

Abstract
Background
Paroxysmal nocturnal hemoglobinuria is an acquired clonal disorder of the hemopoietic stem cells for which the only curative treatment is allogeneic hematopoietic stem cell transplantation.
Design and Methods
The aim of this retrospective study was to assess the long-term clinical and hematologic results in 26 paroxysmal nocturnal hemoglobinuria patients who received hematopoietic stem cell transplantation in Italy between 1988 and 2006. The patients were aged 22 to 60 years (median 32 years). Twenty-three donors were HLA-identical (22 siblings and one unrelated) and 3 were HLA-mismatched (2 related and one unrelated).
Results
Fifteen patients received a myeloablative conditioning consisting of busulfan and cyclophosphamide (in all cases from identical donor) and 11 were given a reduced intensity conditioning (8 from identical donor and 3 from mismatched donor). The cumulative incidence of graft failure was 8% (4% primary and 4% secondary graft failure). Transplant-related mortality for all patients was 42% (26% and 63% for patients transplanted following myeloablative or reduced intensity conditioning, respectively). As of October 31, 2009, 15 patients (11 in the myeloablative conditioning group and 4 in the reduced intensity conditioning group) are alive with complete hematologic recovery and no evidence of paroxysmal nocturnal hemoglobinuria following a median follow-up of 131 months (range 30–240). The 10-year Kaplan-Meier probability of disease-free survival was 57% for all patients: 65% for 23 patients transplanted from identical donor and 73% for 15 patients transplanted with myeloablative conditioning. No thromboembolic event nor recurrence of the disease were reported following transplant.
Conclusions
The findings of this study confirm that most patients with paroxysmal nocturnal hemoglobinuria may be definitively cured with hematopoietic stem cell transplantation.
Keywords: paroxysmal nocturnal hemoglobinuria, hematopoietic stem cell transplantation, conditioning regimen
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a consequence of clonal expansion of one or more hematopoietic stem cells due to a somatic mutation of the PIGA gene located on Xp22.1.1 Progeny of affected stem cells are deficient in glycosyl phosphatidylinositol-anchored proteins (GPI-APs) which are important regulators of the complement system. Although more than 20 functionally diverse GPI-APs are expressed by hematopoietic stem cells, it is deficiency on red blood cells of the two GPI-anchored complement regulatory proteins, CD55 and CD59, that is the leading cause of the intravascular hemolysis that represents the clinical hallmark of paroxysmal nocturnal hemoglobinuria.2,3 The primary clinical manifestations of paroxysmal nocturnal hemoglobinuria are hemolytic anemia, bone marrow failure, and thrombosis. Venous thrombosis may involve unusual sites (hepatic, mesenteric, cerebral, dermal veins) and is the leading cause of death.4
The clinical course of paroxysmal nocturnal hemoglobinuria is usually chronic with frequent exacerbations of the clinical manifestations and sometimes with spontaneous long-term remission. The reported median survival with conventional treatment (red blood cell transfusions, steroids, androgens, growth factors, immunosuppressive therapy) is approximately ten years, ranging from a few months in patients with one or more risk factors (thrombocytopenia at diagnosis, progression to thrombocytopenia, development of thrombosis, pancytopenia, myelodysplasia or acute leukemia, age over 55 years) to many years in patients with no risk factors.5–7 Unfortunately, risk factors have a limited role in predicting individual patient outcome, given that the natural history of paroxysmal nocturnal hemoglobinuria is widely heterogeneous.
In recent years, the introduction of eculizumab, a humanized monoclonal antibody directed against the terminal complement protein C5, has had a big impact on the management of paroxysmal nocturnal hemoglobinuria. This drug has been shown to reduce hemolysis and greatly improve symptoms and quality of life for these patients.8,9
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only potentially curative therapy for paroxysmal nocturnal hemoglobinuria. Eradication of the PNH clone has been achieved with both myeloablative and reduced-intensity conditioning regimens.10–14 Younger patients with severe manifestations of the disease (high transfusion requirement, severe pancytopenia, life-threatening thrombosis) and availability of an HLA-identical sibling donor are the best candidates for hematopoietic stem cell transplantation. There are few reports on the use of allogeneic transplantation for paroxysmal nocturnal hemoglobinuria, and nearly all of them include small numbers of patients with only one large survey (57 consecutive patients) reported by the International Bone Marrow Transplant Registry.15 Here, we describe the outcome of hematopoietic stem cell transplantation in 26 patients with paroxysmal nocturnal hemoglobinuria reported to the Gruppo Italiano Trapianto di Midollo Osseo (GITMO).
