

Abstract
The complex formation between cadmium(II) and the ligands cysteine (H2Cys) or penicillamine (H2Pen = 3, 3′-dimethylcysteine) in aqueous solutions, containing CCd(II) ∼ 0.1 mol dm-3 and CH2L = 0.2 – 2 mol dm-3, was studied at pH = 7.5 and 11.0 by means of 113Cd-NMR and Cd K- and L3-edge X-ray absorption spectroscopy. For all cadmium(II)-cysteine mole ratios the mean Cd-S and Cd-(N/O) bond distances were found in the ranges 2.52 – 2.54 Å and 2.27 – 2.35 Å, respectively. The corresponding cadmium(II)-penicillamine complexes showed slightly shorter Cd-S bonds, 2.50 – 2.53 Å, but with the Cd-(N/O) bond distances in a similar wide range, 2.28 – 2.33 Å. For the mole ratio CH2L / CCd(II) = 2, the 113Cd chemical shifts, in the range 509 – 527 ppm at both pH values, indicated complexes with distorted tetrahedral CdS2N(N/O) coordination geometry. With a large excess of cysteine (mole ratios CH2Cys / CCd(II) ≥ 10) complexes with CdS4 coordination geometry dominate, consistent with the 113Cd NMR chemical shifts, δ ∼ 680 ppm at pH 7.5 and 636 - 658 ppm at pH 11.0, and their mean Cd-S distances of 2.53 ± 0.02 Å. At pH 7.5, the complexes are almost exclusively sulfur-coordinated as [Cd(S-cysteinate)4]n-, while at higher pH the deprotonation of the amine groups promotes chelate formation, and at pH 11.0 a minor amount of the [Cd(Cys)3]4- complex with CdS3N coordination is formed. For the corresponding penicillamine solutions with mole ratios CH2Pen / CCd(II) ≥ 10, the 113Cd-NMR chemical shifts, δ ∼ 600 ppm at pH 7.5 and 578 ppm at pH 11.0, together with the average bond distances Cd-S 2.53 ± 0.02 Å and Cd-O 2.30 – 2.33 Å, indicate that [Cd(penicillaminate)3]n- complexes with chelating CdS3(N/O) coordination dominate already at pH 7.5, and become mixed with CdS2N(N/O) complexes at pH 11.0. The present study reveals differences between cysteine and penicillamine as ligands to the cadmium(II) ion that can explain why cysteine-rich metallothionines are capable of capturing cadmium(II) ions, while penicillamine, clinically useful for treating the toxic effects of mercury(II) and lead(II) exposure, is not efficient against cadmium(II) poisoning.
Keywords: Cadmium(II), cysteine, penicillamine, EXAFS, Cd L3-edge XANES, 113Cd NMR, solution
Introduction
Cadmium(II) is generally known as a non-essential, highly toxic metal ion that acts as a carcinogen in mammals, inhibits growth of plants by interfering with photosynthesis and nitrogen metabolism, and decreases uptake of water and minerals [1]. Recent studies, however, on the marine diatom Thalassiosira weissflogii showed evidence of the first cadmium specific enzyme, cadmium(II)-carbonic anhydrase, which actually has a preliminary function in the diatom's photosynthesis by catalyzing the dehydration of HCO3- to CO2 [2, 3].
A well-known example of cadmium poisoning is the Itai-Itai disease (Itai = pain in Japanese), which was caused by cadmium released from mining waste into the Jinzu river in Japan, contaminating large agricultural areas [4]. Metallothioneins (MTs), which are a family of cysteine-rich polypeptides with low molecular weight [5], are active in vivo in removing heavy metal ions such as Cd2+ and Hg2+ through thiolate coordination from the cysteine residues [6-8]. Even though the toxic effects of cadmium(II) are inhibited when bound to metallothionein (Cd-MT), a sufficient amount of MT must be synthesized in vivo to block cadmium toxicity [5]. Cadmium(II) mainly accumulates in the liver (80-90% as Cd-MT) and to a lesser extent in the kidneys (55-65% as Cd-MT) and other tissues [9].
No effective antidote is known to counteract cadmium poisoning, although to some extent cysteine (H2Cys), homocysteine, N-acetylcysteine and glutathione prevent cell uptake by binding to cadmium(II) through their thiol groups [5, 10]. On the other hand penicillamine (3,3′- dimethylcysteine), commonly used in reducing toxic effects of mercury and lead exposure, is not efficient in cadmium(II) treatments [11]. We have studied the structure and coordination of the cadmium(II) complexes formed with cysteine and penicillamine both at pH 7.5 and 11.0 in aqueous solutions containing CCd(II) ∼ 0.1 mol dm-3 for ligand to metal ratios from 2.0 to 20, to find explanations for the different efficiencies that would allow for more effective detoxifying chelating agents to be designed.
There are numerous reports on formation constants of cadmium(II) cysteine complexes; however, differences in the experimental conditions (e.g. temperature, ionic medium, concentration range) restrict their applicability for the present investigation [12, 13]. We have used the formation constants determined through potentiometric methods by Cole et al. [14] to generate the diagrams showing the distribution of the complexes vs. pH that are displayed in Figure S-1.
In a similar study, Corrie and coworkers reported mononuclear cadmium(II)-penicillamine complex formation in 3 mol·dm-3 NaClO4 as ionic medium [15]. Avdeef and Kearney interpreted alkalimetric titrations of cadmium(II)-penicillamine solutions with protonated polynuclear complexes dominating in the pH range 4 – 8 [16] and suggested that the formation of these complexes was suppressed at high ionic strength. The formation constants from both studies have been used to generate the distribution diagrams shown in Figures S-2a and S-2b.
In the current study, we have combined 113Cd NMR and X-ray absorption spectroscopy (Cd K-edge EXAFS and Cd L3-edge XANES) to investigate the structure of cadmium(II) complexes with cysteine or penicillamine as ligands in aqueous solution. Recent development has made 113Cd NMR a useful technique for classifying the coordination environment in cadmium(II) complexes. The 113Cd NMR chemical shift shows a strong correlation to the type of coordinating ligand atom, with sulfur as the most deshielding, followed by nitrogen and finally oxygen [17, 18]. Chemical shifts reported for several biologically relevant mononuclear cadmium(II) thiolate complexes are collected in Table 1, including solid state δiso (113Cd) for cadmium(II) cysteaminate complex with CdS3N2 coordination geometry for comparison. It should be emphasized, however, that 113Cd NMR chemical shifts cannot only be interpreted based on the type and number of donor atoms (e.g. S, N or O), since cadmium magnetic shielding tensors are sensitive to many other factors such as the type of the ligand, its coordination mode (bridging vs. terminal) and coordination number/geometry of cadmium(II) ions (i.e. 4-, 5- or 6-coordinated) [19].
Table 1.
Reported 113Cd chemical shifts for biologically relevant, mononuclear cadmium(II)-thiolate coordination sites.
| Chemical shift (δ, ppm) |
Ref. | |
|---|---|---|
| CdS4 | 650, 680, 704 – 751 | [20, 21-24] |
| CdS3 | 572, 684 - 690 | [27 - 30] |
| CdS3O | 560 - 645 | [24, 27 - 29, 48] |
| CdS3N | 637 - 659 | [49 - 52] |
| CdS3N2a | 669 | [39] |
| CdS2N2 | 519 | [20] |
| CdS2NOwb | 483 | [20] |
| CdS2NO2 | 442 | [53, 54] |
| CdSS*N2c | 432 | [55] |
Solid state NMR for cadmium(II)-cysteaminate (CdS3N2)
Ow, water
S*, thioether or disulfide.
For CdS4 coordination the observed δ(113Cd) range is rather wide. High frequency δ(113Cd) shifts have been reported for [Cd(S-cysteinate)4]2- in cadmium(II)-substituted LADH (751 ppm) [20], rubredoxin (723 - 732 ppm) [21, 22], and the DNA binding domain of the glucocorticoid hormone receptor (704, 710 ppm) [23]. For a designed cysteine-rich TRI peptide bound to cadmium(II) two signals were observed at 650 and 680 ppm for the distorted tetrahedral CdS4 sites, with the difference originating from “small geometric orientations in the coordination environment” [24]. For CdS4 sites with bridging thiolate groups, the chemical shifts are generally more shielded. Examples are the dinuclear cadmium(II) binding site of the GAL4 protein (669 and 707 ppm) [25, 26], and Cd(II)-loaded metallothionine (Cd7-MT) with several resonances in the 610 – 680 ppm region, which were interpreted as evidence for two sets of clusters, Cd3S9 and Cd4S11, with bridging cysteine sulfur atoms [17].
Pecoraro et al. recently reported 113Cd NMR chemical shifts for the first water-soluble three-coordinated CdS3 structure (δ = 684 - 690 ppm), using designed peptides that specifically bind cadmium(II) ions via bulky Pen residues [27 - 29]. The result calls for re-evaluation of an earlier assignment of the 113Cd chemical shift 572 ppm to a pure CdS3 coordination [30].
The X-ray absorption near edge structure (XANES) region of the cadmium L3-edge has been proposed to be sensitive to the local structure around cadmium, and displays a characteristic pre-edge peak for cadmium complexes with oxygen or nitrogen coordination, while for tetrahedral CdS4 coordination the edge is smooth and almost featureless [31, 32]. We recently measured the Cd L3-edge XANES spectra for a series of crystalline cadmium complexes with CdSx(N/O)y configurations and observed that the distinct pre-edge peak at 3539.1 eV (corresponding to a Cd 2p → 5d transition) in the Cd(ClO4)2·6H2O spectrum (CdO6 model) gradually merges into the absorption edge of the model compounds for CdS2O4, CdS3O3, CdS6, CdS3O and CdS2N2 coordination, and finally disappears in the CdS3N2 and CdS4 spectra [33].
The present study on cadmium(II) complex formation with cysteine and penicillamine is part of a continuing project to obtain structural information on complexes of heavy metals with biomolecules to facilitate understanding of the function of such species in biological systems [34].
Experimental Section
Sample preparation
Cadmium(II) perchlorate hydrate Cd(ClO4)2·6H2O, L-cysteine, D-penicillamine and sodium hydroxide (Sigma Aldrich) were used without further purification. The preparations were performed under argon atmosphere using oxygen-free boiled water to prevent oxidation of the cysteine and penicillamine ligands. The pH of the solutions was monitored with a Corning Semi-Micro electrode.
Cadmium(II) cysteine/ penicillamine solutions
Table 2 presents the compositions of the cadmium(II)-cysteine (A – G) and the cadmium(II)-penicillamine (H – N) solutions, which were prepared with ligand / metal mole ratios CH2L / CCd(II) from 2.0 to 20, and adjusted to different pH values (7.5 and 11.0) in two series. Cysteine or penicillamine (2 – 20 mmol) was dissolved in oxygen-free water (containing 10% D2O) and a weighed amount of Cd(ClO4)2·6H2O (1 mmol) was added. A white precipitate immediately formed with cysteine and the pH ∼1.6 was recorded; no precipitate was formed for penicillamine. Dropwise addition of 6 mol·dm-3 NaOH dissolved the precipitate around pH ∼ 6 – 7 (the lower pH for high L / M ratios), and the clear solutions were collected at pH 7.5 and 11.0. The total cadmium(II) concentration was checked for A2 – E2 and H2 – L2 with a Thermo Jarrell Ash AtomScan 16 inductively coupled plasma atomic emission spectrophotometer (ICP-AES).
