

Abstract
Severe malaria has a high mortality rate (15–20%) despite treatment with effective antimalarial drugs. Adjunctive therapies for severe malaria that target the underlying disease process are therefore urgently required. Adhesion of erythrocytes infected with Plasmodium falciparum to human cells has a key role in the pathogenesis of life-threatening malaria and could be targeted with antiadhesion therapy. Parasite adhesion interactions include binding to endothelial cells (cytoadherence), rosetting with uninfected erythrocytes and platelet-mediated clumping of infected erythrocytes. Recent research has started to define the molecular mechanisms of parasite adhesion, and antiadhesion therapies are being explored. However, many fundamental questions regarding the role of parasite adhesion in severe malaria remain unanswered. There is strong evidence that rosetting contributes to severe malaria in sub-Saharan Africa; however, the identity of other parasite adhesion phenotypes that are implicated in disease pathogenesis remains unclear. In addition, the possibility of geographic variation in adhesion phenotypes causing severe malaria, linked to differences in malaria transmission levels and host immunity, has been neglected. Further research is needed to realise the untapped potential of antiadhesion adjunctive therapies, which could revolutionise the treatment of severe malaria and reduce the high mortality rate of the disease.
Plasmodium falciparum is the causative agent of human falciparum malaria and is responsible for a huge burden of global mortality and morbidity (Ref. 1). The parasite has a complex life cycle involving both human and mosquito hosts (Fig. 1), and despite more than a century of research, has proven recalcitrant to control and eradication measures. The clinical features of malaria occur during the blood stage of infection, when the parasite grows and multiplies within the human host erythrocytes (Fig. 1). The presence of the parasite and the resulting host inflammatory responses lead to high fevers and associated ‘flu’-like symptoms. In 1–2% of infections a life-threatening illness develops, characterised by various clinical features, including impaired consciousness, coma, difficulty breathing, severe anaemia and multi-organ failure (Refs 2, 3). These clinical manifestations of severe malaria are thought to occur because of a combination of a high parasite burden and the sequestration of mature P. falciparum-infected erythrocytes (IEs) in microvascular beds throughout the body (Ref. 4). The sequestered mass of IEs leads to microvascular obstruction (Refs 5, 6), metabolic disturbances, such as acidosis (Ref. 7), and release of damaging inflammatory mediators (Refs 8, 9), which can combine to cause severe disease and death of the human host. Sequestration is thought to benefit the parasite by allowing it to avoid the host's normal splenic clearance mechanisms that remove aged or damaged erythrocytes (Ref. 10).
Figure 1.

Life cycle of Plasmodium falciparum. When an infected female Anopheles mosquito takes a blood meal, sporozoite forms of P. falciparum are injected into the human skin. The sporozoites migrate into the bloodstream and then invade liver cells. The parasite grows and divides within liver cells for 8–10 days, then daughter cells called merozoites are released from the liver into the bloodstream, where they rapidly invade erythrocytes. Merozoites subsequently develop into ring-stage, pigmented-trophozoite-stage and schizont-stage parasites within the infected erythrocyte. P. falciparum-infected erythrocytes express parasite-derived adhesion molecules on their surface, resulting in sequestration of pigmented-trophozoite and schizont stages in the microvasculature. The asexual intraerythrocytic cycle lasts for 48 hours, and is completed by the formation and release of new merozoites that will re-invade uninfected erythrocytes. It is during this asexual bloodstream cycle that the clinical symptoms of malaria (fever, chills, impaired consciousness, etc.) occur. During the asexual cycle, some of the parasite cells develop into male and female sexual stages called gametocytes that are taken up by feeding female mosquitoes. The gametocytes are fertilised and undergo further development in the mosquito, resulting in the presence of sporozoites in the mosquito salivary glands, ready to infect another human host.
The importance of Plasmodium falciparum adhesion
Three major types of Plasmodium falciparum adhesion
Sequestration occurs because parasite-derived adhesins expressed on the surface of mature-IEs bind to receptors on human cells. Three major types of IE adhesion have been described (Fig. 2): (1) cytoadherence to endothelial cells (often referred to simply as cytoadherence or cytoadhesion) (Ref. 11); (2) rosetting with uninfected erythrocytes (Ref. 12); and (3) interactions with platelets that can lead to clumping of IEs in vitro (platelet-mediated clumping) (Ref. 13).
Figure 2.

Adhesion of Plasmodium falciparum-infected erythrocytes to human cells. (Legend; See previous page for figure) (a) Schematic representation of the adhesion properties of P. falciparum-infected erythrocytes to different host cells. Erythrocytes infected with mature forms of P. falciparum parasites (pigmented trophozoites and schizonts) have the ability to bind to a range of host cells, such as endothelium, uninfected erythrocytes (rosetting) and platelets (platelet-mediated clumping). The adhesion of infected erythrocytes to endothelial cells leads to their sequestration in the microvasculature of various organs and tissues such as heart, lung, brain, muscle and adipose tissue. As a result, only erythrocytes carrying young ring forms of the parasite are detected in human peripheral blood samples. Although cytoadherence and sequestration of mature infected erythrocytes in the microvasculature occur in all infections, several specific adhesive phenotypes have been associated with severe pathological outcomes of malaria, such as the formation of rosettes and the adhesion of infected erythrocytes to brain endothelium. Rosetting and platelet-mediated clumping are phenotypes that are displayed by some but not all P. falciparum isolates in vitro. In vivo, it is thought that the formation of rosettes and clumps will be accompanied by adhesion to endothelial cells and sequestration in the microcapillaries (Ref. 115). (b) Cytoadherence of infected erythrocytes to in-vitro-cultured brain endothelial cells, visualised by light microscopy after Giemsa staining. (c) Rosettes detected in in vitro P. falciparum cultures, observed after preparation of Giemsa-stained thin smears and light microscopy. (d) Platelet-mediated clumps of infected erythrocytes formed after in vitro co-incubation of parasite cultures with platelets, observed by Giemsa-stained thin smears and light microscopy.
An additional specialised form of adhesion occurs during malaria in pregnancy, in which IEs adhere to syncytiotrophoblasts to bring about placental sequestration (Ref. 14). The molecular mechanisms of placental sequestration and the drive to develop a vaccine to prevent malaria in pregnancy are covered elsewhere (Refs 15, 16, 17) and are not discussed here. IEs are also known to bind to a variety of immune system cells, which has important immunological consequences. These immunological interactions are considered briefly below; however, the review focuses mainly on the first three major types of adhesion, and considers progress in elucidating the molecular mechanisms of adhesion and the therapeutic implications of understanding these important host–parasite interactions.
Which adhesion phenotypes are important in the pathogenesis of severe malaria?
An important prerequisite for the development of new treatments is an understanding of how different types of adhesion contribute to malaria pathogenesis. All P. falciparum isolates sequester, yet not all infections lead to life-threatening disease. So, are all types of parasite adhesion equally damaging? Or is life-threatening malaria linked to specific binding phenotypes that can target IEs to vital organs such as the brain, or cause particularly severe microvascular obstruction? There are, as yet, no definitive answers to these crucial questions. However, current data suggest that there might be geographic variation in the association between adhesion phenotypes and severe disease (discussed further below).
Discovering which parasite adhesion phenotypes contribute to life-threatening malaria has proved difficult because there is no animal model that reflects the pathogenesis of human malaria. Researchers have therefore used two approaches to investigate parasite adhesion phenotypes in relation to disease severity. The first compares the binding properties of field isolates derived from blood samples of patients with different clinical forms of malaria. Binding of IEs is assessed in static or flow assays using purified host receptors bound to plastic dishes, cell lines, fluorescently labelled receptors or receptor-coated beads. The aim of these studies is to identify parasite adhesion phenotypes that occur at high frequency (or show high levels of binding) in isolates from patients with severe malaria, but are rare (or show low levels of binding) in isolates from patients with uncomplicated disease. A positive correlation between a parasite adhesion phenotype and severe disease supports a role for that phenotype in pathogenesis. A negative result does not, however, prove the phenotype is unimportant, because the assays might not adequately reflect adhesion in vivo. A second approach has been to use human genetic studies to investigate whether receptor polymorphisms that reduce parasite adhesion confer protection against severe malaria. The rationale for these studies is that if an adhesion phenotype is directly involved in causing life-threatening malaria, then any human receptor polymorphism that reduces or abolishes parasite adhesion should confer protection against severe disease and death. Examples of both types of study are given below.
Geographic variation in pathogenic mechanisms linked to malaria transmission intensity and host immunity
There are distinct patterns of severe malaria in different parts of the world linked to differences in malaria transmission intensity. For example, in South East Asia, where transmission is generally low, severe malaria affects all age groups and commonly presents as multiorgan failure (including renal and hepatic failure, pulmonary oedema and impaired consciousness) (Ref. 18). Individuals suffering from severe malaria in SE Asia usually have had few, if any, previous malaria infections. Conversely, in sub-Saharan Africa, transmission levels tend to be higher and more stable, and severe malaria is mainly a disease of children under 5 years that presents as impaired consciousness, severe anaemia or respiratory distress (Ref. 2). In sub-Saharan Africa, patients suffering from severe malaria are likely to have had multiple previous P. falciparum infections (Ref. 19). The distinct clinical features of severe malaria in different parts of the world are probably age-related, because a recent study from a low-transmission area in Asia shows that age has a large effect on presenting syndromes, with seizures, respiratory distress and anaemia being more common in children, whereas renal and hepatic failure are more commonly seen in adults (Ref. 3).
It remains unclear to what extent the different levels of host immunity to malaria that occur under different transmission intensities influences host–parasite interactions. The possibility that parasite phenotypes contributing to severe disease might differ in distinct geographical regions related to transmission intensity has received little attention, but is supported by recent research. In SE Asia, a high parasite multiplication rate in vitro and the ability of parasites to invade erythrocytes nonselectively are linked to severe disease (Refs 20, 21), whereas these factors are not associated with disease severity in Africa (Ref. 22). There is a direct link between total parasite burden and risk of severe malaria and death in SE Asia (Refs 23, 24), whereas the relationship is less clear in sub-Saharan Africa, where some children tolerate extremely high parasitaemia without developing severe clinical complications (Refs 2, 25). In terms of adhesion phenotypes, rosetting is associated with severe malaria in African children (Refs 13, 22, 26, 27, 28, 29, 30, 31, 32, 33), but is not associated with malaria severity in SE Asia (Refs 34, 35, 36, 37). The possible reasons why different parasite properties are linked to severe malaria in different regions are discussed further below. In addition, because of the potential geographic variation in parasite adhesion phenotypes underlying severe malaria, we discuss studies from areas with unstable or low transmission (usually SE Asia) separately to studies from areas with stable or moderate–high transmission (sub-Saharan Africa or Papua New Guinea).
Molecular mechanisms of P. falciparum adhesion
In 1995, the cloning of the var genes encoding the variant surface antigen family P. falciparum erythrocyte membrane protein 1 (PfEMP1) provided essential groundwork for research into the molecular basis of adhesion in falciparum malaria (Refs 38, 39, 40). PfEMP1 variants are expressed on the surface of IEs and are responsible for at least some of the adhesive properties of IEs. Other parasite-derived variant antigens are also present on the IE surface, such as RIFINs (Ref. 41) and STEVORs (Ref. 42), and have the potential to be involved in adhesion; however, the function of these proteins remains unknown. Var genes encode PfEMP1 variants containing extracellular regions consisting of tandemly arranged cysteine-rich domains called duffy-binding-like (DBL), cysteine-rich interdomain regions (CIDR) and C2 domains (Ref. 43) (Fig. 3). Var genes can be divided into three major groups (A, B and C) on the basis of conserved upstream regions, and these groupings have functional and clinical significance (Refs 44, 45, 46). The role of PfEMP1 in different types of adhesion is outlined below, and the structure, functions and diversity of the var gene family are described in more detail in several recent reviews (Refs 47, 48, 49). It is important to appreciate that although each IE is thought to express only one PfEMP1 variant at a time (out of a repertoire of approximately 60 per parasite genome), switching of var gene expression can occur at each new asexual blood stage cycle, giving rise to antigenic variation in malaria (Ref. 50). A switch to an antigenically distinct PfEMP1 variant might result in a switch to a new adhesion phenotype (Ref. 50). The adhesion properties of parasite isolates are therefore not fixed, but can change in subsequent cycles as PfEMP1 expression changes. An individual isolate could express a virulence-associated adhesion phenotype, such as rosetting, in one host, but after transmission to a new host might express a different predominant PfEMP1 variant with a less-damaging adhesion phenotype. The factors that determine which var gene is selected for transcription in each IE are currently unclear. The capacity for phenotypic switching provides an extra level of complexity for researchers studying parasite adhesion properties, and studies using long-term parasite cultures in vitro often require regular selection procedures to maintain the phenotype under investigation.
Figure 3.

Schematic representation of a parasite-derived Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) variant on the surface of an infected erythrocyte. PfEMP1 is a family of proteins encoded by var genes that are transported and expressed on the surface of infected erythrocytes during the mature stages of the intraerythrocytic cycle (pigmented trophozoite and schizont). There are approximately 60 var genes per parasite genome, which encode 60 different variants of PfEMP1; however, only one particular variant of PfEMP1 is expressed per cell at any given time. Switching of var gene expression allows the parasite to modify the antigenic and functional properties of infected erythrocytes, thereby evading immunity and altering adhesion capabilities. The extracellular region of PfEMP1 has an N-terminal segment (NTS) followed by several cysteine-rich domains known as DBL (duffy-binding-like) and CIDR (cystein-rich interdomain regions) that can be classified into distinct types based upon sequence similarity. There are six DBL types, (α, β, γ, δ, ɛ and X) and three CIDR types (α, β and γ). The number, location and type of DBL and CIDR domains vary among PfEMP1 variants, and this variable domain composition and extensive sequence polymorphism is thought to provide great flexibility in binding properties. To date, the binding domains for several host receptors, such as CD36, complement receptor 1 and ICAM1, have been mapped to individual DBL and CIDR domains. This diagram shows a hypothetical model of a PfEMP1 variant. TM, transmembrane region.