Design and Methods
Patients and donors
Between July 1988 and November 2006, 26 patients (16 males, 10 females) affected by paroxysmal nocturnal hemoglobinuria received an allogeneic hematopoietic stem cell transplantation in 10 Italian transplant centers. At time of transplant, median age was 32 years (range 22–60). In all cases, diagnosis was confirmed both by clinical picture and hematologic test (Ham’s test and/or flow cytometry analysis to CD55 and CD59). In one case, paroxysmal nocturnal hemoglobinuria was discovered at first complete remission of acute myeloid leukemia.
Patients’ characteristics at time of transplant are shown in Table 1. Median time from diagnosis to transplant was 33 months (range 3–208). The majority of patients (21 out of 26; 81%) were transfusion-dependent. Median number of red blood cell and platelet transfusions was 30 (range 4–500) and 33 (range 6–86), respectively. Moreover they had received various treatments before transplantation such as steroids, androgens, cyclosporine, anti-thymocyte globulin, and growth factors. Two patients underwent splenectomy before transplant.
Table 1.
Patients’ characteristics at time of transplantation.
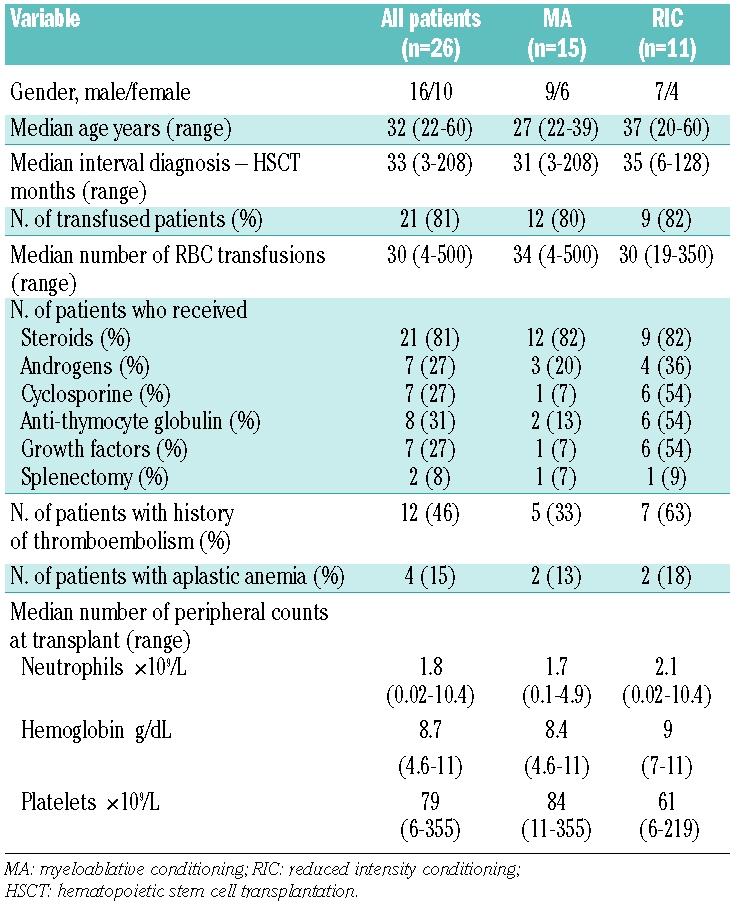
Twelve patients (46%) had a history of pre-transplant thromboembolism involving the venous system in all cases. In particular, 18 events were reported with involvement of the spleno-portal vein in 6, lower limb vein in 4, hepatic vein (Budd-Chiari syndrome) in 3, cerebral vein in 3, and mesenteric vein in 2.
