


Abstract
Dosage compensation in Drosophila melanogaster involves the selective targeting of the male X chromosome by the dosage compensation complex (DCC) and the coordinate, ∼2-fold activation of most genes. The principles that allow the DCC to distinguish the X chromosome from the autosomes are not understood. Targeting presumably involves DNA sequence elements whose combination or enrichment mark the X chromosome. DNA sequences that characterize ‘chromosomal entry sites’ or ‘high-affinity sites’ may serve such a function. However, to date no DNA binding domain that could interpret sequence information has been identified within the subunits of the DCC. Early genetic studies suggested that MSL1 and MSL2 serve to recognize high-affinity sites (HAS) in vivo, but a direct interaction of these DCC subunits with DNA has not been studied. We now show that recombinant MSL2, through its CXC domain, directly binds DNA with low nanomolar affinity. The DNA binding of MSL2 or of an MSL2–MSL1 complex does not discriminate between different sequences in vitro, but in a reporter gene assay in vivo, suggesting the existence of an unknown selectivity cofactor. Reporter gene assays and localization of GFP-fusion proteins confirm the important contribution of the CXC domain for DCC targeting in vivo.
INTRODUCTION
Eukaryotic genomes are balanced systems of gene expression that rely on co-regulation of groups of genes by shared regulatory sequence determinants. A powerful model to study such co-regulation is presented by the process of dosage compensation in Drosophila melanogaster. Female fruit flies have two X chromosomes, whereas males have only a single X in addition to a gene-poor Y chromosome. In the absence of compensation this unequal dose of sex chromosomes leads to a lethal imbalance of gene expression in males (1–3). In order to counteract this imbalance, the transcription output of most genes on the male X chromosome is increased by roughly 2-fold, independent of their actual expression levels, to match the combined transcription from the two female X chromosomes (4,5). The activation is achieved by the action of the dosage compensation complex (DCC, also known as MSL complex), which consists of five male-specific lethal (MSL) proteins and the two non-coding roX RNAs. The DCC almost exclusively associates with the male X chromosome where it activates transcription by specific acetylation of histone H4 at lysine 16 (H4K16ac) via its histone acetyltransferase MOF (6–8).
The mechanism by which the DCC distinguishes the X chromosome from the autosomes and which mediates its specific association with the X is unclear to date. Chromosome-wide high-resolution mapping of DCC through chromatin immunoprecipitation coupled to DNA microarrays (ChIP-chip analysis) showed that the bulk of DCC interacts with the coding sequence of actively transcribed genes. This suggested that dosage compensation acts at the level of transcriptional elongation rather than in initiation (9,10). The DCC binding profile closely follows that of lysine 36 methylation of histone H3 (H3K36me3), a modification placed co-transcriptionally (11). This can be explained, at least in part, by the ability of MSL3 to interact with H3K36me3 through its chromodomain (12,13). In agreement with these findings, genes need to be transcribed in order to be targets for DCC association (14,15). However, transcribed chromatin methylated at H3K36 is not specific for the X chromosome and therefore does not qualify as primary targeting determinant. According to the prevailing concepts, DNA sequences are involved.
First evidence for X chromosomal sites of higher affinity for the DCC has been obtained from the ectopic expression of the male-specific subunit MSL2 in females, which leads to transcription of the roX RNA and—since all other components are expressed to some levels in female cells—to assembly of the DCC. Females are thus transformed into ‘pseudo-males’, which allows assessing the binding of the DCC to larval polytene chromosomes. Importantly, DCC levels and integrity can be tuned by genetic means to determine the requirements for X chromosome binding (16). Early studies of this kind revealed that for complete coating of the X chromosome an intact DCC is required. Nevertheless, the targeting function resides within the protein subunits of the DCC and does not require the roX RNAs (17). Furthemore, a module consisting of only MSL2 and MSL1 sufficed to recognize a subset of sites (16,18,19). The same sites were still occupied when the MSL complex concentrations were limited, leading to the idea of a hierarchy for binding sites (20,21) and a stepwise model for X chromosome targeting and distribution (22–24). Accordingly, the MSL2–MSL1 module recognizes a small number of primary targeting elements on the X that are called the ‘chromosomal entry sites’ (CES) (22–24), or ‘high-affinity sites’ (HAS) (20,25). In subsequent steps the DCC would transfer to secondary sites of lower affinity (including transcribed chromatin) in the vicinity, hence limiting the distribution to the X chromosome (1,26). Notably, efficient distribution over the entire chromosome only occurs if all DCC subunits are intact (27,28).
Common to all high-affinity chromosomal entry sites is their ability to recruit the DCC when inserted into an autosome, which suggests DNA sequence could be a major determinant for a HAS (20,22,23,29). Several studies suggested that the clustering and combination of particular sequence elements, such as degenerate guanine-adenine (GA) repeats, may contribute to defining a HAS (20,23,25,29–31) but so far it is not possible to predict a HAS from DNA sequence alone. It is clear that chromatin organization must also play a role since HAS tend to reside in nucleosome-free regions (22,23,25,29–31). Recently, we showed that HAS also tend to associate in the volume of male nuclei, which hints at a particular conformation of the X chromosome that depends on MSL2–MSL1 (32).
The X chromosomal DNA determinants of DCC targeting are poorly understood, but even less is known about DNA binding domains within the DCC subunits. It seems clear that MSL2 and MSL1 are able to associate with HAS in the absence of other subunits (18,19), but neither of them has an obvious DNA binding domain. In MSL1, a short N-terminal region was suggested to be involved in X chromosome association in vivo (33). MSL2 is characterized by several domains: a RING finger mediates the interaction with MSL1 (18), a conserved cysteine-rich (CXC) domain of unknown function and a basic, proline-rich patch (Pro/Bas patch). Recently, Scott and colleagues (34) have analysed the consequences of C- or N-terminal deletions of MSL2 for chromosomal targeting in transgenic flies and concluded that C-terminal sequences including the Pro/Bas patch may mediate incorporation of roX RNA into the DCC and hence affect chromosomal interactions of MSL2. However, naturally these genetic experiments were unable to reveal direct, molecular interactions.
Employing a biochemical approach, we have now searched for a DNA binding domain within recombinant MSL2 and MSL1 proteins by measuring their affinities to defined DNA sequences, including HAS. We show that the CXC domain of MSL2 can mediate the DNA binding of the MSL2–MSL1 heteromer. The importance of the DNA binding function of the CXC domain and for X chromosome targeting in vivo is confirmed by reporter gene assays and localization studies involving GFP fusion proteins in Drosophila cells.