For each P. falciparum adhesion phenotype, a summary is given below describing what is known about the molecular basis of adhesion, including information on the host receptor, the parasite ligand and the role of the adhesion phenotype in the pathogenesis of severe malaria.
Molecular mechanisms of infected erythrocyte adherence to endothelial cells
The ability of IEs to bind to microvascular endothelial cells and become sequestered from the peripheral blood was described in postmortem studies of patients who died from falciparum malaria in the 1890s (Ref. 51). Since then, cytoadherence has received considerable attention, and although much has been learned, many major questions remain unanswered. P. falciparum IEs have been shown to have the potential for binding to a diverse array of endothelial receptors (Table 1). Evidence for many of these interactions is based on a single, or small number of publications, and only CD36 and intracellular adhesion molecule 1 (ICAM1) have been studied in detail. The neglect of this important area is surprising, as is the fact that it remains unclear which, if any, of these receptors has a pivotal role in the most life-threatening forms of malaria. Each receptor is considered individually below; however, it is important to remember that in vivo, multiple receptors might combine to promote adhesion to endothelial cells (Refs 52, 53). In particular, receptors that promote rolling adhesion [such as ICAM1, vascular cell adhesion molecule 1 (VCAM1) and P-selectin] might act synergistically with static adhesion receptors such as CD36 to enhance the overall degree of sequestration in vivo (Refs 54, 55, 56).
Table 1.
Summary of known receptors for P. falciparum adhesion

aOwing to different clinical and epidemiological features of severe malaria in regions of varying malaria transmission, studies have been separated into ‘Africa’ and ‘Papua New Guinea’ indicating stable, moderate-high transmission areas where severe malaria affects young children and substantial immunity develops in the population, or ‘Asia’ indicating unstable or low transmission areas where severe malaria affects nonimmune individuals of all age groups.
bND, not done.
cSubphenotypes of rosetting and clumping involving individual receptors have not been examined for their relationship with disease severity.
CD36
IEs bind to the scavenger receptor CD36 (Refs 57, 58), which is expressed on endothelial and epithelial cells, macrophages, monocytes, platelets, erythrocyte precursors and adipocytes (Ref. 59). Blocking studies using monoclonal antibodies (Ref. 60) and peptides (Ref. 61) suggest that the binding site for P. falciparum involves amino acids 139-184 of CD36, although involvement of other regions has not been exluded.
The parasite ligands for CD36 binding are PfEMP1 variants (Refs 38, 62) encoded by two major subtypes of var genes (Group B and C) (Ref. 44). The Group B and C var genes comprise approximately 50 of the average repertoire of 60 var genes per parasite genome (Ref. 48). Various PfEMP1 variants have been shown to bind CD36 via the most N-terminal CIDR domain (Refs 63, 64, 65) (Fig. 3), and the structure of this region has been determined (Ref. 66).
CD36 binding is a property of almost all P. falciparum isolates derived from malaria patients (Ref. 30); however, the role of CD36 in malaria pathogenesis remains uncertain (Ref. 67). Studies in Africa have found no difference in CD36-binding ability between parasite isolates from severe and uncomplicated malaria patients (Refs 30, 68, 69, 70), and human genetic studies of CD36-deficient malaria patients have shown conflicting (but mostly negative) results (Table 1). On balance, current evidence does not support a major role for CD36 in severe malaria in sub-Saharan Africa (Table 1).
In Thailand, two small studies showed a significant positive correlation between CD36 binding and severe malaria (Refs 71, 72), but this was not confirmed in a third study (Ref. 35). Only one human genetics study on CD36 polymorphisms and malaria in SE Asia has been reported, and this showed that CD36 deficiency protected against cerebral malaria (Ref. 73). Therefore CD36 might have a role in severe malaria in SE Asia, but further studies would be valuable.
ICAM1
ICAM1 (CD54) is a member of the immunoglobulin superfamily expressed on endothelial cells and leukocytes. Binding of IEs to ICAM1 causes rolling and static adhesion (Refs 74, 75), and ICAM1 might act synergistically with CD36 to enhance static adhesion (Refs 53, 54). The IE-binding site on ICAM1 has been mapped and localises to the opposite face of ICAM1 to that used by its natural ligand LFA-1 (Refs 76, 77, 78). The binding sites for several distinct P. falciparum strains were shown to be overlapping, but not identical (Refs 76, 77, 78).
The parasite ligands for ICAM1 binding are members of the PfEMP1 family that contain a distinct pair of domains found only in a subset of PfEMP1 variants (DBLβ-C2 domains) (Refs 79, 80, 81). Using a genome-wide approach, it was shown that only some PfEMP1 variants containing the DBLβ-C2 domain pair are able to bind to ICAM1, and that the ICAM1-binding variants are all encoded by Group B var genes (Ref. 82).
As with CD36 binding, the pathophysiological significance of ICAM1 binding is unclear. Field-isolate studies have found no statistically significant association between ICAM1 binding and severe malaria in Africa (Refs 30, 69, 70), although increased binding was seen in isolates from patients with clinical malaria (severe and uncomplicated) compared with asymptomatic individuals (Ref. 30). An ICAM1 polymorphism that reduces IE binding to ICAM1 under flow conditions (Ref. 83) occurs at high frequency in African populations (Ref. 84). However, human genetic studies show that this ICAM1 polymorphism does not protect against severe malaria in sub-Saharan Africa (Table 1), but does protect against nonmalarial febrile illness in infants (Ref. 85).
In Asia, ICAM1 binding is not associated with severe malaria in field-isolate studies (Refs 35, 72), and ICAM1 polymorphisms have not been studied. However, histological studies have shown that IEs and ICAM1 colocalise in the brains of patients who died from cerebral malaria (Ref. 86). ICAM1 is widely upregulated on microvascular endothelial cells in the presence of cytokines, such as tumour necrosis factor-α (TNF-α), which reach high levels in severe malaria (Ref. 87). Therefore it is plausible that ICAM1-mediated cytoadherence has the potential to contribute to sequestration throughout the body during severe malaria.
P-selectin
P-selectin (CD62P) is a glycoprotein that is expressed on activated platelets and endothelial cells and is important for leukocyte trafficking. It mediates rolling of IEs on endothelial cells and facilitates adhesion to CD36 in field isolates from Thailand (Refs 54, 56, 88). The parasite-binding site on P-selectin has not been mapped, although it is known that antibodies that inhibit interactions between P-selectin and leukocytes do not affect P. falciparum binding (Ref. 56).
The parasite ligand for P-selectin binding is thought to be PfEMP1, because purified PfEMP1 can bind to P-selectin in vitro (Ref. 89). Specific PfEMP1 variants and binding domains for P-selectin have not yet been identified, and the role of P-selectin in severe malaria is unknown (Table 1).
Thrombospondin
Thrombospondin (TSP) is an adhesive glycoprotein released into plasma in response to platelet activation by thrombin. It was the first molecule identified as a receptor for P. falciparum cytoadherence (Ref. 90), although since then, it has received relatively little attention. IEs bind to purified TSP in static assays (Refs 32, 90) and bind to endothelial cells via TSP under flow conditions (Ref. 91). IEs are thought to bind to the Type 3 repeat regions of TSP (Ref. 92).
The parasite ligand for TSP is controversial, with PfEMP1 (Ref. 62), red-cell-derived phosphatidylserine (a membrane phospholipid) (Ref. 93) and altered Band 3 protein (Ref. 92) as possible candidates. No specific PfEMP1 variants or domains have yet been shown to bind TSP.
Only one study has examined the association of TSP binding with severe malaria, and it was found that although most Kenyan field isolates adhered well to TSP in a static assay, there was no correlation with disease severity (Ref. 32). Whether TSP polymorphisms affect adhesion of P. falciparum or susceptibility to severe malaria is unknown. Hence, a role for TSP binding in severe malaria is not supported by current evidence, but more research is needed to confirm this.
PECAM1
Platelet endothelial cell adhesion molecule 1 (PECAM1 or CD31) is widely expressed on endothelial cells, monocytes, platelets and granulocytes. P. falciparum IEs from laboratory strains (Ref. 94) and field isolates (Ref. 70) bind to PECAM1 on endothelial cells and the binding site has been mapped to the first four immunoglobulin-like domains of PECAM1 (Ref. 94). The parasite ligand for PECAM1 is thought to be PfEMP1, and both the CIDRα and DBL2δ domains of a specific PfEMP1 variant have PECAM1-binding activity (Ref. 95).
Although approximately 50% of field isolates from Kenya adhered well to PECAM1 in some studies (Refs 32, 70), no significant correlation with malaria severity was found (Refs 30, 32). A high-frequency PECAM1 polymorphism did not protect against severe malaria in Kenya or Papua New Guinea (Ref. 96), whereas another PECAM1 polymorphism increased the risk of cerebral malaria in Thailand (Ref. 97).
E-selectin
E-selectin (CD62E) is a glycoprotein that is expressed on endothelial cells at sites of inflammation. Initial work with a P. falciparum laboratory strain showed that it was possible to select parasites for static adhesion to E-selectin (Ref. 98). The parasite-binding site on E-selectin has not been mapped and the parasite ligand is unknown. Studies using multiple Thai field isolates under flow conditions failed to detect significant tethering, rolling or static adhesion on E-selectin (Refs 35, 56). In African isolates, E-selectin binding was extremely low and not associated with disease severity (Ref. 30). The role of E-selectin in cytoadherence is thus probably minor, if any.
VCAM1
VCAM1 (CD106) is a member of the immunoglobulin superfamily and encodes a cell-surface sialoglycoprotein expressed by cytokine-activated endothelium. P. falciparum parasites were selected in vitro for binding to VCAM1 (Ref. 98), and field isolates from Thailand were shown to tether and roll on VCAM1, but static adhesion did not occur (Refs 35, 56). In African isolates, VCAM1 binding was extremely low and not associated with disease severity (Ref. 30). The role of VCAM1 polymorphisms in P. falciparum adhesion and susceptibility to severe malaria has not been investigated.
Heparan sulphate
The glyosaminoglycan heparan sulphate has been shown to mediate binding of rosetting IEs to endothelial cells (Ref. 99) and heparan-sulphate-like molecules on uninfected erythrocytes might have a role in rosetting (described below). It is unclear whether IE binding to heparan sulphate on endothelial cells can occur independently of rosetting, or whether all parasites that bind heparan sulphate form rosettes. Binding of heparin (a highly sulphated form of heparan sulphate produced by mast cells and used as a substitute for endothelial cell heparan sulphate) has been suggested to be dependent on N-sulphation (Ref. 100), and requires a minimal heparin fragment size of 10- or 12-mers (Refs 100, 101). The parasite ligand for heparan sulphate is PfEMP1, and the DBLα domain of a specific PfEMP1 variant is able to bind heparin (Refs 95, 100, 102).
In a Kenyan field-isolate study, binding of fluorescently labelled heparin was significantly higher in isolates from patients with severe malaria than in isolates from patients with uncomplicated disease (Ref. 32), supporting a role for heparan sulphate in severe malaria. Whether there is genetic variation in the human population affecting glycosaminoglycan synthesis that has the potential to affect parasite binding and malaria susceptibility is unknown.
Other potential cytoadherence receptors
A number of other endothelial receptors for P. falciparum cytoadherence have been described, including fractalkine (Ref. 103), integrin αvβ3 (Ref. 104), fibronectin (Ref. 105), NCAM (Ref. 106) and gC1qR–HABP1–p32 (Ref. 107). In all cases, the P. falciparum ligand for these receptors is unknown and any role the receptors have in severe malaria has not been investigated. It is possible that clinically important receptors for P. falciparum cytoadherence remain to be identified.
Effects of cytoadherence on endothelial cell function
There is mounting evidence that adhesion of IEs to endothelium has adverse effects on endothelial cell function. Apoptosis of endothelial cells following interaction with IEs in vitro has been described (Ref. 108, 109). In addition, endothelial function measured by reactive vasodilation following ischaemic stress is impaired in Indonesian adults with severe malaria (Ref. 110). This endothelial dysfunction is linked to a low nitric oxide (NO) level (an important regulator of endothelial cell function) and a low plasma arginine level (the precursor for NO formation in vivo) (Refs 110, 111). NO has been shown to have an antiadhesive effect on cytoadherence in vitro by preventing upregulation of inducible cytoadherence receptors (Ref. 112). Another possible effect of IE cytoadherence to endothelial cells is to induce a procoagulant state (Ref. 113); however, the importance of coagulation in the pathogenesis of severe malaria is currently unknown.
Molecular mechanisms of IE rosetting
The ability of IEs to bind uninfected erythrocytes to form rosettes (Fig. 2C) (Refs 12, 114) varies between isolates, and high levels of rosetting are significantly associated with severe malaria at several sites across sub-Saharan Africa (Table 1). However, such an association between rosetting and severe disease is not seen in SE Asia (Table 1).