In all patients the median blood counts were: hemoglobin 8.7 g/dL (range 4.6–11), polymorphonuclear cells 1,780×109/L (range 20–10,400), platelets 79×109/L (range 6–355). The median value of lactate dehydrogenase was 2940 IU/L (range 1950 to 6800). The main indication for hematopoietic stem cell transplantation was transfusion-dependent hemolytic anemia in 5 patients, aplastic anemia in 4 and recurrent thromboembolic disease in 4. Thirteen patients (50%) fulfilled more than one indication for transplant (hemolytic anemia and thromboembolic disease in 8, hemolytic anemia and bone marrow failure in 5).
Of the 26 donors (12 male, 14 female), 23 were HLA-identical (22 siblings and one unrelated) and 3 were not HLA-identical [antigen mismatched sibling (n=1), haploidentical mother (n=1), antigen mismatched unrelated (n=1)]. Median age of donors was 33 years (range 20–59).
Transplant procedure
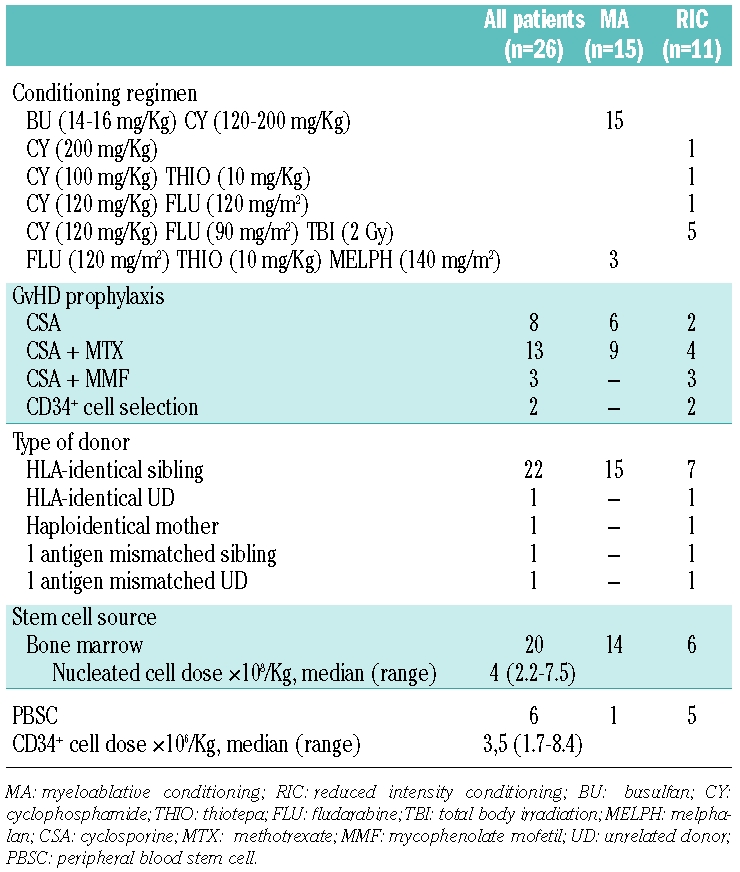
Table 2 shows some details of transplant procedure. Fifteen patients received myeloablative regimen consisting of oral busulfan and cyclophosphamide. Eleven patients received reduced intensity conditioning including cyclophosphamide, thiotepa, fludarabine, total body irradiation and melphalan in different combinations. In addition to reduced intensity conditioning, 3 patients received anti-thymocyte globulin (Thymoglobuline, Genzyme) and 2 patients were given alemtuzumab (Campath-1, Genzyme). Three of these 5 patients were transplanted from a non-HLA identical donor.
Table 2.
Details of transplant procedure.
