

Abstract
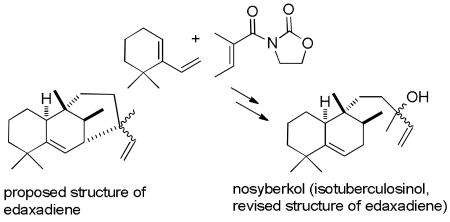
Me2AlCl-catalyzed Diels–Alder reaction of N-tigloyloxazolidinone with 6,6-dimethyl-1-vinylcyclohexene selectively provided the exo adduct, which was converted to nosyberkol (isotuberculosinol) and tuberculosinol. The spectral data for nosyberkol are identical to those reported for edaxadiene, whose structure is revised accordingly.
Some of us recently reported the isolation of a biologically active diterpene edaxadiene (3) from Mycobacterium tuberculosis that is produced from halimadienyl diphosphate (1) by the action of the enzyme encoded by Rv3378c (see Scheme 1).1 Edaxadiene alone arrests maturation of the endocytic phagosomal compartment at an early stage, a result very similar to that observed upon engulfment of M. tuberculosis by macrophage cells of the mammalian immune system.1a M. tuberculosis persists and replicates, causing disease, in such arrested phagosomal organelles, whereas M. tuberculosis mutants unable to make edaxadiene exhibit delayed proliferation, at least in macrophage cell culture.2 Thus, edaxadiene presumably plays an important role early in the M. tuberculosis infection process.
Scheme 1. Conversion of Halimadienyl Diphosphate (1) to Nosyberkol (Isotuberculosinol, Revised Structure of Edaxadiene, 4).

Only 250 μg of edaxadiene was isolated from 1 using purified Rv3378c encoded enzyme. Enzymatic dephosphorylation of 1 afforded tuberculosinol (2),3a which was treated with DCC and CuCl2 in acetone to give 1 mg of edaxadiene in 20% yield. The mass spectrum suggested a molecular formula of C20H32 and structure 3 was proposed from analysis of the NMR spectra.1 However, the formation of 3 would require the insertion of an allylic cation into an allylic C-H bond followed by the loss of a proton. This process is unlikely to occur enzymatically and should not occur by the reaction of 2 with DCC and CuCl2. The 13C NMR spectrum of edaxadiene showed a peak at δ 72.9, which suggested the presence of a tertiary alcohol in edaxadiene, which could readily lose H2O to give the high mass peak corresponding to C20H32. The 1H NMR spectrum of edaxadiene shows doubling of peaks for the vinyl protons indicating the presence of a 1:1 mixture of diastereomers.
This analysis suggested that the structure of edaxadiene is a mixture of stereoisomers of the tertiary alcohol nosyberkol (4), which can be easily formed by hydrolysis of 1 with allylic rearrangement. Nosyberkol (4) was isolated by Kashman from the Nosy be Islands (Madagascar) sponge Raspailia sp in 2004 as a single stereoisomer.4 In 2005, Nakano reported the isolation of a 1:1 mixture of tuberculosinol (2) and 4 (as a 3:1 mixture of stereoisomers), which he called isotuberculosinol, by treatment of 1 with Rv3378c encoded enzyme.3b,c The 1H and 13C NMR spectra of edaxadiene in C6D6 are similar to those reported for nosyberkol in CDCl3, but the different solvents used preclude a definitive comparison. Only partial data were reported by Nakano for isotuberculosinol.3b,c We therefore set out to synthesize nosyberkol (isotuberculosinol, 4) to establish that it is identical to edaxadiene and to provide a ready source of material for further biological evaluation.
Our retrosynthetic analysis is shown in Scheme 2. Nosyberkol (isotuberculosinol, 4) should be available as a mixture of isomers by addition of vinylmagnesium bromide to ketone 5. A Wittig reaction on aldehyde 6 should give an enone that will be reduced to ketone 5 with Li/NH3. An exo Diels–Alder reaction of diene 75 and tiglic aldehyde (8) could give the required bicyclic aldehyde 6.
Scheme 2. Retrosynthesis of Nosyberkol (Isotuberculosinol, Revised Structure of Edaxadiene).

