobalWorks™ software; for complete details see the Supporting Information.
(tBu2Me2eda)Cu(X) (X = BF or nitro-BF)
In a typical procedure, Cu4Mes4 (41.5 mg, 0.055 mmol) and benzoylformic acid (34.1 mg, 0.22 mmol) were combined in CH2Cl2 (5 mL) and stirred for 20 min to give a cloudy yellow solution. tBu2Me2eda (45.5 mg, 0.22 mmol) in CH2Cl2 (1 mL) was added to afford a pale yellow solution. The solvent was removed under vacuum to yield a yellow oil, which was redissolved in a small amount of CH2Cl2 (∼ 0.5 mL). Pentanes (∼ 5 mL) was added to precipitate a bright yellow solid, which was collected by filtration and washed with pentanes (3 × 5 mL) (77.4 mg, 82% for X = BF; 56 mg, 65% for X = nitro-BF). Crystals suitable for X-ray crystallography were obtained by slow diffusion of pentanes into a concentrated CH2Cl2 solution. For X = BF: 1H NMR (CDCl3, 300 MHz): δ 7.92 (d, J = 9 Hz, 2H), 7.53 (t, J = 9 Hz, 1H), 7.42 (t, J = 6.9 Hz, 2H), 2.42 (s, 6H), 2.64 (b), 1.29 (s, 18H) ppm. 13C{1H} NMR (75.0 MHz, CD2Cl2): δ 195.23, 170.94, 134.76, 133.64, 130.02, 58.02, 49.28, 36.46, 26.62 ppm. Anal. Calc. for C20H33CuN2O3: C, 58.16; H, 8.05; N, 6.78. Found: C, 58.21; H, 8.24; N, 6.58. IR (Neat): 1197, 1224, 1393, 1448, 1477, 1617, 1683 cm-1. UV-vis [λmax, nm (ε, M-1 cm-1) in CH2Cl2]: 362 (465). For X = nitro-BF: 1H NMR (CD2Cl2, 300 MHz): δ 8.19 (dd, J = 8.1, 30.3 Hz, 4H), 2.44 (s, 6H), 2.64 (b), 1.29 (s, 18H) ppm. 13C{1H} NMR (75.0 MHz, CD2Cl2): δ 193.19, 169.53, 150.79, 139.64, 131.13, 124.12, 58.09, 49.23, 36.51, 26.64 ppm. Anal. Calc. for C20H33CuN2O3: C, 52.44; H, 7.04; N, 9.17. Found: C, 51.85; H, 6.75; N, 8.79. IR (Neat): 1210, 1348, 1348, 1389, 1473, 1529, 1628, 1695 cm-1. UV-vis [λmax, nm (ε, M-1 cm-1) in CH2Cl2]: 470 (162).
Attempted synthesis of (Me4pda)Cu(BF)
Cu4Mes4 (25 mg, 0.56 mmol) and benzoylformic acid (20 mg 0.14 mmol) were combined in THF (10 ml). The solution was allowed to stir for 20 min to give a cloudy yellow solution. Me4pda (0.022 ml, 0.14 mM) was added via syringe resulting in a golden clear solution. Upon standing (∼10-15min) or reduction in volume, the solution changed to a bright green supernatant with an orange solid deposit typical of a disproportionation. Crystals suitable for X-ray crystallography were obtained by allowing the filtered supernatant to sit at room temperature for several days, and were found to be (Me4pda)Cu(BF)2. Complete characterization of this complex was performed for an independently synthesized sample (see below).
(Me4pda)Cu(BF)2
CuCl2 (124 mg, 0.9 mmol) was suspended in CH2Cl2 (5 mL) and Me4pda (0.128 ml, 0.77 mmol) was added via syringe. After stirring for 2 h, the solution was filtered and TlBF (541.6 mg, 1.5 mmol) was added and the mixture was allowed to stir overnight. The mixture was filtered and solvent was removed under vacuum from the bright blue-green filtrate to leave the product as a blue-green solid (328 mg, 87%). Crystals suitable for X-ray crystallography were obtained allowing a concentrated CH2Cl2 solution to stand at −20 °C. Anal. Calc. for C23H28CuN2O6•0.25CH2Cl2: C, 54.41; H, 5.12; N, 5.46. Found: C, 54.58; H, 5.12; N, 5.31. IR (Neat): 1232, 1405, 1477, 1591, 1685 cm-1. UV-vis [λmax, nm (ε, M-1 cm-1) in CH2Cl2]: 740 (141).
Low Temperature Oxygenations of (tBu2Me2eda)Cu(X) (X = BF or nitro-BF)
Anaerobically prepared CH2Cl2 solutions (0.7 mM) of (tBu2Me2eda)Cu(X) (X = BF or nitro-BF) were cooled to -80 °C under argon in a septum sealed quartz cuvette. Dry O2 was bubbled through the solution with monitoring by UV-vis spectroscopy (data for X= BF shown in Figure 6; X = nitro-BF shown in Figure S1). In some experiments, 1-5 equivalents of cyclooctene or cyclohexene were preloaded into the cuvette prior to cooling and oxygenation. Samples preloaded with substrates were subjected to GC-MS analysis. These solutions were gradually warmed to room temperature, solvent was removed under vacuum, and the resulting residues were washed with 0.1 M HCl (∼5 mL). The aqueous layers were extracted with CH2Cl2 (3 × 5 mL) and the extracts dried with Na2SO4, filtered through Celite, and analyzed by GC-MS. Observed peaks were compared to commercially available oxidized variants of the original alkene (cyclohexene oxide, 2-cyclohexene-1-one, 2-cyclohexene-1-ol, and cyclooctene oxide).
Figure 6.
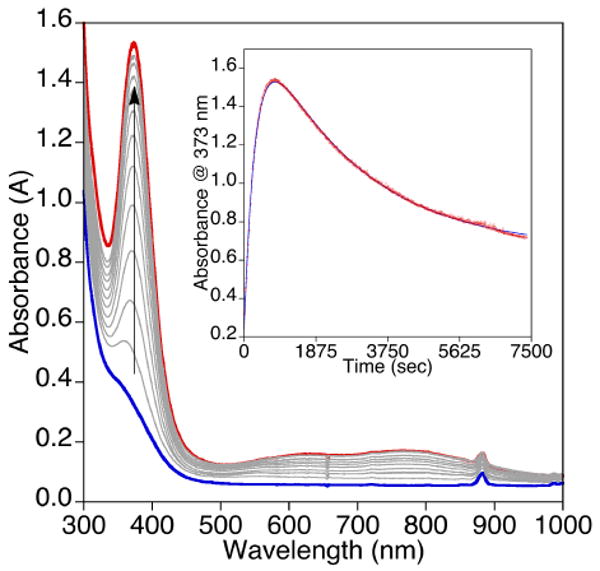
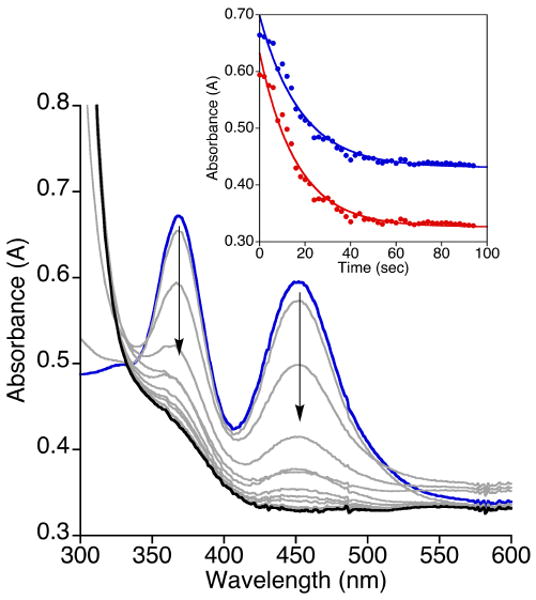
UV-vis spectroscopic data for the reaction of (tBu2Me2eda)Cu(BF) in CH2Cl2 (0.67 mM) (blue spectrum) with O2 at −80 °C with the feature at 373 nm having an approximate ε ∼ 2,300 M-1cm-1. The inset displays the time trace for the formation and decay of the intermediate (red spectrum) with data monitored at 373 nm and fit to a bi-exponential equation [At = A1 + A2*exp(-k2*t) − A3*exp(-k1*t), k1 = 0.0034 s-1 and k2 = 0.00039 s-1; R = 0.999].