Acute and chronic graft versus host disease were diagnosed and graded according to the Seattle criteria.16,17 The date of marrow infusion was designated as day 0. Neutrophil and platelet engraftment was defined as the first of the 3 consecutive days with neutrophils over 0.5×109/L and platelets over 50×109/L, respectively. Chimerism was documented by fluorescent in situ hybridization when recipient and donor were sex-mismatched (n=12) and by analysis of a variable number of tandem repeat polymorphisms and by microsatellite analysis of bone marrow and/or peripheral blood samples in case of matched pairs (n=14). Graft rejection was defined either as the persistence of marrow aplasia at day 28 post-grafting (primary graft failure) or the development of late marrow aplasia following full donor engraftment (secondary graft failure).
The protocol was approved by the Institutional Review Board (IRB) of all the GITMO and participating centers. All treatment complied with the local guidelines; data were collected retrospectively through a GITMO survey, complying with national and local IRB guidelines.
Statistical evaluation
The Kaplan-Meier method was used to evaluate the probability estimates of overall survival and disease-free survival.18 Disease-free survival was defined as the probability of being alive free of paroxysmal nocturnal hemoglobinuria. The reference date for calculating statistics was June 30 2009. The probability of acute and chronic graft versus host disease, graft failure and transplant-related mortality was evaluated applying the cumulative incidence method taking into account the competing risks. Gray’s method was used to compare the cumulative incidence of transplant-related mortality between the 2 conditioning groups. The Kaplan-Meier method was also used to estimate disease-free survival rate and the standard error at five years of follow-up for 23 patients transplanted from identical donor after stratifying for patient-related characteristics (age, number of red blood cell transfusions, grade of neutropenia, history of pre-transplant thromboembolism) and transplant-related factors (intensity of conditioning regimen, interval between diagnosis and transplant, year of transplant). Statistical significance between curves was evaluated using the log-rank test.
All statistical analyses were performed using SPSS® Advanced StatisticalTM 11 (2004, Chicago, IL, USA) software package and the “cmprsk” function of R open source software.
Results
Twenty-five patients (96%) achieved primary sustained engraftment with a median time of 17 days (range 10–38) to reach 0.5×109/L neutrophils and 27 days (range 11–322) to reach 50×109/L platelets. There was no significant difference in the rate of engraftment for patients transplanted following myeloablative conditioning compared to patients who received reduced intensity conditioning.
The cumulative incidence of graft failure was 8% (4% primary and 4% secondary graft failure). One patient showed primary graft failure with no sign of engraftment at day 28 following myeloablative transplant. This patient, who received about 500 red blood cell transfusions before transplant, was given a second transplant from the same HLA-identical sibling donor following conditioning with cyclophosphamide and anti-thymocyte globulin. She regularly engrafted but died from grade IV acute graft versus host disease on day 63 post-transplant.
One patient showed secondary graft failure with loss of engraftment at day 244 following reduced intensity conditioning transplant. She received a second transplant from the same donor following conditioning with fludarabine and 2 Gy total body irradiation and engrafted, but died from infection on day 400 from the first transplant.
The overall cumulative incidence of grade II–IV acute GvHD was 42%. Among the 15 patients in the myeloablative conditioning group, 6 had grade II and 2 grade III–IV acute GvHD. Among the 11 patients in the reduced intensity conditioning group, 2 patients had grade II and one grade III–IV acute GvHD.
The overall cumulative incidence of chronic GvHD in 20 evaluable patients was 50%, whereas the cumulative incidence of extensive chronic GvHD was 16%. In 13 evaluable patients in the myeloablative conditioning group, 7 showed limited and 2 extensive chronic GvHD. Among the patients who received RIC, only one of the 7 evaluable had extensive chronic GvHD.
The cumulative incidence of transplant-related mortality at 12 months was 26% in the myeloablative conditioning group with 4 patients who died because of acute (n=1) and chronic (n=1) GvHD, and infection (n=2). In the reduced intensity conditioning group, the cumulative incidence of transplant-related mortality at 12 months was 63%. The difference is statistically significant (P=0.01). In particular, 4 patients out of 8 transplanted from identical donor died because of multiorgan failure (n=1), infection (n=1), chronic graft versus host disease (n=1), progression of Budd-Chiari syndrome (n=1), whereas all 3 patients transplanted from non-identical donor died because of multiorgan failure (n=1), infection (n=1), Epstein-Barr virus related lymphoma (n=1). Of the 5 patients receiving anti-thymocyte globulin (n=3) or alemtuzumab (n=2) as part of the reduced intensity conditoning therapy, only one who received anti-thymocyte globulin is living and disease-free.