Unfortunately, the Lewis acid catalyzed Diels–Alder reaction of diene 7 with tiglic aldehyde (8) is known to give the expected, but undesired, endo Diels–Alder adduct 9 with 91-96% selectivity (see Scheme 3).6 On the other hand, heating diene 7 in excess methyl tiglate (10) at 170 °C for 1 week afforded 74% of Diels–Alder adducts with 60% selectivity for the exo adduct 12.7 Most significantly, Danishefsky reported that the EtAlCl2-catalyzed reaction of diene 7 with vinyl ketone 13 provided exclusively the desired exo Diels–Alder adduct 14, which was converted to mamanuthaquinone.8 Steric interactions between the aryl group of dienophile 13 and the methyl substituents on the cyclohexene of diene 7 retard the endo Diels–Alder reaction, whereas the methyl groups on the dienophile are small enough so that steric interactions with the methyl group of 7 have no effect on the exo Diels–Alder reaction. Steric interactions are still significant with the smaller methyl ester of 10, which gave 60% of the exo adduct 12, whereas the aldehyde of 8 is small enough that the undesired endo adduct 9 is formed almost exclusively.
Scheme 3. Diels–Alder reactions of Diene 7 with Dienophiles 8, 10, and 13.

A tigloyl dienophile was needed that (1) has a large substituent that would sterically retard the endo Diels–Alder reaction as observed by Danishefsky with the aroyl group of 13 and (2) could be converted to aldehyde 6 after the Diels–Alder reaction. The two methyl groups on the dienophile decrease its reactivity as shown by the harsh conditions needed for the thermal Diels–Alder reaction with methyl tiglate (10). Although exo adduct 14 was isolated in 85% yield by Danishefsky, the yield dropped to 43% without one equivalent of THF and to 0% with other Lewis acids. A substituent was also needed that would enhance the reactivity of the dienophile.
N-Tigloylisoxazolidinone (15)9 seemed well suited for this purpose. Evans developed asymmetric Diels–Alder reactions with chiral acryloyl and crotonyl oxazolidinones catalyzed by two equivalents of Et2AlCl.10 These reactions are not very selective with methacryloyl oxazolidinones and have not been investigated with tigloyl oxazolidinones. The unsubstituted oxazolidinone 15 was chosen to minimize steric interactions between the α-methyl group and the oxazolidinone and because asymmetric induction seemed unlikely in an exo Diels–Alder reaction in which a chiral oxazolidinone would be far away from the diene. Reaction of diene 7 and oxazolidinone 15 (1 equiv) with 1.9 equiv of Me2AlCl in CH2Cl2 at 4 °C for 2 days afforded a difficultly separable ∼10:1 mixture of the desired exo Diels–Alder adduct 16 and the endo adduct in 54% yield.11 We also obtained N-2,3-dimethylbutanoyloxazolidinone12 (20%), which was formed by conjugate addition of a methyl group to the dienophile. The ∼10:1 selectivity for the exo isomer was not quite as good as that observed by Danishefsky with dienophile 13, but was a significant improvement over the 3:2 ratio obtained in the thermal Diels–Alder reaction with methyl tiglate (10).