Table 2.
Composition of the cadmium(II)-cysteine and penicillamine solutions.a
| Solution | H2L/CdII ratio | [Cd2+]totb | [H2L]tot | pH | Solution | H2L/CdII ratio | [Cd2+]totb | [H2L]tot | pH |
|---|---|---|---|---|---|---|---|---|---|
| L = Cys | |||||||||
| A1 | 2.0 | 100 | 200 | 7.5 | A2 | 2.0 | 100 | 200 | 11.0 |
| B1 | 3.0 | 100 | 300 | 7.5 | B2 | 3.0 | 99 | 301 | 11.0 |
| C1 | 4.0 | 100 | 401 | 7.5 | C2 | 4.0 | 100 | 400 | 11.1 |
| D1 | 5.0 | 100 | 500 | 7.5 | D2 | 5.0 | 99 | 499 | 11.0 |
| E1 | 10.0 | 100 | 1000 | 7.5 | E2 | 10.1 | 92 | 927 | 10.9 |
| F1 | 15.0 | 100 | 1498 | 7.5 | F2 | 14.6 | 103 | 1500 | 11.1 |
| G1 | 19.9 | 76 | 1513 | 7.5 | G2 | 19.5 | 93 | 1818 | 11.1 |
| L = Pen | |||||||||
| H1 | 2.0 | 100 | 200 | 7.5 | H2 | 2.0 | 100 | 200 | 11.3 |
| I1 | 3.0 | 100 | 301 | 7.6 | I2 | 3.0 | 100 | 299 | 11.1 |
| J1 | 4.0 | 100 | 399 | 7.5 | J2 | 4.0 | 100 | 399 | 11.0 |
| K1 | 5.0 | 100 | 500 | 7.4 | K2 | 5.0 | 100 | 500 | 11.0 |
| L1 | 10.0 | 87 | 867 | 7.5 | L2 | 10.0 | 87 | 869 | 11.0 |
| M1 | 14.9 | 68 | 1014 | 7.5 | M2 | 14.6 | 103 | 1501 | 11.0 |
| N1 | 20.1 | 46 | 926 | 7.5 | N2 | 19.4 | 89 | 1725 | 11.0 |
Concentrations in mmol·dm-3
The [Cd2+]tot concentrations are within ± 3 mmol·dm-3, according to the ICP analysis.
113Cd NMR measurements
The 113Cd NMR spectra shown in Figures 1 and 2 were collected at 300 K (27 °C) with a Bruker AMX2-300 spectrometer at 66.6 MHz, using a 10 mm broadband (BBO) probe, a 7.0 microsecond 90° pulse and recycle delay of 5.0 seconds. All solutions contained ∼ 10% D2O. A 0.1 mol·dm-3 solution of Cd(ClO4)2·6H2O in D2O was used as external reference (0 ppm) [18]. All spectra were proton decoupled and measured with a sweep width of 850 – 900 ppm. The total number of collected scans for the cadmium(II) cysteine and penicillamine solutions, as well as the FWHH of the NMR signals, are shown in Table S-1.
Figure 1.
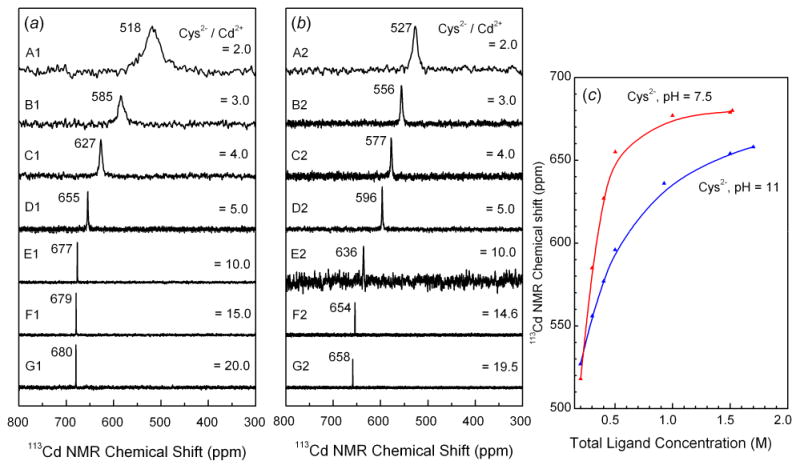
113Cd NMR spectra of ∼ 0.1 mol·dm-3 cadmium(II) solutions with increasing amount of cysteine at pH 7.5 (left) and 11 (middle). The variation of the 113Cd chemical shift vs. total cysteine concentration is shown to the right.
Figure 2.
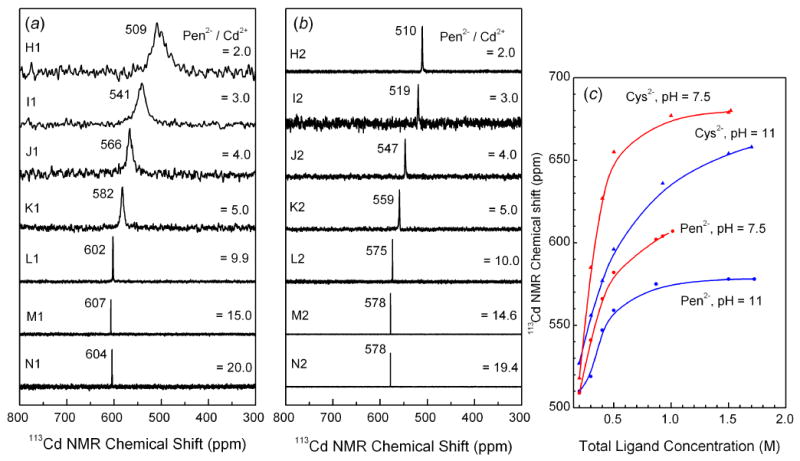
113Cd NMR spectra of cadmium(II) penicillamine solutions at pH 7.5 (left) and 11 (middle). The variation of the 113Cd chemical shift for cadmium(II) cysteine and penicillamine solutions vs. total ligand concentration is shown to the right.
X-ray absorption spectroscopy
Cadmium K-edge extended X-ray absorption fine structure (EXAFS) spectra were collected at BL 2-3 and 7-3 at the Stanford Synchrotron Radiation Lightsource (SSRL) under dedicated conditions of 3.0 GeV and 70-100 mA. Higher harmonics from a Si[220] double crystal monochromator were rejected by detuning to 50% of the maximum incident beam intensity. The spectra were recorded in transmission mode, with argon in the first ion chamber (I0) and krypton in the second (I1) and third (I2) ion chambers. The solutions were enclosed in 10 mm Teflon spacers between 4 μm polypropylene film windows. Three to five scans were collected for each sample. Before averaging, the energy scale was externally calibrated for each scan by assigning the first inflection point of the Cd K-edge of a Cd foil to 26711.0 eV.
The Cd L3-edge XANES measurements were performed at beamline 9-A of the High Energy Accelerator Research Organization (Photon Factory), Tsukuba, Japan. The ring operates under dedicated conditions at 2.5 GeV and 350 – 400 mA. The data were collected in fluorescence mode with helium in the first ion chamber (I0) and an argon-filled Lytle detector (If). Higher harmonics from a Si[111] double crystal monochromator were rejected by means of nickel and rhodium coated mirrors. Solution samples were enclosed in 5 mm Teflon spacers between 4 μm polypropylene windows. For each sample 2-3 scans were collected, externally calibrated by assigning the first inflection point of the Cd L3-edge of a Cd foil to 3537.6 eV, and then averaged.
XAS data analysis
The WinXAS 3.1 program suite was used for the data analysis [35]. The background absorption was subtracted with a first-order polynomial over the pre-edge region, followed by normalization of the edge step. For the Cd K-edge XAS spectra, the energy scale was converted into k-space, where k = (8π2me/h2)(E-E0), using the threshold energy E0 = 26710.0 – 26711.3 eV. The EXAFS oscillation was then extracted using a 7-segment cubic spline to remove the atomic background absorption above the edge.
The EXAFS model functions, χ(k), were constructed by means of the FEFF 8.1 program [36, 37], to obtain ab initio calculated amplitude feff(k)i, phase shift φij(k), and mean free path λ(k) functions (eq. 1). The FEFF input file was generated by means of the ATOMS program [38], using structural information from the crystal structure of the reference compound Cd(SCH2CH2NH2)2 (as CdS3N2 model) with both short and long Cd-S, Cd-N(/O) and Cd-Cd distances [39]. Note that two neighboring elements in the periodic table (such as oxygen and nitrogen) obtain very similar amplitude functions feff(k)i and cannot be distinguished by EXAFS.
| (1) |
The structural parameters were refined by least-squares methods, fitting the k3-weighted model function χ(k) to the experimental unfiltered EXAFS oscillation generally over the k range 3.5 – 12.0 Å -1 (11.2 Å -1 for the solution A2), allowing the bond distance (R), Debye-Waller parameter (σ) and ΔE0 (correlated parameter for all scattering paths) to float, while the amplitude reduction factor (S02) and / or coordination number (N) were fixed. The fitting results are shown in Figures 3, 4, Tables 3 and 4. The estimated errors of the refined coordination numbers, bond distances and their Debye-Waller parameters for the dominating Cd-S path are estimated to be within 20%, ± 0.02 Å and ± 0.001 Å2, respectively, including effects of systematic deviations. The corresponding structural parameters for the Cd-(N/O) path are less accurate, i.e. ± 0.04 Å and ± 0.003 – 0.005 Å2 for bond distances and Debye-Waller parameters, respectively, due to the difficulties associated with separating the EXAFS contribution from the light oxygen and nitrogen atoms from that of the heavier sulfur atom.
Figure 3.
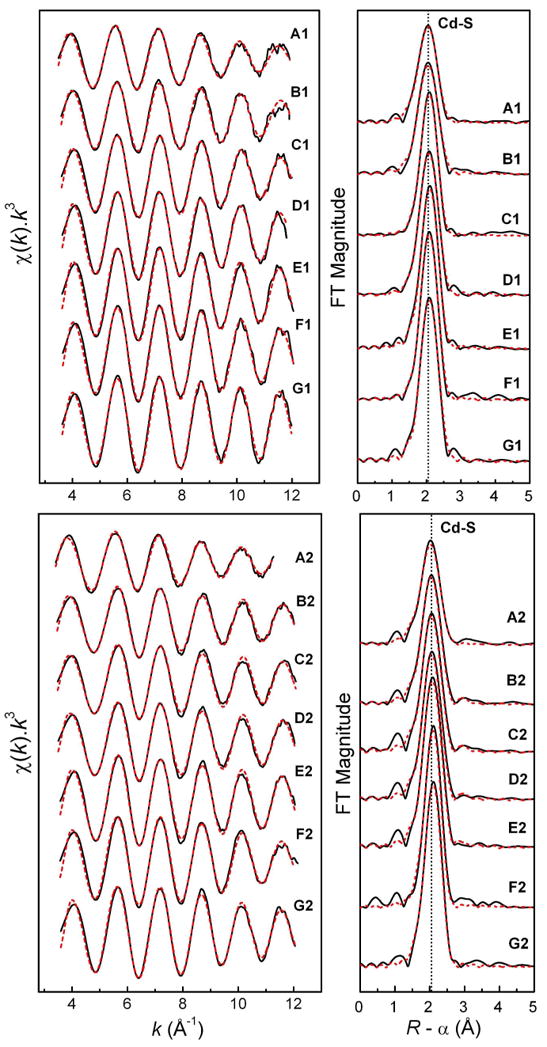
Least-squares curve-fitting of k3-weighted Cd K-edge EXAFS spectra of the cadmium(II)-cysteine solutions at pH = 7.5 (A1 – G1) and pH = 11.0 (A2 – G2), and the corresponding Fourier-transforms, using a model containing both Cd-S and Cd-(N/O) paths (see Table 3).
