

1. Introduction
The structure of a protein complex, together with information about its affinity and other thermodynamic characteristics, provide a “frozen” view of the complex. This picture ignores the kinetic nature of protein-protein association and dissociation, which are of major biological and biophysical interest. This review focuses on recent advances in deciphering the kinetic pathway of protein complex formation, the nature of the pre-complex formed through diffusion (which we have termed the “transient complex”1), the transition state, and other intermediates (such as the so-called encounter complex) along the association pathway.
Protein-protein association is at the center of diverse biological processes ranging from enzyme catalysis/inhibition to regulation of immune response by cytokines. The association rates often play a critical role in such processes, as in situations where speed is of essence.2 For example, the purple cone snail and other venomous animals capture prey with remarkable efficiency and speed by releasing toxins that rapidly bind to ion channels;3 the green mamba achieves a similar feat by targeting acetylcholinesterase (AChE), an enzyme essential for the integrity of neural transmission.4 Bacteria such as Escherichia coli and Bacillus amyloliquefaciens excrete nucleases as weapons against competitors or predators. Defense of the producing cells from damage to their own DNA or RNA by such nucleases requires rapid association with cognate inhibitors.5,6 Indeed, in the last example rapid association is such a priority that the inhibitor barstar has a cluster of acidic residues that facilitate association with the nuclease barnase, even though the clustered charges reduce folding stability.7 In the ruminant gut, RNase A is required for degrading accumulated RNA; potential toxicity of leaked nuclease is prevented by rapid association with a ribonuclease inhibitor.8,9
Reorganization of the actin cytoskeleton provides yet another illustration of the importance of rapid protein association. Reorganization is attained through actin polymerization, which is nucleated by the Arp2/3 complex. The latter is activated by the Wiskott-Aldrich Syndrome protein (WASp), which in turn is released from the auto-inhibited state by the Rho GTPase Cdc42.10 As actin polymerization is initiated with a nucleation process, the speed of upstream signaling has a critical impact on the rate of polymer formation. It is thus not surprising that high association rate constants have been observed between partners along the signaling pathway.11,12 The high association rate constant between Cdc42 and WASp has been found to be essential for the latter to stimulate actin polymerization, as another Rho GTPase sharing 70% sequence identity, TC10, with an identical dissociation rate constant but a 1000-fold lower association rate constant, failed to stimulate actin polymerization.11 The failure to stimulate actin polymerization in patients carrying mutant WAS genes is the root cause of the Wiskott-Aldrich Syndrome.
Several other compelling arguments can be made for the biological roles of rapid protein association.13 (a) Fast association may enhance binding affinity. High affinity can also be achieved through slow dissociation; however, for proteins involved in signaling, slow dissociation is not an option, since it implies a long-lasting bound state, which effectively corresponds to a permanent off- or on-switch. A good example for this is the binding of Ras to its natural affector Raf. This protein dissociates within a fraction of a second, but maintains an affinity in the nM range through fast association. Moreover, the difference between the natural effector, Raf, and the non-natural effector, Ral, lies in their rates of association with Ras.14 Therefore, even if not for a direct reason (such as in stimulation of actin polymerization), the affinity requirement alone may call for fast association. (b) Enzyme-substrate binding is a determining factor for the overall turnover rate and becomes the rate-limiting step for catalytically “perfect” enzymes. Substrate-binding rate constants of such enzymes reach 108 M−1s−1 and beyond, as found for the ribotoxin restrictocin and RNase A.15,16 (c) When several proteins compete for the same receptor or when one protein is faced with alternative pathways, kinetic control, not thermodynamic control, dominates in many cases; this is especially true when dissociation is slow. For example, during protein synthesis cognate and noncognate aminoacyl-tRNA synthetases can potentially compete for the same tRNA. As an additional example, consider newly synthesized proteins, which potentially face aggregation if not isolated by a chaperone. From the point of view of kinetic control, it is easy to see why rapid binding of denatured proteins to the chaperonin GroEL has been observed.17 (d) Differences in binding rate between related proteins may serve as an additional mechanism for specificity, as can be suggested for Rho GTPases Cdc42 and TC10 and for Ras effectors Raf and Ral.
The examples and arguments presented above suggest that rapid binding is as important as high affinity in the proper functioning of proteins. It is now increasingly recognized that proteins function in the context of multi-component complexes. Manipulating association rate constants of various components presents unique opportunities for the control of protein functions. Many interactions between proteins are also targeted for drug development; in designing such drugs, both high affinity and rapid binding should be taken into consideration.
1.1. Overview of Protein Association Kinetics
The observed rate constants of protein association span a wide range, from < 103 M−1s−1 to > 109 M−1s−1 (Figure 1). In comprehending these values, a basic fact is that, for two proteins to recognize each other, their interfaces have to be oriented with high specificity. A relative rotation of as little as a few degrees or a relative translation by a few Angstroms is sufficient to break all specific interactions between the two proteins.18 The rate of association of a protein complex is limited by diffusion and geometric constraints of the binding sites, and may be further reduced by subsequent chemical processes.19
Figure 1.