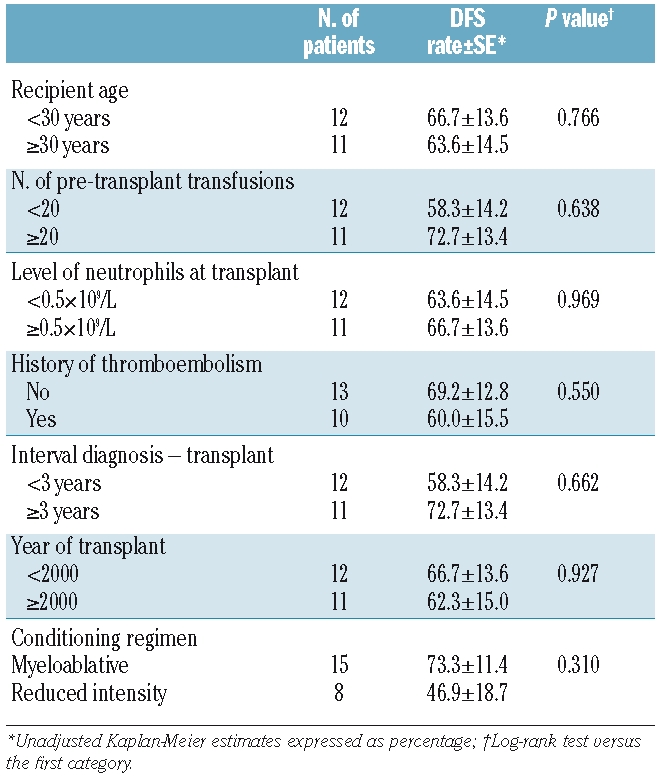
As of June 30 2009, 15 patients (11 in the myeloablative conditioning group and 4 in the reduced intensity conditioning group) are alive with complete hematologic recovery and no evidence of paroxysmal nocturnal hemoglobinuria. The median follow-up is 131 months (range 30–240) for all patients and 152 months (range 36–240) and 57 months (range 30–85) for patients receiving myeloablative conditioning or reduced intensity conditioning respectively. Figure 1 shows the 10-year Kaplan-Meier probability of disease-free survival for all 26 patients (57%) and for the 23 patients transplanted from identical donor (65%). Performance status of surviving patients at last visit was excellent with a Karnofsky score of 90–100% for all patients. No new thromboembolic event was reported following hematopoietic stem cell transplantation. Table 3 describes the univariate analysis restricted to the 23 patients transplanted from identical donor. No significant factor influencing the disease-free survival was identified. However, when patients were analyzed according to type of conditioning regimen, the 5-year Kaplan-Meyer probability of disease-free survival was 73% in patients receiving myeloablative conditioning and only 47% in those transplanted following reduced intensity conditioning.
Figure 1.
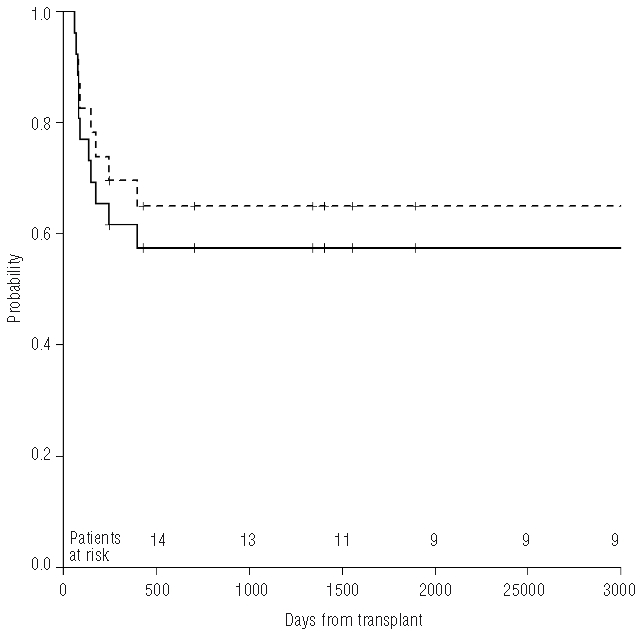
Kaplan-Meier probabilitiy of disease-free survival for 23 patients transplanted from HLA-identical sibling donor (65%, dotted line) and for all 26 patients (57%, continuous line).