Figure 4.
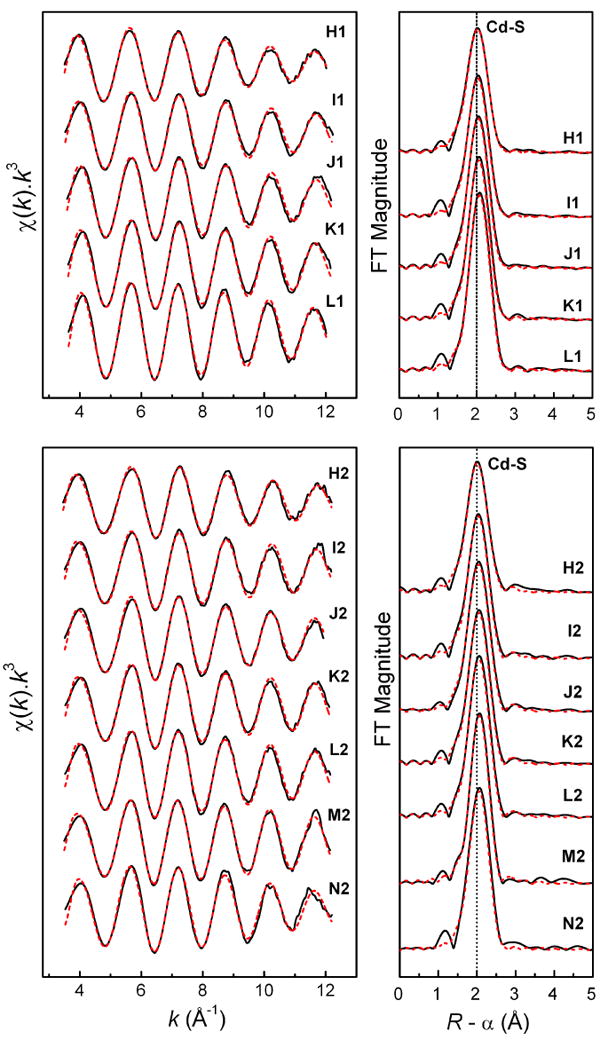
Least-squares curve-fitting of k3-weighted Cd K-edge EXAFS spectra of cadmium(II)-penicillamine solutions at pH = 7.5 (H1 – L1) and pH = 11.0 (H2 – N2), and the corresponding Fourier-transforms (see Table 4).
Table 3.
Cd K-edge EXAFS data analysis for cadmium(II) cysteine solutions at pH = 7.5 (A1 – G1) and pH = 11.0 (A2 – G2, see Figure 3).a
| Solution |
113Cd NMR (δ, ppm) |
Cd-S | Cd-(N/O) | Rb | ||||
|---|---|---|---|---|---|---|---|---|
| N | R (Å) | σ2 (Å2) | N | R (Å) | σ2 (Å2) | |||
| A1 | 518 | 3.6 | 2.52 | 0.0080 | 13.0 | |||
| 2.5 | 2.54 | 0.0056 | 1 f | 2.30 | 0.0053 | 13.8 | ||
| 1.9 | 2.54 | 0.0047 | 2 f | 2.34 | 0.0065 | 13.7 (*) | ||
| B1 | 585 | 3.6 | 2.52 | 0.0065 | 12.8 | |||
| 2.8 | 2.54 | 0.0058 | 1 f | 2.35 | 0.0031 | 13.2 (*) | ||
| 2.1 | 2.55 | 0.0050 | 2 f | 2.36 | 0.0043 | 13.7 | ||
| C1 | 627 | 3.7 | 2.52 | 0.0056 | 9.3 | |||
| 3.7 | 2.52 | 0.0069 | 1 f | 2.41 | 0.0016 | 9.3 | ||
| 3 f | 2.54 | 0.0050 | 0.9 | 2.35 | 0.0030 | 9.5 (*) | ||
| D1 | 655 | 3.9 | 2.53 | 0.0055 | 10.2 (*) | |||
| 2.7 | 2.54 | 0.0033 | 1 f | 2.31 | 0.0012 | 10.6c | ||
| 3.5 f | 2.53 | 0.0049 | 0.5 f | 2.31 | 0.0036 | 10.5 | ||
| E1 | 677 | 4.1 | 2.53 | 0.0053 | 9.9 (*) | |||
| 3.0 | 2.54 | 0.0031 | 1 f | 2.30 | 0.0018 | 10.2c | ||
| 3 f | 2.54 | 0.0030 | 0.8 | 2.29 | 0.0001 | 10.0c | ||
| F1 | 679 | 4.0 | 2.52 | 0.0049 | 10.4 (*) | |||
| 4.0 | 2.53 | 0.0068 | 1 f | 2.39 | -0.0010 | 9.5c | ||
| 3 f | 2.55 | 0.0056 | 2.0 | 2.38 | 0.0014 | 10.1c | ||
| G1 | 680 | 3.8 | 2.53 | 0.0042 | 12.5 (*) | |||
| 3.3 | 2.53 | 0.0035 | 1 f | 2.32 | 0.0111 | 12.6c | ||
| 3 f | 2.54 | 0.0029 | 1.1 | 2.31 | 0.0065 | 12.9c | ||
|
| ||||||||
| A2 | 527 | 3.9 | 2.52 | 0.0095 | 13.3 | |||
| 3.2 | 2.52 | 0.0076 | 1 f | 2.20 | 0.0114 | 11.3 | ||
| 2.9 | 2.52 | 0.0072 | 2 f | 2.25 | 0.0199 | 11.3 | ||
| 2 f | 2.53 | 0.0049 | 2 f | 2.29 | 0.0094 | 12.5 (*) | ||
| B2 | 556 | 3.3 | 2.51 | 0.0063 | 12.6 | |||
| 2.5 | 2.51 | 0.0043 | 1 f | 2.25 | 0.0063 | 10.3 | ||
| 2.2 | 2.52 | 0.0040 | 2 f | 2.30 | 0.0119 | 10.5 (*) | ||
| C2 | 576 | 3.4 | 2.51 | 0.0059 | 13.0 | |||
| 2.8 | 2.51 | 0.0045 | 1 f | 2.24 | 0.0093 | 11.2 | ||
| 2.6 | 2.52 | 0.0045 | 2 f | 2.30 | 0.0179 | 10.5 | ||
| 2.5 f | 2.52 | 0.0041 | 1.5 f | 2.27 | 0.0116 | 11.2 (*) | ||
| D2 | 596 | 3.5 | 2.52 | 0.0057 | 9.5 | |||
| 2.9 | 2.52 | 0.0047 | 1 f | 2.28 | 0.0122 | 9.0 (*) | ||
| E2 | 636 | 3.9 | 2.53 | 0.0054 | 9.7 | |||
| 3.3 | 2.53 | 0.0044 | 1 f | 2.28 | 0.0107 | 9.2 (*) | ||
| F2 | 654 | 4.1 | 2.53 | 0.0060 | 9.1 (*) | |||
| 3.2 | 2.54 | 0.0047 | 1 f | 2.33 | 0.0030 | 8.3c | ||
| G2 | 658 | 3.9 | 2.53 | 0.0052 | 8.3 (*) | |||
| 2.9 | 2.54 | 0.0035 | 1 f | 2.31 | 0.0030 | 8.3c | ||
(*) fits that are compatible with the observed 113Cd NMR chemical shifts and shown in Figure 3; f = fixed; S02 = 0.87 f; N = coordination number/ frequency; k-fitting range = 3.5 - 12.0 Å-1 (11.2 Å-1 for A2);
The residual (%) from the least-squares curve fitting is defined as: where yexp and ytheo are experimental and theoretical data points, respectively.
Attempts to introduce a Cd-(N/O) contribution in the model.
Table 4.
Cd K-edge EXAFS data analysis for cadmium(II) penicillamine solutions at pH = 7.5 (H1-L1) and pH = 11.0 (H2 - N2, see Figure 4)a, b
| Solution |
113Cd NMR (δ, ppm) |
Cd-S | Cd-(N/O) | Rc | ||||
|---|---|---|---|---|---|---|---|---|
| N | R (Å) | σ2 (Å2) | N | R (Å) | σ2 (Å2) | |||
| H1 | 509 | 3.7 | 2.50 | 0.0081 | 11.8 | |||
| 2.7 | 2.51 | 0.0058 | 1 f | 2.25 | 0.0072 | 10.6 | ||
| 2.2 | 2.52 | 0.0052 | 2 f | 2.30 | 0.0098 | 10.6 (*) | ||
| I1 | 541 | 3.5 | 2.50 | 0.0067 | 12.1 | |||
| 2.9 | 2.50 | 0.0055 | 1 f | 2.25 | 0.0123 | 11.1 | ||
| 2 f | 2.52 | 0.0041 | 2 f | 2.31 | 0.0074 | 11.6 (*) | ||
| J1 | 566 | 3.5 | 2.50 | 0.0063 | 10.4 | |||
| 3.3 | 2.50 | 0.0059 | 1 f | 2.28 | 0.0294 | 10.4 | ||
| 2.5 f | 2.52 | 0.0046 | 1.5 f | 2.32 | 0.0083 | 10.6 (*) | ||
| K1 | 582 | 3.7 | 2.51 | 0.0061 | 10.3 | |||
| 3.2 | 2.51 | 0.0052 | 1 f | 2.28 | 0.0145 | 9.6 (*) | ||
| 2.5 f | 2.52 | 0.0040 | 1.5 f | 2.31 | 0.0069 | 10.0 | ||
| L1 | 602 | 4.1 | 2.53 | 0.0061 | 9.0 | |||
| 3.7 | 2.53 | 0.0055 | 1 f | 2.29 | 0.0190 | 8.7 | ||
| 3 f | 2.53 | 0.0041 | 1 f | 2.30 | 0.0046 | 9.4 (*) | ||
|
| ||||||||
| H2 | 510 | 3.0 | 2.48 | 0.0064 | 13.7 | |||
| 2.2 | 2.49 | 0.0044 | 1 f | 2.24 | 0.0069 | 12.2 | ||
| 1.8 | 2.50 | 0.0038 | 2 f | 2.30 | 0.0090 | 11.9 (*) | ||
| I2 | 519 | 3.0 | 2.49 | 0.0057 | 13.7 | |||
| 2.3 | 2.50 | 0.0040 | 1 f | 2.24 | 0.0078 | 11.9 | ||
| 2.1 | 2.50 | 0.0039 | 2 f | 2.30 | 0.0137 | 12.0 (*) | ||
| J2 | 547 | 3.3 | 2.50 | 0.0060 | 9.6 | |||
| 2.1 | 2.52 | 0.0030 | 1 f | 2.27 | 0.0019 | 8.2 | ||
| 2 f | 2.52 | 0.0037 | 2 f | 2.32 | 0.0078 | 8.5 (*) | ||
| K2 | 559 | 3.2 | 2.51 | 0.0056 | 11.1 | |||
| 2.5 | 2.51 | 0.0042 | 1 f | 2.27 | 0.0087 | 10.3 | ||
| 2.5 f | 2.51 | 0.0043 | 1.5 f | 2.31 | 0.0129 | 10.5 (*) | ||
| L2 | 575 | 3.3 | 2.51 | 0.0055 | 9.7 | |||
| 2.9 | 2.51 | 0.0048 | 1 f | 2.32 | 0.0143 | 9.6 | ||
| 2.5 f | 2.52 | 0.0042 | 1.5 f | 2.33 | 0.0100 | 9.8 (*) | ||
| M2 | 578 | 3.0 | 2.51 | 0.0045 | 8.8 | |||
| 2.6 | 2.51 | 0.0037 | 1 f | 2.27 | 0.0146 | 8.1 | ||
| 2.5f | 2.52 | 0.0036 | 1.5 f | 2.31 | 0.0183 | 8.3 (*) | ||
| N2 | 578 | 3.4 | 2.51 | 0.0061 | 12.5 | |||
| 2.5 f | 2.53 | 0.0048 | 1.5 f | 2.33 | 0.0062 | 11.8 (*) | ||
EXAFS spectra of M1 and N1 are not available; (*) fits that are compatible with the observed 113Cd NMR chemical shifts and shown in Figure 4; f = fixed; S02 = 0.87 f; N = coordination number/ frequency; k-fitting range = 3.5 - 12.0 Å-1;
Residual (%).