Rosetting parasites cause enhanced microvascular obstruction compared with isogenic cytoadherent nonrosetting parasites (Ref. 115), providing a plausible mechanism for the pathological effect of rosetting. In an ex vivo model, rosettes were disrupted by the high shear forces in the arterial side of the circulation, but in the postcapillary venules the IEs adhered to the endothelium and the uninfected erythrocytes formed rosettes on top of the adherent cells (Ref. 115) (Fig. 2A). Therefore, rosetting and endothelial cell cytoadherence are intimately linked phenotypes, and because some erythrocyte rosetting receptors are also expressed on endothelial cells, they might have a dual role in endothelial cytoadherence and rosetting (Ref. 99) (Fig. 2).
Current evidence suggests that rosetting requires several interactions between parasite ligands (domains of PfEMP1) and receptors on uninfected erythrocytes. Three distinct receptors have been identified: complement receptor 1 (CR1) (Refs 116, 117), heparan-sulphate-like molecules (Ref. 102) and the A or B blood group antigens (Ref. 118). CD36 (present at very low levels on mature erythrocytes) is a receptor for rosetting in one laboratory strain of P. falciparum (Ref. 119), but does not seem to be important in field isolates (Ref. 117). In addition, the PfEMP1 variants that mediate rosetting are predominantly of the group A type (Refs 116, 120, 121), which do not adhere to CD36 (Ref. 44). Serum factors such as IgM natural antibodies might also have a role in rosette formation (Refs 33, 122, 123, 124). One early report that low molecular mass proteins called ‘rosettins’ (Ref. 125) [which are probably identical to rifins, (Ref. 41)] might be parasite ligands for rosetting has not been confirmed. Whether parasite proteins other than PfEMP1 are involved in rosetting is unknown.
Complement receptor 1 (CD35)
Complement receptor 1 (CR1) is a complement regulatory protein found on erythrocytes, a variety of leukocytes and follicular dendritic cells (Ref. 126). The evidence that CR1 is a rosetting receptor comes from experiments showing that CR1-deficient erythrocytes show greatly reduced rosetting, a monoclonal antibody against CR1 reverses rosetting, and soluble recombinant CR1 reverses rosetting in both laboratory strains and field isolates (Refs 116, 117). Rosetting IEs interact with the C3b-binding site on CR1 (Ref. 117). The parasite ligand for CR1-mediated rosetting is PfEMP1, with the most N-terminal domain of PfEMP1 (DBLα) binding to normal but not CR1-deficient red cells (Ref. 116).
Human genetic studies support a direct role for CR1-mediated rosetting in severe malaria. Human erythrocyte CR1 deficiency, which is known to reduce rosetting (Ref. 116), occurs commonly in high malaria-transmission areas of Papua New Guinea, and confers significant protection against severe malaria, reducing the risk by about two thirds (Ref. 127). In Thailand, however, where rosetting is not associated with severe malaria, polymorphisms affecting erythrocyte CR1 levels might promote susceptibility to severe disease (Refs 128, 129), which is proposed to be due to impaired immune complex clearance (Ref. 129). Some of the Knops blood group polymorphisms, which are due to single nucleotide changes in the CR1 gene (Ref. 222), may affect malaria susceptibility (Refs 223, 224). However, further research is needed to examine this possibility.
A or B blood group antigens
The A and B blood group sugars are trisaccharides attached to a variety of erythrocyte glycoproteins and glycolipids, and are also found on platelets, leukocytes and endothelial cells. Every rosetting isolate has a preference for either A or B cells, and forms larger rosettes with erythrocytes of the preferred blood group (Refs 118, 130, 131). Rosetting does occur in group O cells, but the rosettes are smaller and weaker than those formed in A or B cells (Refs 131, 132). PfEMP1 is thought to bind to A and B sugars and a specific variant from a rosetting parasite clone binds to the group A trisaccharide via the DBLα domain (Ref. 95). Human genetic studies support a direct role for A- and B-mediated rosetting in the pathogenesis of severe malaria, because blood group O reduces rosetting in field isolates (Refs 29, 133) and confers significant protection against severe malaria (Refs 133, 134, 135) (Table 1).
Heparan-sulphate-like molecules
These molecules on erythrocytes might act as rosetting receptors, because rosetting is reduced after treating red cells with an enzyme that degrades glycoasaminoglycans (Ref. 102). However, it is unclear whether erythrocytes express true glycosaminoglycans, and the exact nature of the heparan-sulphate-like molecules on erythrocytes is not yet known (Ref. 136). It has been shown that a specific PfEMP1 variant can bind to heparin (Refs 95, 100, 102), and that this interaction contributes to cytoadherence to endothelial cells (Ref. 99). It is unclear whether rosetting and heparan sulphate binding are independent or identical phenotypes, and further research is needed to fully characterise the role of these molecules in erythrocytes and also to determine their role in rosetting and severe malaria.
Molecular mechanisms of P. falciparum adhesion to platelets
P. falciparum IEs have the capacity to bind platelets and form mixed clumps in vitro, in which platelets act as bridges between the IEs (platelet-mediated clumping, Fig 2D) (Ref. 13). If clumps form in vivo, they could contribute to microvascular obstruction. Platelets might also enhance cytoadherence by acting as bridges between endothelial cells and IEs and so target sequestration to endothelial beds not expressing adhesion receptors such as CD36 (Ref. 137). P. falciparum interaction with platelets might also lead to platelet activation and release of inflammatory mediators (Ref. 138). Accumulation of platelets has been reported in the brains of children dying from cerebral malaria (Ref. 139); however, the precise role of platelets in malaria pathology remains unclear. A recent report highlights the fact that platelets can also have antiparasite effects in vivo, and are able to bind to and kill IEs (Ref. 140).
Similarly to rosetting, platelet-mediated clumping varies between parasite isolates, and a significant association of clumping with severe malaria has been reported from Kenya (Ref. 13), Thailand (Ref. 37) and Malawi (Ref. 141). However, a study in Mali found an association with high parasitaemia, but not severe disease (Ref. 142). The above field-isolate studies each used different experimental methods to assess platelet-mediated clumping, and these different conditions have a profound effect on the outcome of the assay (Ref. 143). To clarify the association between clumping and severe malaria, more field-isolate studies will be necessary using standardised techniques.
The molecular mechanisms of the interaction of P. falciparum with platelets are not fully understood; however, three platelet receptors for clumping have been identified: CD36 (Ref. 13), globular C1q receptor (gC1qR/HABP1/p32) (Ref. 107) and P-selectin (Ref. 141). In all cases, the parasite ligands are unknown, although PfEMP1 is a likely candidate molecule.
CD36
CD36 is constitutively expressed on platelets, and was shown to have a role in clumping, because antibodies to CD36 inhibit clumping and CD36-deficient platelets do not support clumping (Ref. 13). However, although most parasite isolates bind to CD36, they do not all form clumps (Ref. 13, 50). Therefore, it seems likely that an interaction with additional platelet receptors or distinct epitopes on CD36 might differentiate parasite isolates that form platelet-mediated clumps from those that bind to CD36 but do not form clumps. As described in Table 1, human genetic evidence does not support an important role for CD36-mediated adhesion in life-threatening malaria in sub-Saharan Africa, whereas further information is needed for Asia.
gC1qR/HABP/p32
gC1qR/HABP/p32 is a multifunctional protein found on activated platelets and endothelial cells, which was recently shown to act as a receptor for clumping and endothelial cell cytoadherence (Ref. 107). Antibodies to gC1qR/HABP/p32 and soluble recombinant protein inhibit clumping in some parasite isolates (Ref. 107). The importance of this protein in clumping of clinical isolates has not yet been widely tested, nor is it known whether polymorphisms that affect binding and malaria susceptibility occur.
P-selectin
P-selectin is expressed on activated platelets and might have an accessory role in clumping, especially in combination with CD36 (Ref. 141); however, this has not yet been widely tested. There is currently very little evidence to determine whether P-selectin binding is important in severe malaria (Table 1).
Molecular mechanisms of IE interaction with cells of the immune system
Many of the receptors involved in adhesion of P. falciparum to endothelial cells, erythrocytes and platelets are also present on subsets of leukocytes, including CD36, ICAM1, NCAM (CD56), gC1qR, CR1 and the A and B blood group antigens. Therefore the potential exists for parasites to bind to leukocytes and promote immune cell activation and parasite clearance, or lead to immunomodulation and immune evasion. A consensus on whether these interactions are beneficial or detrimental to the human host is still lacking.
Of these immune cell receptors, CD36 is the most well studied, yet it remains the most controversial. Binding of IEs to macrophage CD36 leads to phagocytosis without the production of pro-inflammatory cytokines (Ref. 144), suggesting that CD36 binding could lead to parasite clearance and so favour the host. Other evidence shows that parasite adhesion to CD36 is implicated in the impairment of human dendritic cell function and subsequent inhibition of the adaptive immune response (Refs 145, 146), and so could favour the parasite. However, recent evidence suggests that parasite adhesion to CD36 (or any other receptor) is not required for the modulation of dendritic cell function, and instead a high dose of parasitised red blood cells is sufficient to induce inhibition of dendritic cell maturation (Ref. 147).
Adhesion of malaria parasites to cells of the lymphocyte lineage has also been reported. P. falciparum IEs form large clumps with B cells in vitro, and a domain of PfEMP1 is sufficient to induce B cell proliferation through an unknown host receptor (Ref. 148). Furthermore, a direct interaction between IEs and natural killer cells is thought to be required for optimal initiation of the early inflammatory cytokine response to malaria parasites (Ref. 149). The molecular nature of this interaction remains unknown, although there is evidence against the involvement of PfEMP1 binding to CD36 or ICAM1 (Ref. 150). Indeed, PfEMP1 might actually suppress lymphocyte IFN-γ production (Ref. 151). Interestingly, natural killer cells express the newly identified P. falciparum adhesion receptor NCAM (Ref. 106), although the significance of this in malaria host–parasite interactions is currently unknown.
Adhesion of malaria parasites to leukocytes is complex. Many known P. falciparum receptors with a potential immunomodulatory function have yet to be investigated and even for those receptors that have been studied in detail, the physiological significance of the interaction is largely unresolved. The potential dual role of CD36, both in phagocytic clearance of parasites and in immunosuppression of dendritic cells, serves as a warning that the therapeutic disruption of P. falciparum adhesion could have unintended immunological consequences.
Clinical implications and possible therapeutic applications
Potential for antiadhesion therapies
The discoveries outlined above illuminate some of the adhesion interactions between P. falciparum IEs and human cells and open up the possibility of developing therapeutic interventions aimed at blocking or reversing parasite adhesion. There is good evidence that high parasite burdens and sequestration leading to microvascular obstruction are important in the development of life-threatening malaria (Refs 5, 23, 24, 51, 152, 153), although the precise pathogenic mechanisms leading to death and the relative contributions of physical obstruction and metabolic disturbances versus local release of inflammatory mediators and vasoactive compounds continue to be debated (Refs 6, 8, 9, 154, 155, 156). The importance of organ-specific sequestration (e.g. the brain in cerebral malaria) versus the total sequestered load throughout the body, is also controversial (Ref. 157).
On the basis of current knowledge, any therapeutic intervention able to reverse adhesion of IEs has the potential to relieve microvascular obstruction and could be tested as an adjunct to standard antimalarial drugs in severely ill malaria patients. New treatments are urgently needed because the case mortality rate for severe malaria is 15–20% (Ref. 158), even in well-equipped hospitals with intensive care facilities (Ref. 159). Standard antimalarial drugs take up to 24 hours for their parasite-killing effects to occur, and 85% of malaria–related deaths in hospitalised patients occur in the 24 hour period immediately after hospital admission (Ref. 2). The superior results obtained with artemisinine derivatives over quinine as a first-line antimalarial treatment in SE Asian adults with severe malaria (Ref. 160) might be due to the faster action of artemisinine, which acts on all stages of parasite development, whereas quinine only kills schizonts and mature trophozoites (Ref. 161). Even in artemisinine-treated patients, it is plausible that a therapy that immediately relieves microvascular obstruction might be of clinical benefit. It is less clear whether therapies that are able to block further adhesion but are unable to reverse existing adhesion would be useful, and it seems prudent to suggest that development of adhesion-reversing agents should be given priority.
Adhesion-reversing therapies are likely to be drugs, and ideally should be easy and cheap to manufacture, have minimal side effects and good stability (Ref. 162). Drugs that are already in clinical use for other diseases have an advantage in terms of development time and costs, and some of the current candidate antiadhesion therapies fall into this category. Infusions of monoclonal antibodies or peptides might also have the potential to reverse adhesion, although it seems unlikely that such interventions would be cheap enough to be widely used in developing countries with limited resources for health care. Monoclonal antibody or peptide therapies could, however, provide proof of principal to determine whether adhesion-reversal is of clinical benefit, and might be used in intensive care facilities in more affluent countries.
Potential for antiadhesion vaccines
Knowledge of the molecular mechanisms of parasite adhesion could be used to design vaccines aimed at raising antibodies to block adhesion and prevent sequestration. The spleen would remove nonsequestered mature IEs, and so the build-up of high parasite burdens of avidly sequestering parasites would be avoided and severe malaria prevented. The vaccine approach is problematic because of the variability of the parasite adhesion ligand PfEMP1, although initial exploratory studies are underway (Refs 163, 164, 165), and some preliminary data do support the possibility that crossreactive antibodies can be active against a range of isolates (Refs 166, 167). Another problem would be the logistical difficulty and cost of testing such a vaccine, for which reduced malaria mortality would be the primary endpoint. Although challenging, the development of an adhesion-blocking vaccine would be of great value because it would have the potential to reduce deaths from malaria amongst the many people who currently do not have access to treatment in well-equipped hospitals. For this reason, even if adhesion-reversing adjunctive therapies to be used in hospitals can be developed, research into adhesion-blocking vaccines should also proceed, although the possibility that blood-stage vaccines could drive the evolution of parasite virulence should be considered (Ref. 168).