The wide spectrum of association rate constants. The red vertical line marks the start of the diffusion-controlled regime. The shaded range marks the absence of long-range forces. Adapted with permission from Ref. 1. Copyright 2008 Wiley Interscience..
To better understand the kinetics of association of two proteins (A and B), it is useful to consider the process as going through an intermediate state (A*B), in which the two proteins have near-native separations and orientations.1,20–23 We refer to this intermediate state as the transient complex,1,20 noting that is sometimes also termed the encounter complex.24 A more detailed discussion of terminology, as well as the specification of the ensemble of configurations making up the transient complex is provided in Section 3. From this ensemble, conformational rearrangement can lead to the native complex (C). Accordingly we have the kinetic scheme
| (1) |
While the first step of this scheme depends on relative diffusion between the protein molecules, the second step is akin to an intramolecular chemical reaction, and can therefore be described by the classical transition-state theory25 (with the transition state located at the top of the free energy barrier separating A*B from C26) or by Kramers’ theory.27 The latter theory accounts for barrier recrossing and models motion along the reaction coordinate as diffusive. The overall rate constant of association is
| (2) |
which is bounded by the diffusion-controlled rate constant, kD, for reaching the transient complex. This limit is reached when conformational rearrangement is fast relative to the dissociation of the transient complex (i.e., kc ≫ k−D), leading to
| (3a) |
In the opposite limit kc ≪ k−D, conformational rearrangement or reaction becomes rate-limiting and
| (3b) |
Note that kD/k−D is the equilibrium constant for forming the transient complex.
There is no simple test that would place the association of a given protein complex into the diffusion-controlled, reaction-controlled, or mixed regime. However, two general statements can be made. First, a diffusion-controlled rate constant falls on the high end of the spectrum of observed values and a reaction-controlled rate constant falls on the low end (Figure 1). Second, diffusion-controlled association typically involves only local conformational changes between the unbound proteins and the native complex whereas reaction-controlled association typically involves gross changes such as loop reorganization or domain movement. These points will be further amplified below. For later reference we also introduce here the overall dissociation rate constant, kd, and the overall association constant, Ka = ka/kd.
In the transient complex the two protein molecules must satisfy translational/rotational constraints, which severely hinder the diffusion-controlled rate constant kD. In the absence of any biasing force, theoretical estimates put the resulting “basal” value, kD0, in the range of 105 to 106 M−1s−1 (see below).28–31 Antibody-protein association rate constants are typically observed in this narrow range.32–34 The value 105 M−1s−1 thus may mark the start of the diffusion-controlled regime.
To go beyond the basal rate constant kD0 and reach values in the range of 108 to 109 M−1s−1 as observed for many protein complexes3–6,8,9,15–17,35,36 (Figure 1), intermolecular forces must be present. For a force to speed up a “diffusion-controlled association”, it must be present in the diffusion process that leads to the transient complex. Indeed, analytical results on model systems show that, when the range of the force is reduced, the resulting rate enhancement decreases drastically.29,37,38 For protein-protein association, the dominant long-range force is provided by electrostatic interactions.
Rate constants higher than kD0 are indeed usually correlated with favorable electrostatic interactions, as manifested by complementary charge distributions on the binding partners, which are illustrated in Figure 1 for four protein pairs. RNA and DNA have negative charges from phosphate groups; proteins targeting them (like barnase, RNase A, and DNase E9) generally have enriched distributions of basic residues on the nucleic-acid binding sites.39,40 Electrostatically enhanced protein-nucleic acid association rates can thus be anticipated. The basic residues on the nucleic-acid binding sites can also be easily exploited by inhibitors (like barstar, ribonuclease inhibitor, and Im9): fast inhibition can be achieved through a concentration of acidic residues on the latter molecules. Toxins blocking the Shaker potassium channel follow a similar strategy.23 Apparently for facilitating the conduction of the positively charged potassium ion, the mouth of the channel pore is lined with two rings of acidic residues.41 To complement the resulting negative electrostatic surface, channel toxins have excess basic residues. The charge-complementarity argument can also be made to rationalize the excess basic residues on fasciculin 2 (fas), the snake toxin targeting AChE. AChE uses a negative electrostatic surface around the entrance to the active-site gorge for the fast binding of its positively charged substrate.4,42 In other cases, such as in complexes formed by WASp and Cdc4243 and by interleukin-4 (IL4) and its receptor,44 the reasons for a particular subunit to take up either positive or negative charges are not obvious. That they nonetheless show charge complementarity implicates functional roles of their fast association.
1.2 Analytical Models of Protein Association Rate Constants
The first theoretical result for the association rate constant was obtained by Smoluchowski,45 who found that the diffusion-controlled rate constant for two uniformly reacting spheres to form a complex is
| (4) |
where D is the relative translational diffusion constant and R is the contact distance between the centers of the two spheres. Debye recognized that the association rate between oppositely charged molecules can be increased by electrostatic interactions.46 For two uniformly reacting spheres with a centrosymmetric interaction potential U(r), he found the diffusion-controlled rate constant to be
| (5) |
where r is the inter-sphere distance and kBT is thermal energy. In the following discussion we will use kD0 to denote the diffusion-controlled rate constant in the absence of an interaction potential (also referred to as the basal rate constant), and use kD to denote the counterpart in the presence of an interaction potential. The enhancement over the basal rate constant by an attractive interaction potential in the Debye model is quite modest. For example, for a Coulombic interaction potential U(r) = −Q/r, one finds, upon evaluating the integral in eq 5, kD/4πDR = (Q/kBTR)/(1 − e−Q/kBTR). The rate enhancement, kD/4πDR, is 9-fold when the magnitude of the potential is 9kBT at contact. This modest rate enhancement will be contrasted below with a much greater enhancement predicted on a more realistic model for protein association.
For medium-sized proteins, R and D are of the order of 40 Å and 20 Å2ns−1, respectively. Eq 4 would predict a basal rate constant of 6 × 109 M−1s−1. However, it is important to recognize that the stereospecific association of two proteins involves significant orientational constraints, thus the Smoluchowski formula is of little use. Solc and Stockmayer tackled the problem of orientational constraints through a quasi-chemical approximation.47 For two spheres each with a reactive patch, they found the diffusion-controlled rate constant to be
| (6) |
where F1 and F2 are the surface fractions covered by the reactive patches. Relative diffusion has to bring their separation vector to within both reactive patches, therein the native complex can form instantaneously. Specifying the reactive patches thus amounts to specifying the transient complex. An approximate expression, obtained by Berg,48 for Λi (i = 1 or 2), in the case of a patch spanning polar angles between 0 and δi, is given by
| (7) |
where ξi = [(1 + DiR2/D)/2]1/2 and Di is the rotational diffusion constant. For small patches one finds
| (8) |
The basal rate constant obtained by Brownian dynamics simulations (see Section 2.2),28 with simple structural assumptions about the transient complex (which was modeled by 2–3 correctly-formed bonds between the proteins) is 105 – 106 M−1s−1, which is four orders of magnitude lower than the unrealistic Smoluchowski result. Such a rate constant is predicted by eq 8 for reactive patches at sizes of δ1 ~ δ2 ~ 5° (with R and D at values quoted earlier and Di at 0.02 ns−1). Results similar to eq 8 for the basal rate constant have been obtained by a number of different methods.49–52
Reactive patches at sizes of ~ 5° cover only a fraction of 10−3 of the surface of each sphere. With the inter-sphere vector having to lie within both reactive patches before the native complex can form, the association rate constant is naïvely expected to be lower by a factor of (103)2 = 106. As just stated, the reduction factor predicted by eq 8 is 104. The basal rate constant thus appears to be 100-fold higher than naïve expectation. Northrup and Erickson28 explained this apparent paradox by noting, based on Brownian dynamics simulations, that when two proteins collide there is a high probability that they will re-collide several times before separating again. Further, due to the separation of time scales between rotational and translational diffusion, they may rotate significantly between those collisions.
Within the model of two spheres with reactive patches, the influence of an interaction potential on the association rate constant has also been studied.29,49 The expressions for kD in the presence of a centrosymmetric potential U(r) is
| (9) |
where ρl(r) satisfies
| (10a) |
with the boundary condition ρl(r) → 0 as r → ∞, ci(li) are given by Legendre polynomials,
| (10b) |
and Cll1l2 are given by Wigner 3-j symbols,
| (10c) |
Analytical results such as eq 9 can provide valuable insight on the properties of the association rate constant. In particular, in contrast to the Debye model in which rate enhancement by an attractive interaction potential is quite modest, the rate enhancement in the more realistic patch model can be substantial. For reactive patches with sizes of δ1 ~ δ2 ~ 5°, the rate enhancement is ~5000-fold for a potential with a magnitude of 9kBT at contact.29 More importantly, it is found that, when U(r) is long-ranged and δ1 and δ2 → 0, the dependence of kD on the interaction potential disappears except in the Boltzmann factor e−U(R)/kBT.29,49 Therefore the effect of the interaction potential can be simply captured by the Boltzmann factor. In Section 2.2 we will return to this important result.
In the following sections we give a detailed account of theoretical and experimental studies of protein-protein association kinetics. In Section 2 we outline the main experimental and theoretical methods for studying association kinetics. Section 3 presents experimental and theoretical analyses of the transient complex as well as the transition state for the final step in the association pathway. Section 4 continues with the important subject of electrostatic rate enhancement. In Section 5 we discuss protein-protein association in the membrane environment, a field with growing interest. The subject of Section 6 is the effect of crowding on association. Some final conclusions are drawn in Section 7.
2. Methods for Studying Protein-Protein Association Kinetics
The association kinetics between proteins has been studied vigorously using both experimental and theoretical methods. There are some basic differences in studying binding of proteins and their folding. In folding, the unfolded state is not well defined, while the unbound state of proteins has a well defined structure. This is not true in all cases, as many protein-interaction domains were found in recent years to be unstructured, and thus binding and folding in those cases are coupled.53–56 However, here we review mostly the binding of structured proteins. A second main difference between binding and folding is that folding is a first-order process, where overall translational and rotational diffusion plays only a limited role. Conversely, as discussed in Section 1, for binding overall translational and rotational diffusion play a major role in dictating the rate constant of the reaction. The existence of well-defined unbound structures simplifies the theoretical study of binding. For hetero-complexes, this also simplifies the experimental study, as one has a clear starting point for the reaction, the mixture of the separate components of the complex, and the possibility to follow in real time complex formation. In this review we focus on the experimental and theoretical work done to learn more about association between single globular proteins to form complexes.
2.1 Experimental Methods
Most of the experimental work reviewed here was done with the proteins being in free solution, using methods such as stopped-flow spectrometry and NMR spectroscopy. This excludes the results of many measurements done using surface plasmon resonance (SPR), with one of the proteins attached to a surface and the other one in solution.57 Direct comparisons of association rate constants for a number of protein-protein complexes showed that SPR data may provide different ka values from those obtained in solution.58,59 These differences may be attributed to mass-transport, protein-immobilization effects, surface charges, crowding, etc.60,61 As these factors introduce additional complexity to the analysis of protein association kinetics that is not directly related to the association reaction, we review here only solution data.
2.2 Computer Simulations and Modeling
One can model protein-protein association by simulating the translational and rotational Brownian motion of the subunits. In these Brownian dynamics simulations, conformational fluctuations within the subunits are neglected and hence time scales far beyond those accessible to molecular dynamics simulations can be explored. Brownian dynamics simulations make it possible to calculate the diffusion-controlled rate constant kD for protein shapes and interaction potentials beyond the scope of analytical theories. This approach has been used by many groups.24,28,49,62–71 In such a calculation one must specify a precise set of conditions, which when satisfied signifies the formation of the native complex. This set of conditions, typically implemented as an absorbing boundary in Brownian dynamics simulations, amounts to defining the transient complex. Rather than being guided by any theoretical considerations, the location of the absorbing boundary is usually proposed in an ad hoc way, and often adjusted for best agreement with experiment. For example, in their Brownian dynamics simulations of barnase-barstar association, Gabdoulline and Wade24 tested three different specifications of an absorbing boundary against experimental data. Two of these were based on atom distances and the third was based on electrostatic interaction energy; each specification had an adjustable parameter.
We will give a more detailed account on the specification of the transient complex in Section 3.1. Here we address the effect of interaction potential, particularly one arising from electrostatic interactions between two associating proteins. In Brownian dynamics simulations, the effect of electrostatic interactions can be modeled by accounting for their influence on the translational and rotational Brownian motion of the proteins. In principle, the electrostatic force and torque on the proteins can be calculated by solving the Poisson-Boltzmann equation,
| (11) |
where ε is the dielectric constant, Φ the electrostatic potential, ρ the charge density inside the proteins, M a function with value 1 in all regions where mobile ions in the solvent are accessible and 0 elsewhere, and qi and ci the charges and bulk concentrations of mobile ions, respectively. The solution of the Poisson-Boltzmann equation is simplified by linearization, leading to
| (12) |
where ε s is the dielectric constant of the solvent and κ= (8πe2I/εskBT)1/2 is the Debye-Hückel screening parameter, with the ionic strength. However, even after linearization, solving the Poisson-Boltzmann equation on the fly during a Brownian dynamics simulation is prohibitively expensive. One thus has had to rely on approximations, such as treating one of the proteins as a set of test charges62 (which leads to significant errors from neglecting the low-dielectric region of the protein interior49) or the more elaborate effective-charge model.72 Unfortunately, the approximations are worst when the proteins are in close proximity, precisely where electrostatic interactions are expected to have the strongest influence on kD.
An algorithm is needed to obtain kD from the simulations. A widely used algorithm was developed by McCammon and co-workers.73 According to this algorithm, one protein is traditionally immobilized at the origin and trajectories of the other protein are initiated uniformly on a spherical surface (at a radial distance r = b) (see Figure 2a). This “b” surface should be chosen far away from the immobilized protein such that the electrostatic interaction potential is centrosymmetric. If rotational motion is allowed, then the starting orientations of the two proteins are random. The diffusion-controlled rate constant is given by
Figure 2.