Reaction of [L2Cu2(μ-η2:η2-O2)](OTf)2/[L2Cu2(μ-O)2](OTf)2 (L = tBu2Me2eda) Mixture with Bu4NBF
An anaerobically prepared CH2Cl2 solution (0.1 mM) of [(tBu2Me2eda)Cu(CH3CN)]OTf was cooled to -80 °C under argon in a septum sealed quartz cuvette. Dry O2 was bubbled through the solution, and monitoring by UV-vis spectroscopy revealed the growth of absorption bands (λmax = 365 and 450 nm) indicative of the formation of a mixture of [L2Cu2(μ-η2:η2-O2)](OTf)2/[L2Cu2(μ-O)2](OTf)2 (L = tBu2Me2eda). Once growth of the bands ceased, the solution was purged with Ar (10 min) and a degassed solution of Bu4NBF in dry CH2Cl2 (3 mM, prepared in a glovebox) was added via syringe to the stirred reaction solution, which was monitored by UV-vis spectroscopy. For subsequent GC-MS quantification, samples were allowed to warm to room temperature. Solvent was removed in vacuo and the residue washed with 0.1 M HCl (∼5 ml). The aqueous layer was extracted with CH2Cl2 (3 × 5 ml) to isolate the Cu-free organic products. The solvent was again removed in vacuo and the residue redissolved in acetone. K2CO3 (5 equivs. per added TBABF) and MeI (5 equivs. per added TBABF) were added and the solution was allowed to stir for 2 h under reflux. The acetone was removed in vacuo, the residue was extracted with toluene (∼ 2 ml), and the solution was filtered through a silica plug. The filtrate was diluted to a known volume and analyzed by GC-MS, using a standard curve prepared with commercially available methylbenzoate for comparison.
Reactions of [(Me2pda)2Cu2(μ-O)2](OTf)2 with Bu4NY (Y = BF or BA)
These reactions were performed using the same procedure as described above, except using a 0.2 mM solution of [(Me4pda)Cu(CH3CN)]OTf and involving the intermediacy of [L2Cu2(μ-O)2](OTf)2, as indicated by an intense absorption feature with λmax = 397 nm. Samples for analysis by resonance Raman spectroscopy were prepared using a 20 mM solution (0.8 mL) of [(Me4pda)Cu(CH3CN)]OTf. The solutions were cooled under an argon purge to -78 °C by submerging the flask in a dry ice/acetone bath. Dry O2 was bubbled through the solution forming the characteristic yellow brown color of the bis(μ-oxo)dicopper(III) core. After 30 min, O2 flow was discontinued and the flask was purged for 20 min with argon to remove excess O2. The solution was transferred via a precooled pipette to a NMR tube charged with 60 equivalents of Bu4NBF under an argon purge in a dry ice/acetone bath. This reaction was allowed to sit at -78 °C for 5 s and then rapidly frozen in liquid nitrogen for resonance Raman spectroscopic analysis. For the reaction with Bu4NBA, a portion (58 mg, 1.6 mmol) was dissolved in 0.1 ml of CH2Cl2 and loaded into a syringe in the glovebox. This solution was injected into the Schlenk flask containing the solution of the bis(oxo)dicopper complex and allowed to mix for 5 min before transferring to a precooled (-78 °C) NMR tube. The mixture was then subsequently frozen in liquid N2 for resonance Raman spectroscopic analysis.
Results and Discussion
Synthesis and Characterization of LCu(α-ketocarboxylate) Complexes
Cu(I) complexes supported both by a peralkylated diamine ligand (L = tBu2Me2eda) and an α-ketocarboxylate (BF or nitro-BF) were synthesized by first reacting Cu4Mes4 with the free α-ketocarboxylic acid. Subsequent addition of tBu2Me2eda led to the formation of the complexes (tBu2Me2eda)Cu(X) (X = BF or nitro-BF), which were isolated as air sensitive solids and characterized thoroughly, including by X-ray crystallography.
The complex (tBu2Me2eda)Cu(BF) contains a three coordinate Cu(I) center in a planar geometry (sum of donor-Cu-donor angles = 359.9°) with the α-ketocarboxylate (BF) bound in monodentate fashion via O1 (Figure 3). A relatively insignificant interaction between Cu1 and O2 is suggested by a long Cu1-O2 distance of 2.81 Å. The keto-carboxylate torsion angle, defined by O1-C-C-O3 or O2-C-C-O3, is 104°. This value is closer to that of an ideal perpendicular orientation (90°) than one in which the ketocarbonyl is coplanar with the carboxylate functionality (180°). Overall, the BF binding geometry is similar to that previously reported for Cu(I)-α-ketocarboxylate complexes supported by pyridyl-imine ligands.11
Figure 3.
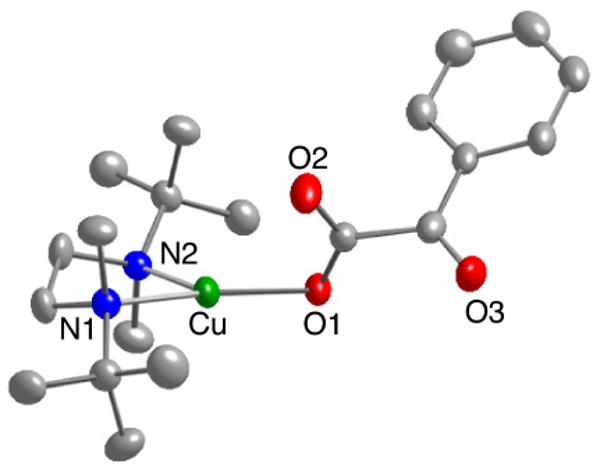
Representation of the X-ray structure of (tBu2Me2)Cu(BF) with all nonhydrogen atoms shown as 50% thermal ellipsoids. Selected interatomic distances (Å) and angles (deg): Cu1-O1, 1.9341(17); Cu1-O2, 2.8096(17); Cu1-N1, 2.0455(19); Cu1-N2, 2.1615(19); N1-Cu1-N2, 87.75(8); O1-Cu1-N1, 150.72(7); O1-Cu1-N2, 121.38(7).