Current antiadhesion drugs under investigation
Drugs that are currently under investigation for their potential as antiadhesion adjunctive therapies are summarised in Table 2.
Table 2.
Current candidate drugs for antiadhesion adjunctive therapy of severe malaria
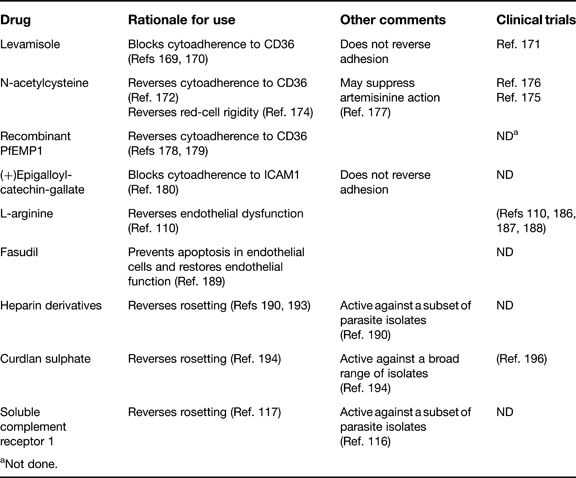
aNot done.
Drugs to inhibit or reverse CD36 binding
Levamisole
Levamisole is an alkaline phosphatase inhibitor that is used as an antihelminth drug in humans. Recent research showed that endothelial CD36 is constitutively phosphorylated and that interaction with IEs leads to phosphatase activity to remove the phosphate group at Thr92 of CD36 (Refs 169, 170). Dephosphorylated CD36 has a higher affinity for IEs under flow conditions than phosphorylated CD36 does, and inhibition of phosphatase activity using levamisole leads to a twofold reduction in IE binding in vitro (Ref. 170). A randomised clinical trial of Thai patients with uncomplicated malaria (12 treated with levamisole and 9 controls) showed that levamisole, used as an adjunctive therapy with quinine and doxycycline, resulted in increased numbers of early–mid trophozoites in the peripheral blood (Ref. 171). It was suggested that levamisole prevented the sequestration of these parasites as they matured from ring stages following treatment. There was no evidence for a reversal of adhesion of existing mature sequestered forms, and schizonts were not seen in the peripheral blood (although it is possible that schizonts were released but were immediately cleared by the spleen). Further trials are awaited to determine whether levamisole will be of clinical benefit.
N-acetylcysteine
N-acetylcysteine (NAC) is an antioxidant drug that is widely used in humans for the treatment of paracetamol (acetaminophen) overdose. In vitro studies showed that IE binding to CD36 was reversed by 72–83% in the presence of NAC (Ref. 172). Further rationale for the use of NAC comes from the suggestion that NAC can reverse the erythrocyte rigidity, which is a characteristic feature of severe malaria (Refs 173, 174) and might be a contributory factor to microvascular obstruction and pathogenesis (Ref. 6). A pilot study of NAC in Thai severe malaria patients found that serum lactate levels (a strong predictor of mortality in severe malaria) normalised significantly faster in 15 patients treated with NAC plus quinine, compared with 15 controls treated with quinine alone (Ref. 175). Another study showed that NAC was safe for use in Thai patients treated with artesunate (Ref. 176). Despite these encouraging preliminary results, a large randomised clinical trial of NAC as an adjunctive treatment for severe malaria, using mortality as an endpoint, has not been reported. Indeed, a recent study showed that NAC can interfere with the action of artesunate during the first 6 hours of co-incubation with P. falciparum in vitro, and cautioned against the use of NAC as an adjunctive treatment with artemisinine derivatives (Ref. 177). The interaction between the two drugs is thought to occur because the parasitocidal effect of artemisinine takes place via oxidative damage, which may be inhibited by the antioxidant effect of NAC. The possibility of antagonism between NAC and the most effective current antimalarial drug might mean that further tests of NAC as an adjunctive therapy will be hard to justify.
Recombinant PfEMP1 peptide
A peptide corresponding to the minimal CD36-binding region of PfEMP1 from the Malayan Camp parasite strain (Refs 65, 178) has been shown to inhibit adhesion of Thai field isolates to an endothelial cell line in vitro under flow conditions, and to reverse adhesion to microvessels in vivo in a human skin graft in SCID mice (Ref. 179). This peptide might have potential as an adhesion-reversing therapy, although it is unclear if it would be practical for widescale use.
Drugs to inhibit ICAM1 binding
( + ) Epigalloyl-catechin-gallate
The best example to date of the rational design of a compound to block cytoadherence comes from Dormeyer and co-workers (Ref. 180), who used the crystal structure of ICAM1 to identify a compound to block parasite binding to ICAM1 in vitro. The team used in silico screening to identify compounds that mimicked the region of ICAM1 that is involved in IE binding (Ref. 76). Thirty-six candidates were identified in an initial screen, and these were then tested in vitro for the ability to inhibit IE adhesion to ICAM1 under flow conditions. One compound, (+)epigalloyl-catechin-gallate [(+)EGCG], was identified that inhibited binding by 50% at micromolar concentrations (Ref. 180). However, this compound did not reverse adhesion. EGCG is a naturally occurring polyphenol compound that is a constituent of green tea and is currently being investigated for its anticancer, anti-inflammatory and anti-infective properties (Refs 181, 182, 183). Substantial further work will be required to translate these promising preliminary findings into a useable adjunctive therapy, and one major question that will need to be addressed is whether the evidence for an involvement of ICAM1 binding in severe malaria is currently strong enough to justify the resources required for drug development. Whether this compound proceeds towards the clinic or not, the study by Dormeyer and colleagues (Ref. 180) remains an excellent illustration of how detailed structural and molecular information can be used for rational drug design in malaria.
Drugs to improve endothelial cell function
L-arginine
L-arginine is the substrate for the synthesis of NO by NO synthase. It is given intravenously and has been used safely in humans for many years, both in endocrine system investigations and as a potential therapeutic agent in cardiovascular diseases. Following on from studies in patients showing low NO production and low plasma arginine levels in severe malaria (Refs 184, 185), and studies in vitro showing that NO has an antiadhesive effect (Ref. 112), researchers have begun to investigate the potential for L-arginine to be used as an adjunctive therapy for severe malaria (Refs 186, 187, 188). They have shown that L-arginine improves endothelial function in adult patients with moderately severe malaria (Ref. 110). Whether this improvement in endothelial function will translate into clinical benefit awaits further trials.
Fasudil
Following on from in vitro studies showing that cytoadherence of IEs induces apoptosis of endothelial cells via induction of the Rho kinase pathway, it has been suggested that the Rho-kinase inhibitor fasudil could be a useful adjunctive therapy (Ref. 189). Fasudil reduced P. falciparum-induced endothelial cell apoptosis in vitro and helped restore endothelial barrier integrity (Ref. 189). Further studies are required to determine the pathophysiological significance of endothelial cell apoptosis in severe malaria, and the therapeutic potential of fasudil.
Drugs to reverse rosetting
Sulphated glycoconjugate compounds
A variety of sulphated glycoconjugate compounds are known to reverse P. falciparum rosetting (Refs 190, 191, 192); however, many of these compounds also have significant anticoagulant properties that could cause side-effects. A heparin derivative with reduced anticoagulant effects has some therapeutic potential (Ref. 193), but the rosette-disrupting effect of heparin and its derivatives is strain-specific (effective on about 30–50% of rosetting isolates) (Ref. 190), which may limit its usefulness.
Another sulfated glycoconjugate that had broader rosette-disrupting activity against a wide range of parasite isolates is curdlan sulphate (Ref. 194) – a drug initially developed as an AIDS therapy (Ref. 195). Curdlan sulphate has been shown to be safe in adult patients with malaria in SE Asia (Ref. 196). However, this is not the most appropriate target population for testing the anti-rosetting effects of the drug and it needs be tested as an adjunctive therapy for severe malaria in African children.
Soluble CR1
Recombinant soluble CR1 (sCR1) is being developed as a drug in humans for ischaemia-reperfusion injuries (such as infarcts and strokes) (Refs 197, 198) or immune-mediated haemolysis and transfusion reactions (Ref. 199). sCR1 disrupts rosettes in some but not all P. falciparum rosetting isolates (Ref. 117); therefore, it is possible that sCR1 could be of benefit as an adjunctive therapy for severe malaria. However, further information is required on the ability of sCR1 to disrupt rosettes in a broad range of clinical isolates.
Drugs to inhibit or reverse platelet binding
Owing to the involvement of platelets in important pathological conditions such as thrombosis and atherosclerotic vascular diseases, there are numerous antiplatelet therapies available for human use [e.g. acetylsalicylic acid (aspirin), dipyridamole, cilostazol, ticlopidin, clopidogrel, abciximab (ReoPro), eptifibatide and tirofiban] (Refs 200, 201). The mechanisms of action of these drugs are well understood; however, because so little is known about the mechanisms and consequences of interactions between P. falciparum and platelets, it is difficult to predict whether any of these compounds might be beneficial in severe malaria. A recent report suggests that some antiplatelet drugs, such as aspirin, could actually be detrimental in malaria, by preventing the parasite-killing effects of platelets (Ref. 140). Further research is needed to clarify the role of platelets in malaria pathophysiology to take advantage of the therapeutic options in this area.
Prospects and outstanding research questions
Assessing which adhesion phenotypes contribute to severe malaria
Currently, our incomplete understanding of the adhesion mechanisms important in the most life-threatening clinical forms of malaria is a major obstacle to the development of adjunctive therapies. Given the huge amount of time, effort and money required for drug development, only those adhesion interactions whose involvement in severe malaria is backed by strong scientific evidence are likely to succeed in raising funding for drug development and clinical trials. The logistic and technical difficulties of carrying out field-isolate adhesion studies might be one reason for the neglect of this important area. For future studies, flow-based systems examining IE binding to endothelial cell lines probably provide the most physiologically relevant approach, and allow investigation of rolling as well as static adhesion, and synergism between receptors that might be important in vivo (Ref. 52).
Another difficulty is the varying definition of severe malaria used in many studies and the choice of a suitable control group for comparison with severe malaria patients (Ref. 202). ‘Severe malaria’ is often considered to be a single disease entity, whereas it is possible that different adhesion phenotypes contribute to distinct clinical syndromes, such as cerebral malaria (unrousable coma), respiratory distress (difficulty breathing) and severe malarial anaemia (haemoglobin levels <5 g/dl). Ideally, studies of parasite adhesion in relation to malaria severity should use clearly defined subtypes of severe disease, although this could be difficult because of overlap in syndromes in many patients, and small numbers in some categories.
Another problem with studies of parasite adhesion phenotypes and severe malaria is the uncertainty as to whether the phenotype of the parasites being tested (derived as ring stages from peripheral blood and matured in vitro for 12–24 hours) truly reflects the phenotype of the sequestered mass of parasites that are not accessible for experimentation, although recent data suggest no substantial genetic differences between the two populations (Ref. 203).
Possibility of geographic variation in parasite adhesion phenotypes causing severe malaria
As described above, there are clear differences in parasite phenotypes linked to severe disease in studies from low-transmission areas such as SE Asia (high multiplication rate and nonselective invasion) compared with moderate–high transmission areas such as sub-Saharan Africa (rosetting). How might these regional differences be explained, and could differences in levels of host immunity be an important factor?
One consistent feature of severe malaria throughout the world is that mortality is linked to markers of metabolic acidosis, such as base excess (Ref. 3) or hyperlactataemia (Ref. 204). This metabolic acidosis is thought to result from microvascular obstruction because of sequestration of large numbers of IEs. We hypothesise that in nonimmune individuals in low-transmission areas (e.g. SE Asia), any parasite isolate that invades red cells efficiently and sequesters adequately can reach a high parasite burden and cause severe disease before the host's immune system mounts a specific antibody response to variant surface antigens to remove IEs. In this case, no specific adhesion phenotype causes severe malaria, but all commonly observed adhesion phenotypes, such as binding of CD36 and ICAM1, are likely to contribute.
In sub-Saharan Africa, however, where individuals are exposed to multiple P. falciparum infections, parasite growth in the human host might occur in the presence of immune responses that reduce parasite proliferation. In particular, initial infections in infants might be modified by in utero exposure to plasmodial antigens or by maternal antibody acquired through the placenta or via breast milk, and entirely immunologically naive individuals in relation to Plasmodium infection may be rare. In this case, it is possible that only parasite isolates expressing adhesion phenotypes that are most effective at promoting parasite growth and survival in the face of host immunity are able to expand rapidly enough to reach high parasite burdens and cause severe disease, before specific antibodies to variant surface antigens develop. Rosetting, for example, is associated with high parasitaemia in vivo (Refs 205, 206), and might act either by promoting red cell invasion (not supported by recent evidence) (Refs 207, 208) or as an immune-evasion mechanism that reduces parasite clearance (R.A.C. and J.A.R., unpublished results). Other adhesion phenotypes might exist that enhance parasite survival in partially immune hosts. In this scenario, specific adhesion phenotypes, such as rosetting, do contribute to severe malaria.
Implications of geographic variation for antiadhesion therapies
The above argument is speculative; however, it does fit with existing data. If such regional differences do exist (linked to malaria transmission intensity and host immunity) this has major therapeutic implications. For example, a rosette-disrupting drug might be of clinical benefit in sub-Saharan Africa but would not be an appropriate treatment for severe malaria in SE Asia. Conversely, a drug that reverses CD36 binding might be more effective in SE Asia. Geographical variation needs to be considered carefully and merits further investigation to ensure that potentially life-saving drugs are tested on the most appropriate patient population. Furthermore, it should not be assumed that an antiadhesion therapy that works in a nonimmune population will be effective in a moderate–high transmission area, and vice versa.