Two algorithms for calculating the diffusion-controlled association rate from Brownian dynamics simulations. (a) Algorithm of Northrup et al.73 (b) Algorithm of Zhou.75 In either algorithm, Brownian trajectories can be terminated by two triggers. In (a), the triggers are absorption by either the inner absorbing boundary or the q surface; in (b) the triggers are either reaction in the reaction region or when the lifetime of the trajectory exceeds a cutoff.
| (13) |
where f∞ is the fraction of trajectories that, instead of escaping to infinity, reach the absorbing boundary specifying the conditions for the formation of the native complex. To find f∞, an outer absorbing boundary at r = q > b, called the q surface, is introduced. The trajectories are terminated when either the absorbing boundary for complex formation or the q surface is reached. From the fraction, f, of trajectories terminated on the inner absorbing boundary, one finds
| (14) |
where kD(b) and kD(q) are given by eq 5 with R replaced by b and q, respectively. It has been shown that eq 14 is strictly valid only when the q surface is far away from the immobilized protein such that the equilibrium distribution of the mobile protein becomes isotropic.74
An alternative algorithm75,76 has also been developed, in which the absorbing boundary is extended into a “reaction region” with a finite volume 𝒱RR (Figure 2b).18,77 A protein pair that has reached the reaction region has a finite rate, γ, to form the native complex. In this treatment of protein association, the reaction rate γ models the conformational rearrangement that brings the protein pair from the transient complex (specified by the reaction region) into the native complex. That is, kc = γ. The equilibrium constant kD/k−D, is given by 𝒱RRe−<U>*/kBT, where <U>* is the average interaction energy within the transient complex. By starting Brownian trajectories from within the reaction region and obtaining the surviving fraction, 𝒮, of the trajectories, the association rate constant is obtained as ka = γ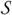
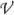 RRe−<U>*/kBT. Substituting the various results into eq 2, one finds that the diffusion-controlled association rate constant is given by
RRe−<U>*/kBT. Substituting the various results into eq 2, one finds that the diffusion-controlled association rate constant is given by
| (15) |
While the surviving fraction 𝒮 depends on the rate γ and on how much the absorbing boundary is extended to form the reaction region, the result for kD given by eq 15 is only determined by the absorbing boundary and the interaction energy. We also note that the reaction-controlled association rate constant (eq 3b) is
| (16) |
in the present treatment.
In a Brownian dynamics study of protein-protein association under the influence of electrostatic interactions, it was discovered that the survival fraction 𝒮 is insensitive to the presence of the electrostatic interaction energy.49 Then one can write eq 15 as
| (17) |
with kD0 as the basal rate constant given by
| (18) |
where 𝒮0 is the survival fraction without any biasing force. Note that a dependence on interaction energy as simple as given by eq 17 is expected for the reaction-controlled rate constant (see eq 16 ) but totally unexpected for the diffusion-controlled rate constant. Later an analytical derivation of eq 17 was presented,78 which confirmed the two requirements for the validity of eq 17 suggested in the original work.49 These requirements are that the association is stereospecific and the interaction energy is long-ranged. Fortuitously (and fortunately!) these conditions are fulfilled by protein-protein association under the influence of electrostatic interactions. The accuracy of eq 17 has been demonstrated against results from Brownian dynamics simulations of protein-ligand binding20,79,80 and against analytical results given by eq 9 for model systems.29 This equation resolves one of the two main obstacles to reliable prediction of protein association rate constants, by making it possible to rigorously treat electrostatic interactions. The effect of electrostatic interactions is captured by the Boltzmann factor e−<U>*/kBT, which can be obtained by averaging over a relatively small number of representative configurations in the transient complex. The basal rate constant kD0 still needs to be obtained through force-free Brownian dynamics simulations, but these simulations are inexpensive.
At first glance, eq 17 looks like the transition-state theory of the Eyring type.25 There is no relation between the two. As already emphasized, eq 17 is for a diffusion-controlled rate constant, but Eyring’s theory is for energy barrier-crossing processes that are activation-controlled. Kramers27 has shown that Eyring’s theory is a very poor approximation when barrier crossing becomes diffusion-controlled. However, there is no link between eq 17 and Kramers’ theory for diffusion-controlled barrier crossing either. In particular, the average interaction energy of the transient complex, <U>*, which superficially resembles the energy barrier in Eyring’s and Kramers’ theories, is typically negative (leading to rate enhancement) instead of positive. Unlike the processes studied by Eyring and Kramers, where the energy barrier is what hinders the rate, the diffusion-controlled rate of protein association is hindered by the translational/rotational constraints of the transient complex. Moreover, eq 17 does not require a reaction coordinate like the theories of Eyring and Kramers. In fact, as will be discussed next, the specification of the transient complex involves at least six degrees of freedom – three for translation and three for rotation.
3. Intermediates along the Association Pathway
Throughout this review we use the word “intermediate” as a generic term for a set of configurations in which there is a degree of correlation in translation and rotation between the associating proteins; it does not necessarily correspond to a local energy minimum or a species trapped in kinetic experiments. We refer to the latter as a kinetic intermediate. As discussed in Section 1.1, the association process starts with the formation of the transient complex (sometimes termed the encounter complex, see Section 3.1 below). The transient complex is close to the native complex in relative separation and relative orientation between the subunits, but still misses a majority of the short-range interactions characterizing the native complex. The formation of these short-range interactions requires passage through another fleeting structure, the transition state for conformational rearrangement.26,81 How can structural information be obtained on these intermediates?
Structural studies are routinely done to elucidate the transition state and kinetic intermediates of protein folding or enzyme catalysis. A range of experimental tools has been developed for this task. NMR is a powerful tool to pin down the residual structures of the unfolded state, as well as to capture transient folding intermediates.82 Φ-value analysis reveals whether specific interactions are formed already during a kinetic intermediate or the transition state of the reaction.56,83,84 Time-resolved spectroscopy and single-molecule spectroscopy are powerful tools, which are frequently applied to investigate kinetic intermediates and transition states in folding.85–88 While these experimental tools provide only a partial view, they are extremely valuable for molecular dynamics simulations and other computational studies, as they provide experimental reference points to benchmark simulations.89–91
In comparison to studies on protein folding, structural studies on intermediates along the pathway of protein-protein association are much less common. This may be partly attributed to the technical difficulties stemming from the low population of the binding intermediates, and the ill-defined nature of the transition state for binding. Still, the development of protein-engineering tools, NMR and time-resolved optical spectroscopy resulted in a number of interesting experimental studies shedding light on the mechanisms of protein-protein association. Valuable guidance has also been provided by theory. In the following we focus on studies characterizing the transient complex and the transition state of association.
3.1 The Transient Complex
In theoretical predictions of association rate constants one must specify the transient complex. In different contexts, the transient complex has appeared as reactive patch, absorbing boundary, reaction region, and encounter complex. The ad hoc way by which the transient complex is specified in Brownian dynamics studies is pointed out in Section 2.2. The situation was no better when eq 17 was first used to predict association rate constants. In one early such study on barnase-barstar association,77 the transient-complex ensemble was specified by adjusting the ranges of relative translation and rotation between the two proteins to match the experimental data at high ionic strength. Miyashita et al.92 directly used experimental data for the association of cytochrome c2 with bacterial reaction center to locate the transient-complex ensemble in the 6-dimensional translation and rotation space. The correlation coefficient between calculated changes in the electrostatic interaction energy by mutations and the experimental counterpart
| (19) |
was used as an effective energy in a Monte Carlo simulation. Schreiber and co-workers93,94 further simplified eq 17 by calculating <U>* from an empirical function simply on the native complex, thus avoiding the specification of the transient complex; the empirical function was parameterized on experimental data. Applications of that approach are presented in Section 4.2.
In order to predict association rate constants from theory alone, the transient-complex ensemble has to be specified without reference to experiment. A solution to this challenging problem was proposed in a recent paper,18 based on analyzing the interaction energy landscape of associating proteins.
In a complete theory, the overall association rate constant ka should not be sensitive to where the transient complex is placed. If it is placed far away from the native complex, then kD will be large but kc will be small. Conversely, if it is placed very close to the native complex, then kD will be reduced but kc will become very large. Either way, eq 2 is expected to give nearly the same result for ka (as explicitly shown for a model system95). However, given the considerable difficulty and uncertainty in the calculation of kc, it is highly desirable to use kD as a close approximation for ka. Then there is an optimal location for placing the transient complex.22 If it is placed too far from the native complex, then the resulting kD would not be a useful approximation for ka. On the other hand, placing the transient complex too close to the native complex would mean that short-range interactions and conformational rearrangement have to be dealt with in calculating kD. The native complex sits in a deep well in the interaction energy landscape.18 The optimal placement for the transient-complex ensemble is at the outer boundary of the native-complex energy well (Figure 3a).18,22
Figure 3.