Table 3.
Univariate analysis of 5-year disease-free survival for patient-related and transplant-related variables in 23 patients transplanted from HLA-identical donor.
Discussion
In this study we reviewed the outcome in 26 patients with paroxysmal nocturnal hemoglobinuria who received an allogeneic hematopoietic stem cell transplantation following either myeloablative (n=15) or reduced intensity (n=11) conditioning. Our survey includes all consecutive cases transplanted in Italy and reported to the GITMO. Seven patients have been described elsewhere.12 Altogether, 15 out of 26 patients became long-term survivors after a median follow-up of more than ten years. The 8% cumulative incidence of graft failure reported in our series seems far lower than that described in other studies.15 Both patients experiencing graft failure were rescued by a second transplant, even if they finally died because of transplant-related mortality (one graft versus host disease and one infection).
In the 15 patients conditioned with busulfan and cyclophosphamide, the 10-year Kaplan-Meyer probability of disease-free survival was 73% with a transplant-related mortality at one year of 26%. The results achieved in the 11 patients transplanted following reduced intensity conditioning have not been as good as those obtained with myeloablative conditioning. Indeed, it is important to note that 3 of these 11 patients were transplanted from non-identical donor and all of them died from transplant-related complications. Moreover, 5 different conditioning regimens have been used with unavoidable negative impact on the survey. Remarkably, the addition of anti-thymocyte globulin or alemtuzumab in the conditioning regimen resulted in severe toxicity considering that 4 of 5 patients died from transplant-related complications, although 3 of them were nonidentical transplants.
The indirect evidence of a real eradication of PNH clone both with myeloablative or reduced intensity conditioning in our study was the disappearance after transplantation of all clinical and laboratory signs of hemolysis as well as the complete absence of thrombotic events in spite of no post-transplant anticoagulant prophylaxis. Available data suggest that PNH clone size is the major risk factor for thrombosis. About half of our patients experienced one or more thrombotic events before transplantation. Hematopoietic stem cell transplantation eliminated the risk of thrombosis with markedly improved quality of life.
There are few reports on the use of allogeneic reduced intensity conditioning transplant in paroxysmal nocturnal hemoglobinuria patients and all include only a few patients.11, 14 These studies demonstrate that a similar approach may be successful to cure the disease and interestingly suggest that an important “graft-versus-PNH” effect with reduced intensity conditioning transplant could be hypothesized.19
The use of hematopoietic stem cell transplantation to treat paroxysmal nocturnal hemoglobinuria has become less frequent now that there is an effective drug therapy such as eculizumab. Eculizumab is a humanized mono-clonal antibody against C5 that inhibits terminal complement activation and is the first effective drug therapy in decreasing intravascular hemolysis. Binding of C3 by paroxysmal nocturnal hemoglobinuria erythrocytes may constitute an additional disease mechanism, strongly enhanced by eculizumab treatment and producing a variable degree of extravascular hemolysis.20 Eculizumab improves quality of life, reduces or eliminates the need for blood transfusions, and may reduce though not abrogate the risk of thrombosis. Nevertheless, not every patient who has a PNH clone is an appropriate candidate for treatment. In particular, patients with small clones whose primary clinical manifestations are a consequence of bone marrow failure are unlikely to benefit. Eculizumab has no effect on the underlying stem cell defect, meaning that treatment will likely continue indefinitely; thus no patient is expected to be cured by the anti-complement treatment.