MATERIALS AND METHODS
Cloning of MSL constructs
For heterologous expression and purification, MSL constructs were cloned with C-terminal FLAG tags into the pFastBac1 vector, which was used in the Bac-to-Bac expression system (Invitrogen) to create recombinant baculoviruses. For transient transfections and reporter gene assays MSL2 constructs were fused to a C-terminal VP16 activation domain (VP16-AD) by cloning the coding sequence into the previously described pVP16 vector (25). For the creation of stable Drosophila SL2 cells and immunofluorescence stainings MSL2 constructs were fused to a C-terminal GFP by subcloning the coding sequence into the previously described pHSP70-EGFP vector (35). Point mutations in MSL2 (C544A/C546A and Y547A) were introduced by site-directed mutagenesis using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). The HsCXC domain from the human protein KIAA1585 was isolated via PCR from cDNA of HeLa cells and cloned into the vectors described above. The CXC domain was additionally cloned into the pGEX-2KG expression vector (Amersham) for expression as a GST fusion protein in Escherichia coli BL21-CodonPlus (DE3)-RIL cells (Stratagene). The identity of all plasmids was confirmed by sequencing.
Heterologous expression of MSL proteins
MSL proteins were expressed in Sf21 cells using recombinant baculoviruses. Wild-type MSL2 and MSL1, as well as all truncated or mutated MSL2 versions contained C-terminal FLAG-tags. The MSL2–MSL1 complex was purified from cells co-expressing untagged MSL1 and FLAG-tagged MSL2. Baculovirus infections were carried out in shaker flasks at a cell density of 1 × 106 cells/ml in Sf-900 II SFM medium supplemented with 9% FBS at 27°C and 75 r.p.m. for 2 days. The expression of the GST tag and the GST-CXC domain in E.coli was induced at OD600 = 0.7 – 0.8 with 0.3 mM IPTG for 2 h at 20°C. Harvested E. coli and Sf21 cells were washed with ice-cold PBS, frozen in liquid nitrogen and stored at −80°C.
Purification of recombinant MSL proteins
Sf21 cell pellets were rapidly thawed and resuspended in ice-cold Extraction Buffer EB (50 mM Hepes/KOH pH 7.6, 5% glycerol, 0.05% NP-40, 0.5 mM EDTA, 1 mM MgCl2, protease inhibitors Aprotinin 1 µg/ml, Leupeptin 1 µg/ml and Pepstatin 0.7 µg/ml) containing 300 mM KCl (EB300). 15 ml EB300 was added to the cell pellet (250 × 106 cells). After 10 min incubation on ice, the suspension was sonicated (4 × 20 s pulses, 20% amplitude, Branson digital sonifier model 250-D) and centrifuged twice (30 and 15 min at 30 000 g at 4°C). The soluble protein fraction was incubated with equilibrated FLAG beads (Anti-FLAG M2 Agarose, Sigma) for 2.5 h at 4°C on a rotating wheel. Two hundred and fifty microliters beads were used per 250 × 106 cells. The beads were washed several times with ice-cold EB300 and high-salt EB1000. The FLAG-tagged MSL proteins were eluted for 2.5 h at 4°C on a rotating wheel in the presence of 0.5 mg/ml FLAG-Peptide (Sigma) in EB100 for MSL2 and in EB300 for MSL1 and the MSL2–MSL1 complex.
The GST-CXC fusion protein was purified from E.coli cells according to standard protocols and finally eluted from glutathione sepharose using 40 mM glutathione in 200 mM Tris/HCl pH 8.0, 150 mM NaCl, 10% glycerol, 0.05% NP-40, 50 µM ZnCl2, 1 mM DTT and protease inhibitors (see above).
The eluted MSL proteins were further concentrated by using Amicon Ultra-4 centrifugal filter devices (50 or 3 kDa exclusion limit, Millipore). Purified proteins were then rapidly frozen in liquid nitrogen and finally stored at −80°C. Protein concentrations were determined via SDS–PAGE and Coomassie staining using BSA (New England Biolabs) as a standard.
Preparation of oligonucleotides for electrophoretic mobility shift assays
Double stranded (ds) DNA and RNA fragments for electrophoretic mobility shift assays were obtained by annealing equimolar concentrations of complementary oligonucleotides in 10 mM Tris/HCl pH 7.5, 50 mM NaCl, 0.1 mM EDTA by slowly cooling from 95°C to room temperature.
DBF12-L15 DNA: 5′-TGCGGCCATCTCTTTCGTTTTGATGTTTCTACGCCATGTG-3′ and 5′-CACATGGCGTAGAAACATCAAAACGAAAGAGATGG-3′.
DBF12-L18 DNA: 5′-TGCGGCCAAAAAATTCGTTTTGATGTTTCTACGCCATGTG-3′ and 5′-CACATGGCGTAGAAACATCAAAACGAATTTTTTGG-3′.
DBF12-L15 RNA:5′-UGCGGCCAUCUCUUUCGUUUUGAUGUUUCUACGCCAUGUG-3′ and 5′-CACAUGGCGUAGAAACAUCAAAACGAAAGAGAUGGCCGCA-3′. The 5′-overhang left by the dsDNA fragments was filled in by the Klenow enzyme with [α-32P]-dCTP. The dsRNA fragments were end-labeled with [γ-32P]-dATP by T4 polynucleotide kinase. The longer DNA fragments 3 × DBF-L15 (163 bp) and the multiple cloning site (170 bp) of pBluescript KS+ (Stratagene) were obtained by digestion with either XhoI/BamHI (pP12eL(15)3) or PvuII/XmaI (pBluescript KS+). 5′-Overhangs of both DNA fragments were filled in by the Klenow enzyme with [α-32P]-dCTP. All radiolabeled duplexes were purified using the QIAquick Nucleotide Removal Kit (Qiagen).
Electrophoretic mobility shift assays
Purified MSL proteins were incubated with sub-saturating concentrations of radiolabeled DNA or RNA fragments (<0.2 nM) in 50 mM Hepes/KOH pH 7.6, 100 mM KCl, 5% glycerol, 0.05% NP-40, 0.5 mM EDTA, 1 mM MgCl2 and 0.1 µg/µl BSA (New England Biolabs) in a total volume of 12 µl. The binding reactions were started by adding the MSL protein and were analyzed after 15 min incubation at 25°C on non-denaturing 1.2% agarose (12 × 7 cm) or 5% polyacrylamide gels (Novex Mini-Cell system, Invitrogen) in 0.5 × TBE at 20°C. Gels were dried and radiolabeled nucleic acids were visualized by using a Phosphor-Imager (FujiFilm FLA-3000). For competition experiments MSL proteins were first incubated at a concentration close to their KD value (50 nM) with sub-saturating concentrations of radiolabeled DNA for 15 min at 25°C. Then unlabeled competitor DNA or RNA was added. After additional 20 min incubation at 25°C the reactions were analyzed by non-denaturing electrophoretic mobility shift assays (EMSA) gels as described above.