Location of the transient complex within the interaction energy landscape. (a) The energy landscape of protein-protein association. Translation (r) and rotation (e,χ) coordinates lie in the horizontal plane. The transient-complex ensemble is indicated by a green ring. The smooth energy landscape results from keeping the two proteins in their native internal conformations. (b) Identification of the transient complex, consisting of configurations with the contact level Nc*, where σχ is poised for a sharp increase with decreasing Nc. (c) Translational volume of the transient complex, which is the projection of the hypersurface Nc = Nc* into the 3 dimensions of translation, for the barnase-barstar pair. A cluster of red dots represent the locations of the displacement vector r (yellow arrow). Parts (a) and (b) were adapted with permission from Ref. 1. Copyright 2008 Wiley Interscience.
The algorithm for identifying the transient complex was based on the following observation: inside the native-complex energy well, translation and rotation are restricted, but once outside the proteins gain significant translational/rotational freedom.18 Thus the outer boundary of the native-complex energy well coincides with the onset of translational/rotational freedom. To simplify the calculations required for determining the transient complex, the short-range interaction energy stabilizing the native complex was modeled by the number of contacts, Nc, formed between the protein partners. Translational/rotational freedom was measured by σχ(Nc), the standard deviation of the rotation angle χ in configurations with a given contact level Nc. A sharp increase in σχ with decreasing Nc marks the onset of translational and rotational freedom and hence the location of the transition complex (Figure 3b). As illustrated in Figure 3c for the barnase-barstar pair, the resulting model for the transient complex is a compact ensemble of configurations which marks the onset of a sharp increase in translational and rotational freedom as the protein partners move out of the native-complex energy well. In the ensemble of transient-complex configurations of the barnase-barstar pair, the displacement vector r is mostly confined in a 4 × 4 × 1 Å3 volume, corresponding to distances of 4.5 ± 0.5 Å between the protein surfaces (allowing for at least one layer of solvent). Within this translational volume, the relative orientations of the two proteins are also severely restricted, with the rotation angle χ mostly confined in the 0° ± 10° range.
Since kD is used as the prediction for ka, eq 17 can be rewritten as an equation for ka:
| (20) |
where ka0 is the rate constant for reaching the transient complex in the absence of any biasing force and can be obtained by Brownian dynamics simulations, and the interaction energy is now denoted with a subscript “el” to signify that only electrostatic interactions are to be included. The neglect of short-ranged non-electrostatic effects from the Boltzmann factor is based on two considerations. First, as Figure 3c shows, the transient-complex configurations are separated by at least one layer of solvent, therefore short-ranged forces such as hydrophobic and van der Waals interactions are relatively weak in the diffusion process leading to the transient complex. Second, short-range interactions, even when present within the transient complex, contribute much less to rate enhancement (i.e., ka/ka0) compared to long-range interactions. However, short-range interactions are essential for determining the location and size of the transient-complex ensemble in configurational space, which in turn affect the magnitude of ka0. A transient-complex ensemble that is less restricted in translation and rotation will lead to a higher ka0. Variation of the restriction in translation and rotation within the transient complex with solvent conditions or among different protein complexes can be viewed as a configurational entropy effect. The basal rate constant ka0 captures all the contributions of short-range interactions and configurational entropy.
The structural model for the transient-complex ensemble presented above along with eq 20 constitutes the transient-complex theory for predicting protein-protein association rate constants. With this theory, the two main obstacles faced by the traditional approach of Brownian dynamics simulations, the necessity for approximate treatment of electrostatic interactions and the ad hoc specification of the transient complex, are both resolved. Applications of this theory are presented in Section 4.3.
3.2 Other Intermediates
The term “encounter complex” has frequently been used in describing the pathway of protein-protein association. It appears that this term is assigned different meanings in different contexts. For example, in calculating association rate constants by Brownian dynamics simulations, Gabdoulline and Wade used this term to refer to “the end-point of diffusional association,”24,96 which would be similar to what we have defined as the transient complex. However, these same authors later also used encounter complex to refer to low free-energy regions in configurational space.70 In the paramagnetic NMR experiments described next, encounter complex refers to a minor, dynamic state that is in equilibrium with a dominant, stereospecific complex.
Using the paramagnetic relaxation enhancement (PRE) NMR technique on the electron transfer pair of yeast cytochrome c peroxidase (CcP) and iso-1-cytochrome c (Cc), Volkov et. al.97 determined the structure of the dominant, well defined complex and delineated the configurational space of the minor, dynamic state. The latter, as just noted, was referred to as encounter complex. The dominant complex is very similar to the X-ray structure of the pair and is occupied for >70% of the time. In the encounter complex, Cc occupies a region around the position in the dominant complex; the negative charges of CcP bordering this region suggest “that electrostatic attraction plays a dominant role in determining the nature of the encounter complex.” However, the data also seem to indicate that the encounter complex contains configurations in which Cc has its “back” side facing CcP.
Tang et al. carried out a similar study for the association between the phosphocarrier protein, Hpr, and three proteins in the bacterial phosphotransferase system.98 They found a rather diffuse encounter complex that qualitatively correlates with the electrostatic surface potentials on the interacting proteins. These experiments were performed in the absence of salt, in which case non-specific electrostatic attraction of these highly charged proteins is strong over a long range.6 In a follow-up paper,99 Tang et al. showed that the non-specific part of the encounter complex is reduced to a large extent by adding salt, while more specific configurations (located near the region of the native complex) were less affected. This study seems to suggest that not all regions of the encounter complex lead to productive association – it is the region which does lead to productive association that we specifically refer to as the transient complex.
Spaar et. al.70 developed a method for mapping the encounter complex along the association path from BD simulations. Applying this method to the barnase-barstar interaction, they suggest two minima along the pathway, one leading (according to an analysis of successful trajectories) to association, while the other does not. Still, they suggest that this second region may be helpful to steer barstar into the region of the transient complex. In a yet unpublished study combining simulation and experiment, GS et.al. aimed to directly evaluate the importance of non-specific encounter regions to association. Using the method developed by Spaar. et. al 70 on TEM1-BLIP association, the BD simulations identified two main regions with low interaction energies for the wild-type complex; however, most trajectories started at these regions did not lead to productive binding. It was found that only mutations designed to increase the size and energy of the encounter region near the binding interface had a major effect on the measured association rate constant. Conversely, mutations increasing or decreasing the occupancy and energy of other intermediates had no effect on the association rate constant. These results indicate that, not only the occupancy or energy of the encounter regions, but also their location is important in determining their effect on the association rate constant. Thus, some encounter regions are fruitful while others are futile. This conclusion was in fact anticipated when the encounter region that does lead to productive association was singled out to define the transient complex (see above discussion illustrated by Figure 3a).1
In some cases, kinetic intermediates in binding have been observed. One such case is the interaction between colicin endonucleases and their cognate and noncognate immunoproteins.100 Stopped-flow spectrofluorimetry shows that binding of both Im9 and Im2 to E9 result in a rapid fluorescence quenching step (pre-equilibrium), with a concentration-dependent rate of formation, followed by a second, concentration-independent slow rate (on a timescale of seconds). The data are best explained by a mechanism where cognate and noncognate complexes alike form a “dynamic encounter complex.” Thereafter the cognate pair transitions to a high-affinity, stereospecific complex, whereas the noncognate pair remains dynamic without achieving high affinity.
A somewhat different mechanism involving a pre-equilibrium complex followed by a reorganization step was suggested for cystatin A binding papain.101,102 The bi-phasic behavior of cystatin A/papain interaction was observed by following independently the inhibition of the catalytic activity of papain (where a linear dependence of kobs on protein concentration was observed), while the spectroscopic probe (which monitors the accumulation of the native complex) showed a hyperbolic dependence of kobs on protein concentration. This example demonstrates that characterization of association behavior may depend on the probe used to monitor it.
3.3 Mapping Intermediates by Mutations
3.3.1 Φ-Value Analysis
Φ-value analysis was successfully applied to map the transition state for protein-folding, and became the golden standard for many simulations.103 It has been demonstrated that this analysis can be used also for studying the rate-determining state (denoted by ‡) for protein-protein association.14,56,104–106 The basic formulation for such analysis for binding is given by
| (21) |
where ΔΔG‡–U is defined by eq 19 and ΔΔGC–U is analogously defined but with the association rate constant ka replaced by the association constant Ka. Mutations that induce a similar effect on state ‡ and on the free energy of binding will have a Φ–value of 1, while mutations that have no effect on ‡ but change the association constant will have a Φ–value of zero. The interpretation of this form of Φ-value analysis is particularly transparent when conformational rearrangement is rate-limiting (cf. eq 3b). In this case ΔΔG‡–U represents the mutational effect on the free energy for forming the transition state between the transient complex and the native complex, and the Φ-value analysis thus reports on the transition state. If, on the other hand, conformational rearrangement is fast (i.e., kc ≫ k−D), then the analysis reports on the formation of the transient complex.
Figure 4 shows a Φ–value analysis for a large number of ‘hotspot’ mutations (those with ΔΔGC–U > 2 kcal/mol in magnitude) collected from TEM1-BLIP, barnase-barstar and Ras-Ral binding.6,14,93,107 The mutations were divided into three groups: one consisting of non-charged residues, the second consisting of charged residues located within the binding interface, and the third consisting of charged residues located outside the binding interface. The reason we show only ‘hotspot’ residues is to avoid erroneous Φ-values, as the experimental error for ΔΔG measurements is in the order of 0.3 kcal/mol. As only very few residues located outside the binding interface pass this criteria, we included in this group also multiple mutations, that were designed to specifically increase the association constant.14,93 The data clearly demonstrate that non-charged mutations always have Φ-values close to zero, while charged residues located outside the physical binding site have Φ–values close to one. Charged residues located within the binding site have mixed values. A simple explanation of these Φ–values is that they report on the transient complexes of the three protein pairs. As noted in the discussion below eq 20, the transient complex is predominantly stabilized by long-range electrostatic interactions, with non-polar interactions playing a minor role. A striking result of the Φ–value analysis is that hydrophobic residues appear to play only a minor role in the formation of the transient complex. As most mutations were made to Ala, which retains a hydrophobic surface, one cannot rule out the possibility that nonspecific hydrophobic interactions are formed at the transient complex also for the mutant, reducing the Φ–value. However, this contribution should be very limited as mutations to Ala create cavities on the surface that are not fully filled up by the incoming protein.
Figure 4.

