

Abstract
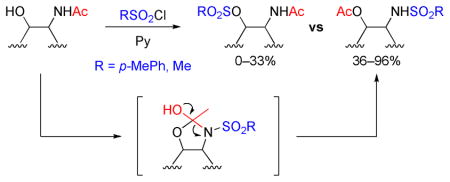
The first example of sulfonylation-induced N- to O-acetyl migration of 2-acetamidoethanol derivatives is described. This type of reaction could happen with any 2-acetamidoethanol derivatives under typical sulfonylation conditions (TsCl or MsCl, pyridine) and might be a common side reaction of significance. Furthermore, the results reveal that 2-acetamidoethanol derivatives with sterically encumbered hydroxyl group result in the migration products in high yields. The mechanism of the migration reaction is discussed.
Sulfonylation of alcohols is a common transformation in numerous synthetic processes. This derivatization is generally performed by utilizing sulfonyl chlorides in the presence of a base (typically, pyridine, triethylamine, DABCO and the like), and gives the desired products in high yield. However, during the course of our efforts to synthesize 3-O-tosyl glucosamine derivative 2a, we identified a significant byproduct (Scheme 1). The byproduct was found to have the same mass as 2a, and the resonances of both the acetyl and tosyl groups were seen in the 1H NMR spectrum. After full characterization, the byproduct was assigned the structure 3a, which disclosed an unusual N- to O-acetyl migration. Our follow up on this reaction revealed that this type of acetyl migration could happen with facility with any number of 2-acetamidoethanol derivatives under typical sulfonylation conditions.
SCHEME 1.
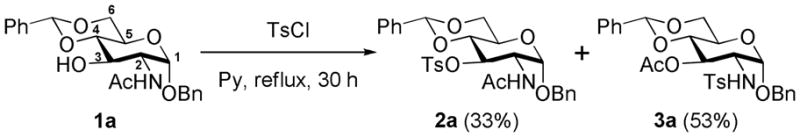
Tosylation of N-acetylglucosamine derivative 1a
For tosylation of 1a, 1 we initially used a mild condition (TsCl, pyridine, CH2Cl2, room temperature). However, the starting material was fully recovered. When heated under reflux in pyridine for a longer duration, two compounds were obtained, as outlined in Scheme 1, with the major component identified as the product of acetyl migration 3a. The products 2a and 3a showed similar 1H and 13C NMR spectra, and had the same mass. Thus, the differentiation of each compound was difficult.
In the 1H NMR spectra (Figure 1), the H-2 resonance of 3a (δ 3.53 ppm) appeared at higher field than that of 2a (δ 4.46 ppm). The H-3 resonance of 3a appeared at δ 5.24 ppm, and the axial orientation for the hydrogen was confirmed by the coupling constants (J2,3 = J3,4 = 10 Hz). Confirmatory evidence for the structure of 3a was the ester carbonyl stretch at 1734 cm−1 in the IR spectrum. The IR resonances of the amide carbonyl groups of 1a and 2a were seen at 1651 and 1656 cm−1, respectively.
FIGURE 1.

1H NMR spectra of the compounds (A) 1a in DMSO-d6, (B) 2a in CDCl3 and (C) 3a in CDCl3. Compound 1a was not soluble in CDCl3.
A similar observation for acetyl migration in compound 1a was reported by Calvo-Mateo et al. (Scheme 2). 2 They obtained the product 6 by heating of the unstable triflate 4 in a mixture of pyridine, water, and DMF (Scheme 2a). It is interesting that an inversion at the C-3 position was observed in their reaction, in contrast to our result. They proposed the transient formation of the oxazoline intermediate 5, with the attendant inversion of configuration at C-3, which then underwent hydrolysis to give 6. The authors indicated that a rearrangement product 9 also formed as a byproduct in small yield after the treatment of 8 with NaN3 (Scheme 2b). To our knowledge, this is the only example that approaches the reaction that we report here.3
SCHEME 2.

The acetyl migration reactions of 2-acetamidoethanol derivatives2
We carried out a series of additional reactions to gain insight into the mechanism of this acetyl migration reaction. Initially, we wondered whether the acetyl group could migrate to the hydroxyl group before the N-tosylation event (Scheme 3, route A). In order to examine this possibility, compound 1a was heated under reflux in pyridine for 48 h. However, in the absence of TsCl, no transformation took place and the starting compound 1a was recovered as the only compound. It is also conceivable that the migration product 3a might be generated from 2a (route D). To confirm this, compound 2a was heated under reflux in pyridine with and without TsCl for 48 h. Again, under these conditions starting 2a was fully recovered. These results, thus, ruled out routes A and D.