Calculation of affinity constants
Quantification of EMSA gels was performed with the Aida Image Analyzer software. The signal of nucleic acid-bound protein (AB) and the signal of total nucleic acid (Atotal) were quantified and the fraction bound (AB/Atotal) calculated. Binding curves were obtained by performing non-linear regression with KaleidaGraph using a standard bimolecular model: 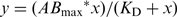 , where y is the fraction bound, x is the concentration of protein, ABmax is the maximum of AB and KD is the affinity constant. For competition experiments, first AB was calculated from the difference between the signal of total nucleic acid (Atotal) and the signal of free nucleic acid (Afree). Then the fraction bound was calculated (AB/Atotal) and normalized to the fraction bound measured at 0 nM competitor. Competition curves were obtained with the following competition model:
, where y is the fraction bound, x is the concentration of protein, ABmax is the maximum of AB and KD is the affinity constant. For competition experiments, first AB was calculated from the difference between the signal of total nucleic acid (Atotal) and the signal of free nucleic acid (Afree). Then the fraction bound was calculated (AB/Atotal) and normalized to the fraction bound measured at 0 nM competitor. Competition curves were obtained with the following competition model: 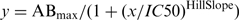 , where y is the normalized fraction bound, x is the concentration of competitor, ABmax is the maximum of AB at 0 nM competitor, IC50 is the half maximal inhibitory concentration and Hill Slope describes the steepness of the curve. In Table 1 affinity constants and IC50 values are shown as the mean value plus and minus the standard deviation from several replicates obtained with at least two independent protein preparations.
, where y is the normalized fraction bound, x is the concentration of competitor, ABmax is the maximum of AB at 0 nM competitor, IC50 is the half maximal inhibitory concentration and Hill Slope describes the steepness of the curve. In Table 1 affinity constants and IC50 values are shown as the mean value plus and minus the standard deviation from several replicates obtained with at least two independent protein preparations.
Table 1.
Comparison of binding affinities of different recombinant MSL2 derivatives to 40 bp DBF12-L15 dsDNA and RNA
| MSL protein | KD dsDNA [nM] | KD dsRNA [nM] |
|---|---|---|
| MSL2 | 33 ± 13 | 21 ± 4 |
| ΔCXC | 189 ± 18 | 29 ± 12 |
| ΔRING | 51 ± 5 | 23 ± 5 |
| ΔPro / Bas | 61 ± 5 | 37 ± 10 |
| C544A/C546A | 82 ± 4 | |
| Y547A | 81 ± 5 | |
| HsCXC | 33 ± 9 | |
| MSL2-MSL1 | 26 ± 15 | |
| MSL1 | no binding |
KD values were calculated from binding curves obtained from quantification of EMSA gels and fitting to a standard bimolecular model. Mean values are shown plus and minus the standard deviation from several replicates obtained with at least two independent protein preparations. For details see the ‘Materials and Methods’ section.
Reporter gene assays
Reporter gene assays in male D. melanogaster SL2 cells were performed as described (25). In brief, 0.5 × 106 cells were transfected with 15 ng of a renilla luciferase construct, 315 ng reporter gene construct carrying the DBF12 binding site attached to an inducible firefly luciferase reporter gene, and 160 ng of a MSL2-VP16 activator construct using Effectene (Qiagen). After 2 days the luciferase activities were measured by using the Dual Luciferase Kit (Promega) and normalized to renilla activity (normalized RLU). Induction was calculated from the ratio of normalized RLU of MSL2-VP16 to that of the VP16 activation domain alone. Fold induction is shown as the mean value plus and minus the standard deviation from several replicates obtained with at least two independent plasmid preparations. The expression of the MSL2-VP16 constructs was verified by Western blots using a rabbit anti-MSL2 antibody (Supplementary Figure S7).
Generating stable cell lines
Drosophila SL2 cells (0.5 × 106 cells) were cotransfected with 400 ng of an pHSP70-EGFP-MSL construct and 20 ng of the pCoBlast selection vector (Invitrogen) using Effectene (Qiagen). Stable cells were selected using Blasticidin (25 ng/µl) and were kept under constant selection pressure (20 ng/µl Blasticidin) at 26°C in Schneider’s Drosophila medium (Invitrogen) supplemented with l-glutamine and 9% FBS. The expression of the MSL2-GFP constructs was checked by Western blots using a rabbit anti-MSL2 antibody (Supplementary Figure S7).
Immunofluorescence staining
Immunofluorescence on Drosophila SL2 cells that express a GFP-MSL2 construct was performed as described (36). An anti-GFP antibody (Molecular Probes) was used to visualize the MSL2-GFP constructs. Localization of endogenous MSL1 was detected by rabbit anti-MSL1 serum (kindly provided by E. Schulze). Pictures were taken at 1200 × magnification using a Zeiss Axiovert microscope coupled to a CCD Camera (AxioCamMR, Zeiss). Images were level adjusted in Adobe Photoshop CS4.
RESULTS
MSL2 is the DNA binding factor in the MSL2–MSL1 complex
Following the suggestions from the genetic studies (18,19), we hypothesized that MSL2–MSL1 may directly bind to DNA sequences that characterize HAS. As a candidate high-affinity sequence, we used the DCC-binding fragment (DBF) DBF12-L15, which we had previously isolated as a minimal sequence necessary for recruitment of MSL2 in a reporter gene assay (25). The 40 bp element contains a GA-rich sequence that conforms to the consensus sequences defined by chromosome-wide analyses (30,31). Insertion of a trimerized element into an autosome created one of the strongest DCC recruitment sites seen so far (25). Mutating the GA motif to a stretch of thymidines (DBF12-L18) destroyed the function as a DCC recruitment site (25).