Results
113Cd NMR spectroscopy
The 113Cd-NMR spectra obtained for the cadmium(II)-cysteine solutions containing CCd(II) ∼ 0.1 mol·dm-3 at pH 7.5 (A1 – G1) and 11.0 (A2 – G2) are shown in Figure 1. The solutions contain several cadmium(II) cysteine species, as indicated by the distributions of complexes calculated for compositions corresponding to solutions A, B, D and E, with the use of the equilibrium constants in Ref. 14; see Figure S-1. The increase in the total cysteine concentration in solutions B – G, resulted in more deshielded 113Cd chemical shifts, indicating a high degree of thiol coordination in the cadmium(II) complexes [17]. For solutions A - C, chemical exchange reactions with intermediate rate (on the NMR time scale) between the several Cd(II)-species in equilibrium resulted in an averaged broad signal for each solution. Considerably sharper NMR signals were obtained for solutions D - G with high total cysteine concentration, which may be due to a single dominating cadmium(II) complex, and/or faster ligand exchange between different cadmium(II) species in the solution. The alkaline solutions B2 - G2 showed somewhat more shielded chemical shifts than the corresponding solutions B1 - G1 at pH = 7.5, probably due to an increase in chelate Cd(II)-(S,N-Cys) coordination of the cysteinate ligands (Cys2-) when the amine group deprotonates at higher pH. The NMR signals were generally narrower for alkaline solutions than for the corresponding neutral ones, especially for A2 - C2, which indicates a faster ligand exchange process, probably promoted by the increasing availability of -NH2 groups or OH- ions.
The 113Cd NMR spectra for the cadmium(II)-penicillamine solutions (H – N) with CH2Pen / CCd(II) ratios from 2 to 20 are shown in Figure 2, and the distributions of the cadmium(II)-penicillamine complexes for solutions H, I, K and L according to the available stability constants [15] are presented in Figure S-2. The observed chemical shifts for solutions H1 and H2 containing CH2Pen = 0.2 mol·dm-3 were close to those of the corresponding cadmium(II) cysteine solutions A1 and A2, and therefore similar coordination environments are expected around the cadmium(II) ions.
As for the cadmium(II)-cysteine solutions, the increase in total concentration of penicillamine for solutions H to N, resulted in more deshielded NMR signals, even though the range of Δδ(113Cd) was considerably more limited. At pH 7.5 the NMR peak for cadmium(II)-penicillamine solutions shifts from 509 ppm to 607 ppm for H1 to M1 (∼ 100 ppm), while for the corresponding cysteine solutions the shift is from 518 ppm to 679 ppm (∼ 160 ppm) for A1 to F1. A similar decrease was observed for the alkaline solutions, with the difference ∼ 70 ppm between the 113Cd chemical shifts for the Cd(II)-penicillamine solutions H2 and N2, at 510 and 578 ppm, respectively, compared with the difference ∼ 130 ppm between the Cd(II)-cysteine solutions A2 and G2 at 527 and 658 ppm, respectively. This indicates a higher tendency for Cd(II) ions to coordinate to the thiolate groups from cysteine than from penicillamine.
The NMR peaks for all the alkaline cadmium(II)-penicillamine solutions (H2 - N2) were sharp, while at pH 7.5 the peaks were broader, especially for solutions H1 - K1, indicating ligand exchange with intermediate rate (on the NMR time scale) between cadmium(II) penicillamine complexes. For solution H1 (H2Pen / Cd(II) = 2.0, pH = 7.5), the broad 113Cd resonance became much sharper as the solution pH is increased to 11.0 in H2, while remaining in the same position at 510 ppm. This signal is even sharper than that of the corresponding cadmium(II)-cysteine solution A2, indicating that a single stable cadmium(II) complex with penicillamine is formed in H2, probably [Cd(Pen)2]2- according to the calculated distribution diagram in Figure S-2.
X-ray absorption spectroscopy - Cd K-edge EXAFS
The least squares curve-fitting results for the k3-weighted Cd K-edge EXAFS spectra of the cadmium(II) cysteine and penicillamine solutions are shown in Tables 3 and 4, Figures 3 and 4. Since coordination number, amplitude reduction factor (S02) and Debye-Waller parameters (σ2), all contribute to the amplitude of the EXAFS oscillation and are strongly correlated, the S02 value was kept constant at 0.87 in all refinements to facilitate comparisons. This value was chosen by calibrating the amplitude reduction factors to 0.87 and 0.85 for two crystalline cadmium(II) complexes, imidazolium tris(thiosaccharinato)aqua cadmate(II) (HIm)[Cd(tsac)3(H2O)] (CdS3O model) and bis(thiosaccharinato)bis(imidazole) cadmium(II) [Cd(tsac)2(Im)2] (CdS2N2 model), respectively; see Figures S-3a and S-3b [40]. The estimated error in the coordination numbers obtained in the refinement procedure is ∼ 20%. For each solution two fitting models were applied: one with only a single Cd-S shell, and the other including both Cd-S and Cd-(N/O) scattering paths. Often the fitting residuals had very minor differences, and only by combining with information from the 113Cd NMR chemical shifts, the more appropriate model could be chosen. For most cadmium(II)-cysteine and penicillamine solutions, the mean Cd-S and Cd-(N/O) bond distances were obtained within the ranges 2.52 – 2.54 Å and 2.28 – 2.35 Å, respectively, which are consistent with what is expected for cadmium(II) complexes with tetrahedral CdS2(N/O)2, CdS3(N/O) and CdS4 configuration [Supporting Material in Ref. 33]. However, the contribution from the light coordinated atoms (oxygen or nitrogen) to the EXAFS oscillation is difficult to separate from the dominating backscattering of the sulfur atoms, and therefore, in the model refinements the coordination number for the Cd-(N/O) scattering pathway often was fixed at N = 1 or 2, based on the observed 113Cd chemical shift values.
X-ray absorption spectroscopy - Cd L3-edge XANES
The normalized Cd L3-edge XANES spectra and the corresponding smoothed 2nd derivatives for the cadmium(II)-cysteine solutions A2 – G2 (pH 11.0), as well as those of a few related crystalline compounds with CdSx(N/O)y coordination, are shown in Figure 5. The XANES spectra of solutions A2 – G2 were rather similar with only a gradual change in the second derivatives. For solutions A2 – D2 the XANES spectra and their 2nd derivatives were intermediate to the spectra of Cd(cysteaminate)2 (as CdS3N2 model) and bis(thiosaccharinato)bis(imidazole) cadmium(II) [Cd(tsac)2(Im)2] (as CdS2N2 model) (Figures 5 and S-4) [39, 40]. As the amount of cysteine in solutions E2 – G2 increased to a 10 – 20 fold excess of the ligand, the relative intensity of the two main features in the 2nd derivative gradually became almost equal. For solution G2 both the Cd L3-edge XANES spectrum and its 2nd derivative are quite similar to those for the CdS4 standard complex, (Et3NH)4[S4Cd10(SPh)16] (Figures 5 and S-4).
Figure 5.
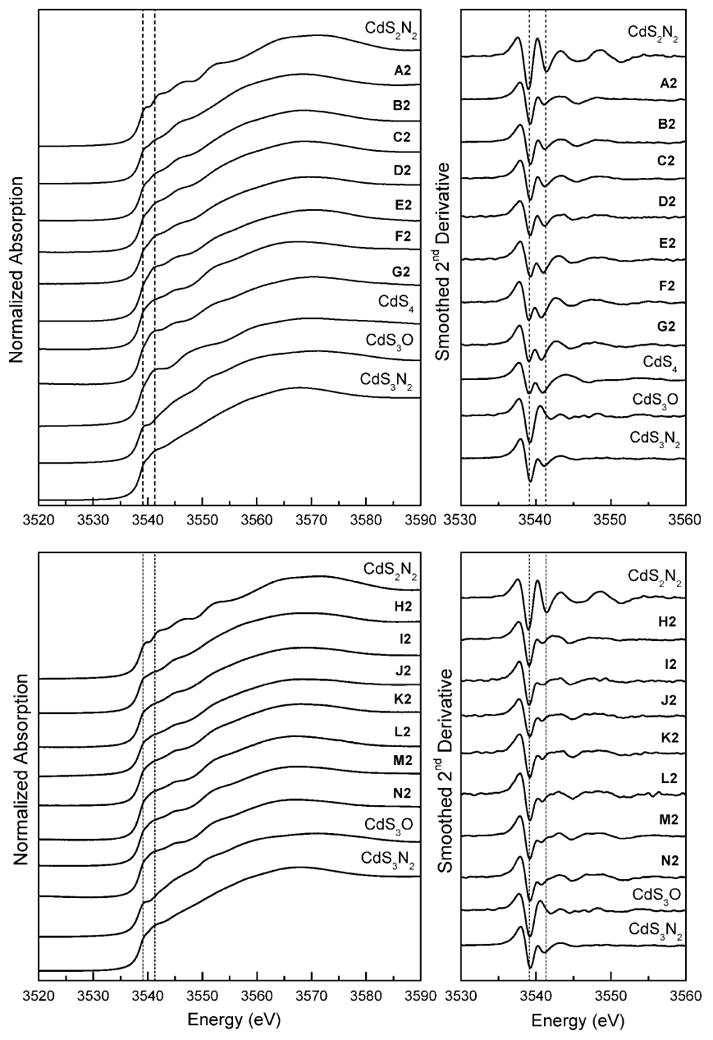
Normalized Cd L3-edge XANES spectra and corresponding smoothed 2nd derivatives for the cadmium(II)-cysteine (A2 – G2) and cadmium(II)-penicillamine (H2 – N2) solutions (pH = 11.0), and for crystalline compounds with CdSx(N/O)y coordination (Ref. 33). Dashed lines are at 3539.1 and 3541.3 eV.
For the cadmium(II) penicillamine solutions H2 – N2, the Cd L3-edge XANES spectra and corresponding smoothed 2nd derivatives appeared quite similar, as expected from the small difference, 68 ppm, between the 113Cd NMR chemical shifts of solutions H2 and N2, and no further structural information was gained from the comparison with L3-edge spectra of standard models (Figure 5).