SCHEME 3.

Mechanistic possibilities of N- to O-acetyl migration of 1a
Two additional possibilities, routes B and C, can be envisioned. That is to say that product 3a could be formed via an intermediate ortho amide, 12, which would be generated from tosylation of 10 (route B) or through N-tosylation of the acetamido group of 1a (route C). However, N-tosylation of an amide moiety (as in route C) generally requires a strong base such as n-BuLi or LiHMDS. 4 Tosylation of a simple acetamido group in a compound such as 14 under the same condition as Scheme 1 resulted only in the recovery of the starting material (Scheme 4). For this reason, route C might be ruled out. The most probable mechanism would then be through route B.
SCHEME 4.
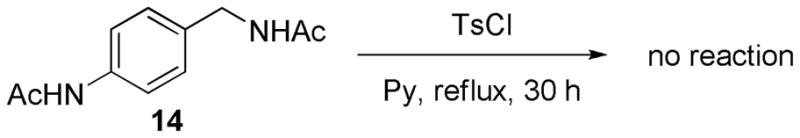
Tosylation of the compound 14
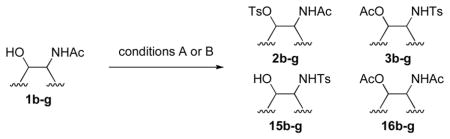
To explore the scope of this transformation, we investigated tosylations of several additional examples (Table 1). Substrates 1b, 1d and 1g were prepared from the corresponding commercially available amines (see Supporting Information). Tosylation of 1b, with the flexible cyclohexane skeleton, also afforded the acetyl migration compound 3b as the major product, and O-tosyl product 2b was present in smaller yield at 9% (entry 1). Unexpectedly, N-tosyl cyclohexanol 15b and N,O-diacetyl product 16b were present in 25% and 19%, respectively. We believe that the activated acetyl group of 18 probably migrated via an intermolecular reaction to afford compounds 15b and 16b (Scheme 5). The structures of products 3b, 15b and 16b were confirmed by comparison to the reported data. 5 In contrast to this result, tosylation of 1g gave O-tosyl compound 2g as the major product, and the migration product 3g was not seen, as to be expected (entry 6). This result supports that the migration products 3a and 3b are produced by intramolecular acetyl migration. The case of the simple 2-acetamidoethanol (1c) is of interest (entry 2), as this too gave the migration product 3c, in the highest yield among the three products, whereas O-tosyl product 2c was not obtained. We conclude that acetyl migration can happen with any kind of 2-acetamidoethanol derivatives under typical tosylation condition. Compound 1d, with a hindered hydroxyl group, afforded the migration product 3d in 52% yield (entry 3, condition A). Moreover, under condition B, 3d was obtained in 96% yield. It is possible that the tertiary alcohol in this example might have played an entropic role as the intramolecular nucleophile. 1-Deoxy-N-acetylglucosamine 1e, prepared from an oxazoline derivative,6 was also allowed to react with TsCl (entry 4). In this case, the migration product 3e was produced in 94%. Compared to compound 1a having the α-benzyloxy group at C-1, the less-hindered 1e gave the migration product in higher yield. Meanwhile, tosylation of 2-acetamidophenol (1f) gave O-tosyl product 2f exclusively in 98% yield (entry 5, conditions A or B). This result might be due to different reactivity of phenol 1f compared to alcohols. The structure of 2f was confirmed by X-ray structure analysis (see Supporting Information).
TABLE 1.
Tosylation of various 2-acetamidoethanol derivativesa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | substrates | conditions | products (yield, %) |
||||
| 2b-g | 3b-g | 15b-g | 16b-g | 1b-gb | |||
| 1 |
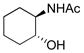 1b |
A | 9 | 36 | 25 | 19 | — |
| 2 |
1c |
A | — | 36 | 32 | 29 | — |
| 3 |
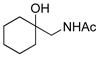 1d |
A B |
— — |
52 96 |
— — |
— — |
46 — |
| 4 |
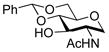 1e |
B | — | 94 | — | — | — |
| 5 |
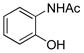 1f |
A B |
98 98 |
— — |
— — |
— — |
— — |
| 6c |
 1g |
A | 41 | — | 9 | 8 | 39 |
The tosylation was performed under condition A or B. Condition A: TsCl (1.05 equiv) and pyridine (3 equiv) were added to a solution of substrate in CH2Cl2 at 0 °C, and the mixture was stirred at room temperature for 24 h. Condition B: TsCl (2 equiv) was added to a solution of substrate in pyridine at room temperature, and the mixture was heated under reflux for 30 h.