We expressed MSL2, MSL1 and the MSL2–MSL1 complex from baculovirus vectors in Sf21 cells, purified the proteins via a C-terminal FLAG tag (Figure 1A and C) and tested their ability to bind to DBF12-L15 in electrophoretic mobility shift assays (EMSAs). MSL2 bound to DBF12-L15 with an affinity in the low nanomolar range (33 ± 13 nM) as derived from the binding curve (Figure 2A and C; Table 1). In contrast, MSL1 did not form a stable complex with this DNA fragment (Figure 2A and C; Table 1). Since MSL1 and MSL2 may cooperate for DNA binding in vivo we also assayed an MSL2–MSL1 complex (Figure 2B). The affinity of the MSL2–MSL1 complex to DBF12-L15 was similar to the affinity of MSL2 alone (Figure 2C and Table 1). Clearly, the DNA binding potential of the MSL2–MSL1 complex is contained within MSL2.
Figure 1.

Recombinant MSL derivatives investigated in this study. (A) Schematic representation of MSL2 domain organization and various MSL2 expression constructs. All MSL2 constructs contain a C-terminal FLAG tag. Numbers correspond to the amino acid positions in full-length MSL2. (B) Alignment of orthologue CXC domains from the Drosophila melanogaster MSL2 protein (DmCXC) and from the H. sapiens protein KIAA1585 (HsCXC). Black boxes highlight the conserved cysteines (37). Arrows indicate the introduced point mutations. (C) Coomassie-stained SDS–polyacrylamide gel of purified recombinant MSL proteins.
Figure 2.

Binding of recombinant MSL proteins to a DNA high-affinity site in vitro. Electrophoretic mobility shift assay. Increasing concentrations (from 5 to 250 nM) of (A) MSL2 and MSL1 or (B) MSL2 and the MSL2–MSL1 complex were incubated with radiolabeled 40 bp DBF12-L15 dsDNA and protein–DNA complexes were separated from unbound DNA in non-denaturing agarose gels. (C) Binding curves obtained from quantification of (A) and (B) and fitting to a standard bimolecular model (see ‘Materials and Methods’ section).
The CXC domain of MSL2 is necessary but not sufficient for DNA binding
Since MSL2 does not contain a typical DNA binding motif, we explored whether the known MSL2 domains are involved in DNA binding. We focused on three candidate domains: The RING finger, the CXC domain and the Pro/Bas patch, a C-terminal sequence rich in prolines and basic amino acids. We constructed and purified MSL2 derivatives that lack each of these domains (Figure 1A and C) and measured their affinity to the DBF12-L15 fragment by EMSA (Figure 3 and Table 1). Deletion of the RING finger or the Pro/Bas patch reduced the affinity of MSL2 to the DNA only modestly (<2-fold). On the other hand the deletion of the CXC domain strongly reduced the affinity to the DNA (189 ± 18 nM versus 33 ± 13 nM, see Table 1). To explore whether the CXC domain alone would be able to bind DNA we fused the domain to glutathione-S-transferase (GST) or to a FLAG-tag and tested the fusion protein for DNA binding. The CXC-GST and CXC-FLAG bound DNA at least a 1000-fold less well than full length MSL2 (Supplementary Figure S1). These data suggest that the CXC domain is necessary but not sufficient to endow MSL2 with robust DNA binding and that additional regions of MSL2 are required.
Figure 3.

Binding of different recombinant MSL2 versions to a DNA high-affinity site in vitro. Electrophoretic mobility shift assay as in Figure 2 with increasing concentrations of MSL2 and MSL2-ΔCXC (A) or MSL2 and MSL2-ΔRING (B) or MSL2 and MSL2-ΔPro/Bas (C). (D) Binding curves obtained from quantification of (A–C) and fitting to a standard bimolecular model.
Point mutations in the CXC domain had been shown earlier to delay the development and to reduce viability of male transgenic flies (19). When the corresponding cysteine to alanine mutations (Figure 1B) were introduced into the CXC domain the affinity of recombinant MSL2 for DNA dropped from 33 to 82 nM, suggesting that DNA binding of MSL2 is an important aspect of DCC function. A similar drop in affinity was also observed if tyrosine 547, adjacent to C546, was replaced by alanine (Table 1 and Supplementary Figure S2). This latter substitution leaves all zinc-coordinating cysteines intact and thus minimizes the risk of a general unfolding of the structure.
An important DNA binding function may be conserved through evolution. Dosage compensation mechanisms are poorly conserved, however, an MSL2 homolog of unknown function is present in Homo sapiens (37). The HsMSL2 also contains the CXC scaffold and several conservative amino acids changes, yet most amino acids between the CXC domains differ (Figure 1B). To assess the DNA binding properties of the human CXC domain, we generated a chimeric MSL2 protein where the CXC domain in the fly MSL2 was replaced by the corresponding human sequence (HsCXC, Figure 1B and C). Strikingly, the HsCXC protein exhibited a similar affinity for DNA as Drosophila MSL2 (Table 1 and Supplementary Figure S3). This functional conservation despite of very limited sequence similarity suggests that DNA binding is an important property of MSL2.
MSL2 has a higher affinity for DNA over RNA
The in vivo cross-linking analyses had pointed to sequence motifs that are enriched in HAS, but they could not distinguish whether these sequences attract the DCC as dsDNA, as melted, single stranded (ss) DNA or as RNA. We therefore determined the relative affinities of MSL2 for the DBF12-L15 sequence presented in the form of ssDNA, dsDNA, ssRNA or dsRNA in a series of competition assays (Figure 4). Neither ssDNA nor ssRNA competed efficiently with the MSL2-bound dsDNA. In contrast, dsRNA was able to compete for the binding, albeit with a lower relative affinity compared to dsDNA (IC50 dsDNA of 13.0 ± 6 nM versus IC50 dsRNA of 36 ± 8 nM). The competition curve for dsRNA reveals a slight sigmoid shape, raising the possibility that there might be more than one binding site for RNA.
Figure 4.

Competition assays to determine the binding preference of MSL2 for different nucleic acids. (A) Electrophoretic mobility shift assay. Fifty nanomolar of MSL2 was incubated with the radiolabeled 40 bp DBF12-L15 dsDNA and then increasing concentrations (from 5 to 2080 nM) of unlabeled competitor nucleic acids were added. Representative examples of competition with ds- and ssDNA are shown. (B) Competition curves obtained from quantification of data such as in (A), but also including ssRNA and dsRNA as competitors and fitting to the model as described in ‘Materials and Methods’ section.