Reduction of acyloxazolidinone 16 proceeded uneventfully with LiBH4 in 10:1 THF/MeOH for 2 h at 25 °C to give alcohol 17 in 82% yield. Oxidation of 17 with Dess-Martin periodinane in CH2Cl2 for 2 h at 25 °C provided the desired exo aldehyde 6 in 94% yield. A similar sequence on a 9:1 mixture of 16 and the undesired endo isomer afforded a 9:1 mixture of aldehyde 6 and the undesired endo aldehyde 9, which has spectral data identical to those previously reported.6
The equatorial aldehyde of 6 is very hindered by the flanking methyl group and cyclohexane methylene group. Reaction of 6 with diethyl 2-oxopropylphosphonate and LiCl, i-Pr2EtN in CH3CN (Roush-Masamune conditions)13 did not proceed at 25 °C. At reflux, enone 18 was formed in 15-20% yield, but the yield could not be improved because the Horner-Emmons Wittig reagent decomposed at a comparable rate. Other Wittig reaction conditions gave even lower yields of 18. Fortunately, aldehyde 6 cannot undergo aldol dimerization and is stable in base so 6 was converted to 18 in 60% yield by treating it with 1 equiv of NaOMe in acetone for 20 h at 25 °C to effect a mixed aldol reaction.14
The β-position of enone 18 is very hindered for the reasons discussed above for the aldehyde carbonyl of 6. Nickel boride reduction of 18 to give 5 was unsuccessful.15 Reduction with Li in NH3/THF/t-BuOH (10 equiv) gave a mixture of ketone 5, the corresponding saturated alcohol and what may be the unsaturated pinacol reduction product.16 Jones oxidation of this mixture afforded a 3:1 mixture of the desired saturated ketone 5 and unsaturated ketone 18, which was presumably regenerated by oxidative cleavage of the pinacol product. Formation of pinacol byproducts is not usually a major side reaction in the reduction of conjugated enones with lithium in ammonia, but steric hindrance at the β-position of 18 will retard protonation of the radical anion so that dimerization at the unhindered carbonyl carbon to give the pinacol product is a significant side reaction. This problem was solved by using more of the less hindered alcohol EtOH. Reduction with Li in NH3/THF/EtOH (40 equiv) at -78 °C, followed by oxidation of the mixture of saturated ketone and alcohol gave saturated ketone 5 in 90% yield. Addition of ketone 5 to vinylmagnesium bromide in THF at 0 °C provided nosyberkol (isotuberculosinol, 4) in 88% yield as a 2:1 mixture of stereoisomers.
The 400 MHz 1H and 100 MHz 13C NMR spectra in CDCl3 of synthetic 4 correspond well to those of nosyberkol. Some carbons appear as closely spaced doublets in synthetic 4. The presence of stereoisomers is detectable in the 1H NMR spectrum only by the two methyl doublets. The major isomer absorbs downfield (δ 0.790 vs 0.783). Because the data for the two isomers are so similar, it can't be determined whether nosyberkol corresponds to the major or minor synthetic isomer.
Both the retention time and fragmentation pattern in the GCMS of synthetic 4 and edaxadiene are identical. The 700 MHz 1H and 175/100 MHz 13C NMR spectra in C6D6 of synthetic 4 (2:1 mixture of isomers) and edaxadiene (1:1 mixture of isomers) are identical except for differences due to the differing isomer ratios. Therefore, the structure of edaxadiene should be revised from 3 to that of nosyberkol (isotuberculosinol, 4). The vinyl protons of the two diastereomers are slightly separated at 700 MHz. The methyl doublets are better separated in C6D6 (major at δ 0.839 vs minor at δ 0.813) than in CDCl3. The major methyl doublet of the 3:1 mixture of isotuberculosinol isomers obtained by Nakano absorbs downfield in C6D6 indicating that Nakano's major isomer is identical to the major synthetic isomer.3c
Reaction of saturated ketone 5 with triethyl phosphonoacetate and NaH in THF at 95 °C17 afforded ethyl enoate 1918 in 84% yield and the Z-isomer in 12% yield. Reduction of 19 with DIBAL-H in CH2Cl2 afforded tuberculosinol (2) in 92% yield. The spectral data are identical to those reported for the natural product.3a
In conclusion, we have revised the structure of edaxadiene from 3 to nosyberkol (isotuberculosinol, 4). The biological activity previously reported for edaxadiene by some of us1 is still valid, but should be ascribed to nosyberkol (4, isotuberculosinol). The efficient routes to isotuberculosinol (4) (six steps, 19% overall yield) and tuberculosinol (2) (seven steps, 17% overall yield) make these compounds readily available for further biological evaluation.
Supplementary Material
Scheme 4. Synthesis of Nosyberkol (Isotuberculosinol, Revised Structure of Edaxadiene, 4) and Tuberculosinol (2).

Acknowledgments
We gratefully acknowledge support for this work from the National Institutes of Health (GM050151 to B.B.S. and GM076324 to R.J.P.).