Recovery of starting substrate.
4-(Acetamido)cyclohexanol was used as a substrate in this case.
SCHEME 5.

The proposed mechanistic outcome for transformations of 1b
We also tested the scope of this type of acetyl migration in a mesylation reaction. As shown in Scheme 6, mesylation of compound 1d efficiently afforded migration product 19. Therefore, the mesylation reaction also can afford the N- to O-acetyl migration.
SCHEME 6.
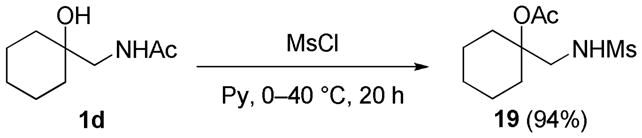
Mesylation of the compound 1d
In summary, we have demonstrated sulfonylation-induced N- to O-acetyl migration of 2-acetamidoethanol derivatives. The acetyl migration proceeds even in a simple compound such as 2-acetamidoethanol (1c), although the sterically hindered hydroxyl group is found to be an excellent substrate for acetyl migration. Furthermore, it is also noteworthy that the unusual “N- to O-acetyl migration” proceeds under typical sulfonylation conditions in 2-acetamidoethanol. Since the 2-acetamidoethanol motif is common in both natural products and synthetic samples, this type of acetyl migration might be generally useful. Also significantly, the type of acetyl (acyl) migration could be happening as a side product of consequence in reactions of this type.
Experimental Section
General procedures for the tosylation of 2- acetamidoethanol derivatives
Condition A: p-Toluenesulfonyl chloride (1.05 equiv) and pyridine (3.0 equiv) were added to a solution of substrate in CH2Cl2 at 0 °C, and the mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure, and the crude product was purified by silica gel column chromatography. Condition B: p-Toluenesulfonyl chloride (2.0 equiv) was added to a solution of substrate in pyridine, and the mixture was heated under reflux for 30 h. The mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography.
Sample procedure and data for 2a and 3a
Condition B was used for compound 1a (100 mg, 0.25 mmol), and the crude product was purified by silica gel column chromatography (AcOEt/hexane, 1:3) to afford 2a (46 mg, 33%) and 3a (74 mg, 53%) as white solids. 2a: 1H NMR (500 HMz, CDCl3) δ 2.02 (s, 3H, CH3CO), 2.28 (s, 3H, CH3Ph), 3.68 (dd, J = 9.6, 9.6 Hz, 1H, H-4), 3.72 (dd, J = 10.7, 10.7 Hz, 1H, H-6a), 3.88 (ddd, J = 4.8, 9.8, 9.8 Hz, 1H, H-5), 4.21 (dd, J = 5.0, 10.4 Hz, 1H, H-6b), 4.46 (ddd, J = 3.6, 9.2, 10.4 Hz, 1H, H-2), 4.53 and 4.73 (AB, J = 11.9 Hz, 2H, OCH2Ph), 4.92 (dd, J = 10.0, 10.0 Hz, 1H, H-3), 5.02 (d, J = 3.8 Hz, 1H, H-1), 5.37 (s, 1H, CHPh), 6.00 (d, J = 9.0 Hz, 1H, NH), 6.97 (d, J = 8.0 Hz, 2H, Ts), 7.19–7.44 (m, 10H), 7.67 (d, J = 8.4 Hz, 2H, Ts); 13C NMR (126 MHz, CDCl3) δ 21.9 (CH3Ph), 23.4 (CH3CO), 52.5 (C-2), 63.6 (C-5), 68.9 (C-6), 70.6 (OCH2Ph), 78.4 (C-3), 79.2 (C-4), 98.0 (C-1), 101.8 (CHPh), 126.4, 128.1, 128.3, 128.4, 128.6, 128.9, 129.2, 129.5, 133.9, 136.7, 136.8, 144.6, 170.6 (C=O); IR 3324, 1656, 1547, 1362, 1182, 1123, 1097 cm−1; HRMS (FAB) calcd for C29H32NO8S (M + H+), 554.1849, found 554.1848. 