The possibility that MSL2 might directly bind dsRNA was new and unexpected given that the association of MSL2 with the X chromosome resists extensive RNase treatment of nuclei (38,39). To determine whether the RNA binding function mapped to any of the domains in question, we measured the affinity of the different MSL2 deletion variants to dsRNA (Figure 5 and Supplementary Figure S4). Interestingly, the affinity of MSL2 for RNA was not affected significantly by deletion of the RING or CXC domains. This highlights the function of the CXC domain as a DNA binding domain. On the other hand, the region(s) responsible for RNA binding must reside elsewhere in the protein. The Pro/Bas patch may contribute to this as its deletion led to a modest 1.5-fold reduced affinity for RNA.
Figure 5.

Binding of different recombinant MSL2 derivatives to RNA. Electrophoretic mobility shift assays. Increasing concentrations of MSL2 versions were incubated with radiolabeled dsRNA of DBF12-L15 and protein–RNA complexes were separated from unbound RNA in non-denaturing agarose gels. A representative example of MSL2-ΔCXC binding to dsRNA is shown. (B) Binding curves obtained from quantification of EMSAs as in (A), but including MSL2-ΔRING and MSL2-ΔPro/Bas and fitting to a standard bimolecular model.
DNA binding of MSL2 is sequence-independent but length-dependent
The observation that the CXC domain enables MSL2 to bind DNA with nanomolar affinity suggests that it could be directly involved in targeting the DCC to the X chromosome. To explore this possibility, we tested whether MSL2 was able to discriminate between DBF12-L15 and a derivative, DBF12-L18, which can no longer recruit MSL2 in a cell-based reporter gene assay due to mutation of the GA-rich consensus element (25). The affinity of MSL2 for both DNA fragments was in the same range (KD of 33 ± 13 nM versus 23 ± 8 nM, Figure 6), indicating that MSL2 did not bind the HAS sequence preferentially. An MSL2–MSL1 complex also bound the two fragments with similar affinity (data not shown), which shows that the MSL1 interaction does not increase MSL2’s discriminative power.
Figure 6.

Binding of recombinant MSL2 to different DNA fragments in vitro. Electrophoretic mobility shift assay. Increasing concentrations of MSL2 were incubated with radiolabeled dsDNA of (A) the high-affinity DBF12-L15 element and the mutated version DBF12-L18 or (C) the DBF12-L15 trimer and a random fragment of similar length derived from the multiple cloning site of a vector (mcs). Protein–DNA complexes were separated from unbound DNA in non-denaturing agarose gels. (B) and (D) Binding curves obtained from quantification of (A) and (C) and fitting to a standard bimolecular model.
We had found previously that a single copy of the 40 bp DBF12-L15 fragment had only a weak ability to recruit the DCC to both, a reporter gene in cells and to an autosomal insertion site in flies, but trimerization of this sequence resulted in a very strong recruitment site (25). We, therefore, tested whether trimerization of DBF12-L15 increases the affinity and selectivity of MSL2 for this sequence. MSL2 bound the trimeric element with a dramatically improved affinity (KD of 0.59 ± 0.18 nM, as opposed to 33 ± 13 nM for the single site). However, a synthetic control sequence of identical length (the multiple cloning site of a vector) was bound with the same affinity (Figure 6). MSL2 also bound a 226 bp fragment derived from the roX1 gene and known to contain a strong HAS (22) with similar affinity (Supplementary Figure S5).
Binding of MSL2 to DNA obviously is very length dependent. The EMSA patterns derived from analysis of the DNA elements tested so far suggest the possibility that more than one MSL2 protein might be bound, either as a complex of unknown stoichiometry or occupying neighboring binding sites. To clarify this point we performed EMSAs with progressively shorter fragments of DBF12-L15 and L18 (30 and 20 bp, Supplementary Figure S6). Reducing the length of the DNA fragments, the affinity of MSL2 for DNA decreased such that binding to a 20 bp fragment was not detectable anymore. This placed the minimum length of a target sequence between 20 and 30 bp. Comparison of 30 bp fragments containing the ‘core’ of the DBF12-L15 and L18 sequences (Supplementary Figure S6A) revealed no binding preference for the HAS sequence. The affinities were similar within error for both 30 bp long fragments (KD of 116 ± 22 nM versus 87 ± 15 nM, Supplementary Figure S6); if anything, the control sequence bound slightly better. In summary, we conclude that the affinity of MSL2 for DNA increases with the length of the fragment, but we have no evidence for sequence discrimination that is based on differential binding affinities of MSL2 in vitro.
The CXC domain is necessary for targeting MSL2 to a reporter gene
As shown above, the CXC domain is necessary for DNA binding of MSL2 in vitro. To explore the relevance of this domain in vivo, we used the previously described reporter gene assay in D. melanogaster SL2 cells (25). In this assay, the 40 bp DBF12-L15 element precedes a minimal promoter driving transcription of a luciferase reporter gene. The plasmid is transfected into SL2 cells together with a vector that directs expression of an MSL2-VP16 fusion protein. Binding of MSL2 to the candidate targeting element tethers VP16, which then leads to strong activation of luciferase transcription. Importantly, all transfected MSL2 constructs were expressed at similar levels (Supplementary Figure S7A). Recruitment of MSL2-VP16 to the DBF12-L15 element activated the luciferase reporter roughly 5-fold [Figure 7 and reference (25)]. Deletion of the RING domain did not affect reporter gene activation indicating that MSL1, which interacts with the RING domain of MSL2 (18), is not involved in targeting MSL2 to DBF12-L15. In contrast, MSL2 lacking the CXC domain or carrying point mutations therein were unable to activate. Deletion of the Pro/Bas patch had an intermediate effect. Surprisingly, even though the chimeric HsCXC protein bound DNA with wild-type affinity in vitro, it was unable to activate luciferase expression (Figure 7). Evidently, an unknown Drosophila factor that synergises with Drosophila MSL2 but does not match the human counterpart confers selectivity of DNA binding in vivo. This notion is further supported by the finding that the mutated DBF12-L18 sequence was unable to attract any of the MSL2-VP16 proteins, including wild-type, to the reporter gene [Figure 7 and reference (25)]. We conclude that the CXC domain is required for DNA binding in general, but that sequence discrimination requires an as yet unkown principle present in Drosophila cells.
Figure 7.

Reporter gene assay to measure transactivation potential of different MSL2 constructs in vivo. (A) Schematic representation of the cotransfected plasmids that constitute the reporter gene system (drawing is not to scale). Drosophila SL2 cells were transiently cotransfected with different MSL2-VP16 constructs together with luciferase reporter gene constructs (pGL3-TK) carrying the model DBF12-L15 or the mutated site DBF12-L18 (40 bp elements). A renilla luciferase expression vector (pRL-TK) served as normalization control. (B) Transactivation potential of each MSL2 derivative is displayed as fold activation (for details see ‘Materials and Methods’ section). The black bars indicate a reporter gene bearing MSL2 binding sites (DBF12-L15), the white bars represent (lack of) activation in the presence of DBF12-L18.