Φ-value analysis of the transient complex/transition state for association determined for ‘hotspot’ mutations (affecting the binding affinity by > 2 kcal/mol) collected from TEM1-BLIP, Barnase-barstar and Ras-Ral association.6,14,93 Φ-values close to 1 suggest that the involved residues have similar interactions in the transient complex/transition state and in the native complex, while values close to 0 indicate the residues do not form any inter-protein contacts in the transient complex/transition state.
A hallmark of a partially reaction-limited reaction (i.e. not purely diffusion-controlled) is a non-linear relation between the rate of association and the concentrations of reactants. This has been observed for enzyme-substrate interactions, but rarely for protein-protein association. However, in the case of the association between RalGDS-RBD and Ras this non-linearity could be experimentally demonstrated, presenting direct evidence for the transient complex as a kinetic intermediate for association.14 The non-linearity was observed despite a high ka value (107 M−1s−1) and a putative basal rate of 6 × 105 M−1s−1, which should in principle place this association in the diffusion-controlled regime (see Section 1.1). Using the program PARE (see 4.2.1),93 mutations of charged residues located at the periphery of the binding interface were designed, which specifically increased ka, and did not affect kd. Stopped-flow measurements showed that the increase in ka of a +7 mutant was a result of an increased rate of formation of the transient complex, while the rate of conversion to the native complex was unchanged at a value of ~400 s−1.14 From these data a Φ-values of 1.0 and 0.9 for the transient complex and transition state, respectively were calculated. This demonstrates that favorable electrostatic interactions introduced by mutation stabilize the transient complex and the transition state to the same extent as they stabilize the native complex. This study also points to the difficulty in assigning a particular association process as diffusion-limited or reaction-limited. At low protein concentrations, the overall association rate may appear to be diffusion-limited, but at high protein concentrations it may appear to be reaction-limited.
The +7 RalGDS-RBD variant (obtained by electrostatic design) binds Ras with an association constant similar to that measured for the native effector, Raf. Interestingly, the electrostatic potential map of the RalGDS-RBD variant is also similar to that of Raf (Figure 5a), despite the very different sequences of the two effectors (<15% homology).14 The initial aim of this project was to optimize through mutation the electrostatic interactions between RalGDS-RBD and Ras. The similarity of the electrostatic potential maps of the +7 Ral mutant and Raf suggests that the natural complex between Ras and Raf is optimized by natural selection for fast binding.
Figure 5.

(a) Electrostatic potential maps of Ras, Raf-RBD, and Ral-RBD. The Ral + 7 mutant contains the mutations M26K/D47K/E54K/D90K, which were predicted by the program PARE to significantly increase the rate constant of association of Ral-RBD to Ras. These four mutations are located outside the Ras-binding site on Ral-RBD. The figure was drawn using the program GRASP, with the contours drawn at 2kBT/e (blue for positive and red for negative). (b) Plot of experimental values of the association rate constants for mutants of Ral-RBD to Ras vs. values calculated using PARE. Adapted with permission from Ref. 14. Copyright 2004 The National Academy of Sciences of the United States of America.
pKa shifts during the association process can also provide structural information about the transient complex.108,109 The pKa of His102 in unbound barnase was measured to be 6.3, while in the native complex a pKa of < 5 was measured. The pH dependence of ka showed a pKa value similar to that in the unbound protein, therefore the shift in pKa upon binding barstar occurs after the transient complex. The X-ray structure of the native complex and electrostatic calculations110 indicate that the shift in pKa is attributable to the burial of His102 in the interface with barstar and interactions with surrounding barstar residues; the pH dependence of ka suggests that the burial and interactions are not fully formed within the transient complex. A similar behavior was observed for the association of R67 DHFR, with a pKa of 6.6 that was assigned to H62, but a dissociation reaction with a pKa of under 5.5, with the pKa shift being attributed to specific short-range interactions that are not formed within the transient complex/transition state for association.111 These studies provide a clear indication that short-range interactions are mostly formed late along the association pathway.
3.3.2 Double-Mutant Cycle Analysis
Double-mutant cycles measure the coupling energy between a pair of residues from the difference in binding free energy between a double mutant and the two single mutations. For binding, the coupling energy defined by108,112–114
| (22) |
provides a measure of the interaction between two residues, X and Y, in the transient complex or the transition state (depending on which dictates the rate-limiting step). Like Φ-value analysis, this technique was also first applied successfully to protein-folding studies.112,113,115,116
From a large number of double-mutant cycles studied concerning the association rate constant of barnase and barstar, significant coupling energies were found only between charged residues separated by < 10 Å from one another in the native complex.6 None of the non-charged residues had a significant value with any other residue. A similar experiment was done on the interaction between cytochrome C2 and the bacterial reaction center, but only between charged residues. values, as calculated from the measured electron transfer rate constant, which is closely related to ka,92 again showed that significant interactions exist in the transient complex/transition state only between residues which are < 10 Å away from each other in the native complex.117 A similar result was obtained also for the association of TEM1-β-lactamase and its protein inhibitor BLIP.118 Repeating the double-mutant cycles at up to 1 M salt (which masks the effects of charge-charge interactions) showed that, for barnase-barstar and for the complex between P. laminosum Cyt f and plastocyanin, some, but not all, pairwise charge-charge interactions were maintained, suggesting that structural specificity of the transient complex/transition state is preserved even at high salt, but its size may shrink.119,120
The experimental mutant and double-mutant cycle data measured on the association rate constants were further used to model the structures of the transient complex/transition state. In the study of Harel et. al.,118 the transient complex/transition state was modeled from the experimental values by searching for those inter-protein orientations that best account for the experimental ΔΔG‡int values (Figure 6). Similarly, Miyashita92 related the experimental ka values of mutant proteins to differences in the calculated electrostatic energies for a wide range of cytochrome C2-reaction center (Cyt-RC) configurations. Both studies gave a very similar description of the transient complex/transition state. In both cases, the transient complex/transition state was stabilized by electrostatic interactions, with the ensemble of configurations spread out around the native complex, but in neither case were short-range interactions formed, suggesting a solvated transient complex/transition state. The average structure of the transient complex/transition state was not necessarily located exactly at the center of the binding interface, but could be shifted towards one side of the interface. This was observed for both the Cyt-RC and the electrostatically optimized TEM1-BLIP pairs, while for the barnase-barster pair the average structure of the transient complex/transition state overlapped the native complex.92,94,118 These characteristics bear striking resemblance to the compact structural model of the transient complex derived from theory (Section 3.1). They suggest a certain pathway for association (down an energy funnel), which would help in speeding up the association.18 Contrary to these results, no indication for a transient complex with specific structures was found for the association between wild-type TEM1-BLIP and BLIP or between IFNα2 and IFNAR2 (Figure 6). Therefore, a diffusive transient complex/transition state was suggested for these protein pairs. What distinguishes a diffusive transient complex from a compact one is the absence of defined inter-protein orientations. As was clearly shown for the TEM1-BLIP complex, mutations introduced through rational design can change the transient complex/transition state from diffusive to compact and vice versa,.118
Figure 6.

Mapping the transient complex/transition state for protein-protein association for the TEM1-BLIP and barnase-barstar complexes using double-mutant cycle data as constraints. Each point represents the center of mass of one of 2220 configurations of TEM1 or barnase perturbed from the native complex. (the mobile proteins in the simulations). BLIP and barstar are the fixed proteins and are represented as ribbon. The point in the middle of each cap represents the X-ray structure of the native complex. The different colors represent configurations selected by different filtering cutoffs; colder colors designate a configuration that passes a more stringent cutoff (thus, has a higher probability of occupancy in the transient complex/transition state). The TEM1-BLIP complex was electrostatically optimized using the program PARE, by introducing mutations located outside the binding interface.
All the evidence described above suggests the existence of a transient complex/transition state along the association pathway. The main points presented are: 1. The binding of Ras to either Ral or Raf is bi-phasic, with the first phase being diffusion-limited and the second phase reaction-limited; 2. Increasing the electrostatic complementarity between Ras and Ral specifically affects the rate of the first phase and does not affect the second phase of association; 3. Increasing electrostatic complementarity affects only the association but not dissociation rate constant; 4. Double-mutant cycle experiments as well as Φ-value analyses clearly show that only charged residue pairs form interactions in the transient complex, while short-range interactions are largely unformed in the transient complex. Further formation of such short-range interactions, which are important for stabilizing the native complex, may encounter a transition state. In the case of a compact transient complex, the transient complex already has inter-protein orientations similar to those in the native complex. The transition state will at least inherit such an orientational similarity. Therefore, in terms of gross structure, the transient complex and the transition state seem to be close.
4. Probing Electrostatic Enhancement of Association Rates
As discussed in previous sections, electrostatic interactions have long been recognized as the dominant factor used by Nature in order to enhance protein association beyond the basal rates dictated by diffusion.13,49,121 In this section we discuss experimental and theoretical methods to probe and analyze the effect of electrostatic attraction on association kinetics, as well as protein engineering studies which enable us to modify and control this effect. The preceding sections have already presented a glimpse into some of these issues.
4.1 Ionic Strength Dependences of ka and kd
The magnitude of electrostatic attraction between two proteins can be modulated most simply by changing the ionic strength of the solution. It has been recognized that, when the association between two proteins is diffusion-controlled, the modulation of electrostatic attraction leads to a universal phenomenon related to the association and dissociation rate constants ka and kd which show very disparate dependences on ionic strength.22,122 The association rate constant decreases significantly with increasing ionic strength, whereas the dissociation rate constant is only modestly affected by ionic strength. The disparate ionic-strength effects have been observed on a number of the protein-protein complexes discussed above.4,5,11,35,109 Many other proteins conform to the same behavior.123–133
The compact structural model of the transient complex derived from theory, presented in Section 3.1, provides a nice explanation for the disparate effects of ionic strength on association and dissociation rate constants when association is diffusion-controlled.22,122 As the transient complex lies at the outer boundary of the interaction energy well and hence is close to the native complex, ionic strength is expected to screen electrostatic interactions in the two types of complexes to nearly the same extent. Hence the association constant and the association rate constant are expected to have nearly the same dependence on ionic strength and the dissociation rate would be little affected by ionic strength.
The relation between ionic strength and ka can be quantitatively predicted from the solution of the Poisson-Boltzmann equation (eq 12), as will be discussed in Section 4.3. Empirically, it has been shown that this relation can be described quite well by a Debye-Hückel-like approximation,77,93
| (23) |
where U0 and a are fitting parameters. Comparison of eq 23 with eq 20 shows that U corresponds to the electrostatic interaction energy of the transient complex at κ = 0 (i.e., in the absence of salt). According to eq 23, a plot of lnka versus (1 + κa)−1 is linear. The intercept of the line at (1 + κa)−1 = 0 is lnka0, from which the basal rate constant ka0 can be determined (Figure 7). The y value at (1 + κa)−1 = 1 is lnka in the absence of salt, wherein the electrostatic attraction is maximized.134 This linear relation was shown to hold for the association of the TEM1-BLIP, interferon-receptor, hirudin-thrombin, and barnase-barstar pairs and a heterodimeric leucine zipper for all salt concentrations tested.93,129,135
Figure 7.