In the X-ray crystal structure of (tBu2Me2eda)Cu(nitro-BF) (Figure 4), a highly disordered O2 atom was modeled by splitting the α-ketocarboxylate ligand over two positions, one with monodentate (minor, 25% occupancy) and the other with bidentate carboxylate coordination (75% occupancy). The former, monodentate geometry is similar to that in the parent BF complex (Figure 3), with Cu1-O1 and –O2 distances of 1.942(5) Å and 2.981(6) Å, respectively, and a planar coordination geometry (sum of donor-Cu-donor angles = 356.2°). In the latter, bidentate structure, the Cu-O distances are more similar (2.028(18) Å and 2.344(16) Å), with an overall copper coordination geometry best described as distorted tetrahedral (τ4 = 0.63, where τ4 = 0 corresponds to a square planar and τ4 = 1 corresponds to a tetrahedral geometry).19 While bidentate carboxylate coordination has been reported for simple carboxylate complexes of copper,20 to the best of our knowledge such a binding mode in an α-ketocarboxylate complex has not been reported for complexes of any metals.16,21-23 The existence of both monodentate and bidentate carboxylate binding modes within the same crystal implies similar stabilities for these structures, as noted in previously reported computations on analogous compounds.11
Figure 4.
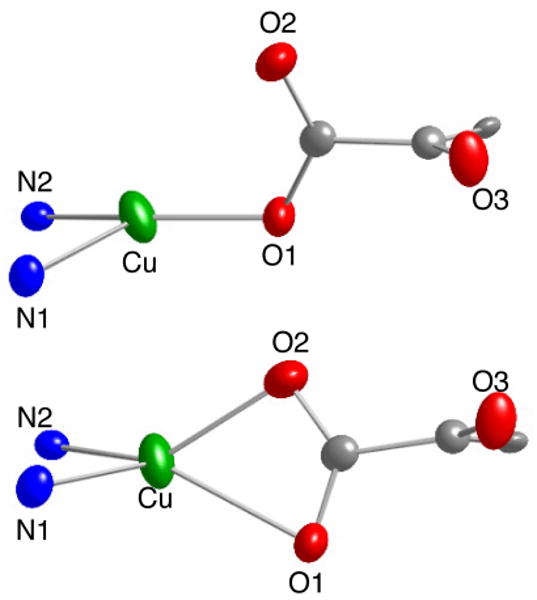
Representation of the X-ray structure of (tBu2Me2eda)Cu(nitro-BF) with nonhydrogen atoms shown as 50% thermal ellipsoids and tBu2Me2eda carbon atoms omitted for clarity. The monodentate (top) and bidentate (bottom) α-ketocarboxylate bound forms are shown. Selected interatomic distances (Å) and angles (deg): [Monodentate] Cu1-O1, 1.942(5); Cu1-O2, 2.981(6); Cu1-N1, 2.094(4); Cu1-N2, 2.087(4); O1-Cu-N1, 138.30(19)°; O1-Cu-N2, 119.1(5)°; N1-Cu-N2, 88.07(14)°; [Bidentate] Cu1-O1, 2.028(18); Cu1-O2, 2.344(16); Cu1-N1, 2.094(4); Cu1-N2, 2.087(4); N1-Cu-N2, 88.07(14); O1-Cu-N1, 139.1(5); O1-Cu-N2, 119.1(5); O2-Cu-N1, 124.9(5); O2-Cu-N2, 132.6(5).
Attempts to isolate Cu(I) complexes of BF or nitro-BF utilizing Me4pda as supporting ligand where unsuccessful due to problems with disproportionation. Thus, upon addition of Me4pda to solutions of CuBF (i.e., from mixing Cu4Mes4 and benzoylformic acid) an initial change from cloudy yellow to clear gold was observed, but the solutions soon turned bright green and deposited an insoluble dark solid (presumably Cu metal). From these solutions a small amount of crystals of the Cu(II) complex (Me4pda)Cu(BF)2 were isolated and characterized by X-ray crystallography (Figure 5). More substantive samples of the complex were independently synthesized and fully characterized (unit cell analysis, elemental analysis, FTIR, and UV-vis spectrosocpy) by reacting (Me4pda)CuCl2 with 2 equivalents of TlBF. As shown in Figure 5, both α-ketocarboxylates are bound by carboxylate O-atoms to a copper ion displaying a distorted square planar geometry (τ4 = 0.36).19 The atoms O2 and O5 display an elongated, but still significant, interaction to with the Cu(II) center (∼2.5 Å). The Cu1-O1 and Cu1-O4 distances observed in this complex are longer than those observed for a previously reported Cu(II)BF complex supported by a tris(methylpyridyl)amine ligand (Cu-O = 1.932(2) Å).21f Both coordinated α-ketocarboxylates have torsion angles (107.12° and 93.25°) that indicate significant deviation of the carboxylate and keto-carbonyl from coplanarity.
Figure 5.
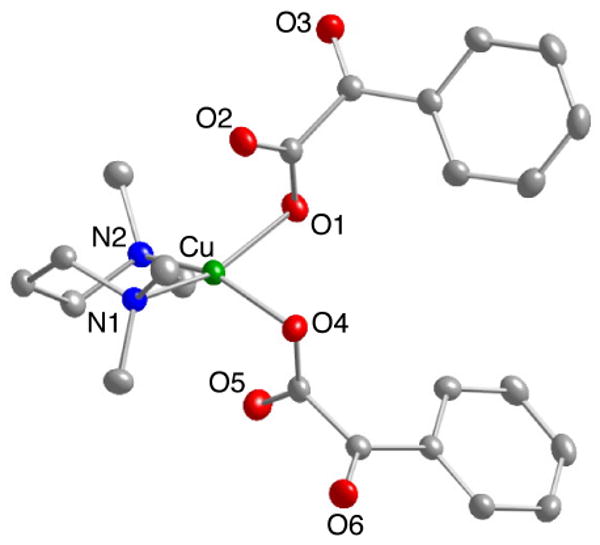
Representation of the X-ray structure of (Me4pda) Cu(BF)2 with all nonhydrogen atoms shown as 50% thermal ellipsoids. Selected interatomic distances (Å) and angles (deg): Cu(1)-N(1), 2.035(2); Cu(1)-N(2), 2.031(2); Cu(1)-O(1), 2.014(2) Cu(1)-O(3), 1.9966(19); N(2)-Cu(1)-N(1), 97.47(8); O(3)-Cu(1)-O(1), 88.50(7).
In addition to determining their structures by X-ray crystallography, all of the copper-α-ketocarboxylate complexes were characterized by UV-vis and 1H NMR spectroscopy in solution and by FTIR spectroscopy as solids and in solution. For (tBu2Me2eda)Cu(X) (X = BF and nitro-BF), the lowest energy features in UV-vis spectra are weak absorptions with λmax = 362 nm (ε ∼ 465 M-1 cm-1) and λmax = 470 nm (ε ∼ 162 M-1 cm-1), respectively. Drawing analogies to what is known for Fe(II)-α-ketocarboxylate complexes,22,24 the lack of intense absorptions in the visible region for these Cu(I) compounds is consistent with monodentate and/or bidentate coordination of the α-ketocarboxylate via the carboxylate group. Coordination via both the carboxylate and ketone units to Fe(II) results in intense absorptions in the 500-700 nm region that have been assigned as Fe(II) → BF charge transfer bands, their low energy being a result of the co-planarity and π conjugation between the α-ketocarboxylate moiety and the aryl ring.24 The absence of such features in the Cu(I) complexes argues against such a ketocarboxylate binding mode in solution and is in agreement with the solid-state structures delineated by X-ray crystallography.