In the eculizumab era, the role of hematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuria patients has changed. Before eculizumab, each clinical hallmark of the disease (hemolytic anemia, thrombosis and marrow failure) may have qualified one of these patients for transplant, especially younger patients. Eculizumab is now considered the therapy of choice for treating intravascular hemolysis and related manifestations in transfusion-dependent or symptomatic patients with a large percentage of paroxysmal nocturnal hemoglobinuria cells. Given its efficacy in preventing thromboembolic events, eculizumab may also be considered for PNH patients with recurrent thrombosis, irrespective of the symptoms of hemolysis and even of the PNH clone size. As outlined by Parker et al.,6 hematopoietic stem cell transplantation should be restricted to patients with an underlying bone marrow disease (e.g. aplastic anemia/PNH syndrome), while transfusion-refractory hemolytic anemia should no longer be an indication for transplantation, given that it may be effectively managed by anti-complement therapy. Life-threatening thrombotic disease might still lead to consideration of hematopoietic stem cell transplantation, even if it increases the transplant-related risks by itself. Hematopoietic stem cell transplantation is potentially curative for paroxysmal nocturnal hemoglobinuria patients, but the decision on its use must be evaluated on a case-by-case basis because of the heterogeneous natural history of the disease and of alternative treatments available. To date, there are no data which evaluate the possible impact of a previous eculizumab treatment on transplant outcome. In fact, one can hypothesize that its use may reduce transfusions and thromboembolic risk, possibly leading to a better outcome. However, it has to be said that such possible benefits may be different in case of a short-term pre- (or even peri-) transplant treatment, or in case of a long-term treatment, which delays the transplant procedure (which has been proven detrimental in related disorders, such as aplastic anemia).
Some transplant related issues specific to this disease remain: a) the optimal time for hematopoietic stem cell transplantation; b) the best conditioning regimen, myeloablative or reduced intensity; c) the role for transplantation using an alternative donor. While the optimal time for transplant may be changing in the eculizumab era (at least for patients likely benefiting from anti-complement therapy), as well as the indication for a high-risk transplant procedure (i.e. alternative donors), no conclusion may be drawn about the best conditioning regimen. Taking into account the limited sample size, our study suggest that using a reduced intensity conditioning regimen did not result in any improvement in transplant outcome for paroxysmal nocturnal hemoglobinuria patients, in comparison to standard myeloablative conditioning regimen. Thus, reduced intensity conditioning regimen should be restricted to prospective clinical trials; perhaps some specific reduced intensity conditioning regimens which seem to be as effective as myeloablative conditioning in a single center experience.14
In conclusion, our survey shows that hematopoietic stem cell transplantation may lead to a long-term cure rate as high as 60% in a heterogeneous cohort of seriously ill paroxysmal nocturnal hemoglobinuria patients but improvements in hematopoietic stem cell transplantation procedure are still desirable. While the recent availability of targeted therapy has certainly reduced the indication for hematopoietic stem cell transplantation, its potentially curative use remains a valid therapeutic option for patients candidate to a low-risk transplant procedure.21 Given the rarity of the disease, worldwide cooperative studies are desirable to compare hematopoietic stem cell transplantation with conservative treatments, including newer therapeutic options, as well as to investigate specific issues concerning the transplant procedure in paroxysmal nocturnal hemoglobinuria patients.
Footnotes
Authorship and Disclosures
SS and PDB designed the study, coordinated the data collection, interpreted the results, and wrote the paper; AB, AMR, ET, EDB, API, AR, EA, AS, FP, and ST participated in the result interpretation and revised the paper; MDN performed all the statistical analysis.
All the authors were involved in analysis and interpretation of the results. All authors read, gave comments, and approved the final version of the manuscript.
The authors reported no potential conflicts of interest.