Discussion
Cadmium(II) Cysteine Solutions
Solution A1 containing CH2Cys = 0.2 mol·dm-3 was obtained by dissolving the Cd(HCys)2 precipitate by adding NaOH. While the 113Cd NMR spectrum of the solid [Cd(HCys)2]·H2O compound showed a broad signal with peak maximum at ∼640 ppm [33], in solution the resonance shifts to 518 ppm (pH = 7.5), and then to 527 ppm at pH = 11.0 (A2). We recently proposed an oligomeric, “cyclic / cage” type of structure for the solid [Cd(HCys)2]·H2O compound with the cadmium(II) ions in CdS3O and / or CdS4 coordination sites, similar to a in Scheme 1 [33]. When it dissolves in solution A1, several species may exist in equilibrium (Scheme 1, b – e), including [Cd(HCys)(Cys)]- and [Cd(Cys)2]2- complexes, as indicated in the reported formation constants (Figure S-1, top left) [14]. However, any appreciable amount of an oligomeric complex similar to a does not seem likely in solution, because of the shift of the 113Cd-NMR signal from ∼ 640 ppm for the [Cd(HCys)2]·H2O compound to a more shielded region (∼ 520 ppm) for solution A1, which corresponds to two sulfur atoms in the coordination sphere of cadmium(II) ion. Neither is complex b with CdS2O2 coordination likely to be present. The only reported CdS2O2 complexes, cadmium(II) thio-β-diketonate in acetone, 191 ppm [41], and two bis(phenoxide) bis(tetrahydrothiphene) cadmium(II) complexes, 76 and 144 ppm [42], show considerably higher shielding than that of solution A1 (518 ppm). However, these complexes contain S-donor ligands other than thiolates and as discussed elsewhere [33], for a cadmium(II) thiolate complex with a stable CdS2O2 coordination environment, a 113Cd chemical shift of ∼ 400 ppm would be expected.
Scheme 1.
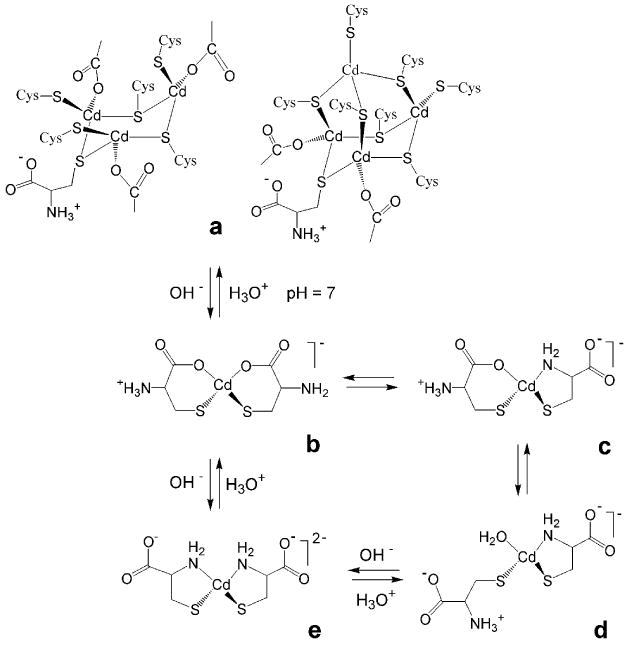
Transformations between possible types of coordination for mononuclear cadmium(II)-cysteine [Cd(HCys)(Cys)]- (b – d) and [Cd(Cys)2]2- (e) complexes. The species c - e with CdS2N(N/O) coordination may exist in comparable amount in solution A1 (pH 7.5), prepared by dissolving the solid Cd(HCys)2·H2O compound. Structures (a) are two of the possible structures for this compound (Ref. 33), with the coordinated COO- groups from cysteine ligands.
The coordination site for d is similar to that of cadmium(II)-substituted horse liver alcohol dehydrogenase (LADH), with the 113Cd chemical shift 483 ppm for CdS2NOwater coordination [20]. In a large excess of imidazole, the 113Cd chemical shift for Cd(II)-LADH was observed at 519 ppm, which has been assigned to CdS2N2 coordination (see Table 1), similar to the coordination site of e in Scheme 1. Based on recent theoretical calculations of 113Cd chemical shifts for proteins and model systems, it was proposed that the contribution for each type of ligand in a “tetrahedral” coordination geometry is: δS = 187 ppm, δN = 77 ppm, δO(COO-) = -25 ppm and δO(H2O) = -53 ppm [43], i.e., the carboxylate oxygen is somewhat less shielding than water. Therefore, the 113Cd chemical shift for complex c is expected to be more deshielded than that of complex d, i.e. ∼ 500 ppm.
Hence, the broad peak observed at 518 ppm in the 113Cd NMR spectrum of solution A1 (pH = 7.5) is proposed to result from a ligand exchange with intermediate rate (on the NMR time scale) between species c, d and e with CdS2N(N/O) coordination, with estimated 113Cd chemical shifts of ∼ 500 ppm (CdS2NOCOO-), ∼ 480 ppm (CdS2NOwater) and ∼ 520 ppm (CdS2N2), respectively. When raising the pH to 11.0 (solution A2) the 113Cd NMR signal shifts slightly downfield to 527 ppm, probably due to the complete deprotonation of the amine group, which allows the [Cd(Cys)2]2- chelate complex (e) with CdS2N2 coordination to dominate in the solution (Scheme 2).
Scheme 2.
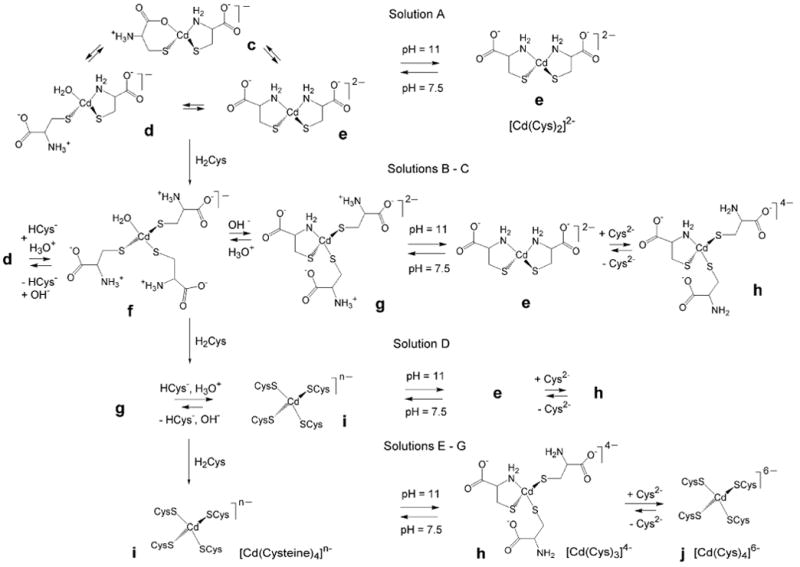
An overview of the dominating mononuclear species present in the cadmium(II)-cysteine solutions (A – G) at pH 7.5 and 11.0.
Least-squares curve-fitting of the Cd K-edge EXAFS spectrum of A1 shows the minimum residual for a single Cd-S shell model with refined coordination number of ∼ 3.6 (Table 3). However, such a high number of sulfur backscatters should correspond to a δ(113Cd) value of at least 600 ppm (see Table 1), and also is not consistent with the stoichiometric ratio of H2Cys / Cd(II) = 2.0 in solution A1. Although the fitted two-shell model shows slightly higher residuals, the differences between the fits are insignificant. The model including two Cd-(N/O) resulted in a coordination number of 1.9 for the Cd-S path. The Cd-S and Cd-(N/O) bond distances were 2.54 ± 0.02 and 2.34 ± 0.04 Å, respectively, which fits well with a mixture of [Cd(HCys)(Cys)]- (CdS2NO) and [Cd(S,N-Cys)2]2- (CdS2N2) species (c - e, Scheme 1) with distorted tetrahedral geometries.
EXAFS curve-fitting for solution A2 using the same CdS2(N/O)2 model results in a similar mean Cd-S distance, 2.53 ± 0.02 Å, while the average Cd-(N/O) distance, 2.29 ± 0.04 Å, is slightly shorter than that of solution A1. This is consistent with an increase of the dominating [Cd(S,N-Cys)2]2- (CdS2N2) chelate complex (Scheme 1 e) with stronger bonds between the cadmium(II) ions and the deprotonated cysteine amine groups (-NH2), and the observed 113Cd NMR chemical shift at 527 ppm. For ten structurally known cadmium(II) complexes with CdS2N2 configuration, the average Cd-S and Cd-N distances are 2.473 and 2.288 Å, respectively [Supporting Material in Ref. 33], with the former slightly shorter than that of solution A2. Figure 6 presents the separate contributions to the fitted EXAFS model for solution A2.
Figure 6.
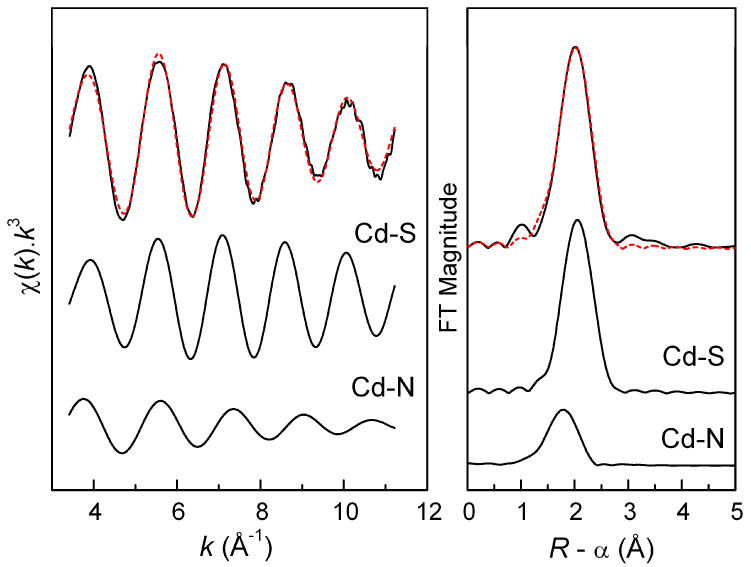
Least-squares k3-weighted curve fitting for a CdS2N2 coordination model to the Cd K-edge EXAFS oscillation of the cadmium(II) cysteine solution A2 (pH = 11.0) and the corresponding Fourier-transform (solid line, exp.; red dash line, fit), with the separate contributions below (see Table 3).
For solutions F1 and G1 with large cysteine excess (CH2Cys ∼ 1.5 mol dm-3), probably with partially protonated amino groups (HCys-) at pH = 7.5, the 113Cd chemical shift is ∼ 680 ppm, close to that of solution E1 (677 ppm). These chemical shifts are higher than the δ(113Cd) ranges for CdS3O and CdS3N, but rather similar to those recently reported for CdS3 configurations (Table 1). However, the mean Cd-S bond distances, 2.52 - 2.53 Å, obtained from EXAFS spectra of these solutions (Table 3) are much longer than the average Cd-S bond distance in three crystalline CdS3 thiolate complexes (2.446 Å; Supporting Material in Ref. 33). For cadmium(II)-substituted rubredoxin from Clostridium pasteurianum, a crystal structure determination at 1.5 Å resolution resulted in an average Cd-S distance of ∼ 2.5 Å for a CdS4 center [44]. For [Cd(S-cysteinate)4]2- complexes, there are several reports of higher frequency δ(113Cd) shifts, e.g. for cadmium(II)-substituted LADH (751 ppm) [20], rubredoxin (723 - 732 ppm) [21, 22], and the DNA binding domain of the glucocorticoid hormone receptor (704, 710 ppm) [23]. However, recently the chemical shifts from a designed cysteine-rich TRI peptide at δ(113Cd) = 650, 680 ppm could, with support from perturbed angular correlation (PAC) spectroscopy, be attributed to distorted tetrahedral [Cd(S-cysteinate)4]2- complexes [24]. Therefore, based on the 113Cd NMR chemical shift, solutions E1 - G1 may contain 100% [Cd(S-cysteinate)4]n- (with the cysteine ligands in HCys- or Cys2- forms), or a combination of CdS4 and CdS3(N/O) species.