Possible problems with antiadhesion therapies
One unanswered question in the approach of reversing adhesion is whether the release of large numbers of mature IEs into the circulation could be damaging. Would the spleen be able to cope with removing millions of IEs, or could it lead to potentially catastrophic side effects such as splenic rupture? In a saimiri monkey model of falciparum malaria, immune serum was used to reverse sequestration, without any damaging effect to the animals, supporting the safety of this approach (Ref. 209).
Another problem is the lack of an animal model that truly reflects the pathophysiology of severe falciparum malaria in humans, because it is not currently possible to test antiadhesion therapies in a meaningful way (none of the primate or rodent models develop clinical and pathological features similar to those in humans). Attempts have been made to develop animal models of sequestration (Refs 210, 211); however, their relevance to human disease mechanisms is unclear and unproven. Human vasculature grafted onto immunodeficient mice has been used successfully to investigate sequestration and antiadhesion drugs (Refs 55, 179). Further research in this area would clearly be of benefit, and the development of humanised animal models and transgenic parasites to enable study of specific human: P. falciparum receptor–ligand interactions might be one way forward (Ref. 212).
A further potential problem with adhesion-reversing therapies is the possibility that many severe malaria patients might be too far down the ‘pathogenesis pathway’ by the time they reach hospital to derive benefit from treatment. Reversing IE adhesion in moribund patients could amount to shutting the stable door after the horse has bolted. However, this cannot be predicted in advance and only carefully designed and adequately powered clinical trials will provide the answer to whether adhesion-reversing therapies can save lives.
Conclusion
Despite the above problems, the pressing need for novel adjunctive therapies to lower the mortality rate from severe malaria argues strongly for further research in this area. Antiadhesion therapies have great potential for saving lives (Ref. 213) and further research to clarify the adhesion phenotypes causing severe malaria and development of interventions to reverse adhesion should be a priority for malaria research in the next decade.
Acknowledgements
J.A.R. and M.A. are funded by a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science to J.A.R. (grant no. 084226) and A.C. is funded by a Wellcome 4 year PhD studentship. R.C. is funded by a BBSRC studentship. We are grateful to Clare Fennell and Ash Ghumra for reading the manuscript, and to the reviewers for helpful comments.
References
- 1.Snow R.W.. et al. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marsh K.. et al. Indicators of life-threatening malaria in African children. New England Journal of Medicine. 1995;332:1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
- 3.Dondorp A.M.. et al. The relationship between age and the manifestations of and mortality associated with severe malaria. Clinical Infectious Diseases. 2008;47:151–157. doi: 10.1086/589287. [DOI] [PubMed] [Google Scholar]
- 4.Miller L.H.. et al. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 5.Dondorp A.M.. et al. Direct in vivo assessment of microcirculatory dysfunction in severe falciparum malaria. Journal of Infectious Diseases. 2008;197:79–84. doi: 10.1086/523762. [DOI] [PubMed] [Google Scholar]
- 6.Dondorp A.M., Pongponratn E., White N.J.. Reduced microcirculatory flow in severe falciparum malaria: pathophysiology and electron-microscopic pathology. Acta Tropica. 2004;89:309–317. doi: 10.1016/j.actatropica.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Planche T., Krishna S.. Severe malaria: metabolic complications. Current Molecular Medicine. 2006;6:141–153. doi: 10.2174/156652406776055177. [DOI] [PubMed] [Google Scholar]
- 8.Schofield L.. Intravascular infiltrates and organ-specific inflammation in malaria pathogenesis. Immunology and Cell Biology. 2007;85:130–137. doi: 10.1038/sj.icb.7100040. [DOI] [PubMed] [Google Scholar]
- 9.van der Heyde H.C.. et al. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends in Parasitology. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Mebius R.E., Kraal G.. Structure and function of the spleen. Nature Reviews Immunology. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 11.Udeinya I.J.. et al. Falciparum malaria-infected erythrocytes specifically bind to cultured human endothelial cells. Science. 1981;213:555–557. doi: 10.1126/science.7017935. [DOI] [PubMed] [Google Scholar]
- 12.Udomsangpetch R.. et al. Plasmodium falciparum-infected erythrocytes form spontaneous erythrocyte rosettes. Journal of Experimental Medicine. 1989;169:1835–1840. doi: 10.1084/jem.169.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pain A.. et al. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1805–1810. doi: 10.1073/pnas.98.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bray R.S., Sinden R.E.. The sequestration of Plasmodium falciparum infected erythrocytes in the placenta. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1979;73:716–719. doi: 10.1016/0035-9203(79)90028-2. [DOI] [PubMed] [Google Scholar]
- 15.Rogerson S.J.. et al. Malaria in pregnancy: pathogenesis and immunity. Lancet Infectious Diseases. 2007;7:105–117. doi: 10.1016/S1473-3099(07)70022-1. [DOI] [PubMed] [Google Scholar]
- 16.Duffy P.E., Fried M.. Malaria in the pregnant woman. Current Topics in Microbiology and Immunology. 2005;295:169–200. doi: 10.1007/3-540-29088-5_7. [DOI] [PubMed] [Google Scholar]
- 17.Rowe J.A., Kyes S.A.. The role of Plasmodium falciparum var genes in malaria in pregnancy. Molecular Microbiology. 2004;53:1011–1019. doi: 10.1111/j.1365-2958.2004.04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White N.J.. Clinical and pathological aspects of severe malaria. Acta Leidensia. 1987;56:27–47. [PubMed] [Google Scholar]
- 19.Erunkulu O.A.. et al. Severe malaria in Gambian children is not due to lack of previous exposure to malaria. Clinical and Experimental Immunology. 1992;89:296–300. doi: 10.1111/j.1365-2249.1992.tb06948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chotivanich K.. et al. Parasite multiplication potential and the severity of Falciparum malaria. Journal of Infectious Diseases. 2000;181:1206–1209. doi: 10.1086/315353. [DOI] [PubMed] [Google Scholar]
- 21.Simpson J.A.. et al. Red cell selectivity in malaria: a study of multiple-infected erythrocytes. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:165–168. doi: 10.1016/s0035-9203(99)90295-x. [DOI] [PubMed] [Google Scholar]
- 22.Deans A.M.. et al. Low multiplication rates of African Plasmodium falciparum isolates and lack of association of multiplication rate and red blood cell selectivity with malaria virulence. American Journal of Tropical Medicine and Hygiene. 2006;74:554–563. [PMC free article] [PubMed] [Google Scholar]
- 23.Field J.W., Niven J.C.. A note on prognosis in relation to parasite counts in acute subtertian malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1937;30:569–574. [Google Scholar]
- 24.Dondorp A.M.. et al. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Medicine. 2005;2:e204. doi: 10.1371/journal.pmed.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyke K.E.. et al. Association of intraleukocytic Plasmodium falciparum malaria pigment with disease severity, clinical manifestations, and prognosis in severe malaria. American Journal of Tropical Medicine and Hygiene. 2003;69:253–259. [PubMed] [Google Scholar]
- 26.Carlson J.. et al. Human cerebral malaria: association with erythrocyte rosetting and lack of anti-rosetting antibodies. Lancet. 1990;336:1457–1460. doi: 10.1016/0140-6736(90)93174-n. [DOI] [PubMed] [Google Scholar]
- 27.Treutiger C.J.. et al. Rosette formation in Plasmodium falciparum isolates and anti-rosette activity of sera from Gambians with cerebral or uncomplicated malaria. American Journal of Tropical Medicine and Hygiene. 1992;46:503–510. doi: 10.4269/ajtmh.1992.46.503. [DOI] [PubMed] [Google Scholar]
- 28.Ringwald P.. et al. Parasite virulence factors during falciparum malaria: rosetting, cytoadherence, and modulation of cytoadherence by cytokines. Infection and Immunity. 1993;61:5198–5204. doi: 10.1128/iai.61.12.5198-5204.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowe A.. et al. Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infection and Immunity. 1995;63:2323–2326. doi: 10.1128/iai.63.6.2323-2326.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newbold C.. et al. Receptor-specific adhesion and clinical disease in Plasmodium falciparum. American Journal of Tropical Medicine and Hygiene. 1997;57:389–398. doi: 10.4269/ajtmh.1997.57.389. [DOI] [PubMed] [Google Scholar]
- 31.Kun J.F.. et al. Merozoite surface antigen 1 and 2 genotypes and rosetting of Plasmodium falciparum in severe and mild malaria in Lambarene, Gabon. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1998;92:110–114. doi: 10.1016/s0035-9203(98)90979-8. [DOI] [PubMed] [Google Scholar]
- 32.Heddini A.. et al. Fresh isolates from children with severe Plasmodium falciparum malaria bind to multiple receptors. Infection and Immunity. 2001;69:5849–5856. doi: 10.1128/IAI.69.9.5849-5856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowe J.A.. et al. Nonimmune IgM, but not IgG binds to the surface of Plasmodium falciparum-infected erythrocytes and correlates with rosetting and severe malaria. American Journal of Tropical Medicine and Hygiene. 2002;66:692–699. doi: 10.4269/ajtmh.2002.66.692. [DOI] [PubMed] [Google Scholar]
- 34.Ho M.. et al. Rosette formation of Plasmodium falciparum-infected erythrocytes from patients with acute malaria. Infection and Immunity. 1991;59:2135–2139. doi: 10.1128/iai.59.6.2135-2139.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Udomsangpetch R.. et al. Receptor specificity of clinical Plasmodium falciparum isolates: nonadherence to cell-bound E-selectin and vascular cell adhesion molecule-1. Blood. 1996;88:2754–2760. [PubMed] [Google Scholar]
- 36.Angkasekwinai P., Looareesuwan S., Chaiyaroj S.C.. Lack of significant association between rosette formation and parasitized erythrocyte adherence to purified CD36. Southeast Asian Journal of Tropical Medicine and Public Health. 1998;29:41–45. [PubMed] [Google Scholar]
- 37.Chotivanich K.. et al. Platelet-induced autoagglutination of Plasmodium falciparum-infected red blood cells and disease severity in Thailand. Journal of Infectious Diseases. 2004;189:1052–1055. doi: 10.1086/381900. [DOI] [PubMed] [Google Scholar]
- 38.Baruch D.I.. et al. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitised human erythrocytes. Cell. 1995;82:77–87. doi: 10.1016/0092-8674(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 39.Smith J.D.. et al. Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. 1995;82:101–110. doi: 10.1016/0092-8674(95)90056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su X.-Z.. et al. A large and diverse family gene family (var) encodes 200-350 kD proteins implicated in the antigenic variation and cytoadherence of Plasmodium falciparum-infected erythocytes. Cell. 1995;82:89–99. doi: 10.1016/0092-8674(95)90055-1. [DOI] [PubMed] [Google Scholar]
- 41.Kyes S.A.. et al. Rifins: A second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9333–9338. doi: 10.1073/pnas.96.16.9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blythe J.E.. et al. Plasmodium falciparum STEVOR proteins are highly expressed in patient isolates and located in the surface membranes of infected red blood cells and the apical tips of merozoites. Infection and Immunity. 2008;76:3329–3336. doi: 10.1128/IAI.01460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith J.D.. et al. Classification of adhesive domains in the Plasmodium falciparum erythrocyte membrane protein 1 family. Molecular and Biochemical Parasitology. 2000;110:293–310. doi: 10.1016/s0166-6851(00)00279-6. [DOI] [PubMed] [Google Scholar]
- 44.Robinson B.A., Welch T.L., Smith J.D.. Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Molecular Microbiology. 2003;47:1265–1278. doi: 10.1046/j.1365-2958.2003.03378.x. [DOI] [PubMed] [Google Scholar]
- 45.Kaestli M.. et al. Virulence of malaria is associated with differential expression of Plasmodium falciparum var gene subgroups in a case-control study. Journal of Infectious Diseases. 2006;193:1567–1574. doi: 10.1086/503776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kyriacou H.M.. et al. Differential var gene transcription in Plasmodium falciparum isolates from patients with cerebral malaria compared to hyperparasitaemia. Molecular and Biochemical Parasitology. 2006;150:211–218. doi: 10.1016/j.molbiopara.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kyes S.A., Kraemer S.M., Smith J.D.. Antigenic Variation in Plasmodium falciparum: Gene Organization and Regulation of the var Multigene Family. Eukaryotic Cell. 2007;6:1511–1520. doi: 10.1128/EC.00173-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraemer S.M., Smith J.D.. A family affair: var genes, PfEMP1 binding, and malaria disease. Current Opinion in Microbiology. 2006;9:374–380. doi: 10.1016/j.mib.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Smith J.D.. et al. Decoding the language of var genes and Plasmodium falciparum sequestration. Trends in Parasitology. 2001;17:538–545. doi: 10.1016/s1471-4922(01)02079-7. [DOI] [PubMed] [Google Scholar]
- 50.Roberts D.J.. et al. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature. 1992;357:689–692. doi: 10.1038/357689a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchiafava E., Bignami A. On Summer-Autumnal Fever. The New Sydenham Society; London: 1894. [Google Scholar]