1H NMR spectra of (tBu2Me2eda)Cu(X) (X = BF or nitro-BF) displayed peaks between 7 and 9 ppm with chemical shifts and splitting patterns consistent with assignment as the aryl protons of the respective α-ketocarboxylate ligand (Figures S2-S3). The peak pattern is consistent with either retention of a single binding mode or rapid fluxionality between two or more binding geometries in solution. In addition to the aryl peaks, two singlets assignable to methyl and t-butyl groups on the tBu2Me2eda ligand for both complexes were observed at 2.4 and 1.3 ppm, respectively. A well resolved peak or set of peaks was not observed for the ethyl bridge in the tBu2Me2eda ligand, although a broad baseline feature centered at ∼2.64 ppm was observed in the expected region for these protons in both samples. Upon cooling to −60 °C this broad, undefined feature converted to relatively sharp features at 3.06 and 2.07 ppm that integrated to two protons each and were modeled as an A2X2 spin system (Figures S2 and S4). We attribute this behavior to conformational fluxionality of the chelate ring that is sufficiently slowed at low temperature to result in the observation of distinct peaks for the diastereotopic ethylene hydrogens.
All Cu(I) and Cu(II) complexes were further characterized by Fourier transform infrared spectroscopy (FTIR) both in the solid and solution state. Data were also acquired for the respective free α-ketocarboxylate salt or free acid for comparison (Figures S5-S8). All complexes containing an α-ketocarboxylate displayed two peaks between 1600 and 1800 cm-1 attributable to carboxylate and/or ketone C-O vibrations (e.g., vsym), as well as a complex set of peaks between 1300 and 1500 cm-1 that resemble those typically assigned to vsym vibrations from the carboxylate group.25 The complex (tBu2Me2eda)Cu(nitro-BF) also displayed a feature at ∼1530 cm-1 that was not observed in samples with BF, suggesting that this feature is due to a N-O vibration from the nitro functionality.26 Overall, the data are consistent with the solid state structures determined by X-ray crystallography.
Oxygenation of (tBu2Me2eda)Cu(X) (X = BF or nitro-BF)
Room temperature reactions of (tBu2Me2eda)Cu(X) (X = BF or nitro-BF) in CH2Cl2 with excess O2 led to a rapid change of the initially pale yellow solutions to bright green. ESI-MS analysis of these solutions displayed a peak assignable only to free diamine [tBu2Me2eda + H+]+. Attempts to crystallize any compounds out of the reaction mixture were unsuccessful and led to green oils. After a mild acidic workup and extraction to remove copper and isolate organic products, 1H NMR spectroscopic analysis revealed < 1% decarboxylation (conversion of BF to benzoic acid, BA) with the only observed peaks being assigned to the benzoylformic acid and tBu2Me2eda. The extent of decarboxylation increased slightly (to ∼5%) when the oxygenation was performed at −80 °C and the solution subsequently allowed to warm to room temperature. Monitoring of the reaction at −80 °C by UV-vis spectroscopy revealed the formation of a transient feature at λmax ∼ 373 nm for X = BF and λmax ∼ 371 nm for X = nitro-BF (Figures 6 and S1, respectively). The growth and decay of these features could be fit to a simple biexponential expression, consistent with first-order rate laws for both processes (presumably, the growth of the intermediate is pseudo-first order with respect to excess O2). For X = BF, the feature reached a maximum intensity at ∼14 min with rate constants for growth and decay being k1 = 0.0034 s-1 and k2 = 0.00039 s-1, respectively. Both processes were faster for X = nitro-BF, as the feature reached a maximum intensity at ∼6.7 min with k1 = 0.0062 s-1 and k2 = 0.0010 s-1. Unfortunately, attempts to identify these intermediates through resonance Raman spectroscopy were unsuccessful, as peaks that could be attributed to oxygen-containing species could not be identified (λex = 406.7 nm, CH2Cl2, 20 mM, −196 °C).
Reasoning that the small amount of decarboxylation seen in the −80 °C reactions implicated the intermediacy of an oxidizing intermediate that might be trapped, we performed the oxygenation using solutions of (tBu2Me2eda)Cu(BF) containing excess cyclohexene or cis-cyclooctene (5 equivalents). GC-MS analysis of the organic products after removing copper via an acidic workup did not show any appreciable amounts of oxidized substrates such as epoxides, alcohols, enones, or coupled products. However, 1H NMR spectroscopic analysis revealed a significant increase in the extent of decarboxylation when cyclohexene (11.5%) and cyclooctene (16%) were present. A similar increase in decarboxylation yield (12%) also was observed when the oxygenation was conducted in the presence of acetonitrile (5 equiv.). Taken together, these results suggest that added alkene or acetonitrile coordinates to the initial copper(I)-ketocarboxylate complex, and that this ligand binding somehow facilitates the decarboxylation reaction.
To test this notion, 1H NMR spectra of either (tBu2Me2eda)Cu(BF) or (tBu2Me2eda)Cu(nitro-BF) with 5 equiv. of cyclohexene at room temperature or −60 °C were acquired. The data did not indicate any significant interaction between the alkene and the metal complex; the peaks assigned to the BF and tBu2Me2eda ligands and the added cyclohexene remained unaffected (Figures S9-S10). We conclude from these results that if alkene binding occurs, the equilibrium constant for such a reaction must be small.
To further examine if the α-ketocarboxylate remains bound to the Cu-center in the presence of the additives cyclohexene, cyclooctene, and acetonitrile, conductivity measurements were explored. We reasoned that while the starting Cu(I) complex, (tBu2Me2eda)Cu(BF), is neutral, displacement of the anionic α-ketocarboxylate ligand by an alkene or acetonitrile would result in the formation of a salt that would behave as a 1:1 electrolyte solution. The conductivity of solutions of (tBu2Me2eda)Cu(BF) both with and without additives and of an ideal 1:1 electrolyte ([Bu4N]PF6) over various concentrations (c) are compared in the Onsager plots shown in Figure 7. A linear fit with a similar slope to that for ([Bu4N]PF6) would indicate the displacement of the BF ligand to form a 1:1 electrolyte. However, the Onsager plot for (tBu2Me2eda)Cu(BF) in the absence and presence of additives has a small, invariant slope (∼0.9) that is significantly smaller than that for [Bu4N]PF6 (8.3), suggesting that the additives do not displace the α-ketocarboxylate from the copper atom to a significant degree. On the other hand, the data do not rule out an equilibrium between species with bound and unbound α-ketocarboxylate with only a small, experimentally unobservable amount of the latter (i.e., small Keq).
Figure 7.