Footnotes
Supporting Information Available: Complete experimental procedures, comparison of the spectral data of synthetic and natural isobuterculosinol (4) and tuberculosinol (2), and copies of 1H and 13C NMR spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Mann FM, Xu M, Chen X, Fulton DB, Russell DG, Peters RJ. J Am Chem Soc. 2009;131:17526–17527. doi: 10.1021/ja9019287. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mann FM, Prisic S, Hu H, Xu M, Coates RM, Peters RJ. J Biol Chem. 2009;284:23574–23579. doi: 10.1074/jbc.M109.023788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. Proc Natl Acad Sci U S A. 2004;101:13642–13647. doi: 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Nakano C, Okamura T, Sato T, Dairi T, Hoshino T. Chem Commun. 2005:1016–1018. doi: 10.1039/b415346d. [DOI] [PubMed] [Google Scholar]; (b) Nakano C, Sato T, Hoshino T. Koryo, Terupen oyobi Seiyu Kagaku ni kansuru Toronkai Koen Yoshishu. 2005;49:247–249. [Google Scholar]; (c) Nakano C, Okamura T, Sato T, Hara T, Hoshiono T, Dairi T, Toyomasu T, Sassa T. Symposium on the Chemistry of Natural Products; 2006. pp. 19–24. http://ci.nii.ac.jp/naid/110006682624/en. [Google Scholar]
- 4.Rudi A, Aknin M, Gaydou E, Kashman Y. J Nat Prod. 2004;67:1932–1935. doi: 10.1021/np049834b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Knapp S, Sharma S. J Org Chem. 1985;50:4996–4998. [Google Scholar]; (b) Tanis SP, Abdallah YM. Synth Commun. 1986;16:251–259. [Google Scholar]
- 6.(a) Brohm D, Waldmann H. Tetrahedron Lett. 1998;39:3995–3998. [Google Scholar]; (b) Brohm D, Philippe N, Metzger S, Bhargava A, Müller O, Lieb F, Waldmann H. J Am Chem Soc. 2002;124:13171–13178. doi: 10.1021/ja027609f. [DOI] [PubMed] [Google Scholar]
- 7.de Miranda DS, da Conceição GJA, Zukerman-Schpector J, Guerrero MA, Schuchardt U, Pinto AC, Rezende CM, Marsaioli AJ. J Braz Chem Soc. 2001;12:391–402. [Google Scholar]
- 8.Yoon T, Danishefsky SJ, de Gala S. Angew Chem, Int Ed. 1994;33:853–855. [Google Scholar]
- 9.(a) Miyata O, Shinada T, Nimomiya I, Naito T, Date T, Okamura K, Inagaki S. J Org Chem. 1991;56:6556–6564. [Google Scholar]; (b) Sibi MP, Prabagaran N, Ghorpade SG, Jasperse CP. J Am Chem Soc. 2003;125:11796–11797. doi: 10.1021/ja0372309. [DOI] [PubMed] [Google Scholar]
- 10.Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1988;110:1238–1256. [Google Scholar]
- 11.Use of other Lewis acids or thermal conditions gave significantly poorer results with 7 and the more readily available analogous diene 5,5-dimethyl-1-ethenylcyclohexene.
- 12.Murray RS, Boyd VA, Lynch VM, Negrete GR. ARKIVOC. 2001;(xi):58–73. [Google Scholar]
- 13.Blanchette MA, Choy W, Davis JT, Essenfeld AP, Masamune S, Roush WR, Sakai T. Tetrahedron Lett. 1984;25:2183–2186. [Google Scholar]
- 14.For related aldol reactions, see: Kumar K, Wang SS, Sukenik CN. J Org Chem. 1984;49:665–670.Cockerill GS, Kocienski P, Treadgold R. J Chem Soc, Perkin Trans 1. 1985:2101–2108.Parry AD, Neill SJ, Horgan R. Phytochemistry. 1990;29:1033–1039.Yamamoto T, Shimada A, Ohmoto T, Matsuda H, Ogura M, Kanisawa T. Flavour Frag J. 2008;19:121–133.
- 15.Khurana JM, Sharma P. Bull Chem Soc Jpn. 2004;77:549–552. [Google Scholar]
- 16.Caine D. Org React. 1976;23:1–258. [Google Scholar]
- 17.Diaz S, Cuesta J, González A, Bonjoch J. J Org Chem. 2003;68:7400–7406. doi: 10.1021/jo034838r. [DOI] [PubMed] [Google Scholar]
- 18.Ester 19 was prepared by an unrelated route as an intermediate in the synthesis of agelasimines-A and B: Ohba M, Kawase N, Fujii T. J Am Chem Soc. 1996;118:8250–8257.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.