References
- 1.Miyata T, Takeda J, Iida Y, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259(5099):1318–20. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 2.Rosse WF, Ware RE. The molecular basis of paroxysmal nocturnal hemoglobinuria. Blood. 1995;86(9):3277–86. [PubMed] [Google Scholar]
- 3.Brodsky RA. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 2008;22(2):65–74. doi: 10.1016/j.blre.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Socié G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, et al. Paroxysmal nocturnal hemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996;348(9027):573–7. doi: 10.1016/s0140-6736(95)12360-1. [DOI] [PubMed] [Google Scholar]
- 5.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Eng J Med. 1995;333(19):1253–8. doi: 10.1056/NEJM199511093331904. [DOI] [PubMed] [Google Scholar]
- 6.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099–106. doi: 10.1182/blood-2008-01-133918. [DOI] [PubMed] [Google Scholar]
- 8.Hillmen P, Hall C, Marsh JC, Elebute M, Bombara MP, Petro BE, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–9. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 9.Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–43. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- 10.Bemba M, Guardiola P, Garderet L, Devergie A, Ribaud P, Esperou H, et al. Bone marrow transplantation for paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1999;105(2):366–8. [PubMed] [Google Scholar]
- 11.Hegenbart U, Niederwieser D, Forman S, Holler E, Leiblein S, Johnston L, et al. Haematopoietic cell transplantation from related and unrelated donors after minimal conditioning as curative treatment modality for severe paroxysmal nocturnal hemoglobinuria. Biol Blood Marrow Transplant. 2003;9(11):689–97. doi: 10.1016/s1083-8791(03)00264-7. [DOI] [PubMed] [Google Scholar]
- 12.Raiola AM, Van Lint MT, Lamparelli T, Gualandi F, Benvenuto F, Figari O, et al. Bone marrow transplantation for paroxysmal nocturnal hemoglobinuria. Haematologica. 2000;85(1):59–62. [PubMed] [Google Scholar]
- 13.Woodard P, Wang W, Pitts N, Benaim E, Horwitz E, Cunningham J, Bowman L. Successful unrelated donor bone marrow transplantation for paroxysmal nocturnal hemoglobinuria. Bone Marrow Transplant. 2001;27(6):589–92. doi: 10.1038/sj.bmt.1702827. [DOI] [PubMed] [Google Scholar]
- 14.Srinivasan R, Takahashi Y, McCoy JP, Espinoza-Delgado I, Dorrance C, Igarashi T, et al. Overcoming graft rejection in heavily transfused and allo-immunised patients with bone marrow failure syndromes using fludarabine-based haematopoietic cell transplantation. Br J Haematol. 2006;133 (3):305–14. doi: 10.1111/j.1365-2141.2006.06019.x. [DOI] [PubMed] [Google Scholar]
- 15.Saso R, Marsh J, Cevreska L, Szer J, Gale RP, Rowlings PA, et al. Bone marrow transplants for paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1999;104(2):392–6. doi: 10.1046/j.1365-2141.1999.01195.x. [DOI] [PubMed] [Google Scholar]
- 16.Nash RA, Pepe MS, Storb R, Longton G, Pettinger M, Anasetti C, et al. Acute graft-versus-host disease: analysis of risk factors after allogeneic marrow transplantation and prophylaxis with cyclosporine and methotrexate. Blood. 1992;80(7):1838–45. [PubMed] [Google Scholar]
- 17.Sullivan KM, Agura E, Anasetti C, Appelbaum F, Badger C, Bearman S, et al. Chronic graft-versus-host disease and other late complications of bone marrow transplantation. Semin Hematol. 1991;28(3):250–9. [PubMed] [Google Scholar]
- 18.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 19.Brodsky RA. How I treat paroxysmal nocturnal emoglobinuria. Blood. 2009;113(26):6522–7. doi: 10.1182/blood-2009-03-195966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal emoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–100. doi: 10.1182/blood-2008-11-189944. [DOI] [PubMed] [Google Scholar]
- 21.Matos-Fernandez NA, Abou Mourad YR, Caceres W, Kharfan-Dabaja MA. Current status of allogenic hematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuria. Biol Blood Marrow Transplant. 2009;15(6):656–61. doi: 10.1016/j.bbmt.2008.12.507. [DOI] [PubMed] [Google Scholar]