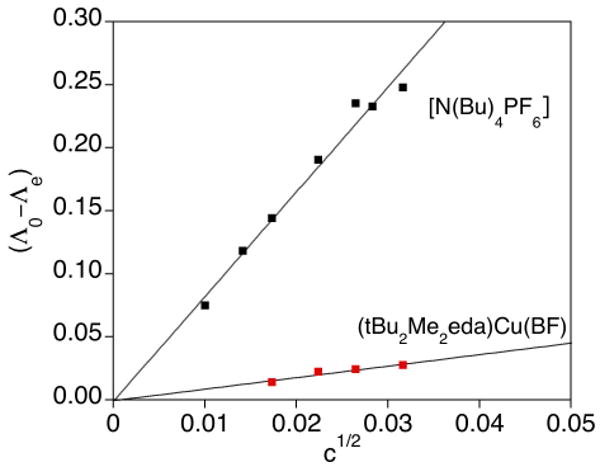
Onsager plot of [N(Bu)4PF6] compared to (tBu2Me2eda)Cu(BF) in CH2Cl2 at −78 °C. The best fit line for [N(Bu)4PF6] data has a slope of 8.31 versus that for (tBu2Me2eda)Cu(BF) with a slope of 0.91. Slopes for (tBu2Me2eda)Cu(BF) + 5 eq. S (S = Additive): cyclohexene = 0.87; cyclooctene = 0.99; acetonitrile = 0.88.
Taking into account both the 1H NMR spectroscopy and conductivity data, two possible reaction pathways can be envisioned to explain the enhanced decarboxylation in the presence of additives “S” (Figure 8). Both presume rapid equilibration between mono- and bidentate carboxylate binding modes in solution for the starting LCuBF complexes; adoption of just one of these modes in solution is also consistent with the available data. One pathway involves the binding of an additive to the Cu(I)-complex with the α-ketocarboxylate ligand remaining bound (path A). This binding of S would somehow prime the complex to react with O2 and facilitate decarboxylation relative to the complex lacking S. A second possible route (path B) involves displacement of the α-ketocarboxylate by S to a small extent in an unfavorable equilibrium process, generating a small amount of a cation/anion pair. Such a displacement is consistent with the relatively low basicity of BF; benzoylformic acid has a lower pKa of 1.21 than that of benzoic acid (pKa = 4.20).27 The cationic Cu(I) fragment supported by tBu2Me2eda is known to be highly reactive with O2, ultimately yielding bis(μ-oxo)dicopper(III) and μ-η2:η2-peroxodicopper(II) cores that can be observed by spectroscopy at low temperatures.13 According to this hypothetical pathway, these species, or a 1:1 Cu/O2 precursor (not shown), would then react with the displaced α-ketocarboxylate to generate the observed benzoate. To our knowledge, however, there is no precedent for decarboxylation of an α-ketocarboxylate by a copper-oxygen species such as a bis(oxo)- or peroxodicopper complex. Thus, we decided to test the viability of path B by independently generating such species and exploring their reactions with added α-ketocarboxylate anions.
Figure 8.
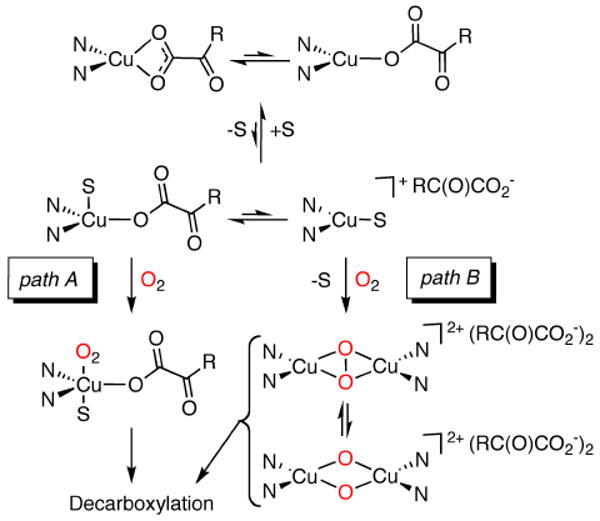
Proposed mechanisms for decarboxylation of Cu(I)-α-ketocarboxylate complexes in the presence of additives “S” (R = aryl group).
Reactivity of [Cu2O2]2+ With Exogenous BF
Following the published procedure,13 we oxygenated [(tBu2Me2eda)Cu(NCCH3)]OTf in CH2Cl2 at −80 °C and observed both bis(μ-oxo)dicopper(III) and μ-η2:η2-peroxodicopper(II) by UV-vis spectroscopy (Figure 9). Thus, the absorption band with λmax = 455 matches that reported for [(tBu2Me2eda)2Cu2(μ-O)2](OTf)2 (λmax = 455 nm; ε = 14,000 M-1cm-1), while the feature with λmax = 369 matches that for [(tBu2Me2eda)2Cu2(μ-η2:η2-O2)](OTf)2 (λmax = 369 nm; ε = 15,000 M-1cm-1).12,13 Also, the ratio of the two species matches that reported for this particular counterion and solvent. After purging with argon to remove unreacted O2 (which did not affect the UV-vis spectrum), Bu4NBF (10 equivalents) was added. Both oxygenated species decayed rapidly (Figure 9), but at slightly different rates. Fitting the decay at the λmax values of 369 nm and 455 nm to simple exponential functions as appropriate for pseudo-first order processes yielded k = 0.055 s-1 and 0.067 s-1, respectively. It is not obvious how to interpret these slightly different rate constants in view of the rapid equilibration between the bis(μ-oxo)dicopper(III) and μ-η2:η2-peroxodicopper(II) isomers under these conditions (CH2Cl2, OTf- counterion); either or both species could be reacting with the added α-ketocarboxylate.13 The yield of decarboxylation was determined by GC-MS analysis of the organic fraction remaining after warming the reaction solution to room temperature, extracting the copper ions with aqueous base, and performing GC/MS analysis of the organic fraction after methylation with MeI/K2CO3 (i.e., determination of the amounts of methyl esters of BF and BA; see Experimental Section); we found it to be 19% based on the amount of BA produced relative to the total amount of [Cu2O2]2+ species reacting. In sum, either or both of the dicopper-oxygen complexes react with an α-ketocarboxylate and induce its decarboxylation (as hypothesized for path B, Figure 8), but the facile isomerization between these cores complicates more detailed analysis of the reaction pathway.
Figure 9.
UV-Vis spectra of the reaction of the mixture of bis(μ-oxo)dicopper(III) and μ-η2:η2-peroxodicopper(II) complexes resulting from oxygenation of [(tBu2Me2eda)Cu(NCCH3)]OTf (0.1 mM) (blue) with TBABF (10 eq) at −80 °C in CH2Cl2, with intermediate spectra shown every 6 s. The inset shows the time trace for the decay of the μ-η2:η2-peroxodicopper(II) (red) and bis(μ-oxo)dicopper(III) (blue) cores at 369 and 455 nm, respectively. The data were fit to exponential equations [At = A1 + A2exp(-kt)] (see text for k values).