The EXAFS spectra of solutions E1 – G1 overlap (see Figure S-5), as expected from the similarity of their 113Cd chemical shifts (677 – 680 ppm). Least-squares curve-fittings of these EXAFS spectra using only a single Cd-S shell resulted in a refined coordination number of 3.8 - 4.1. When the Cd-(N/O) path with a fixed contribution N = 1 is included in the fitting model, the frequency/ coordination number for the Cd-S path refined to N ∼ 3 for solutions E1 and G1. Both models yielded similar residuals and reasonable distances (except F1), but too low / high Debye-Waller parameters for Cd-(N/O) path. Therefore, the information from Cd K-edge EXAFS data analyses for solutions E1 - G1 does not confirm whether or not these solutions contain CdS4 species exclusively.
The k3-weighted EXAFS oscillations of the corresponding alkaline (pH = 11.0) solutions F2 – G2 containing deprotonated Cys2- virtually overlap (see Figure S-6). However, their increasing 113Cd chemical shifts, 636 ppm (E2), 654 ppm (F2) and 658 ppm (G2), are more sensitive to small changes in the distribution of the complexes than the mean Cd-S bond distances from EXAFS spectroscopy (Table 3). The 113Cd chemical shifts for F2 and G2 are in between the values reported for CdS3N configuration (see Table 1) and the distorted [Cd(S-cysteinate)4]2- complexes in the TRI peptide. The Cd K-edge EXAFS model fittings for these solutions resulted in very similar residuals for the CdS4 or CdS3N models (Table 3). However, the features in the Cd L3-edge XANES spectra of F2 and G2, and their corresponding 2nd derivatives, are almost identical to those of the CdS4 model compound (see Cd L3-edge XANES section above). Therefore, with emphasis on the Cd L3-edge XANES spectra, we propose that at pH = 11 the dominating complex is [Cd(S-Cys)4]6- with fully deprotonated Cys2- ligands in the cadmium(II) cysteine solutions with CH2Cys > 1.0 mol dm-3 (F2 – G2; δ(113Cd) = 654 – 658 ppm), together with a minor amount of the [Cd(Cys)3]4- (CdS3N) complex. Those species (h and j in Scheme 2) are in equilibrium with fast ligand-exchange, which results in one averaged signal in their NMR spectra. In the corresponding solutions at pH = 7.5 (E1 – G1), with 113Cd NMR signals at 677 – 680 ppm and partially protonated amine groups, [Cd(S-cysteinate)4]n- (CdS4) species are predominantly formed.
In the solution E2, the [Cd(Cys)3]4- (CdS3N) complex is dominating, as shown by the shift of the 113Cd NMR signal upfield to 636 ppm. The mean Cd-S and Cd-(N/O) distances of 2.53 ± 0.02 and 2.28 ± 0.04 Å for solution E2 are comparable to the corresponding average distances for the only structurally known cadmium(II) complex with CdS3N configuration (2.522 and 2.207 Å) [Supporting Material in Ref. 33], and are consistent with our proposed structure h for the [Cd(Cys)3]4- complex in Scheme 2. Formation of a [Cd(Cys)3]4- complex with CdS3N2 coordination (Scheme S-1) can be excluded, since the average Cd-S and especially the Cd-(N/O) bond distances for solution E2 are appreciably shorter than the mean Cd-S and Cd-N distances for five crystalline cadmium(II) complexes with CdS3N2 coordination (2.551 and 2.386 Å, respectively), which all are dinuclear complexes with long, bridging Cd-S bonds [Supporting Material in Ref. 33]. As a specific example, the Cd(cysteaminate)2 complex with CdS3N2 coordination (solid state 113Cd NMR δiso = 669 ppm) could be considered with one short (2.534 Å) Cd-S bond distance and two longer bridging Cd-S distances at 2.572 and 2.620 Å, and a mean Cd-N distance of 2.376 Å [45], which is ∼ 0.1 Å longer than the mean Cd-(N/O) distances obtained for solution E2.
In solution D1 (pH = 7.5) with δ(113Cd) = 655 ppm the [Cd(S-cysteinate)4]n- complex is expected to be the dominating species as for F2 and G2, together with a minor amount of [Cd(cysteinate)3]2-(CdS3N) (i and g in Scheme 2). The EXAFS model fitting for solution D1 resulted in similar residuals for three different models, i.e. CdS4, CdS3N and a mixture of CdS4 + CdS3N (50 : 50) (Table 3), all with the average Cd-S distance of 2.53 ± 0.02 Å.
Curve-fitting of the EXAFS spectra for solutions B1 and C1 (pH = 7.5), again resulted in the minimum residual for a single Cd-S shell model (Table 3); however, the 113Cd chemical shifts of 585 - 627 ppm show that these solutions contain mixtures of cadmium(II)-cysteine complexes that are in equilibrium with intermediate ligand-exchange rate, with mainly CdS3O and CdS3N geometries (f and g in Scheme 2), for which the reported ranges of chemical shifts are 560 – 645 ppm and 637 - 659 ppm, respectively (Table 1). EXAFS model fitting using both Cd-S and Cd-(N/O) shells resulted in average bond distances of 2.54 ± 0.02 and 2.35 ± 0.04 Å, respectively (Table 3), which are close to the corresponding mean Cd-S and Cd-O distances, 2.53 and 2.30 Å, for the crystalline cadmium(II) complex (HIm)[Cd(tsac)3(H2O)], with CdS3O coordination and a coordinated water molecule [40].
The 113Cd chemical shifts for solutions B2 – G2 are generally lower than those of solutions B1 – G1 with comparable ligand / metal ratios (Figure 1). The partial protonation of the amine groups (-NH3+) in the solutions B1 and C1 at pH = 7.5, favors the formation of cadmium(II) cysteine complexes with CdS3O coordination (from water). By increasing the cysteine concentration in the solutions D1 – G1, another cysteine thiolate group can substitute the water and promote formation of the [Cd(S-cysteinate)4]n- complex. By raising the pH to 11.0, i.e. deprotonating all the amine groups, the chelate complexes [Cd(S,N-Cys)2]2- and [Cd(Cys)3]4- (e and h in Scheme 2) gain stability, which is reflected in the lower chemical shifts for the alkaline solutions (B2 – G2), relative to those at pH 7.5 (B1 – G1). These species are in fast ligand-exchange equilibrium, resulting in a single averaged peak in NMR.
The curve-fitting of EXAFS models for solutions A2 – G2 and the corresponding Fourier-transforms are shown in Figure 3. For solutions B2 – E2, where CH2Cys increases from 0.3 to 1.0 mol dm-3, the refinement of the Cd-S contribution shows a gradual increase in the coordination number from N = 2.2 to 3.3 (Table 3), indicating an increasing concentration of the [Cd(Cys)3]4- complex.
For solutions A2 – D2, the Cd L3-edge absorption spectra and their 2nd derivatives are intermediate to the spectra of Cd(cysteaminate)2 (as CdS3N2 model) and bis(thiosaccharinato)-bis(imidazole) cadmium(II) [Cd(tsac)2(Im)2] (as CdS2N2 model) (Figures 4 and S-4) [39, 40]. This is consistent with a mixture of [Cd(Cys)2]2- and [Cd(Cys)3]4- complexes in the solutions A2 – D2. No standard complex with CdS3N coordination was available for a more direct comparison.
Cadmium(II) Penicillamine Solutions
The 113Cd chemical shifts for solutions H1 and H2 with CH2Pen = 0.2 mol·dm-3 are comparable (Figure 2) with those of corresponding cadmium(II) cysteine solutions A1 and A2 (Figure 1), and therefore, similar CdS2N(N/O) coordination environments are expected (like c – e, Scheme 2). The distribution diagram of the cadmium(II)-penicillamine complexes (Figure S-2a, top left) supports this conclusion, indicating that solution H1 (pH = 7.5) contains a mixture of [Cd(HPen)(Pen)]- (CdS2NO) and [Cd(Pen)2]2- (CdS2N2) complexes, while in solution H2 at pH = 11.0, the [Cd(Pen)2]2- complex is the dominating species. This is also reflected in the broadness of 113Cd NMR signals for H1 and H2, where the broad signal for H1 indicates an intermediate ligand-exchange between the cadmium(II) penicillamine complexes, and the narrow signal for H2 is interpreted as an indication for presence of one dominating species.
For solution H1 the EXAFS curve-fitting resulted in the minimum residual for a two-shell model. When the contribution of Cd-(N/O) path is fixed at N = 1.0, the Cd-S coordination number is refined to 2.7 (Table 4). For such a CdS3(N/O) coordination, however, a 113Cd chemical shift higher than 560 ppm would be expected. A model with a fixed Cd-(N/O) contribution at 2.0 resulted in similar residual, and corresponds better to the observed δ(113Cd) = 509 ppm. The average Cd-S and Cd-(N/O) bond distances 2.52 ± 0.02 and 2.30 ± 0.04 Å are slightly shorter than for the corresponding cysteine solution A1 (2.54 ± 0.02 and 2.34 ± 0.04 Å), indicating stronger Cd-S bonding for the penicillamine complexes (like c – e in Scheme 1), a result of the inductive effect of the two methyl groups adjacent to the thiolate sulfur atom. When increasing the pH to 11.0 (solution H2), a good fit is obtained to the EXAFS oscillation for a model with two Cd-S distances at 2.50 ± 0.02 Å and two Cd-(N/O) at 2.30 ± 0.04 Å (Table 4). The similarity to the average Cd-S (2.473 Å) and Cd-N (2.288 Å) bond distances for ten crystalline CdS2N2 complexes [Supporting Material in Ref. 33], supports a dominating [Cd(S,N-Pen)2]2- complex in solution H2, with CdS2N2 coordination as for e in Scheme 2. In the corresponding cadmium(II)-cysteine solution A2, the average Cd-S bond distance of 2.53 ± 0.02 Å is somewhat longer.