Association rate constants of wild-type and mutant TEM1-BLIP complexes determined at different salt concentrations, with lnka plotted against (1 + κa)−1. The data can be fitted to a line according to eq 23.
Figure 7 clearly shows that electrostatic attraction may be very strong at low salt. This phenomenon is well known for proteins, and is the basis of ion exchange purification. To avoid strong, non-specific electrostatic attraction, the ionic strength in the cell is ~150 mM. At this ionic strength charge-charge interactions are partially shielded, reducing the negative effect of non-specific interactions by allowing for specific rate enhancement. A good example for this was reported for the complex of barnase-barstar in the presence of the poly-ion hirudin.136 The association rate at low salt was actually lower than in higher salt, due to non-specific interactions of barnase with hirudin, which effectively removed free barnase from the system. The rate peaked at 150–200 mM salt, and slowed down at higher salt (this time due to masking of the charges). Thus, the physiological ionic strength is optimal to obtain fast specific binding, yet reduce non-specific binding.
4.2 Probing the Contributions of Individual Residues Towards ka by Mutation
Measuring the effects of mutations on the rate constants of association is a powerful tool to decipher the mechanism of association. As already discussed in Section 3.2, extensive site-directed mutagenesis of surface residues of the TEM1-BLIP, barnase-barstar, interferon-receptor, growth-hormone-receptor, IL4-receptor, and other protein pairs has demonstrated that mutations involving charged residues have the largest effect on ka, while mutations of uncharged resides are much less important.6,93,107,108,135,137–141 This point is further illustrated in Figure 8, which presents the analysis on the effects on ka and kd by a set of 55 single mutations located in the binding interfaces of the TEM1-BLIP, barnase-barstar, and interferon-receptor pairs. Except for the A19W mutation in interferon, none of the neutral mutations had a large effect on ka, while many of them had major effects on kd. This points towards a clear distinction between the process of association and dissociation. Hotspot residues affecting dissociation rates are both of charged and uncharged nature, while hotspots for association are almost always charged residues. Moreover, the energy gained or lost though a mutation is of a different scale, while for ka it is rare to find a residue that alters the rate by over 10 fold, for kd it is common that >5 such residues are found within an interface. While charge is important for association, the magnitude of perturbation of ka is not simply a measure of the change in charge, but rather relates to the specific location of the mutation and its contribution to the electrostatic energy of interaction between the two proteins in the transient complex (eq 20).1,13,77,93,94 For example, charged mutations located far away from the binding interface will not affect association, even if these mutations are drastic.6
Figure 8.

The changes in the rate constants of (a) association and (b) dissociation plotted against the change in charge resulting from a mutation. The rate constants were measured for 55 mutations in the barnase-barster, TEM1-BLIP, and IFNα2-IFNAR2 pairs on stopped-flow instruments.
4.2.1 Altering Association Kinetics by Protein Design
As will be discussed in Section 4.3, eq 20 allows for a rigorous prediction of the association rate constant. The prediction requires the generation of the transient complex. For design purpose, a simplified approach is called for. In the simplified approach taken by Schreiber and co-workers, the electrostatic interaction energy <Uel>* was calculated on the native complex itself:93,94
| (24) |
Here UX, X = A, B, or C, refers to the electrostatic energy of two proteins or their native complex, given by
| (25) |
where qi are the charges of the atoms in a protein or the protein complex and rij are the distances between the charges. The parameter a is the same as the one appearing in eq 23, and a value of 5.6 Å was found experimentally to give the best results in the calculations.93,94 Despite their simplicity, and the use of the native complex instead of the transient complex, eqs 24 and 25 were shown to give good estimates of electrostatic contributions to the rate constants of association, and of the way mutations will affect the rate constants for diverse systems such as RNaseA-RI, AChE-fas, TEM1-BLIP, hiruding-thrombin, CheY-CheA, Ras-Raf and Ras-Ral (Figure 5b).9,14,93,94,132,142 This was found to be true whether mutations were placed within, or outside the binding interfaces. Most informative were the cases where charged mutations were engineered outside the binding interfaces of protein-protein complexes, such as for the TEM1-BLIP and Ras-Ral pairs.14,93 In these cases, strong increases in the rate constants of association were achieved (250- and 17-fold, respectively), in agreement with the calculated values. These increased ka values were however, not accompanied by a change in kd, leading to increased association constants of the magnitudes described. This observation has far-reaching implications for our understanding of the transient complex and transition state for association, as will be described below. It is important to note that eqs 24 and 25 successfully predict the changes in association rate constant also for mutations located within the binding interfaces of protein-protein complexes, as was shown for the association of the barnase-barstar, TEM1-BLIP, Ras-Ral, AChE-fas, hirudin-thrombin, CheY-CheA, and other protein pairs.6,132
Based on eqs 24 and 25, two web servers for calculating the changes in ka upon mutations were set up: PARE (http://www.weizmann.ac.il/home/bcges/PARE.html) and HyPare (http://bip.weizmann.ac.il/HyPare). The first provides a more exact calculation for single mutations, while the second provides a full analysis of the entire protein surface, including the automated identification of “hotspots” for association.94,143
According to eq 20, basal rate constants are the rate constants of association in the absence of electrostatic attraction. These can be calculated either by extrapolating the values of ka to the limit of infinite salt concentration, or by introducing mutations that reduce the electrostatic interaction energy to zero. From such calculations the basal rate constants were found to be 4.4 × 104 or 4 × 104 M−1s−1 for thrombin-hirudin association, 4.2 × 104 or 2.3 × 104 M−1s−1 for TEM1-BLIP association, 1.4 × 107 or 1.5 × 105 M−1s−1 for barnase-barstar association (the two values are extrapolated from mutant or salt data, respectively -- see Figure 7 and eq 23). Similarly, the values of ka0 were 6.6 × 105 M−1s−1 for Ras-Raf association and 2 × 105 M−1s−1 for AChE-fas association using mutation data. Except for barnase-barstar association, the basal rate constants were the same whether calculated from salt or mutant data. The reason for the large difference between the two methods in predicting the basal rate constant for barnase-barstar association is currently not clear;143 possibly this is an indication of the limitation of the simplification in eqs 24 and 25 (see Section 4.3 for further discussion).
It is important to note that eq 25 does not treat explicitly the contributions of non-charged residues to ka. While indeed the contributions are small, they were found to be significant in a number of cases. For example, the A19W mutation on IFNα2 reduced ka by 4-fold, a reduction that clearly relates to conformational rearrangement during the process of association (as verified using double-mutant cycle analysis with the W100A mutation on IFNAR2).144 Another example for conformational rearrangement rather than electrostatics affecting association rates upon mutation was shown for MICA (a major histocompatibility complex-like protein) undergoing a structural transition from disorder to order upon binding its immuno-receptor, NKG2D.145 In contrast, the rate-limiting step for the human transcription factor pKID domain of CREB, which also undergoes a folding transition upon binding to the KIC domain of the co-activator CBP, appears to be the formation of a transient complex.146,147 Stabilizing the structure of the pKID domain prior to binding does not increase the association rate constant. Also for the complex formation between fas and AChE, molecular dynamics simulations suggested that fas undergoes a conformational rearrangement during the binding to AChE, but the rearrangement does not appear to slow down the association rate.148,149
4.2.2 A Proteomics View of ‘Hotspots’ for Association
As discussed above, the association rate constant of a protein complex depends on a basal rate constant and on the magnitude of electrostatic attraction, which can be calculated in a simplified fashion from the structure of the native complex. In an analysis on a database of 68 hetero-dimeric complexes using HyPare, it was found that in about half of the complexes electrostatic contributions to the association rate constants were small (< 10-fold effects on ka); in one quarter of the complexes electrostatic attraction had a major effect on ka (> 100-fold increases). Defining a residue as a hotspot for association if it changes ka by over ten-fold leaves about half of the complexes without any potential hotspot, and a few hotspots per complex in the other complexes. Of these putative hotspot residues, about 40% are calculated to increase the rate of association upon mutation, and thus increase binding affinity. These data suggest that a majority of protein-protein complexes are not optimized for fast association. Moreover, about 40% of the hotspots for association are located outside the binding interfaces, making them ideal candidates for protein engineering to achieve faster and tighter binding protein complexes. Hotspot residues are not evenly distributed among the 20 types of amino acids. About 75% of all hotspots are charged residues. This is understandable, as a charge-reversal mutant changes the total charge by two. More intriguing is the small number of hydrophobic residues that are hotspots, in comparison to polar residues. For 18 out of the 68 complexes in the database, experimental values of ka were available, from which basal rate constants were calculated to be in the range of 104 M−1s−1 to 107 M−1s−1. Some of these basal rate constants were corroborated by both salt and mutant data (see above). The basal rate constants seem to correlate with the sizes of the proteins and the shapes of the interfaces (see next Section).
4.3 Prediction of Electrostatic Rate Enhancement by the Transient-Complex Theory
Let us now discuss the rigorous prediction of the association rate constant from the transient-complex theory. The theory is given by eq 20, along with a structural model for the ensemble of the transient complex, which is described in Section 3.1. The transient-complex theory offers two important advantages. First, unlike in previous theoretical approaches, the model for the transient complex is based on rigorous theoretical considerations, and is uniquely defined. Second, electrostatic interactions between two associating proteins are treated directly by the Poisson-Boltzmann equation without approximations.
The transient-complex theory was put to a comprehensive test against experimental data4–6,128 for the association rate constants of four protein pairs (shown in Figure 1) and 23 of their mutants over wide ranges of ionic strength.13 For each protein pair, 100 configurations were randomly selected from the transient-complex ensemble to calculate the average electrostatic interaction energy, <Uel>*. Eq 20 was then used to predict the association rate constant. As shown in Figure 9a, the ionic strength dependences of the association rates for all four protein pairs are predicted well by the transient-complex theory. Moreover, the predictions for 23 mutants at various ionic strengths agree closely with experimental results (Figure 9b). In all there are 81 data points in the latter comparison, spanning four orders of magnitude in association rate. The theory thus appears to fulfill the promise of having truly predictive power. It reveals that, among the protein pairs and their mutants studied, the basal rate ka0 can differ by ~20-fold, but the bulk of the variations in ka is due to the variations in <Uel>*, which ranges from 0 to −6 kcal/mol (the last value translates into a 104-fold rate enhancement).
Figure 9.