In order to simplify matters and enable deeper mechanistic evaluation, we turned to a system supported by a bidentate diamine, Me4pda, that only yields a bis(μ-oxo)dicopper(III) complex and no discernable μ-η2:η2-peroxodicopper(II) isomer.12 As described previously, bubbling dry O2 through a solution of [(Me4pda)Cu(NCCH3)]OTf in CH2Cl2 at −80 °C results in the formation of intense UV-vis absorption features with λmax = 297 nm and 397 nm corresponding to a bis(μ-oxo)dicopper(III) complex (Figure 10, blue spectrum).12 Addition of Bu4NBF (1-60 equivalents) caused the feature at λmax = 397 nm to rapidly bleach concomitant with generation of an intermediate spectrum characterized by an absorption at λmax = 352 nm and weaker shoulder ∼420 nm (Figure 10, green spectrum). This spectrum subsequently decayed to yield a spectrum with a feature at λmax = 364 nm (Figure 10, red spectrum). Approximate first order rate constants for formation and decay of the intermediate were obtained through global fitting of the UV-vis spectroscopic data, although the fast rate of formation of the intermediate in these batch experiments limits the accuracy of the analysis (see Supporting Information for details, Figure S11). For the reaction with 60 equiv. Bu4NBF, values of kformation = 5-7 × 10-1 s-1 and kdecay = 4-7 × 10-1 s-1 were found, with the latter decreasing slightly to 1 × 10-2 s-1 when 2 equiv. Bu4NBF was used.
Figure 10.
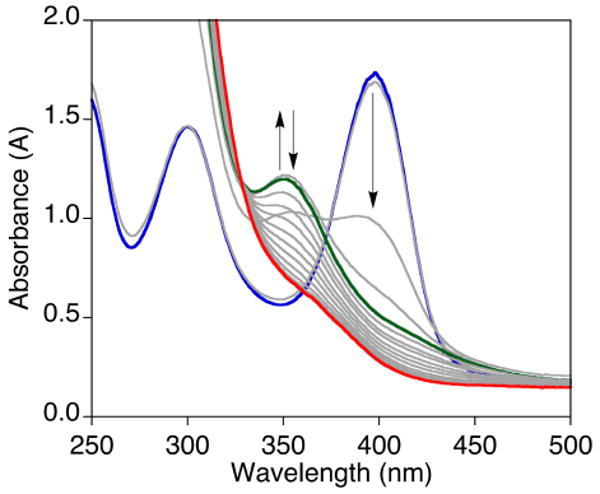
UV-Vis spectra of the reaction of [(Me4pda)2Cu2(O)2](OTf)2 in CH2Cl2 (0.1 mM) at −80 °C (blue) with 60 equivalents of Bu4NBF to form a transient species (green) and then the final product (red), with intermediate spectra at 0.5 s intervals shown in gray.
The decarboxylation yield after warming of the solution was evaluated by GC-MS analysis of organic fractions after work-up (see Experimental Section). The results as a function of equivalents of Bu4NBF added are plotted in Figure 11; the decarboxylation yield increases to a maximum of ∼ 30% at ca. 5 equivalents of Bu4NBF added. Importantly, the data confirm the viability of path B (Figure 8), wherein a dicopper-oxygen complex induces the decarboxylation of exogenous BF.
Figure 11.
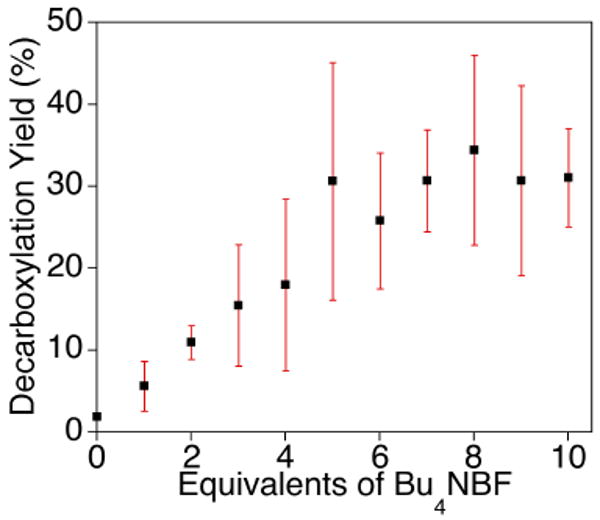
Yields of decarboxylation for the reaction of [(Me4pda)2Cu2(O)2](OTf)2 with various amounts of Bu4NBF. The data are plotted as averages of 3 replicate determinations with standard deviation indicated as error bars. The yields were determined by GC-MS analysis of methylbenzoate (see Experimental Section) and are reported relative to the amount of starting complex, [(Me4pda)2Cu2(O)2](OTf)2.
Resonance Raman spectroscopy was used in an effort to identify the intermediate species (Figure 10, green spectrum) observed in the reaction of [(Me4pda)2Cu2(O)2](OTf)2 with Bu4NBF. A sample for such analysis was prepared by mixing a solution of [(Me4pda)2Cu2(16O)2](OTf)2 (20 mM in CH2Cl2) with a Bu4NBF (60 equiv.), followed within a few seconds by rapid freezing by immersion into liquid N2. Excitation at 406.7 nm yielded a Raman spectrum (Figure 12a) with non-solvent features at 609 (not shown) and 764 cm-1. Due to difficulties trapping the short-lived intermediate, we were unable to obtain reliable data with 18O-labeled material, so unambiguous identification of the Raman features was not possible. We speculate that the 609 cm-1 peak is a core breathing mode of the bis(μ-oxo)dicopper(III) starting material28 and that the feature at 764 cm-1 is an O-O stretching vibration of a μ-η2:η2-peroxodicopper(II) core formed upon coordination of the BF anion. Precedent for this notion comes from observations of isomerization of bis(μ-oxo)dicopper(III) cores to μ-η2:η2-peroxodicopper(II) units from interactions with basic anions or other substrates.29,30
Figure 12.
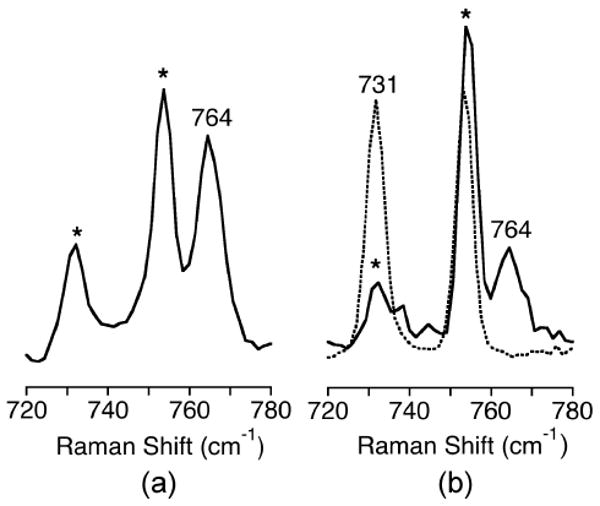
Resonance Raman spectra of [(Me4pda)2Cu2(O)2](OTf)2 (20 mM in CH2Cl2) after addition of (a) Bu4NBF (60 eq.) or (b) Bu4NBA (10 eq.), for samples prepared from 16O2 (solid) or 18O2 (dotted). Conditions: λex = 406.7 nm, -196 °C. Solvent peaks are marked with an asterisk.