For the solutions L1 – N1 (pH = 7.5) with large excess of penicillamine (CH2Pen ∼ 0.87 - 1.0 mol dm-3), the 113Cd NMR chemical shifts are quite close, 602 – 607 ppm, in the ranges expected for CdS3O and CdS3N coordination (see Table 1), indicating mainly trithiolate [Cd(penicillaminate)3]m- species with deprotonated HPen- or Pen2- penicillamine ligands (similar to f and g in Scheme 2), for which no stability constants have been reported. These species are in fast ligand-exchange equilibrium. Their composition is probably comparable to that of the cadmium(II)-cysteine solution C1 (Scheme 2), with a rather similar 113Cd chemical shift of 627 ppm. The enhanced amplitude of the EXAFS oscillation for L1 relative to H1, indicates an increase in the Cd-S coordination number (Figure S-7). EXAFS model fitting for the solution L1 yielded average Cd-S and Cd-(N/O) distances of 2.53 ± 0.02 and 2.30 ± 0.04 Å, respectively (Table 4). For the only reported crystalline cadmium(II) complex with CdS3N coordination, the average bond distances are Cd-S 2.522 Å and Cd-N 2.207 Å [Supporting Material in Ref. 33], and for CdS3O coordination in the thiosaccharinato complex (HIm)[Cd(tsac)3(H2O)], the mean bond distances are Cd-S 2.532 Å and Cd-O 2.304 Å [40], in very good agreement with those for L1 (Table 4). Solutions I1 – K1 with chemical shifts (541 – 582 ppm) between those of H1 and N1 would contain mixtures of cadmium(II)-penicillamine complexes with CdS2(N/O)2 and CdS3(N/O) coordination, similar to c - g in Scheme 2, that are in ligand-exchange equilibrium with intermediate rate. EXAFS model fittings for solutions I1 - K1 using different models, i.e. CdS3(N/O), CdS2(N/O)2 or a mixture of CdS2(N/O)2 + CdS3(N/O) (50 : 50), result in equally good fits, with the Cd-S distance 2.50 - 2.52 Å, and the Cd-(N/O) distance varying between 2.28 - 2.32 Å.
The stability constants reported by Avdeef et al. [16], propose polynuclear cadmium(II) penicillamine complexes in the pH range 4 - 8. According to the distribution diagram in Figure S-2b (top left), solution H1 (pH 7.5) would contain almost equal amounts (∼ 40%) of the [Cd3(HPen)4(Pen)2]2- and [Cd(Pen)2]2- (CdS2N2) complexes and a minor amount of the [Cd2(HPen)3(Pen)2]3- complex. We expect that the polynuclear species would have structures similar to those shown in Scheme S-2 (see Supporting Information), with CdS4 and/or CdS3(N/O) coordination site(s). However, polynuclear cadmium(II) complexes seem unlikely in this solution (H1) for the following reason. For the two bridged CdS4 groups forming the dinuclear cadmium(II) binding site of the GAL4 protein [25], two 113Cd NMR signals were observed at 669 and 707 ppm [26]. The reported 113Cd chemical shifts for CdS2N2, CdS3O and CdS3N coordination are 519 ppm, 560 – 645 ppm and 637 – 659 ppm, respectively (Table 1). Thus, the expected δ(113Cd) for a mixture of [Cd(Pen)2]2- and [Cd3(HPen)4(Pen)2]2- complexes should be close to ∼ 600 ppm (for the coordination sites CdS2N2 + 2 × CdS3(N/O) + CdS4) (similar to the [Cd(HCys)2] solid), rather than the experimental value of 509 ppm for solution H1.
By increasing the pH of the solutions containing a large excess of penicillamine to 11.0 in L2 - N2 (CH2Pen ∼ 0.87 - 1.7 mol dm-3), the 113Cd chemical shifts become more shielded, moving to 575 - 578 ppm. Recently, chemical shifts of 574 – 588 ppm have been reported for a few members of the TRI family of peptides at pH 8.5 – 9.5, and were attributed to CdS3O coordination [27 - 29]. In an earlier study on cadmium(II) thiolate complexes [46], 113Cd chemical shifts of 623 and 577 ppm were observed for alkaline cadmium(II) cysteine and penicillamine solutions (pH = 13, CCd(II) = 0.05 mol dm-3, CH2L / CCd(II) = 12). While the former value was attributed to the formation of the tetra-thiolate [Cd(Cys)4]6- complex, the upfield shift of the corresponding penicillamine solution was interpreted as a result of the steric effect from the methyl groups, preventing ligation through the sulfur atom alone [46], or causing weaker Cd-S bonding and therefore poorer deshielding of the thiolate groups [18].
We may interpret the 113Cd chemical shifts of L2 – N2 in two different ways: 1) either these solutions exclusively contain the [Cd(S-Pen)3]4- complex with CdS3O coordination, where the O-donor ligand is water (or OH-); or 2) a mixture of [Cd(Pen)2]2- (CdS2N2) and [Cd(Pen)3]4- (CdS3N) complexes are present in a fast ligand-exchange equilibrium. In the first case the downfield shift of the NMR signal to 602 – 607 ppm for the corresponding L1 – N1 solutions would be difficult to explain. If we assume that the solutions L2 – N2 would contain the [Cd(Pen)3(H2O)]4- (CdS3O) complex, the composition should not change at pH = 7.5, when most of the coordinated cysteine amine groups are protonated. Assuming the existence of a hydroxo complex [Cd(Pen)3(OH)]5- (CdS3O) in alkaline solutions L2 – N2 (as shown in Figure S-2a, b), would require a hydrated [Cd(Pen)3(H2O)]4- complex at pH = 7.5. Since H2O is a more shielding ligand than OH- [47], the NMR signal for [Cd(Pen)3(H2O)]4- would be more shielded than for [Cd(Pen)3(OH)]5- in alkaline solution. However, this is opposite to the observed trend for the 113Cd chemical shift for solution L1 (pH 7.5), which is more deshielded than L2 (pH 11.0). Hence, a hydroxo complex in L2 does not seem to be feasible, and therefore, we conclude that the solutions L2 – N2 (CH2Pen ≥ 0.9 mol dm-3) contain mixtures of [Cd(Pen)2]2- and [Cd(Pen)3]4- complexes, similar to the cadmium(II) – cysteine solution C2 with a 113Cd NMR chemical shift of 577 ppm (see Figure 1 and e, h in Scheme 2).
The EXAFS spectra of solutions L2 - N2 almost overlap (Figure S-8), as would be expected from the similarity of their 113Cd NMR spectra. The single-shell Cd-S model refinements of these spectra resulted in coordination numbers between 3.0 - 3.4 and a mean Cd-S distance of 2.51 ± 0.02 Å, which is longer than the average Cd-S bond distance in the crystalline trithiolate CdS3 complexes (2.446 Å; Supporting Material in Ref. 33). Adding Cd-(N/O) backscattering to the fitting model slightly improved the residual for L2 and M2. The model fitted to the EXAFS spectra of solutions L2 - N2, assuming a 50:50 mixture of the [Cd(Pen)2]2- (CdS2N2) and [Cd(Pen)3]4- (CdS3N) complexes by fixing the coordination numbers to CdS2.5N1.5, resulted in mean Cd-S and Cd-(N/O) distances of 2.52 ± 0.02 and 2.31 - 2.33 Å, respectively (Table 4).
The 113Cd chemical shifts for M1 and N1 (604 - 607 ppm) are upfield relative to those of the corresponding cadmium(II) –cysteine solutions F1 and G1 (679 – 680 ppm) with similar ligand / metal mole ratios (CH2Cys / CCd(II) = 15 – 20). This upfield shift is probably an effect of the steric hindrance from the two methyl groups close to the thiolate group, preventing the formation of [Cd(S-penicillamine)4]n- (CdS4) species in these solutions. We also observe that the 113Cd chemical shifts for the cadmium(II) cysteine solutions F2 and G2 at pH 11.0 (654 – 658 ppm), are considerably more deshielded than those of the corresponding penicillamine solutions M2 and N2 (578 ppm). According to the Cd L3-edge XANES spectra, the solutions F2 and G2 with comparable ligand excess (CH2Cys ≥ 1.5 mol dm-3) mainly contain the [Cd(Cys)4]6- complex, possibly with some minor amount of [Cd(Cys)3]4- but not [Cd(Cys)2]2-. One reason is the fact that the cysteine thiolate group does not experience the steric hindrance problem that the penicillamine thiolate has. Therefore, in the presence of an excess amount of cysteine in the solution the formation of cadmium(II) complexes with higher thiolate coordination number is facilitated. Another reason is probably related to the lower stability of the [Cd(Cys)2]2- complex in comparison with [Cd(Pen)2]2-, as indicated by its slightly shorter mean Cd-S bond distance, 2.50 ± 0.02 Å (solution H2) vs. 2.53 ± 0.02 Å for [Cd(Cys)2]2- (in solution A2); see Tables 3 and 4.
Conclusion
The cadmium(II) complex formation with cysteine or penicillamine (3, 3′-dimethylcysteine) has been studied at the pH values (7.5 and 11.0) using 113Cd NMR and Cd K and L3-edge X-ray absorption spectroscopy, for solutions with CCd(II) ∼ 0.1 mol dm-3 and ligand / metal mole ratios varied from CH2L / CCd(II) = 2.0 to 20. At CH2L / CCd(II) = 2.0 both ligands form complexes with distorted tetrahedral CdS2N(N/O) coordination geometries, which correspond to a single 113Cd NMR resonance at 509 – 527 ppm. For the [Cd(cysteinate)2]k- species at pH 7.5, the average Cd-S and Cd-(N/O) bond distances from Cd K-edge EXAFS spectra, 2.54 ± 0.02 and 2.34 ± 0.04 Å, respectively, show a slight tendency to become shorter for the dominating [Cd(S,N-Cys)2]2- complex formed when the amine groups deprotonate at pH = 11.0, to 2.53 ± 0.02 and 2.29 ± 0.04 Å. The [Cd(S,N-Pen)2]2- complex that forms in the corresponding penicillamine solution at pH 11.0 has a slightly shorter Cd-S bond distance, 2.50 ± 0.02 Å, but the Cd-(N/O) distance remains similar, 2.30 ± 0.04 Å.
For solutions with higher ligand concentration, the 113Cd resonances shift downfield, which indicates an increasing number of thiolate ligands in the cadmium(II) complexes. For solutions containing a large excess of cysteine (CH2Cys / CCd(II) = 10 – 20), the 113Cd chemical shifts of ∼ 680 ppm at pH = 7.5, and the average Cd-S bond distance of 2.53 ± 0.02 Å, were attributed to a predominant [Cd(S-cysteinate)4]n- complex, with the cysteine ligands in HCys- or Cys2- forms. The average Cd-S distance does not change at pH = 11, and the Cd L3-edge XANES spectra for alkaline solutions with CH2Cys / CCd(II) = 15 – 20 show similar features as in the spectrum of the CdS4 model compound. However, the 113Cd resonances of the solutions shift upfield to 636 - 658 ppm, indicating that when all thiol and amine groups of the cysteine ligands are deprotonated a minor amount of the [Cd(Cys)3]4- (CdS3N) complex is present together with the dominating [Cd(Cys)4]6- complex in these solutions.
For cadmium(II)-penicillamine solutions with similar ligand excess, at pH 7.5 the average Cd-S and Cd-(N/O) bond distances are 2.53 ± 0.02 and 2.30 ± 0.04 Å (for CH2Pen / CCd(II) = 10), while their 113Cd resonance (at ∼ 600 ppm) indicates that [Cd(penicillaminate)3]m- complexes with CdS3(N/O) geometry are dominating. That upfield shift of ∼ 80 ppm relative to the corresponding cadmium(II)-cysteine solutions is probably an effect of the steric hindrance by the two methyl groups in penicillamine, which obstructs formation of the [Cd(S-penicillaminate)4]n- complex. At pH = 11.0, the average Cd-S bond distances remain unchanged, while the 113Cd chemical shifts are found to be ∼ 578 ppm. Those signals, again about 60 – 80 ppm upfield relative to similar cadmium(II)-cysteine solutions, indicate that these solutions contain a mixture of [Cd(Pen)3]4- and [Cd(S,N-Pen)2]2- complexes, with the latter being more stable than the corresponding [Cd(S,N-Cys)2]2- complex, consistent with its shorter Cd-S bond distance (see above).