3a: 1H NMR (500 HMz, CDCl3) δ 1.72 (s, 3H, CH3CO), 2.37 (s, 3H, CH3Ph), 3.53 (ddd, J = 3.8, 10.2, 10.2 Hz, 1H, H-2), 3.56 (dd, J = 9.7, 9.7 Hz, 1H, H-4), 3.67 (dd, J = 10.3, 10.3 Hz, 1H, H-6a), 3.83 (ddd, J = 4.8, 9.8, 9.8 Hz, 1H, H-5), 4.15 (dd, J = 4.8, 10.4 Hz, 1H, H-6b), 4.37 and 4.62 (AB, J = 11.6 Hz, 2H, OCH2Ph), 4.71 (d, J = 3.8 Hz, 1H, H-1), 5.00 (d, J = 10.2 Hz, 1H, NH), 5.24 (dd, J = 10.1, 10.1 Hz, 1H, H-3), 5.42 (s, 1H, CHPh), 7.21 (d, J = 8.4 Hz, 2H, Ts), 7.26–7.39 (m, 10H), 7.62 (d, J = 8.4 Hz, 2H, Ts); 13C NMR (126 MHz, CDCl3) δ 20.8 (CH3CO), 21.7 (CH3Ph), 56.8 (C-2), 63.1 (C-5), 68.9 (C-6), 69.4 (C-3), 70.5 (OCH2Ph), 79.4 (C-4), 98.0 (C-1), 101.7 (CHPh), 126.3, 127.1, 128.4, 128.6, 128.6, 128.9, 129.3, 129.9, 136.4, 137.0, 138.4, 143.7, 170.8 (C=O); IR 3356, 1734, 1498, 1337, 1234, 1163, 1090 cm−1; HRMS (ESI) calcd for C29H31NNaO8S (M + Na+), 576.1659, found 576.1662.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health.
Footnotes
Supporting Information Available: General experimental procedures, characterization data for all new compounds, including copies of 1D NMR spectra (1H, 13C NMR, and DEPT) and 2D NMR spectra (H-H COSY and C-H HETCOR), and crystallographic information file (CIF) of compound 2f. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Compound 1a was prepared from D-glucosamine in 3 steps with an overall yield of 77%, by a small variation of a known procedures: Berger I, Nazarov AA, Hartinger CG, Groessl M, Valiahdi SM, Jakupec MA, Keppler BK. ChemMedChem. 2007;2:505–514. doi: 10.1002/cmdc.200600279.Babic A, Pecar S. Tetrahedron: Asymmetry. 2008;19:2265–2271.
- 2.Calvo-Mateo A, Fiandor J, De Las Heras FG. An Quím. 1987;83:77–80. [Google Scholar]
- 3.The same type of acyl migration to Scheme 2a is known: D’hooghe M, Vervisch K, Van Nieuwenhove A, De Kimpe N. Tetrahedron Lett. 2007;48:1771–1774.Feng Z, Mohapatra S, Klimko PG, Hellberg MR, May JA, Kelly C, Williams G, McLaughlin MA, Sharif NA. Bioorg Med Chem Lett. 2007;17:2998–3002. doi: 10.1016/j.bmcl.2007.03.073.Gornostaev LM, Sokolova MS. Russ J Org Chem. 2006;42:1473–1476.Avenoza A, Busto JH, Canal N, Peregrina JM. J Org Chem. 2005;70:330–333. doi: 10.1021/jo048943s.
- 4.For n-BuLi: Harling JD, Steel PG, Woods TM, Yufit DS. Org Biomol Chem. 2007;5:3472–3476. doi: 10.1039/b708967h.For LiHMDS: Hong S, Yang J, Weinreb SM. J Org Chem. 2006;71:2078–2089. doi: 10.1021/jo052504r.
- 5.For compound 3b: Liu YK, Li R, Yue L, Li BJ, Dhen YC, Wu Y, Ding LS. Org Lett. 2006;8:1521–1524. doi: 10.1021/ol0529905.For compound 15b: Wang Z, Cui YT, Xu ZB, Qu J. J Org Chem. 2008;73:2270–2274. doi: 10.1021/jo702401t.Fan RH, Hou XL. Org Biomol Chem. 2003;1:1565–1567. doi: 10.1039/b301081c.For compound 16b: Miller SJ, Copeland GT, Papaioannou N, Horstmann TE, Ruel EM. J Am Chem Soc. 1998;120:1629–1630.Tabanella T, Valancogne I, Jackson RFW. Org Biomol Chem. 2003;1:4254–4261. doi: 10.1039/b308750f.
- 6.Hesek D, Lee M, Yamaguchi T, Noll BC, Mobashery S. J Org Chem. 2008;73:7349–7352. doi: 10.1021/jo801037z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.