To test this idea experimentally, we treated [(Me4pda)2Cu2(O)2](OTf)2 with Bu4NBA (BA = benzoate), which we reasoned would be capable of binding to the dicopper core in a manner akin to BF, but due to the absence of the α-ketocarbonyl moiety would not be susceptible to decarboxylation and thus would yield a more stable and readily characterized species. Addition of Bu4NBA (10 equiv.) to a solution of [(Me4pda)2Cu2(O)2](OTf)2 (0.1 mM in CH2Cl2) at −80 °C resulted in a rapid decay of the feature at λmax = 397 nm to a new species with UV-Vis features at λmax = 352, 413, and 587 nm (Figure 13) similar to those of the intermediate observed in the reaction with Bu4NBF.31 Excitation at 406.7 nm led to enhancement of a feature in the Raman spectrum at 764 cm-1 (Figure 12b) identical to that observed for the transient intermediate produced from Bu4NBF (Figure 12a), but in the case of the product of the reaction with Bu4NBA data for the 18O isotopomer was obtained (Δ18O = 33 cm-1). Taken together, the UV-vis, kinetic, and resonance Raman data for the reactions of [(Me4pda)2Cu2(O)2](OTf)2 with Bu4NBA or Bu4NBF support carboxylate binding that induces isomerization of the bis(μ-oxo)dicopper(III) unit to a μ-η2:η2-peroxodicopper(II) core. We speculate on the basis of the spectroscopic data and precedent29 that the latter species has the structure drawn in Figure 14. In the case of BA this core is stable at low temperature, but for the case of BF it reacts further to yield BA as a result of decarboxylation.
Figure 13.
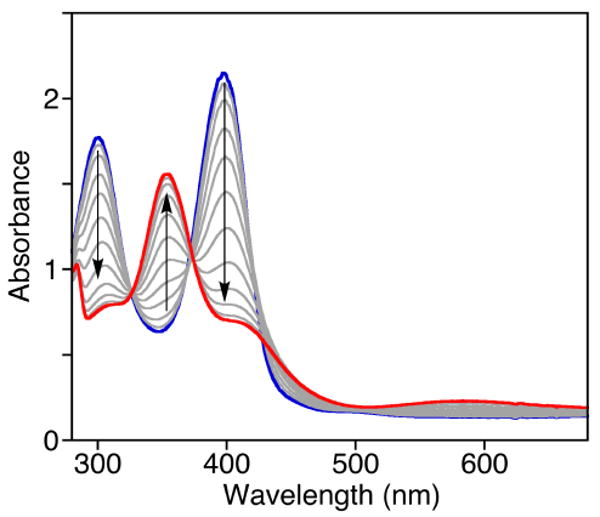
UV-vis spectroscopic changes for the reaction of [(Me4pda)2Cu2O2]OTf2 in CH2Cl2 (0.1 mM; blue) at −80 °C with Bu4NBA (10 equiv.), with spectra displayed every 4 sec.
Figure 14.
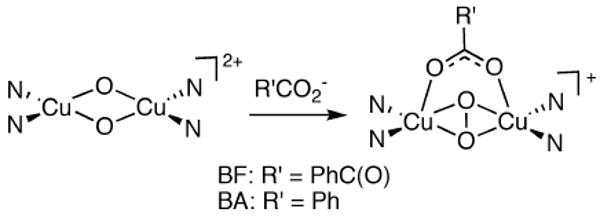
Proposed isomerization of a bis(μ-oxo)dicopper(III) unit to a μ-η2:η2-peroxodicopper(II) core upon binding of BF or BA (N = N-donor atoms of Me4pda).
Summary and Conclusions
While disproportionation plagued attempts to synthesize a Cu(I)-α-ketocarboxylate complex supported by Me4pda, such complexes were successfully isolated and fully characterized using tBu2Me2eda as auxilliary ligand. The data support coordination via the carboxylate, but not the ketone functional group of the α-ketocarboxylate to the Cu(I) center in the complexes of BF or nitro-BF. Unlike previously reported Cu(I)-α-ketocarboxylate complexes with pyridyl-imine ligands,11 oxygenation of the complexes with tBu2Me2eda did not result in significant decarboxylation, except when alkenes or acetonitrile were present. We hypothesize that these additives coordinate to (tBu2Me2eda)Cu(BF) and that the binding enhances the decarboxylation reaction, but the equilibrium constant for binding must be small in order to explain the NMR spectroscopy and conductivity data. A plausible mechanism (Figure 8) involves generation of a small amount of a reactive copper-oxygen species that reacts with the α-ketocarboxylate. On the basis of precedent for Cu(I)/O2 reactivity supported by tBu2Me2eda,13 we suggest that bis(μ-oxo)dicopper(III) and/or μ-η2:η2-peroxodicopper(II) species are likely candidates for the responsible reactant.
In support of this idea, an independently generated mixture of bis(μ-oxo)dicopper(III) and/or μ-η2:η2-peroxodicopper(II) complexes supported by tBu2Me2eda was found to decarboxylate added Bu4NBF. Similarly, a bis(μ-oxo)dicopper(III) complex supported by Me4pda also effected the decarboxylation of exogenous Bu4NBF. In this instance, an intermediate was identified, and on the basis of UV-vis, kinetic, and resonance Raman spectroscopy and parallel experiments using Bu4NBA, we hypothesize that it features a μ-η2:η2-peroxodicopper(II) core bridged by the carboxylate unit (BA or BF). Taken together, the data support the hypothesis that dicopper-oxygen species are capable of inducing decarboxylation of α-ketocarboxylates, thus providing supporting evidence for the mechanism proposed (Figure 8) for decarboxylation of (tBu2Me2eda)Cu(BF). Such a pathway differs from that likely for the Cu(I)-α-ketocarboxylate complexes supported by pyridyl-imine ligands, which because of their poorer electron-donating capabilities relative to peralkylated diamines do not appear to be capable of supporting the generation of dicopper-oxygen complexes.11 More broadly, our findings suggest that metal-α-ketocarboxylate reactivity10 may involve multiple pathways, including ones whereby the α-ketocarboxylate is displaced prior to formation of a metal-oxygen reagent capable of inducing decarboxylation.
Supplementary Material
Acknowledgments
We thank the University of Minnesota Department of Chemistry (Graham N. Gleysteen Fellowship to A.K.G) and the NIH (GM47365 to W.B.T.) for financial support of this work and L. Que, Jr., for access to the resonance Raman spectroscopy facility.
Footnotes
Supporting Information Available. X-ray crystallographic data in cif format with parameters listed in Table S1; UV-vis, FTIR, variable temperature 1H NMR spectra and global fitting details shown in Figures S1-S11. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 2.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 3.Hatcher L, Karlin KD. J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 4.Itoh S. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki M. Acc Chem Res. 2007;40:609–617. doi: 10.1021/ar600048g. [DOI] [PubMed] [Google Scholar]
- 6.Cramer CJ, Tolman WB. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Kamachi T, Kihara N, Shiota Y, Yoshizawa K. Inorg Chem. 2005;44:4226–4236. doi: 10.1021/ic048477p. [DOI] [PubMed] [Google Scholar]; (b) Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]; (c) Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]; (d) Decker A, Solomon EI. Curr Opin Chem Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]; (e) Schröder D, Holthausen MC, Schwarz H. J Phys Chem B. 2004;108:14407–14416. [Google Scholar]; (f) Gherman BF, Tolman WB, Cramer CJ. J Comp Chem. 2006;27:1950–1961. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]; (g) Comba P, Knoppe S, Martin B, Rajaraman G, Rolli C, Shapiro B, Stork T. Chem Eur J. 2008;14:344–357. doi: 10.1002/chem.200700865. [DOI] [PubMed] [Google Scholar]
- 8.(a) Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. J Am Chem Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]; (b) Prigge ST, Mains RE, Eipper BA, Amzel LM. Cell Mol Life Sci. 2000;57:1236–1259. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Evans JP, Ahn K, Klinman JP. J Biol Chem. 2003;278:49691–49698. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]; (d) Balasubramanian R, Rosenzweig A. Acc Chem Res. 2007;40:573–580. doi: 10.1021/ar700004s. [DOI] [PubMed] [Google Scholar]
- 9.Selected examples: Maiti D, Narducci Sarjeant AA, Karlin KD. Inorg Chem. 2008;47:8736–8747. doi: 10.1021/ic800617m.Maiti D, Lucas HR, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2007;129:6998–6999. doi: 10.1021/ja071704c.Capdevielle P, Sparfel D, Baranne-Lafont J, Cuong NK, Maumy M. J Chem Soc, Chem Commun. 1990:565–566.Reinaud O, Capdevielle P, Maumy M. J Chem Soc, Chem Commun. 1990:566–568.