Comparison of predicted and experimental results for association rate constants. (a) Ionic-strength dependences of four protein pairs. (b) Twenty-three mutants at various ionic strengths. Reprinted with permission from Ref. 13. Copyright 2007 Elsevier.
In the rigorous calculation of ka, it was found that the same basal rate constant, 1.4 × 106 M−1s−1, explains both salt and mutation data on barnase-barstar association. The relatively high basal rate constant for the barnase-barstar complex compared to other protein pairs is reproduced by Brownian dynamics simulations.1,13 The variations of basal rate constants among protein complexes can be explained by the structures of the transient complexes. A higher ka0 value, as found for barnase-barstar association, corresponds to a more open ensemble of the transient complex.1
The question of whether the predicted ka is sensitive to the precise specification of the transient complex was also addressed.13 As explained in Section 3.1, the transient complex is specified by the contact level, Nc*, at the onset of a sharp increase in transltional/rotational freedom (Figure 3b). For the barnase-barstar complex, the value of Nc* was determined to be 14. The value of Nc* was artificially varied from 10 to 18, and at each value, ka0 and <Uel>* were calculated. When combined according to eq 20, the predicted ka was found to vary little (< 2-fold), due the opposite dependences on Nc* of the two contributing factors, ka0 and exp(−<Uel>*/kBT), to ka.
The comparison of theory against experimental data presented above was based on calculating the electrostatic interaction energy from the linearized Poisson-Boltzmann equation (eq 12). It has been found that, when the full Poisson-Boltzmann equation (eq 11) was used, agreement with experiment improved, albeit modestly.1 This underscores the point that a rigorous treatment of electrostatic interactions is essential for the accuracy of calculated ka.
For the binding between a protein and an RNA, the difference between the full Poisson-Boltzmann equation and the linearized version is no longer modest because of the large charge density on the nucleic acid. Then use of the full Poisson-Boltzmann equation becomes a necessity. The transient-complex theory has made it possible to realistically model protein-RNA binding rate constants for the first time.26 In that work the binding of the spliceosomal protein U1A and its target on the U1 small nuclear RNA (Figure 10a) was studied. The binding and dissociation rate constants of this and other protein-RNA systems exhibit the disparate dependences on salt familiar to proteins,150–153 indicating that the structural model for the transient complex developed for protein-protein association is applicable to protein-RNA binding. Representative configurations in the transient complex of the U1 system are shown in Figure 10b. As Figure 11 shows, the binding rate constants of the wild-type system and eight of its mutants are in close agreement with experimental data.150,151
Figure 10.

The U1 protein-RNA system. (a) The native complex. (b) Representative configurations in the transient complex. The U1A protein is represented by the electrostatic surface. Notice that the RNA is moved away from the protein in the transient complex. Reprinted with permission from Ref. 26. Copyright 2008 American Chemical Society.
Figure 11.

Comparison of predicted and experimental results for the binding rates of the wild-type U1 system and eight of its mutants. Locations of the mutated residues are shown in the inset. Reprinted with permission from Ref. 26. Copyright 2008 American Chemical Society.
Comparison of predicted and experimental association rates also helps settling an important technical detail in the calculation of the electrostatic interaction energy. The detail in question is the definition of the boundary between the protein low dielectric and solvent high dielectric. In many Poisson-Boltzmann calculations this dielectric boundary is specified by the molecular surface. An alternative choice is the van der Waals surface.154,155 The difference between the two surfaces lies in the many crevices accessible to a spherical solvent probe, which are treated as part of the solute dielectric in the molecular surface protocol but as part of the solvent dielectric in the van der Waals surface protocol. It is known that the sign of the electrostatic interaction energy of two oppositely charged subunits can be reversed between the two protocols, positive in the former and negative in the latter.110,156–158
The results shown earlier for the average electrostatic interaction energies in the transient complexes were all obtained with the van der Waals surface protocol. With the molecular surface protocol, the same sign reversal mentioned above was seen on <Uel>*.1,26 Hence electrostatic rate enhancement now turned into rate retardation. For example, for the barnase-barstar pair, when the ionic strength was varied from 13 mM to 2000 mM, <Uel>* calculated with the van der Waals surface protocol varied from −3.30 to −0.82 kcal/mol. Correspondingly, <Uel>* calculated with molecular surface protocol varied from 2.50 to 5.13 kcal/mol. For the latter results to be consistent with the experimental data for the association rate constant would require a basal rate constant in the order of 1010 – 1011 M−1s−1, which is clearly unrealistic.
The transient-complex theory lays out a framework for understanding the wide variation in association rate constants (Figure 1) and for elucidating the pathways of protein association and dissociation. As documented above, the theory rationalizes the insensitivity of kd to ionic strength and has achieved great success in making quantitative predictions of ka for association reactions where conformational rearrangements are not rate-limiting. However, quantitative calculations for rate constants that involve the formation or breakup of short-range stereospecific interactions are beyond the scope of the transient-complex theory.
5. Interaction Dynamics of Membrane-Anchored Proteins
Key processes in the initiation of transmembrane signaling involve ligand-induced interactions between receptor proteins, which are anchored to the membrane. Compared to interaction kinetics in solution, anchoring of proteins into the membrane dramatically changes the mobility of the interaction partners, both in terms of diffusion constants (which are reduced by two orders of magnitude) and in restricting the movements to two dimensions (thus actually making the mutual search problem simpler). In general, reduction in dimensionality as well as potentially favorable pre-orientation has been proposed to explain the rapid initiation of signaling.37,159–161 However, few quantitative experimental studies of 2-dimensional protein interactions have been reported. For IL-4-induced crosslinking of the IL-4 Rα with the common γ-chain on live cells a surprisingly low efficiency of association of the low-affinity γ-chain subunit with the highly stable complex of IL-4 and IL-4 Rα on the membrane was observed.162 Recent systematic studies of ternary cytokine-receptor complexes tethered on artificial membranes have provided some quantitative understanding of 2-dimensional interactions. These data confirmed the regulatory role of binding affinities for complex formation on membranes. Equilibrium dissociation constants in the order of 1–10 μM for the interaction in solution yielded 2-dimensional equilibrium dissociation constants in the order of 10–100 molecules/μm2.163 Kinetic analysis of such complexes revealed that the probability of successful collisions is indeed higher on membranes, which can be ascribed to the pre-orientation and the longer lifetime of the transient complex. This effect is, however, largely compensated by the decreased diffusion rate, which reduces the number of collisions in the same order of magnitude. Orientation of the proteins significantly affects the rate constants of association,164 though proteins domains interacting outside the membrane are often rather flexible. Overall, the association rates at a given average distance of the molecules in solution and on the surface are surprisingly similar.165 In the dissociation process, the separation of the interacting species by diffusion plays a critical role. This agrees with the notion that the association kinetics is highly diffusion-controlled. As a consequence, local membrane fluidity in membrane microdomains may play a critical role for stabilizing membrane protein complexes. Thus, the energy landscapes of cellular membrane protein complexes are highly influenced by the micro-compartmentalization of the membranes. Crowding by by-stander proteins in the membranes may also have a significant effect (cf. Section 6).
Single-molecule fluorescence techniques may contribute to the study of association within membranes. The process of dimerization of epidermal growth factor receptors (EGFR) in cellular membranes and binding of their substrate, epidermal growth factor (EGF), was studied recently using single-molecule fluorescence.166 Fluorescent EGF molecules bound to EGFR molecules were tracked. Association of a second EGF molecule to a preformed EGF-EGFR complex was seen as a sudden doubling of the fluorescence intensity. It was concluded that EGFR dimerization preceded binding of a second EGF molecule to a complex. The kinetics of the dimerization process itself, however, is yet to be studied.
6. Roles of the Depletion Effect and Crowding in Association Kinetics
Since protein-protein association mostly takes place within cells, it is important to understand the particular properties of the cellular environment which may have a profound effect on such reactions.167 Most importantly, the cytoplasm of cells is a dense solution of macromolecules of many sizes and shapes. In fact, macromolecules may occupy as much as 40% of the total intracellular volume.168 These “crowding agents”, as they are sometimes called, include mostly folded proteins and nucleic acids. Structural proteins form dynamic networks of filaments, creating the cytoskeleton and thus dividing the cell into discrete bulk areas of cytoplasm, where soluble proteins can interact. In addition, there are many ‘natively unfolded proteins’, whose random-coil like conformations can add complexity to the cytoplasmic melieu.169
There are two major outcomes of the high intracellular concentration of macromolecules in their various forms. First, the macroscopic viscosity of the cytoplasm is significantly higher than the viscosity of a dilute aqueous solution. This higher viscosity should affect long-range translational diffusion of proteins. On small length scales there should be a large heterogeneity in the density of macromolecules, leading to microscopic viscosity which depends strongly on position. The crowded cytoplasm may also affect rotational motion of proteins, which is also crucial for protein-protein association. Since rotational diffusion is by nature more local than translational diffusion, and occurs on faster time scales, it is likely to be affected differently by the crowded cytoplasm.170,171 A second effect of the crowded cytoplasmic solution is depletion, which can be seen as an effective osmotic pressure exerted on all diffusing molecules. This phenomenon, which is entropic in nature, is due to the fact that protein solutes exclude (or deplete) some of the solution volume from other solutes. These effects are the subjects of the following sections.
6.1 Translational and Rotational Diffusion of Proteins in Concentrated Solutions and in Cells
Dense macromolecular solutions should be discussed more appropriately in terms of their viscoelasticity, rather than their viscosity.172 Viscoeleastic solutions possess time-scale and length-scale dependent viscosity. The effective viscosity sensed by a tracer molecule undergoing fast, short-range motion is therefore different than that sensed by a particle moving slowly over large distances. Further, the cytoplasmic environment is not homogenous, and the density of macromolecules and other intracellular components may vary from one spot to another within the cell. Thus diffusion times may scale with distance in an unexpected way. Recent experiments have probed the viscoelasticity of cellular interiors using microrheology techniques (for reviews see Refs. 173 and 174). Of most interest to the present review are experiments that directly measure diffusion coefficients inside cells, or in complex solutions that may mimic the cytoplasm.
A useful probe for studying translational and rotational diffusion inside cells is green fluorescent protein (GFP), which can be expressed alone or in tandem with other proteins. Verkman and coworkers have used GFP and GFP-labeled proteins to study diffusion in the cytoplasm170 and inside mitochondria.175 They found that translational diffusion is more retarded than rotational diffusion, an observation which is in line with studies in model solutions described below. Particles larger than the ~28 kDa GFP may show an even more complex behavior. Golding and Cox176 studied a complex of ~100 nm in size formed by a messenger RNA molecule with a large number of GFP-labeled proteins. The slow motion of this complex allowed the tracking of individual copies using time-lapse microphotography. The complex exhibited anomalous sub-diffusive behavior, with a mean-squared displacement proportional to tα, where t is the time and α≈ 0.7. This behavior suggests that the molecules are hindered in their motion, perhaps due to transient interactions with other components of the cell. Fluorescence correlation spectroscopy (FCS) of dextran molecules in cytoplasms also showed anomalous sub-diffusion.177
To gain a broader view of diffusion in cell-like environments, it is useful to conduct experiments in solutions of polymers, in which the composition and concentration of co-solutes can be systematically changed.178 Motion of proteins in solutions of large polymers is expected to be sub-diffusive, and this behavior has been observed by Banks and Fradin,179 who used FCS to study the motion of tracer proteins in solutions of dextran molecules of various sizes. The diffusion became more anomalous as the size of the polymer molecules and their concentration increased. This behavior was seen even in the solutions of the smaller dextrans, with molecular weights <10 kDa. In contrast, in a study of diffusion of proteins in polyethylene glycol (PEG) solutions of molecular weights up to 8000 by FCS, it was found that normal diffusion (i.e. mean-squared displacement linear with time) could well-account for the data.180 It is likely that the size of the crowding agents relative to the tracer protein size is the key parameter that determines the functional dependence of diffusive motion on time.
The relation of diffusion coefficients to viscosity can be another useful indicator of the degree of retardation of diffusive motion by polymeric solutions. The translational diffusion coefficients of small proteins in PEG solutions of molecular weights up to 8000 obeyed the Stokes-Einstein relation, i.e., in inverse proportion to solution macroscopic viscosity.180 In stark contrast, rotational diffusion correlation times in the same solution were not proportional to macroscopic viscosity,180 in agreement with other experiments.181 The results for diffusion in PEG8000 are shown in Figure 12. Rotational motion occurs on a much faster time scale than translational motion, and therefore rotation probes only the local viscosity at the position of a protein molecule, which experiences rather little friction from the polymer molecules themselves. This effect of microviscosity on protein rotational motion has strong impact on protein-protein association in polymer solutions, as will be seen below.
Figure 12.