- 52.Yipp B.G.. et al. Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum-infected erythrocytes to microvascular endothelial cells under flow. Blood. 2000;96:2292–2298. [PubMed] [Google Scholar]
- 53.McCormick C.J.. et al. Intercellular adhesion molecule-1 and CD36 synergize to mediate adherence of Plasmodium falciparum-infected erythrocytes to cultured human microvascular endothelial cells. Journal of Clinical Investigation. 1997;100:2521–2529. doi: 10.1172/JCI119794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yipp B.G.. et al. Differential roles of CD36, ICAM-1, and P-selectin in Plasmodium falciparum cytoadherence in vivo. Microcirculation. 2007;14:593–602. doi: 10.1080/10739680701404705. [DOI] [PubMed] [Google Scholar]
- 55.Ho M.. et al. Visualization of Plasmodium falciparum-endothelium interactions in human microvasculature: mimicry of leukocyte recruitment. Journal of Experimental Medicine. 2000;192:1205–1211. doi: 10.1084/jem.192.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Udomsangpetch R.. et al. Promiscuity of clinical Plasmodium falciparum isolates for multiple adhesion molecules under flow conditions. Journal of Immunology. 1997;158:4358–4364. [PubMed] [Google Scholar]
- 57.Oquendo P.. et al. CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell. 1989;58:95–101. doi: 10.1016/0092-8674(89)90406-6. [DOI] [PubMed] [Google Scholar]
- 58.Barnwell J.W.. et al. A human 88-kD membrane glycoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum-infected erythrocytes. Journal of Clinical Investigation. 1989;84:765–772. doi: 10.1172/JCI114234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greenwalt D.E.. et al. Membrane glycoprotein CD36: a review of its roles in adherence, signal transduction and transfusion medicine. Blood. 1992;80:1105–1115. [PubMed] [Google Scholar]
- 60.Daviet L.. et al. Characterization of two vaccinia CD36 recombinant-virus-generated monoclonal antibodies (10/5, 13/10): effects on malarial cytoadherence and platelet functions. European Journal of Biochemistry. 1997;243:344–349. doi: 10.1111/j.1432-1033.1997.0344a.x. [DOI] [PubMed] [Google Scholar]
- 61.Baruch D.I.. et al. CD36 peptides that block cytoadherence define the CD36 binding region for Plasmodium falciparum-infected erythrocytes. Blood. 1999;94:2121–2127. [PubMed] [Google Scholar]
- 62.Baruch D.I.. et al. Plasmodium falciparum erythocyte membrane protein 1 is a parasitised erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:3497–3502. doi: 10.1073/pnas.93.8.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller L.H.. et al. Definition of the minimal domain of CIDR1alpha of Plasmodium falciparum PfEMP1 for binding CD36. Molecular and Biochemical Parasitology. 2002;120:321–323. doi: 10.1016/s0166-6851(02)00011-7. [DOI] [PubMed] [Google Scholar]
- 64.Smith J.D.. et al. Analysis of adhesive domains from the A4VAR Plasmodium falciparum erythrocyte membrane protein-1 identifies a CD36 binding domain. Molecular and Biochemical Parasitology. 1998;97:133–148. doi: 10.1016/s0166-6851(98)00145-5. [DOI] [PubMed] [Google Scholar]
- 65.Baruch D.I.. et al. Identification of a region of PfEMP1 that mediates adherence of Plasmodium falciparum infected erythrocytes to CD36: conserved function with variant sequence. Blood. 1997;90:3766–3775. [PubMed] [Google Scholar]
- 66.Klein M.M.. et al. The cysteine-rich interdomain region from the highly variable Plasmodium falciparum erythrocyte membrane protein-1 exhibits a conserved structure. PLoS Pathogens. 2008;4:e1000147. doi: 10.1371/journal.ppat.1000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serghides L.. et al. CD36 and malaria: friends or foes? Trends in Parasitology. 2003;19:461–469. doi: 10.1016/j.pt.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 68.Marsh K.. et al. Plasmodium falciparum: the behavior of clinical isolates in an in vitro model of infected red blood cell sequestration. Experimental Parasitology. 1988;65:202–208. doi: 10.1016/0014-4894(88)90123-3. [DOI] [PubMed] [Google Scholar]
- 69.Rogerson S.J.. et al. Cytoadherence characteristics of Plasmodium falciparum-infected erythrocytes from Malawian children with severe and uncomplicated malaria. American Journal of Tropical Medicine and Hygiene. 1999;61:467–472. doi: 10.4269/ajtmh.1999.61.467. [DOI] [PubMed] [Google Scholar]
- 70.Heddini A.. et al. Binding of Plasmodium falciparum-infected erythrocytes to soluble platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): frequent recognition by clinical isolates. American Journal of Tropical Medicine and Hygiene. 2001;65:47–51. doi: 10.4269/ajtmh.2001.65.47. [DOI] [PubMed] [Google Scholar]
- 71.Ho M.. et al. Clinical correlates of in vitro Plasmodium falciparum cytoadherence. Infection and Immunity. 1991;59:873–878. doi: 10.1128/iai.59.3.873-878.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ockenhouse C.F.. et al. Molecular basis of sequestration in severe and uncomplicated Plasmodium falciparum malaria: differential adhesion of infected erythrocytes to CD36 and ICAM-1. Journal of Infection Dis. 1991;164:163–169. doi: 10.1093/infdis/164.1.163. [DOI] [PubMed] [Google Scholar]
- 73.Omi K.. et al. CD36 polymorphism is associated with protection from cerebral malaria. American Journal of Human Genetics. 2003;72:364–374. doi: 10.1086/346091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berendt A.R.. et al. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341:57–59. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- 75.Chakravorty S.J., Craig A.. The role of ICAM-1 in Plasmodium falciparum cytoadherence. European Journal of Cell Biology. 2005;84:15–27. doi: 10.1016/j.ejcb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 76.Berendt A.R.. et al. The binding site on ICAM-1 for Plasmodium falciparum-infected erythrocytes overlaps, but is distinct from, the LFA-1-binding site. Cell. 1992;68:71–81. doi: 10.1016/0092-8674(92)90207-s. [DOI] [PubMed] [Google Scholar]
- 77.Ockenhouse C.F.. et al. Plasmodium falciparum-infected erythrocytes bind ICAM-1 at a site distinct from LFA-1, Mac-1, and human rhinovirus. Cell. 1992;68:63–69. doi: 10.1016/0092-8674(92)90206-r. [DOI] [PubMed] [Google Scholar]
- 78.Tse M.T.. et al. Divergent binding sites on intercellular adhesion molecule-1 (ICAM-1) for variant Plasmodium falciparum isolates. Molecular Microbiology. 2004;51:1039–1049. doi: 10.1046/j.1365-2958.2003.03895.x. [DOI] [PubMed] [Google Scholar]
- 79.Smith J.D.. et al. Identification of a Plasmodium falciparum intercellular adhesion molecule-1 binding domain: a parasite adhesion trait implicated in cerebral malaria. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1766–1771. doi: 10.1073/pnas.040545897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chattopadhyay R.. et al. Molecular analysis of the cytoadherence phenotype of a Plasmodium falciparum field isolate that binds intercellular adhesion molecule-1. Molecular and Biochemical Parasitology. 2004;133:255–265. doi: 10.1016/j.molbiopara.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 81.Springer A.L.. et al. Functional interdependence of the DBLbeta domain and c2 region for binding of the Plasmodium falciparum variant antigen to ICAM-1. Molecular and Biochemical Parasitology. 2004;137:55–64. doi: 10.1016/j.molbiopara.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 82.Howell D.P.. et al. Mapping a common interaction site used by Plasmodium falciparum Duffy binding-like domains to bind diverse host receptors. Molecular Microbiology. 2008;67:78–87. doi: 10.1111/j.1365-2958.2007.06019.x. [DOI] [PubMed] [Google Scholar]
- 83.Craig A.. et al. A functional analysis of a natural variant of intercellular adhesion molecule-1 (ICAM-1Kilifi) Human Molecular Genetics. 2000;9:525–530. doi: 10.1093/hmg/9.4.525. [DOI] [PubMed] [Google Scholar]
- 84.Fernandez-Reyes D.. et al. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Human Molecular Genetics. 1997;6:1357–1360. doi: 10.1093/hmg/6.8.1357. [DOI] [PubMed] [Google Scholar]
- 85.Jenkins N.E.. et al. A polymorphism of intercellular adhesion molecule-1 is associated with a reduced incidence of nonmalarial febrile illness in Kenyan children. Clinical Infectious Diseases. 2005;41:1817–1819. doi: 10.1086/498156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turner G.D.H.. et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. American Journal of Pathology. 1994;145:1057–1069. [PMC free article] [PubMed] [Google Scholar]
- 87.Kwiatkowski D.. et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336:1201–1204. doi: 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 88.Ho M.. et al. Characterization of Plasmodium falciparum-infected erythrocyte and P-selectin interaction under flow conditions. Blood. 1998;91:4803–4809. [PubMed] [Google Scholar]
- 89.Senczuk A.M.. et al. Plasmodium falciparum erythrocyte membrane protein 1 functions as a ligand for P-selectin. Blood. 2001;98:3132–3135. doi: 10.1182/blood.v98.10.3132. [DOI] [PubMed] [Google Scholar]
- 90.Roberts D.D.. et al. Thrombospondin binds falciparum malaria parasitized erythrocytes and may mediate cytoadherence. Nature. 1985;318:64–66. doi: 10.1038/318064a0. [DOI] [PubMed] [Google Scholar]
- 91.Rock E.P.. et al. Thrombospondin mediates the cytoadherence of Plasmodium falciparum-infected red cells to vascular endothelium in shear flow conditions. Blood. 1988;71:71–75. [PubMed] [Google Scholar]
- 92.Eda S., Lawler J., Sherman I.W.. Plasmodium falciparum-infected erythrocyte adhesion to the type 3 repeat domain of thrombospondin-1 is mediated by a modified band 3 protein. Molecular and Biochemical Parasitology. 1999;100:195–205. doi: 10.1016/s0166-6851(99)00058-4. [DOI] [PubMed] [Google Scholar]
- 93.Eda S., Sherman I.W.. Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cellular Physiology and Biochemistry. 2002;12:373–384. doi: 10.1159/000067908. [DOI] [PubMed] [Google Scholar]
- 94.Treutiger C.J.. et al. PECAM-1/CD31, an endothelial receptor for binding Plasmodium falciparum-infected erythrocytes. Nature Medicine. 1997;3:1405–1408. doi: 10.1038/nm1297-1405. [DOI] [PubMed] [Google Scholar]
- 95.Chen Q.. et al. The semiconserved head structure of Plasmodium falciparum erythrocyte membrane protein 1 mediates binding to multiple independent host receptors. Journal of Experimental Medicine. 2000;192:1–10. doi: 10.1084/jem.192.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Casals-Pascual C.. et al. Short report: codon 125 polymorphism of CD31 and susceptibility to malaria. American Journal of Tropical Medicine and Hygiene. 2001;65:736–737. doi: 10.4269/ajtmh.2001.65.736. [DOI] [PubMed] [Google Scholar]
- 97.Kikuchi M.. et al. Association of adhesion molecule PECAM-1/CD31 polymorphism with susceptibility to cerebral malaria in Thais. Parasitology International. 2001;50:235–239. doi: 10.1016/s1383-5769(01)00082-4. [DOI] [PubMed] [Google Scholar]
- 98.Ockenhouse C.F.. et al. Human vascular endothelial cell adhesion receptors for Plasmodium falciparum-infected erythrocytes: roles for endothelial leukocyte adhesion molecule 1 and vascular cell adhesion molecule 1. Journal of Experimental Medicine. 1992;176:1183–1189. doi: 10.1084/jem.176.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vogt A.M.. et al. Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the DBL1alpha domain of PfEMP1. Blood. 2003;101:2405–2411. doi: 10.1182/blood-2002-07-2016. [DOI] [PubMed] [Google Scholar]
- 100.Barragan A.. et al. The duffy-binding-like domain 1 of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) is a heparan sulfate ligand that requires 12 mers for binding. Blood. 2000;95:3594–3599. [PubMed] [Google Scholar]
- 101.Skidmore M.A.. et al. Disruption of rosetting in Plasmodium falciparum malaria with chemically modified heparin and low molecular weight derivatives possessing reduced anticoagulant and other serine protease inhibition activities. Journal of Medicinal Chemistry. 2008;51:1453–1458. doi: 10.1021/jm701337t. [DOI] [PubMed] [Google Scholar]
- 102.Chen Q.. et al. Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum. Journal of Experimental Medicine. 1998;187:15–23. doi: 10.1084/jem.187.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hatabu T.. et al. Binding of Plasmodium falciparum-infected erythrocytes to the membrane-bound form of Fractalkine/CX3CL1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15942–15946. doi: 10.1073/pnas.2534560100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siano J.P.. et al. Short report: Plasmodium falciparum: cytoadherence to alpha(v)beta3 on human microvascular endothelial cells. American Journal of Tropical Medicine and Hygiene. 1998;59:77–79. doi: 10.4269/ajtmh.1998.59.77. [DOI] [PubMed] [Google Scholar]
- 105.Eda S., Sherman I.W.. Plasmodium falciparum-infected erythrocytes bind to the RGD motif of fibronectin via the band 3-related adhesin. Experimental Parasitology. 2004;107:157–162. doi: 10.1016/j.exppara.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 106.Pouvelle B.. et al. Neural cell adhesion molecule, a new cytoadhesion receptor for Plasmodium falciparum-infected erythrocytes capable of aggregation. Infection and Immunity. 2007;75:3516–3522. doi: 10.1128/IAI.01852-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Biswas A.K.. et al. Plasmodium falciparum uses gC1qR/HABP1/p32 as a receptor to bind to vascular endothelium and for platelet-mediated clumping. PLoS Pathogens. 2007;3:1271–1280. doi: 10.1371/journal.ppat.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pino P.. et al. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. Journal of Infectious Diseases. 2003;187:1283–1290. doi: 10.1086/373992. [DOI] [PubMed] [Google Scholar]