- 10.(a) Purpero V, Moran GR. J Biol Inorg Chem. 2007;12:587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]; (b) Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]; (c) Abu-Omar MM, Loaiza A, Hontzeas N. Chem Rev. 2005;105:2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]; (d) Costas M, Mehn MP, Jensen MP, Que L. J Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 11.Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. J Am Chem Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahadevan V, DuBois JL, Hedman B, Hodgson KO, Stack TDP. J Am Chem Soc. 1999;121:5583–5584. [Google Scholar]
- 13.Mahadevan V, Henson MJ, Solomon EI, Stack TDP. J Am Chem Soc. 2000;122:10249–10250. [Google Scholar]
- 14.Tsuda T, Yazawa T, Watanabe K, Fujii T, Saegusa T. J Org Chem. 1981;46:192–194. [Google Scholar]
- 15.Hayoz P, Ilg S. WO Patent 2006067061. 2006:92.
- 16.Friese SJ, Kucera BE, Young VG, Que L, Jr, Tolman WB. Inorg Chem. 2008;47:1324–1331. doi: 10.1021/ic701823y. [DOI] [PubMed] [Google Scholar]
- 17.Geary WJ. Coord Chem Rev. 1971;7:81–122. [Google Scholar]
- 18.Casella L, Ibers JA. Inorg Chem. 1981;20:2438–2448. [Google Scholar]
- 19.Yang L, Powell D, Houser R. Dalton Trans. 2007:955–964. doi: 10.1039/b617136b. [DOI] [PubMed] [Google Scholar]
- 20.For a pertinent example, see: Reyes-Ortega Y, Alcantara-Flores JL, Hernandez-Galindo MdC, Gutierrez-Perez R, Ramirez-Rosales D, Bernes S, Cabrera-Vivas BM, Duran-Hernandez A, Zamorano-Ulloa R. J Mol Struct. 2006;788:145–151.
- 21.Selected examples of complexes of Cu(II): Arnaud C, Faure R, Loiseleur H. Acta Crystallogr, Sect C: Cryst Struct Commun. 1986;42:814–816.Nakasa K, Nakagawa H, Kani Y, Tsuchimoto M, Ohba S. Acta Crystallogr, Sect C: Cryst Struct Commun. 1999;55:513–517.Ovcharenko VI, Vostrikova KE, Podoplelov AV, Romanenko GV, Ikorskii VN, Reznikov VA. Polyhedron. 1997;16:1279–1289.Kaizer J, Csonka R, Speier G, Giorgi M, Reglier M. J Mol Catal A: Chem. 2005;236:12–17.Lippai I, Speier G, Huttner G, Zsolnai L. Chem Commun. 1997:741–742.Zheng H, Que L., Jr Inorg Chim Acta. 1997;263:301–307.
- 22.Selected examples of complexes of Fe(II): Reference 16 and Mehn MP, Fujisawa K, Hegg EL, Que L., Jr J Am Chem Soc. 2003;125:7828–7848. doi: 10.1021/ja028867f.Paine TK, Zheng H, Que L., Jr Inorg Chem. 2005;44:474–476. doi: 10.1021/ic048427k.Chiou YM, Que L., Jr J Am Chem Soc. 1992;114:7567–7568.Hegg EL, Ho RYN, Que L., Jr J Am Chem Soc. 1999;121:1972–1973.Hikichi S, Ogihara T, Fujisawa K, Kitajima N, Akita M, Moro-oka Y. Inorg Chem. 1997;36:4539–4547. doi: 10.1021/ic960903m.
- 23.Selected examples of complexes of other metal ions other than Cu or Fe: Law GL, Wong KL, Lau KK, Tam HL, Cheah KW, Wong WT. Eur J Inorg Chem. 2007:5419–5425.Muller R, Hubner E, Burzlaff N. Eur J Inorg Chem. 2004:2151–2159.Ruman T, Ciunik Z, Szklanny E, Wolowiec S. Polyhedron. 2002;21:2743–2753.Tekeste T, Vahrenkamp H. Eur J Inorg Chem. 2006:5158–5164. doi: 10.1021/ic061667a.Teoh SG, Looi ES, Teo SB, Ng SW. J Organomet Chem. 1996;509:57–61.Sharutin VV, Sharutina OK, Bondar' EA, Pakusina AP, Krivolapov DB, Gubaidullin AT, Litvinov IA. Russ J Gen Chem. 2002;72:245.Krogmann K. Z Anorg Allg Chem. 1968;358:67–81.Ng SW. Z Kristallogr-New Cryst Struct. 1997;212:279–281.
- 24.Chiou YM, Que L., Jr J Am Chem Soc. 1995;117:3999–4013. [Google Scholar]
- 25.Deacon GB, Phillips RJ. Coord Chem Rev. 1980;33:227–250. [Google Scholar]
- 26.Pretsch E, Bühlmann P, Affolter C. Structure Determination of Organic Compounds: Tables of Spectral Data. 2003 [Google Scholar]
- 27.Scherrer RA, Donovan SF. Anal Chem. 2009;81:2768–2778. doi: 10.1021/ac802729k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J Am Chem Soc. 1999;121:10332–10345. [Google Scholar]
- 29.Funahashi Y, Nishikawa T, Wasada-Tsutsui Y, Kajita Y, Yamaguchi S, Arii H, Ozawa T, Jitsukawa K, Tosha T, Hirota S, Kitagawa T, Masuda H. J Am Chem Soc. 2008;130:16444–16445. doi: 10.1021/ja804201z. [DOI] [PubMed] [Google Scholar]
- 30.Ottenwaelder X, Rudd DJ, Corbett MC, Hodgson KO, Hedman B, Stack TDP. J Am Chem Soc. 2006;128:9268–9269. doi: 10.1021/ja061132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.In contrast to this result, the analogous reaction of an equilibrium mixture of [(tBu2Me2eda)2Cu2(μ-O)2](OTf)2 and [(tBu2Me2eda)2Cu2(μ-η2:η2-O2)](OTf)2 with Bu4NBA resulted in the decay of both observed features. It is also noteworthy that oxygenation of a premixed solution of [(tBu2Me2eda)Cu(NCCH3)]OTf + Bu4NBA (1-10 eqvuivalents) at −80 °C did not result in the formation of any observable intermediates.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.