The CXC domain is necessary for targeting the DCC to the X chromosome
As a final test for the role of the CXC domain of MSL2, we explored its contributions to targeting a GFP-tagged MSL2 to the X chromosome in male SL2 cells. Vectors were created that code for MSL2, MSL2-ΔCXC and MSL2-C544A/C546A fused to the Green Fluorescent Protein (GFP), transfected into SL2 cells and stable lines were selected for several weeks. All cell lines expressed roughly similar amounts of endogenous MSL2 and GFP-MSL2 transgenes (Supplementary Figure S7B). The localization of MSL2-GFP and of endogenous MSL1 was then assessed by parallel immunofluorescence microscopy employing antibodies directed against GFP and MSL1 (Figure 8). Wild-type MSL2-GFP co-localized perfectly with endogenous MSL1 at a defined chromosomal territory that corresponds to the X chromosome (35). In contrast, when the DNA binding function of MSL2 was impaired by deletion of the CXC domain or mutation of its critical cysteines the X chromosomal targeting was disturbed: in ∼75% of the cells the MSL2-ΔCXC and MSL2-C544A/C546A GFP fusion proteins did not associate with well defined X chromosome territories, but were delocalized to many, dispersed foci in the nucleus. Interestingly, endogenous MSL1 was delocalized to these ectopic sites as well, which shows that the impairment of the CXC domain did not perturb the MSL1 interaction. Clearly, MSL2 was revealed as the primary targeting determinant, whose delocalization moved MSL1 with it and MSL1 was unable to target MSL2 to the X chromosome. We conclude that for the proper targeting of the DCC in vivo the functional DNA binding domain of MSL2—the CXC domain—is required.
Figure 8.

In vivo localization of different MSL2 derivatives. Stable Drosophila SL2 cell lines, which express different MSL2 versions fused to GFP, were analyzed by immunofluorescence staining. Localization of MSL2-GFP derivatives were visualized using anti-GFP antibodies (α-GFP). Endogenous MSL1 was detected using anti-MSL1 antibodies (α-MSL1). DNA was counterstained with Hoechst 33258 (DNA). (A) Examples showing single nuclei with proper targeting of MSL2 to the X chromosomal territory (left) or dispersed, non-physiological distribution of the GFP fusion proteins. (B) Representative fields of cells. Cells that express the MSL2-GFP transgene were analyzed for proper X territory staining. The percentage of cells, which show mislocalized, dispersed GFP signals from the total number of GFP positive cells counted are displayed to the right.
DISCUSSION
Dosage compensation is appreciated as a model for chromosome-wide, coordinate fine-tuning of gene expression. Understanding how the DCC discriminates between the X chromosome and autosomes for selective activation of X-linked genes is, therefore, of general interest. The recent identification of DNA sequence motifs enriched within HAS for the DCC in vivo have given new support to the idea that an X-specific DNA sequence signature must be involved. However, so far no DNA binding surface has been identified within the DCC subunits that may serve to recognize and interpret such a signature. We have taken a biochemical approach to assaying direct protein–DNA interactions in a fully defined system. We identified the first DNA binding function, the CXC domain of MSL2, within the MSL2-MSL1 module of the DCC and showed that it is important not only for in vitro binding but also for proper X chromosomal targeting. A heteromeric assembly of MSL2-MSL1 is thought to be minimally required for initial recognition of a relatively small number of HAS or chromosomal entry sites (19,20,23,30,31). We came to conclude that all DNA binding potential resides within MSL2 and that the important role of MSL1 cannot be explained by contributions to DNA binding.
MSL2 binds DNA with high-affinity and low sequence discrimination
We quantitatively measured the binding of MSL proteins to a number of DNA fragments that vary in length and sequence. The affinity constant KD of MSL2 to the short 40 bp DBF12-L15 DNA element is around 33 nM. Increasing the size of the target DNA to 167 bp led to a robust improvement of affinity (KD around 1 nM). Such low nanomolar affinities are typical for sequence-specific DNA binding proteins. For example, the glucocorticoid receptor bound with a KD of 21 nM to a glucocorticoid response element in the context of a 33 bp DNA fragment and with a lower affinity of 165 nM to a mutated DNA element (40). To our surprise, we found that the high-affinity of MSL2 for DNA did not involve sequence discrimination in vitro, yet MSL2 binding was sensitive to mutation of the HAS in the reporter gene assay in Drosophila cells. Apparently, a crucial cellular selectivity factor is still missing. Several scenarios are conceivable. MSL2 may synergize with an abundant sequence-specific DNA binding protein. Inspired by the frequent occurrence of GAGA motifs in HAS an involvement of the GAGA factor had been considered earlier, however, interfering with GAGA factor function does not affect the majority of binding of MSL2 to the X chromosome (41). DNA recognition by MSL2 may be modulated through allosteric interactors within the DCC (such as roX RNA) or yet unknown factors. It is also possible that the sequences that characterize HAS have to be presented in a non-B form secondary conformation or in the context of chromatin [discussed in (42)]. We did not explore a nucleosomal organization of the candidate sequence since HAS tend to reside in nucleosome-free regions (30,31). DNA sequences themselves may function as allosteric effectors that modulate the properties of interacting factors, including co-regulator recruitment. This was recently shown for the examples of the transcription factors NF-κB and glucocorticoid receptor (43,44). Moreover, selectivity for target sequences do not need to be based on binding affinity, but rather on conformational changes subsequent to binding, as shown for DNA topoisomerase II (45). Considering these cases, one may hypothesize that target and non-target DNA sequences for MSL2 are not distinguished by their different affinities, but by their effects on the conformation of MSL2 upon binding and DCC assembly. Accordingly, only interactions with the HAS sequence would be productive for the assembly of a DCC.
Recently, Scott and colleagues hypothesized that the N-terminus of MSL1 may be involved in DNA binding (33), since deletion of the N-terminal 26 amino acids prevented the association of MSL1 with the X chromosome. Our biochemical analysis now shows that MSL1 by itself does not have DNA binding potential and that it does not influence the sequence-independent binding of MSL2 to B-form DNA. However, given that we found strong association of MSL1 with chromatin in vitro in the presence of MOF (36), it is possible that MSL1 contributes to targeting the complex in a chromatin context.