- 109.Wilson N.O.. et al. Soluble factors from Plasmodium falciparum-infected erythrocytes induce apoptosis in human brain vascular endothelial and neuroglia cells. Molecular and Biochemical Parasitology. 2008;162:172–176. doi: 10.1016/j.molbiopara.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yeo T.W.. et al. Impaired nitric oxide bioavailability and L-arginine reversible endothelial dysfunction in adults with falciparum malaria. Journal of Experimental Medicine. 2007;204:2693–2704. doi: 10.1084/jem.20070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weinberg J.B.. et al. Arginine, nitric oxide, carbon monoxide, and endothelial function in severe malaria. Current Opinion in Infectious Diseases. 2008;21:468–475. doi: 10.1097/QCO.0b013e32830ef5cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Serirom S.. et al. Anti-adhesive effect of nitric oxide on Plasmodium falciparum cytoadherence under flow. American Journal of Pathology. 2003;162:1651–1660. doi: 10.1016/S0002-9440(10)64299-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Francischetti I.M.. et al. Plasmodium falciparum-infected erythrocytes induce tissue factor expression in endothelial cells and support the assembly of multimolecular coagulation complexes. Journal of Thrombosis and Haemostasis. 2007;5:155–165. doi: 10.1111/j.1538-7836.2006.02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.David P.H.. et al. Rosetting: a new cytoadherence property of malaria-infected erythrocytes. American Journal of Tropical Medicine and Hygiene. 1988;38:289–297. doi: 10.4269/ajtmh.1988.38.289. [DOI] [PubMed] [Google Scholar]
- 115.Kaul D.K.. et al. Rosetting of Plasmodium falciparum-infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood. 1991;78:812–819. [PubMed] [Google Scholar]
- 116.Rowe J.A.. et al. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388:292–295. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- 117.Rowe J.A.. et al. Mapping of the region of complement receptor (CR) 1 required for Plasmodium falciparum rosetting and demonstration of the importance of CR1 in rosetting in field isolates. Journal of Immunology. 2000;165:6341–6346. doi: 10.4049/jimmunol.165.11.6341. [DOI] [PubMed] [Google Scholar]
- 118.Carlson J., Wahlgren M.. Plasmodium falciparum erythrocyte rosetting is mediated by promiscuous lectin-like interactions. Journal of Experimental Medicine. 1992;176:1311–1317. doi: 10.1084/jem.176.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Handunnetti S.M.. et al. Involvement of CD36 on erythrocytes as a rosetting receptor for Plasmodium falciparum-infected erythrocytes. Blood. 1992;80:2097–2104. [PubMed] [Google Scholar]
- 120.Russell C.. et al. Further definition of PfEMP-1 DBL-1alpha domains mediating rosetting adhesion of Plasmodium falciparum. Molecular and Biochemical Parasitology. 2005;144:109–113. doi: 10.1016/j.molbiopara.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 121.Vigan-Womas I.. et al. An in vivo and in vitro model of Plasmodium falciparum rosetting and autoagglutination mediated by varO, a group A var gene encoding a frequent serotype. Infection and Immunity. 2008;76:5565–5580. doi: 10.1128/IAI.00901-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Scholander C.. et al. Novel fibrillar structure confers adhesive property to malaria-infected erythrocytes. Nature Medicine. 1996;2:204–208. doi: 10.1038/nm0296-204. [DOI] [PubMed] [Google Scholar]
- 123.Treutiger C.J.. et al. Rouleaux-forming serum proteins are involved in the rosetting of Plasmodium falciparum-infected erythrocytes. Experimental Parasitology. 1999;93:215–224. doi: 10.1006/expr.1999.4454. [DOI] [PubMed] [Google Scholar]
- 124.Ghumra A.. et al. Identification of residues in the Cmu4 domain of polymeric IgM essential for interaction with Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) Journal of Immunology. 2008;181:1988–2000. doi: 10.4049/jimmunol.181.3.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Helmby H.. et al. Rosetting Plasmodium falciparum-infected erythrocytes express unique strain-specific antigens on their surface. Infection and Immunity. 1993;61:284–288. doi: 10.1128/iai.61.1.284-288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Khera R., Das N.. Complement Receptor 1: Disease associations and therapeutic implications. Molecular Immunology. 2009;46:761–772. doi: 10.1016/j.molimm.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cockburn I.A.. et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:272–277. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nagayasu E.. et al. CR1 density polymorphism on erythrocytes of falciparum malaria patients in Thailand. American Journal of Tropical Medicine and Hygiene. 2001;64:1–5. doi: 10.4269/ajtmh.2001.64.1.11425154. [DOI] [PubMed] [Google Scholar]
- 129.Teeranaipong P.. et al. A functional single-nucleotide polymorphism in the CR1 promoter region contributes to protection against cerebral malaria. Journal of Infectious Diseases. 2008;198:1880–1891. doi: 10.1086/593338. [DOI] [PubMed] [Google Scholar]
- 130.Udomsangpetch R.. et al. The effects of hemoglobin genotype and ABO blood group on the formation of rosettes by Plasmodium falciparum-infected red blood cells. American Journal of Tropical Medicine and Hygiene. 1993;48:149–153. doi: 10.4269/ajtmh.1993.48.149. [DOI] [PubMed] [Google Scholar]
- 131.Barragan A.. et al. Blood group A antigen is a coreceptor in Plasmodium falciparum rosetting. Infection and Immunity. 2000;68:2971–2975. doi: 10.1128/iai.68.5.2971-2975.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Carlson J.. et al. Natural protection against severe Plasmodium falciparum malaria due to impaired rosette formation. Blood. 1994;84:3909–3914. [PubMed] [Google Scholar]
- 133.Rowe J.A.. et al. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17471–17476. doi: 10.1073/pnas.0705390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fry A.E.. et al. Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Human Molecular Genetics. 2008;17:567–576. doi: 10.1093/hmg/ddm331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pathirana S.L.. et al. ABO-blood-group types and protection against severe, Plasmodium falciparum malaria. Annals of Tropical Medicine and Parasitology. 2005;99:119–124. doi: 10.1179/136485905X19946. [DOI] [PubMed] [Google Scholar]
- 136.Vogt A.M.. et al. Heparan sulphate identified on human erythrocytes: a Plasmodium falciparum receptor. Biochemical Journal. 2004;381:593–597. doi: 10.1042/BJ20040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wassmer S.C.. et al. Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion to activated endothelial cells. Journal of Infectious Diseases. 2004;189:180–189. doi: 10.1086/380761. [DOI] [PubMed] [Google Scholar]
- 138.Srivastava K.. et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host and Microbe. 2008;4:179–187. doi: 10.1016/j.chom.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grau G.E.. et al. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. Journal of Infectious Diseases. 2003;187:461–466. doi: 10.1086/367960. [DOI] [PubMed] [Google Scholar]
- 140.McMorran B.J.. et al. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science. 2009;323:797–800. doi: 10.1126/science.1166296. [DOI] [PubMed] [Google Scholar]
- 141.Wassmer S.C.. et al. Platelet-induced clumping of Plasmodium falciparum-infected erythrocytes from Malawian patients with cerebral malaria-possible modulation in vivo by thrombocytopenia. Journal of Infectious Diseases. 2008;197:72–78. doi: 10.1086/523761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Arman M.. et al. Platelet-mediated clumping of Plasmodium falciparum infected erythrocytes is associated with high parasitemia but not severe clinical manifestations of malaria in African children. American Journal of Tropical Medicine and Hygiene. 2007;77:943–946. [PMC free article] [PubMed] [Google Scholar]
- 143.Arman M., Rowe J.A.. Experimental conditions affect the outcome of Plasmodium falciparum platelet-mediated clumping assays. Malaria Journal. 2008;7:243. doi: 10.1186/1475-2875-7-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McGilvray I.D.. et al. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum-parasitized erythrocytes: a role for CD36 in malarial clearance. Blood. 2000;96:3231–3240. [PubMed] [Google Scholar]
- 145.Urban B.C.. et al. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400:73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 146.Urban B.C., Willcox N., Roberts D.J.. A role for CD36 in the regulation of dendritic cell function. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8750–8755. doi: 10.1073/pnas.151028698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Elliott S.R.. et al. Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infection and Immunity. 2007;75:3621–3632. doi: 10.1128/IAI.00095-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Donati D.. et al. Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infection and Immunity. 2004;72:5412–5418. doi: 10.1128/IAI.72.9.5412-5418.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Artavanis-Tsakonas K.. et al. Activation of a subset of human NK cells upon contact with Plasmodium falciparum-infected erythrocytes. Journal of Immunology. 2003;171:5396–5405. doi: 10.4049/jimmunol.171.10.5396. [DOI] [PubMed] [Google Scholar]
- 150.Baratin M.. et al. Dissection of the role of PfEMP1 and ICAM-1 in the sensing of Plasmodium falciparum-infected erythrocytes by natural killer cells. PLoS ONE. 2007;2:e228. doi: 10.1371/journal.pone.0000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.D'Ombrain M.C.. et al. Plasmodium falciparum erythrocyte membrane protein-1 specifically suppresses early production of host interferon-gamma. Cell Host and Microbe. 2007;2:130–138. doi: 10.1016/j.chom.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 152.MacPherson G.G.. et al. Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. American Journal of Pathology. 1985;119:385–401. [PMC free article] [PubMed] [Google Scholar]
- 153.Taylor T.E.. et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nature Medicine. 2004;10:143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 154.Dondorp A.M.. Clinical significance of sequestration in adults with severe malaria. Transfusion Clinique et Biologique. 2008;15:56–57. doi: 10.1016/j.tracli.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 155.Artavanis-Tsakonas K., Tongren J.E., Riley E.M.. The war between the malaria parasite and the immune system: immunity, immunoregulation and immunopathology. Clinical and Experimental Immunology. 2003;133:145–152. doi: 10.1046/j.1365-2249.2003.02174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Francischetti I.M.. Does activation of the blood coagulation cascade have a role in malaria pathogenesis? Trends in Parasitology. 2008;24:258–263. doi: 10.1016/j.pt.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Seydel K.B.. et al. The distribution and intensity of parasite sequestration in comatose Malawian children. Journal of Infectious Diseases. 2006;194:208. doi: 10.1086/505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.WHO. Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2000;94(Suppl 1):S1–90. [PubMed] [Google Scholar]
- 159.Bruneel F.. et al. The clinical spectrum of severe imported falciparum malaria in the intensive care unit: report of 188 cases in adults. American Journal of Respiratory and Critical Care Medicine. 2003;167:684–689. doi: 10.1164/rccm.200206-631OC. [DOI] [PubMed] [Google Scholar]
- 160.Dondorp A.. et al. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366:717–725. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- 161.Skinner T.S.. et al. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. International Journal for Parasitology. 1996;26:519–525. doi: 10.1016/0020-7519(96)89380-5. [DOI] [PubMed] [Google Scholar]
- 162.Wang C.C.. Validating targets for antiparasite chemotherapy. Parasitology. 1997;114(Suppl):S31–44. [PubMed] [Google Scholar]
- 163.Duffy P.E., Craig A.G., Baruch D.I.. Variant proteins on the surface of malaria-infected erythrocytes–developing vaccines. Trends in Parasitology. 2001;17:354–356. doi: 10.1016/s1471-4922(01)02022-0. [DOI] [PubMed] [Google Scholar]
- 164.Baruch D.I.. et al. Immunization of Aotus monkeys with a functional domain of the Plasmodium falciparum variant antigen induces protection against a lethal parasite line. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3860–3865. doi: 10.1073/pnas.022018399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen Q.. et al. Immunization with PfEMP1-DBL1alpha generates antibodies that disrupt rosettes and protect against the sequestration of Plasmodium falciparum-infected erythrocytes. Vaccine. 2004;22:2701–2712. doi: 10.1016/j.vaccine.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 166.Gamain B., Miller L.H., Baruch D.I.. The surface variant antigens of Plasmodium falciparum contain cross-reactive epitopes. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2664–2669. doi: 10.1073/pnas.041602598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baruch D.I., Gamain B., Miller L.H.. DNA immunization with the cysteine-rich interdomain region 1 of the Plasmodium falciparum variant antigen elicits limited cross-reactive antibody responses. Infection and Immunity. 2003;71:4536–4543. doi: 10.1128/IAI.71.8.4536-4543.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gandon S.. et al. Imperfect vaccination: some epidemiological and evolutionary consequences. Proceedings of the Royal Society B Biological Sciences. 2003;270:1129–1136. doi: 10.1098/rspb.2003.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yipp B.G.. et al. Src-family kinase signaling modulates the adhesion of Plasmodium falciparum on human microvascular endothelium under flow. Blood. 2003;101:2850–2857. doi: 10.1182/blood-2002-09-2841. [DOI] [PubMed] [Google Scholar]