The differences revealed between cysteine and penicillamine as ligands to cadmium(II) ions in the present study can be linked to the fact that the toxicity of cadmium(II) is reduced when captured in vivo by cysteine-rich metallothionines in CdS4 coordination sites, while penicillamine, which has been clinically used for treating the toxic effects of mercury(II) and lead(II) exposure, is not an efficient antidote against cadmium(II) poisoning.
Supplementary Material
Acknowledgments
We are grateful to Qiao Wu and Dorothy Fox at the instrument facility at the Department of Chemistry, University of Calgary, for their skilful assistance in measuring the NMR spectra. Beamtime was allocated for X-ray absorption measurements at the Photon Factory, Tsukuba, Japan (proposal No. 2005G226), and SSRL (proposal No. 2848), which is operated by the Department of Energy, Office of Basic Energy Sciences, USA. The SSRL Biotechnology Program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and by the Department of Energy, Office of Biological and Environmental Research. We gratefully acknowledge the Natural Sciences and Engineering Research Council (NSERC) of Canada, Canadian Foundation for Innovation (CFI), Alberta Science and Research Investments Program (ASRIP), Alberta Synchrotron Institute (ASI) and the University of Calgary for providing financial support. F.J. is recipient of NSERC University Faculty Award (UFA).
Footnotes
Supporting Information Available. Diagrams for the distribution of cadmium(II) cysteine and penicillamine complexes, EXAFS curve-fitting results for CdS3O and CdS2N2 model compounds, comparison of the EXAFS spectra for solutions E – G, H and L (pH 7.5 and 11), and L2 - N2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Deckert J. Biometals. 2005;18:475–481. doi: 10.1007/s10534-005-1245-0. [DOI] [PubMed] [Google Scholar]
- 2.Lane TW, Saito MA, George GN, Pickering IJ, Prince RC, Morel FFM. Nature. 2005;435:42. doi: 10.1038/435042a. [DOI] [PubMed] [Google Scholar]
- 3.Lane TW, Morel FMM. Proc Natl Acad Sci USA. 2000;97:4627–4631. doi: 10.1073/pnas.090091397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishihara T, Kobayashi E, Okubo Y, Suwazono Y, Kido T, Nishijyo M, Nakagawa H, Nogawa K. Toxicology. 2001;163:23–28. doi: 10.1016/s0300-483x(01)00367-5. [DOI] [PubMed] [Google Scholar]
- 5.Shaikh ZA, Vu TT, Zaman K. Toxicol Appl Pharmacol. 1999;154:256–263. doi: 10.1006/taap.1998.8586. [DOI] [PubMed] [Google Scholar]
- 6.Vašák M, Kägi JHR, Hill HAO. Biochemistry. 1981;20:2852–2856. doi: 10.1021/bi00513a022. [DOI] [PubMed] [Google Scholar]
- 7.Boulanger Y, Goodman CM, Forte CP, Fesik SW, Armitage IM. Proc Natl Acad Sci USA. 1983;80:1501–1505. doi: 10.1073/pnas.80.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henkel G, Krebs B. Chem Rev. 2004;104:801–824. doi: 10.1021/cr020620d. [DOI] [PubMed] [Google Scholar]
- 9.Goyer RA, Miller CR, Zhu SY, Victery W. Toxicol Appl Pharmacol. 1989;101:232–244. doi: 10.1016/0041-008x(89)90272-x. [DOI] [PubMed] [Google Scholar]
- 10.Fotakis G, Timbrell JA. Toxicol in Vitro. 2006;20:641–648. doi: 10.1016/j.tiv.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Shibasaki T, Matsumoto H, Gomi H, Ohno I, Ishimoto F, Sakai O. Biol Trace Elem Res. 1996;52:1–9. doi: 10.1007/BF02784085. [DOI] [PubMed] [Google Scholar]
- 12.Berthon G. Pure Appl Chem. 1995;67:1117–1240. [Google Scholar]
- 13.Bottari E, Festa MR. Talanta. 1997;44:1705–1718. doi: 10.1016/S0039-9140(97)00015-5. [DOI] [PubMed] [Google Scholar]
- 14.Cole A, Furnival C, Huang ZX, Jones DC, May PM, Smith GL, Whittaker J, Williams DR. Inorg Chim Acta. 1985;108:165–171. [Google Scholar]
- 15.Corrie MA, Walker MD, Williams DR. J Chem Soc Dalton Trans. 1976:1012–1015. [Google Scholar]
- 16.Avdeef A, Kearney DL. J Am Chem Soc. 1982;104:7212–7218. [Google Scholar]
- 17.Öz G, Pountney DL, Armitage IM. Biochem Cell Biol. 1998;76:223–234. doi: 10.1139/bcb-76-2-3-223. [DOI] [PubMed] [Google Scholar]
- 18.Summers MF. Coord Chem Rev. 1988;86:43–134. [Google Scholar]
- 19.Eichele K, Wasylishen RE. Inorg Chem. 1994;33:2766–2773. [Google Scholar]
- 20.Bobsein BR, Myers RJ. J Am Chem Soc. 1980;102:2454–2455. [Google Scholar]
- 21.Henehan CJ, Pountney DL, Zerbe O, Vašák M. Protein Sci. 1993;2:1756–1764. doi: 10.1002/pro.5560021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HJ, Lian LY, Scrutton NS. Biochem J. 1997;328:131–136. doi: 10.1042/bj3280131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan T, Freedman LP, Coleman JE. Biochemistry. 1990;29:9218–9225. doi: 10.1021/bi00491a016. [DOI] [PubMed] [Google Scholar]
- 24.Luczkowski M, Stachura M, Schirf V, Demeler B, Hemmingsen L, Pecoraro VL. Inorg Chem. 2008;47:10875–10888. doi: 10.1021/ic8009817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baleja JD, Marmorstein R, Harrison SC, Wagner G. Nature. 1992;356:450–453. doi: 10.1038/356450a0. [DOI] [PubMed] [Google Scholar]
- 26.Gardner KH, Pan T, Narula S, Rivera E, Coleman JE. Biochemistry. 1991;30:11292–11302. doi: 10.1021/bi00111a015. [DOI] [PubMed] [Google Scholar]
- 27.Lee KH, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew Chem Int Ed. 2006;45:2864–2868. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]
- 28.Iranzo O, Cabello C, Pecoraro VL. Angew Chem Int Ed. 2007;46:6688–6691. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc Natl Acad Sci USA. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Suzuki K, Kanaori K, Tajima K, Kashiwada A, Hiroaki H, Kohda D, Tanaka T. Protein Sci. 2000;9:1327–1333. doi: 10.1110/ps.9.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pickering IJ, Prince RC, George GN, Rauser WE, Wickramasinghe WA, Watson AA, Dameron CT, Dance IG, Fairlie DP, Salt DE. Biochim Biophys Acta. 1999;1429:351–364. doi: 10.1016/s0167-4838(98)00242-8. [DOI] [PubMed] [Google Scholar]
- 32.Isaure MP, Fayard B, Sarret G, Pairis S, Bourguignon J. Spectrochim Acta B. 2006;61:1242–1252. [Google Scholar]
- 33.Jalilehvand F, Mah V, Leung BO, Mink J, Hajba L. Inorg Chem. 2009 doi: 10.1021/ic900145n. in press. [DOI] [PubMed] [Google Scholar]
- 34.Jalilehvand F, Leung BO, Izadifard M, Damian E. Inorg Chem. 2006;45:66–73. doi: 10.1021/ic0508932. [DOI] [PubMed] [Google Scholar]
- 35.Ressler T. J Synchrotron Rad. 1998;5:118–122. doi: 10.1107/S0909049597019298. [DOI] [PubMed] [Google Scholar]
- 36.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Phys Rev B. 1995;52:2995–3009. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 37.Ankudinov AL, Rehr JJ. Phys Rev B. 1997;56:R1712–R1716. [Google Scholar]
- 38.Ravel B. J Synchrotron Rad. 2001;8:314–316. doi: 10.1107/s090904950001493x. [DOI] [PubMed] [Google Scholar]
- 39.Bharara MS, Kim CH, Parkin S, Atwood DA. Polyhedron. 2005;24:865–871. [Google Scholar]
- 40.Tarulli SH, Quinzani OV, Baran EJ, Piro OE, Castellano EE. J Mol Struct. 2003;656:161–168. [Google Scholar]
- 41.Maitani T, Suzuki KT. Inorg Nuclear Chem Lett. 1979;15:213–217. [Google Scholar]
- 42.Darensbourg DJ, Niezgoda SA, Draper JD, Reibenspies JH. J Am Chem Soc. 1998;120:4690–4698. [Google Scholar]
- 43.Hemmingsen L, Olsen L, Antony J, Sauer SPA. J Biol Inorg Chem. 2004;9:591–599. doi: 10.1007/s00775-004-0553-0. [DOI] [PubMed] [Google Scholar]
- 44.Maher M, Cross M, Wilce MCJ, Guss JM, Wedd AG. Acta Cryst. 2004;D60:298–303. doi: 10.1107/S090744490302794X. [DOI] [PubMed] [Google Scholar]
- 45.Fleischer H, Dienes Y, Mathiasch B, Schmitt V, Schollmeyer D. Inorg Chem. 2005;44:8087–8096. doi: 10.1021/ic050814m. [DOI] [PubMed] [Google Scholar]
- 46.Carson GK, Dean PAW, Stillman MJ. Inorg Chim Acta. 1981;56:59–71. [Google Scholar]
- 47.Jonsson NBH, Tibell LAE, Evelhoch JL, Bell SJ, Sudmeier JL. Proc Natl Acd Sci USA. 1980;77:3269–3272. doi: 10.1073/pnas.77.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao Z, Lavery MJ, Ayhan M, Scrofani SDB, Wilce MCJ, Guss JM, Tregloan PA, George GN, Wedd AG. J Am Chem Soc. 1998;120:4135–4150. [Google Scholar]
- 49.Giedroc DP, Johnson BA, Armitage IM, Coleman JE. Biochemistry. 1989;28:2410–2418. doi: 10.1021/bi00432a011. [DOI] [PubMed] [Google Scholar]
- 50.Roberts WJ, Pan T, Elliott JI, Coleman JE, Williams KR. Biochemistry. 1989;28:10043–10047. doi: 10.1021/bi00452a024. [DOI] [PubMed] [Google Scholar]
- 51.South TL, Kim B, Summers MF. J Am Chem Soc. 1989;111:395–396. [Google Scholar]
- 52.Fitzgerald DW, Coleman JE. Biochemistry. 1991;30:5195–5201. doi: 10.1021/bi00235a012. [DOI] [PubMed] [Google Scholar]
- 53.Bobsein BR, Myers RJ. J Biol Chem. 1981;256:5313–5316. [PubMed] [Google Scholar]
- 54.Meijers R, Morris RJ, Adolph HW, Merli A, Lamzin VS, Cedergren-Zeppezauer ES. J Biol Chem. 2001;276:9316–9321. doi: 10.1074/jbc.M010870200. [DOI] [PubMed] [Google Scholar]
- 55.Engeseth HR, McMillin DR, Otvos JD. J Biol Chem. 1984;259:4822–4826. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.