The CXC domain—a conserved DNA binding module
Our studies highlight the important contribution of the conserved CXC domain for direct association of MSL2 with DNA. Deletion of the CXC domain also impaired the targeting of MSL2 to a HAS in the reporter gene assay and to the X chromosomal territory in male cells. Importantly, deletion of or point mutations in the CXC domain did not abolish the interaction of MSL2 with MSL1 in vitro or in vivo, nor did it affect the RNA binding of MSL2, which shows that the folding of MSL2 was not generally perturbed. Our finding that the CXC domain from the H. sapiens MSL2 homolog has similar DNA binding properties suggests that this function is evolutionary conserved, despite of different dosage compensation strategies in flies and humans. Our results provide a mechanistic explanation for the earlier observation of Kuroda and colleagues (19), who had shown that the very same mutations in the CXC domain that in our hands diminish DNA binding caused a developmental delay and reduced viability (to 16–64% of wild-type) in male flies. The phenotype described in this study qualifies as a partial loss-of-function, which suggests that in addition to the CXC domain, other structures may contribute to targeting. A targeting influence of the Pro/Bas domain had recently been suggested by Scott and colleagues (34). These authors hypothesized that the C-terminus of MSL2 may be involved in roX RNA binding. Our study unveiled the RNA binding potential of recombinant MSL2, and although we were unable to attribute the RNA binding to any specific domain, a contribution of non-coding RNA to MSL2 targeting remains an interesting possibility.
The CXC domain had not been identified as particularly important in the previous structure–function analysis in transgenic flies by Scott and colleagues, but in this case the domain was only deleted as part of an N- or C- terminal truncation series, of which most MSL2 derivatives did not localize properly (34). CXC domains are frequent protein structure modules that occur in a variety of different arrangements, however, for none of the CXC domains the structure has been solved. According to Marin, a common denominator is the presence of one to three CXC motifs N-terminal to the general C-X4-CXC-X6-C-X4-5-C-X2-C formula (37). Like for other cysteine-rich structures, it is likely that the structuring of the domain involves zinc coordination (46). Two recent studies on novel Drosophila testis-specific protein complexes identified proteins of the tesmin/TSO1-family (Mip120 and Tomb), which share one or two CXC domains. A common role for all proteins of this family in DNA binding via their CXC domains has been proposed (47,48) but so far only for one related member of the tesmin/TSO1-family, the CPP1 protein of the soybean, in vitro DNA binding of two clustered CXC domains was shown (49). It is possible that in MSL2 the CXC cooperates with other structures, such as the Pro/Bas patch, the deletion of which led to a slightly lower affinity of MSL2 for nucleic acids and a modest reduction of MSL2 targeting to a HAS in cells. Further evaluation of these hypotheses will require knowledge of the MSL domain structures at atomic resolution.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the Deutsche Forschungsgemeinschaft through SFB-TR5. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank E. Schulze for a kind gift of MSL1 antibody and members of the Becker lab for stimulating discussions.
REFERENCES
- 1.Gelbart ME, Kuroda MI. Drosophila dosage compensation: a complex voyage to the X chromosome. Development. 2009;136:1399–1410. doi: 10.1242/dev.029645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucchesi JC, Kelly WG, Panning B. Chromatin remodeling in dosage compensation. Annu. Rev. Genet. 2005;39:615–651. doi: 10.1146/annurev.genet.39.073003.094210. [DOI] [PubMed] [Google Scholar]
- 3.Straub T, Dahlsveen IK, Becker PB. Dosage compensation in flies: mechanism, models, mystery. FEBS Lett. 2005;579:3258–3263. doi: 10.1016/j.febslet.2005.03.050. [DOI] [PubMed] [Google Scholar]
- 4.Hamada FN, Park PJ, Gordadze PR, Kuroda MI. Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev. 2005;19:2289–2294. doi: 10.1101/gad.1343705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Straub T, Gilfillan GD, Maier VK, Becker PB. The Drosophila MSL complex activates the transcription of target genes. Genes Dev. 2005;19:2284–2288. doi: 10.1101/gad.1343105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akhtar A, Becker PB. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell. 2000;5:367–375. doi: 10.1016/s1097-2765(00)80431-1. [DOI] [PubMed] [Google Scholar]
- 7.Bone JR, Lavender J, Richman R, Palmer MJ, Turner BM, Kuroda MI. Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev. 1994;8:96–104. doi: 10.1101/gad.8.1.96. [DOI] [PubMed] [Google Scholar]
- 8.Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC. The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell Biol. 2000;20:312–318. doi: 10.1128/mcb.20.1.312-318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 2006;20:848–857. doi: 10.1101/gad.1400206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B, Becker PB. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev. 2006;20:858–870. doi: 10.1101/gad.1399406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol. Cell. 2007;28:121–133. doi: 10.1016/j.molcel.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 12.Bell O, Conrad T, Kind J, Wirbelauer C, Akhtar A, Schubeler D. Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol. Cell Biol. 2008;28:3401–3409. doi: 10.1128/MCB.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sural TH, Peng S, Li B, Workman JL, Park PJ, Kuroda MI. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat. Struct. Mol. Biol. 2008;15:1318–1325. doi: 10.1038/nsmb.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI. Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev. 2009;23:2266–2271. doi: 10.1101/gad.1840409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kind J, Akhtar A. Cotranscriptional recruitment of the dosage compensation complex to X-linked target genes. Genes Dev. 2007;21:2030–2040. doi: 10.1101/gad.430807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, Kuroda MI. Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell. 1995;81:867–877. doi: 10.1016/0092-8674(95)90007-1. [DOI] [PubMed] [Google Scholar]