- 170.Ho M.. et al. Ectophosphorylation of CD36 regulates cytoadherence of Plasmodium falciparum to microvascular endothelium under flow conditions. Infection and Immunity. 2005;73:8179–8187. doi: 10.1128/IAI.73.12.8179-8187.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Dondorp A.M.. et al. Levamisole inhibits sequestration of infected red blood cells in patients with falciparum malaria. Journal of Infectious Diseases. 2007;196:460–466. doi: 10.1086/519287. [DOI] [PubMed] [Google Scholar]
- 172.Gruarin P.. et al. Cytoadherence of Plasmodium falciparum-infected erythrocytes is mediated by a redox-dependent conformational fraction of CD36. Journal of Immunology. 2001;167:6510–6517. doi: 10.4049/jimmunol.167.11.6510. [DOI] [PubMed] [Google Scholar]
- 173.Dondorp A.M.. et al. Prognostic significance of reduced red blood cell deformability in severe falciparum malaria. American Journal of Tropical Medicine and Hygiene. 1997;57:507–511. doi: 10.4269/ajtmh.1997.57.507. [DOI] [PubMed] [Google Scholar]
- 174.Nuchsongsin F.. et al. Effects of malaria heme products on red blood cell deformability. American Journal of Tropical Medicine and Hygiene. 2007;77:617–622. [PubMed] [Google Scholar]
- 175.Watt G., Jongsakul K., Ruangvirayuth R.. A pilot study of N-acetylcysteine as adjunctive therapy for severe malaria. Quarterly Journal of Medicine. 2002;95:285–290. doi: 10.1093/qjmed/95.5.285. [DOI] [PubMed] [Google Scholar]
- 176.Treeprasertsuk S.. et al. N-acetylcysteine in severe falciparum malaria in Thailand. Southeast Asian Journal of Tropical Medicine and Public Health. 2003;34:37–42. [PMC free article] [PubMed] [Google Scholar]
- 177.Arreesrisom P.. et al. Suppressive effects of the anti-oxidant N-acetylcysteine on the anti-malarial activity of artesunate. Parasitology International. 2007;56:221–226. doi: 10.1016/j.parint.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 178.Cooke B.M.. et al. A recombinant peptide based on PfEMP-1 blocks and reverses adhesion of malaria-infected red blood cells to CD36 under flow. Molecular Microbiology. 1998;30:83–90. doi: 10.1046/j.1365-2958.1998.01040.x. [DOI] [PubMed] [Google Scholar]
- 179.Yipp B.G.. et al. Recombinant PfEMP1 peptide inhibits and reverses cytoadherence of clinical Plasmodium falciparum isolates in vivo. Blood. 2003;101:331–337. doi: 10.1182/blood-2002-06-1725. [DOI] [PubMed] [Google Scholar]
- 180.Dormeyer M.. et al. Rational design of anticytoadherence inhibitors for Plasmodium falciparum based on the crystal structure of human intercellular adhesion molecule 1. Antimicrobial Agents and Chemotherapy. 2006;50:724–730. doi: 10.1128/AAC.50.2.724-730.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Benelli R.. et al. Anti-invasive effects of green tea polyphenol epigallocatechin-3-gallate (EGCG), a natural inhibitor of metallo and serine proteases. Biological Chemistry. 2002;383:101–105. doi: 10.1515/BC.2002.010. [DOI] [PubMed] [Google Scholar]
- 182.Fassina G.. et al. Mechanisms of inhibition of tumor angiogenesis and vascular tumor growth by epigallocatechin-3-gallate. Clinical Cancer Research. 2004;10:4865–4873. doi: 10.1158/1078-0432.CCR-03-0672. [DOI] [PubMed] [Google Scholar]
- 183.Yoda Y.. et al. Different susceptibilities of Staphylococcus and Gram-negative rods to epigallocatechin gallate. Journal of Infection and Chemotherapy. 2004;10:55–58. doi: 10.1007/s10156-003-0284-0. [DOI] [PubMed] [Google Scholar]
- 184.Anstey N.M.. et al. Nitric oxide in Tanzanian children with malaria: inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. Journal of Experimental Medicine. 1996;184:557–567. doi: 10.1084/jem.184.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lopansri B.K.. et al. Low plasma arginine concentrations in children with cerebral malaria and decreased nitric oxide production. Lancet. 2003;361:676–678. doi: 10.1016/S0140-6736(03)12564-0. [DOI] [PubMed] [Google Scholar]
- 186.Yeo T.W.. et al. Pharmacokinetics of L-arginine in adults with moderately severe malaria. Antimicrobial Agents and Chemotherapy. 2008;52:4381–4387. doi: 10.1128/AAC.00421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yeo T.W.. et al. Recovery of endothelial function in severe falciparum malaria: relationship with improvement in plasma L-arginine and blood lactate concentrations. Journal of Infectious Diseases. 2008;198:602–608. doi: 10.1086/590209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yeo T.W.. et al. Safety profile of L-arginine infusion in moderately severe falciparum malaria. PLoS ONE. 2008;3:e2347. doi: 10.1371/journal.pone.0002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Taoufiq Z.. et al. Rho kinase inhibition in severe malaria: thwarting parasite-induced collateral damage to endothelia. Journal of Infectious Diseases. 2008;197:1062–1073. doi: 10.1086/528988. [DOI] [PubMed] [Google Scholar]
- 190.Carlson J.. et al. Disruption of Plasmodium falciparum erythrocyte rosettes by standard heparin and heparin devoid of anticoagulant activity. American Journal of Tropical Medicine and Hygiene. 1992;46:595–602. doi: 10.4269/ajtmh.1992.46.595. [DOI] [PubMed] [Google Scholar]
- 191.Rowe A.. et al. Plasmodium falciparum: a family of sulphated glycoconjugates disrupts erythrocyte rosettes. Experimental Parasitology. 1994;79:506–516. doi: 10.1006/expr.1994.1111. [DOI] [PubMed] [Google Scholar]
- 192.Rogerson S.J.. et al. Sulfated glycoconjugates as disrupters of Plasmodium falciparum erythrocyte rosettes. American Journal of Tropical Medicine and Hygiene. 1994;51:198–203. doi: 10.4269/ajtmh.1994.51.198. [DOI] [PubMed] [Google Scholar]
- 193.Vogt A.M.. et al. Release of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathogens. 2006;2:e100. doi: 10.1371/journal.ppat.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kyriacou H.M.. et al. In vitro inhibition of Plasmodium falciparum rosette formation by Curdlan sulfate. Antimicrobial Agents and Chemotherapy. 2007;51:1321–1326. doi: 10.1128/AAC.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kaneko Y.. et al. Inhibition of HIV-1 infectivity with curdlan sulfate in vitro. Biochemical Pharmacology. 1990;39:793–797. doi: 10.1016/0006-2952(90)90161-d. [DOI] [PubMed] [Google Scholar]
- 196.Havlik I.. et al. Curdlan sulphate in human severe/cerebral Plasmodium falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2005;99:333–340. doi: 10.1016/j.trstmh.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 197.Weisman H.F.. et al. Soluble human complement receptor type I: In vivo inhibitor of complement suppressing post-ischaemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 198.Keshavjee S.. et al. A randomized, placebo-controlled trial of complement inhibition in ischemia-reperfusion injury after lung transplantation in human beings. Journal of Thoracic and Cardiovascular Surgery. 2005;129:423–428. doi: 10.1016/j.jtcvs.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 199.Yazdanbakhsh K.. Review: complement receptor 1 therapeutics for prevention of immune hemolysis. Immunohematology. 2005;21:109–118. [PubMed] [Google Scholar]
- 200.Ahrens I., Bode C., Peter K.. Inhibition of platelet activation and aggregation. Handbook of Experimental Pharmacology. 2005:443–462. doi: 10.1007/3-540-27661-0_16. [DOI] [PubMed] [Google Scholar]
- 201.Bennett J.S.. Novel platelet inhibitors. Annual Review of Medicine. 2001;52:161–184. doi: 10.1146/annurev.med.52.1.161. [DOI] [PubMed] [Google Scholar]
- 202.Nacher M.. et al. Case-control studies on host factors in severe malaria. Trends in Parasitology. 2001;17:253–254. doi: 10.1016/s1471-4922(01)01926-2. [DOI] [PubMed] [Google Scholar]
- 203.Montgomery J.. et al. Genetic Analysis of Circulating and Sequestered Populations of Plasmodium falciparum in Fatal Pediatric Malaria. Journal of Infectious Diseases. 2006;194:115–122. doi: 10.1086/504689. [DOI] [PubMed] [Google Scholar]
- 204.Krishna S.. et al. Lactic acidosis and hypoglycaemia in children with severe malaria: pathophysiological and prognostic significance. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1994;88:67–73. doi: 10.1016/0035-9203(94)90504-5. [DOI] [PubMed] [Google Scholar]
- 205.Rowe J.A.. et al. Positive correlation between rosetting and parasitemia in Plasmodium falciparum clinical isolates. American Journal of Tropical Medicine and Hygiene. 2002;66:458–460. doi: 10.4269/ajtmh.2002.66.458. [DOI] [PubMed] [Google Scholar]
- 206.Le Scanf C.. et al. Rosetting is associated with increased Plasmodium falciparum in vivo multiplication rate in the Saimiri sciureus monkey. Microbes and Infection. 2008;10:447–451. doi: 10.1016/j.micinf.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 207.Clough B., Atilola F.A., Pasvol G.. The role of rosetting in the multiplication of Plasmodium falciparum: rosette formation neither enhances nor targets parasite invasion into uninfected red cells. British Journal of Haematology. 1998;100:99–104. doi: 10.1046/j.1365-2141.1998.00534.x. [DOI] [PubMed] [Google Scholar]
- 208.Deans A.M., Rowe J.A.. Plasmodium falciparum: Rosettes do not protect merozoites from invasion-inhibitory antibodies. Experimental Parasitology. 2006;112:269–273. doi: 10.1016/j.exppara.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.David P.H.. et al. Parasite sequestration in Plasmodium falciparum malaria: spleen and antibody modulation of cytoadherence of infected erythrocytes. Proceedings of the National Acadamy of Science of the United States of America. 1983;80:5075–5079. doi: 10.1073/pnas.80.16.5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Willimann K.. et al. In vivo sequestration of Plasmodium falciparum-infected human erythrocytes: a severe combined immunodeficiency mouse model for cerebral malaria. Journal of Experimental Medicine. 1995;182:643–653. doi: 10.1084/jem.182.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pettersson F.. et al. Whole-body imaging of sequestration of Plasmodium falciparum in the rat. Infection and Immunity. 2005;73:7736–7746. doi: 10.1128/IAI.73.11.7736-7746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.McIntosh R.S.. et al. The importance of human FcgammaRI in mediating protection to malaria. PLoS Pathogens. 2007;3:e72. doi: 10.1371/journal.ppat.0030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Simmons D.L.. Anti-adhesion therapies. Current Opinion in Pharmacology. 2005;5:398–404. doi: 10.1016/j.coph.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 214.Aitman T.J.. et al. Malaria susceptibility and CD36 mutation. Nature. 2000;405:1015–1016. doi: 10.1038/35016636. [DOI] [PubMed] [Google Scholar]
- 215.Ayodo G.. et al. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. American Journal of Human Genetics. 2007;81:234–242. doi: 10.1086/519221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Amodu O.K.. et al. Plasmodium falciparum malaria in south-west Nigerian children: is the polymorphism of ICAM-1 and E-selectin genes contributing to the clinical severity of malaria? Acta Tropica. 2005;95:248–255. doi: 10.1016/j.actatropica.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 217.Pain A.. et al. A non-sense mutation in Cd36 gene is associated with protection from severe malaria. Lancet. 2001;357:1502–1503. doi: 10.1016/S0140-6736(00)04662-6. [DOI] [PubMed] [Google Scholar]
- 218.Bellamy R., Kwiatkowski D., Hill A.V.. Absence of an association between intercellular adhesion molecule 1, complement receptor 1 and interleukin 1 receptor antagonist gene polymorphisms and severe malaria in a West African population. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1998;92:312–316. doi: 10.1016/s0035-9203(98)91026-4. [DOI] [PubMed] [Google Scholar]
- 219.Ndiaye R.. et al. Genetic study of ICAM1 in clinical malaria in Senegal. Tissue Antigens. 2005;65:474–480. doi: 10.1111/j.1399-0039.2005.00388.x. [DOI] [PubMed] [Google Scholar]
- 220.Fry A.E.. et al. Variation in the ICAM1 gene is not associated with severe malaria phenotypes. Genes and Immunity. 2008;9:462–469. doi: 10.1038/gene.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kun J.F.. et al. Association of the ICAM-1Kilifi mutation with protection against severe malaria in Lambarene, Gabon. American Journal of Tropical Medicine and Hygiene. 1999;61:776–779. doi: 10.4269/ajtmh.1999.61.776. [DOI] [PubMed] [Google Scholar]
- 222.Moulds J.M.. et al. Molecular identification of Knops blood group polymorphisms found in long homologous region D of complement receptor 1. Blood. 2001;97:2879–2885. doi: 10.1182/blood.v97.9.2879. [DOI] [PubMed] [Google Scholar]
- 223.Thathy V.. et al. Complement receptor 1 polymorphisms associated with resistance to severe malaria in Kenya. Malaria Journal. 2005;4:54. doi: 10.1186/1475-2875-4-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zimmerman P.A.. et al. CR1 Knops blood group alleles are not associated with severe malaria in the Gambia. Genes and Immunity. 2003;4:368–373. doi: 10.1038/sj.gene.6363980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further reading, resources and contacts
Books
- D.A. Warrell, H.M. Gilles. 4th Edition. Oxford University Press; 2002. eds ( ) Essential Malariology, [Google Scholar]; This book provides a broad background to malariology.
- I.W. Sherman. ASM Press; 2005. ed. ( ) Molecular Approaches to Malaria, [Google Scholar]; A good overview of current molecular approaches to malaria research.
Websites
For up-to-date information on malaria risks, epidemiology and prevention see:
- http://www.cdc.gov/Malaria/ http://www.cdc.gov/Malaria/; For general information on malaria:
- http://www.who.int/topics/malaria/en/ http://www.who.int/topics/malaria/en/; For information on malaria parasite biology, biochemistry and physiology see:
- http://sites.huji.ac.il/malaria/ http://sites.huji.ac.il/malaria/