- 17.Oh H, Park Y, Kuroda MI. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev. 2003;17:1334–1339. doi: 10.1101/gad.1082003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, Kuroda MI. Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. EMBO J. 1998;17:5409–5417. doi: 10.1093/emboj/17.18.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI. Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics. 1997;147:1743–1753. doi: 10.1093/genetics/147.4.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlsveen IK, Gilfillan GD, Shelest VI, Lamm R, Becker PB. Targeting determinants of dosage compensation in Drosophila. PLoS Genet. 2006;2:e5. doi: 10.1371/journal.pgen.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demakova OV, Kotlikova IV, Gordadze PR, Alekseyenko AA, Kuroda MI, Zhimulev IF. The MSL complex levels are critical for its correct targeting to the chromosomes in Drosophila melanogaster. Chromosoma. 2003;112:103–115. doi: 10.1007/s00412-003-0249-1. [DOI] [PubMed] [Google Scholar]
- 22.Kageyama Y, Mengus G, Gilfillan G, Kennedy HG, Stuckenholz C, Kelley RL, Becker PB, Kuroda MI. Association and spreading of the Drosophila dosage compensation complex from a discrete roX1 chromatin entry site. EMBO J. 2001;20:2236–2245. doi: 10.1093/emboj/20.9.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh H, Bone JR, Kuroda MI. Multiple classes of MSL binding sites target dosage compensation to the X chromosome of Drosophila. Curr. Biol. 2004;14:481–487. doi: 10.1016/j.cub.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Park Y, Kelley RL, Oh H, Kuroda MI, Meller VH. Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science. 2002;298:1620–1623. doi: 10.1126/science.1076686. [DOI] [PubMed] [Google Scholar]
- 25.Gilfillan GD, Konig C, Dahlsveen IK, Prakoura N, Straub T, Lamm R, Fauth T, Becker PB. Cumulative contributions of weak DNA determinants to targeting the Drosophila dosage compensation complex. Nucleic Acids Res. 2007;35:3561–3572. doi: 10.1093/nar/gkm282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straub T, Becker PB. Dosage compensation: the beginning and end of generalization. Nat. Rev. Genet. 2007;8:47–57. doi: 10.1038/nrg2013. [DOI] [PubMed] [Google Scholar]
- 27.Gu W, Szauter P, Lucchesi JC. Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev. Gene. 1998;22:56–64. doi: 10.1002/(SICI)1520-6408(1998)22:1<56::AID-DVG6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Meller VH, Rattner BP. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J. 2002;21:1084–1091. doi: 10.1093/emboj/21.5.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park Y, Mengus G, Bai X, Kageyama Y, Meller VH, Becker PB, Kuroda MI. Sequence-specific targeting of Drosophila roX genes by the MSL dosage compensation complex. Mol. Cell. 2003;11:977–986. doi: 10.1016/s1097-2765(03)00147-3. [DOI] [PubMed] [Google Scholar]
- 30.Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet. 2008;4:e1000302. doi: 10.1371/journal.pgen.1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grimaud C, Becker PB. The dosage compensation complex shapes the conformation of the X chromosome in Drosophila. Genes Dev. 2009;23:2490–2495. doi: 10.1101/gad.539509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F, Parry DA, Scott MJ. The amino-terminal region of Drosophila MSL1 contains basic, glycine-rich, and leucine zipper-like motifs that promote X chromosome binding, self-association, and MSL2 binding, respectively. Mol. Cell Biol. 2005;25:8913–8924. doi: 10.1128/MCB.25.20.8913-8924.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li F, Schiemann AH, Scott MJ. Incorporation of the noncoding roX RNAs alters the chromatin-binding specificity of the Drosophila MSL1/MSL2 complex. Mol. Cell Biol. 2008;28:1252–1264. doi: 10.1128/MCB.00910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Straub T, Neumann MF, Prestel M, Kremmer E, Kaether C, Haass C, Becker PB. Stable chromosomal association of MSL2 defines a dosage-compensated nuclear compartment. Chromosoma. 2005;114:352–364. doi: 10.1007/s00412-005-0020-x. [DOI] [PubMed] [Google Scholar]
- 36.Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, Becker PB. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J. 2004;23:2258–2268. doi: 10.1038/sj.emboj.7600235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marin I. Evolution of chromatin-remodeling complexes: comparative genomics reveals the ancient origin of “novel” compensasome genes. J. Mol. Evol. 2003;56:527–539. doi: 10.1007/s00239-002-2422-1. [DOI] [PubMed] [Google Scholar]
- 38.Akhtar A, Zink D, Becker PB. Chromodomains are protein-RNA interaction modules. Nature. 2000;407:405–409. doi: 10.1038/35030169. [DOI] [PubMed] [Google Scholar]
- 39.Buscaino A, Kocher T, Kind JH, Holz H, Taipale M, Wagner K, Wilm M, Akhtar A. MOF-regulated acetylation of MSL-3 in the Drosophila dosage compensation complex. Mol. Cell. 2003;11:1265–1277. doi: 10.1016/s1097-2765(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 40.Schoenmakers E, Alen P, Verrijdt G, Peeters B, Verhoeven G, Rombauts W, Claessens F. Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem. J. 1999;341(Pt 3):515–521. [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg AJ, Yanowitz JL, Schedl P. The Drosophila GAGA factor is required for dosage compensation in males and for the formation of the male-specific-lethal complex chromatin entry site at 12DE. Genetics. 2004;166:279–289. doi: 10.1534/genetics.166.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Straub T, Becker PB. DNA sequence and the organization of chromosomal domains. Curr. Opin. Genet. Dev. 2008;18:175–180. doi: 10.1016/j.gde.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 43.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mueller-Planitz F, Herschlag D. DNA topoisomerase II selects DNA cleavage sites based on reactivity rather than binding affinity. Nucleic Acids Res. 2007;35:3764–3773. doi: 10.1093/nar/gkm335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersen SU, Algreen-Petersen RG, Hoedl M, Jurkiewicz A, Cvitanich C, Braunschweig U, Schauser L, Oh SA, Twell D, Jensen EO. The conserved cysteine-rich domain of a tesmin/TSO1-like protein binds zinc in vitro and TSO1 is required for both male and female fertility in Arabidopsis thaliana. J. Exp. Bot. 2007;58:3657–3670. doi: 10.1093/jxb/erm215. [DOI] [PubMed] [Google Scholar]
- 47.Beall EL, Lewis PW, Bell M, Rocha M, Jones DL, Botchan MR. Discovery of tMAC: a Drosophila testis-specific meiotic arrest complex paralogous to Myb-Muv B. Genes Dev. 2007;21:904–919. doi: 10.1101/gad.1516607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang J, Benson E, Bausek N, Doggett K, White-Cooper H. Tombola, a tesmin/TSO1-family protein, regulates transcriptional activation in the Drosophila male germline and physically interacts with always early. Development. 2007;134:1549–1559. doi: 10.1242/dev.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cvitanich C, Pallisgaard N, Nielsen KA, Hansen AC, Larsen K, Pihakaski-Maunsbach K, Marcker KA, Jensen EO. CPP1, a DNA-binding protein involved in the expression of a soybean leghemoglobin c3 gene. Proc. Natl Acad. Sci .USA. 2000;97:8163–8168. doi: 10.1073/pnas.090468497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.