Protein translational and rotational correlation times in solutions of PEG of MW 8000, plotted as a function of solution viscosity. Translational correlation times (red circles) were measured by fluorescence correlation spectroscopy, while rotational correlation times (green circles) were measured by fluorescence polarization spectroscopy. All plotted values are normalized by water values. Reprinted with permission from Ref. 180. Copyright 2008 American Chemical Society.
6.2 The Depletion Effect
The interaction between solutes in a concentrated solution can be understood within the framework of the classical McMillan-Mayer theory of multicomponent systems.182 In the simplest form of this theory, solute molecules are represented as hard spheres that cannot penetrate each other, but there is no direct interaction between them when they are separated. Their interactions and reaction equilibria, as they are modified due to the excluded volume effects of other solutes, can then be calculated.183 Asakura and Oosawa realized, in a terse letter to the Journal of Chemical Physics written in 1954,184 that the excluded volume effect on two particles immersed in a macromolecular solution can be cast in terms of an effective distance-dependent attractive potential, which is operating when their distance is smaller than the size of a macromolecule. This attractive potential 185 can be readily attributed to the inability of the macromolecular solute molecules to enter the region between the two particles (Figure 13). This region is said to be ‘depleted’ of solute molecules, hence the name ‘depletion effect’ to this phenomenon. The depletion potential, entropic in nature, will prove useful for calculating the effect of polymer co-solutes on protein-protein association kinetics (Section 6.3).
Figure 13.

The depletion effect. (a) An osmotic pressure is exerted on two particles (proteins) immersed in a macromolecular solution, when their separation does not allow the solute macromolecules to penetrate the volume between them. (b) This entropic effect is minimal when the two particles are in contact (e.g. when two proteins associate).
The depletion effect is similar to the situation occurring in an osmotic pressure experiment, where a semi-permeable membrane precludes macromolecular solutes from entering one region of the solution. In the Asakura and Oosawa (AO) theory, the depletion potential is indeed proportional to the osmotic pressure of the solution, as well as to the depleted volume. The AO theory is formally correct only in the colloid limit, when the macromolecules exerting the depletion force are much smaller than the particles feeling it.186 It also does not take into account the polymeric nature of macromolecular solutions, modeling the macromolecules as hard spheres. Schweizer and coworkers187,188 have developed an integral equation theory, called the Polymer Reference Interaction Site Model (PRISM), which attempts to properly treat macromolecular solutions, and is also applicable beyond the colloid limit.
The excluded-volume interactions underlying the depletion potential manifest themselves in many equilibrium processes.183 Indeed, precipitation and crystallization of proteins by polymeric additives have been studied for many years.189 Minton and coworkers developed a sedimentation equilibrium method190 to study excluded volume-driven association between proteins. In particular, inert proteins, acting as crowders, were shown to promote specific self-association of the cell division protein FtsZ into rod-like oligomers.191
Theoretically, equilibrium effects of excluded-volume interactions on a solution of proteins can be quantified through the second virial coefficient, B2, which is related to the depletion potential, U (r), via
| (26) |
The effect of crowding and depletion on the second virial coefficient of protein solutions was directly probed by scattering experiments.192–195 Kulkarni et al. performed a series of experiments on the proteins lysozyme and bovine serum albumin in PEG solutions of several molecular weights.193 The dependence of B2 on polymer concentration was extracted and compared to models, in order to understand non-specific, solute-induced interactions between protein molecules. Since PEG is a flexible polymer, the AO theory of depletion did not describe well the results. It was found that the PRISM theory, which takes into account the flexibility of polymer, accounts better for the experimental results (Figure 14). Vivares et al. extracted the full U (r) from small-angle X-ray scattering experiments of urate oxidase in PEG solutions.195 By comparison to several models they also reached the conclusion that PEG-induced depletion has to be described by models that take into account its polymeric nature.
Figure 14.

B2 (normalized by the hard-sphere value) for lysozyme and BSA as a function of concentration (wt%) of PEG of molecular weight 12000. The dotted and solid lines are AO and PRISM predictions, respectively. Adapted with permission from Ref. 193. Copyright 2000 American Institute of Physics.
6.3 Crowding and Protein-Protein Association Kinetics
The kinetics of protein-protein association in a crowded environment may be affected by both effects discussed above.196 First, the increase in solution viscosity should slow-down diffusion, which is what brings two associating proteins toward their transient complex. Since translational and rotational diffusion may be differently affected by the presence of macromolecules (as discussed in Section 6.2), modeling the viscosity effect on association rates may not be trivial. Second, the effective attraction induced by the depletion interaction may not only affect the equilibrium of protein solutions, but also the kinetics of their interactions. Just as an electrostatic potential enhances the association rate (see Section 1.1 and Section 4), so is the depletion potential expected to speed up association. When two protein molecules are close enough to each other, the osmotic pressure exerted by macromolecular co-solutes should lead to a significant increase in the probability of reaching the transient complex. A simple, albeit approximate description of the effect of depletion on association kinetics can be obtained by using eq 17, where now <U>* is interpreted as the average depletion interaction potential in the transient complex, in which the two proteins are essentially at contact. Illustrative calculations in Ref. 196 show that, when the effect of slowed-down diffusion and the contribution of the attractive depletion potential are combined, “cancellation of the two opposing effects leaves a modest overall effect of crowding on the binding rate.”
Several studies of protein-protein association kinetics in macromolecular solutions were published.197–199 The common denominator of all these experiments is that the effect of polymers on association kinetics is small, which, as just noted, is precisely what is predicted by theory.196 The first experimental evidence for the underlying reason in that prediction was obtained in a recent study in which association rate was dissected into its various components.180 This study is discussed next.
6.3.1 The Case of TEM-BLIP
The association kinetics of the protein TEM1-β-lactamase (TEM1) and its protein inhibitor BLIP was studied in macromolecular solution, in an attempt to mimic the crowded intracellular milieu and systematically probe its effects on the process. A stopped-flow apparatus was used to measure the millisecond kinetics of association of these proteins in solutions of polyethylene glycol molecules of various molecular weights, from 200 to 8000 (designated as PEG200, PEG8000, etc.).198 Measuring the translational and rotational diffusion coefficients in some of these solutions180 allowed the use of eq 8 to provide an estimate for the change in the association rate constant due to changes in the diffusion coefficients. The reader is reminded that in this equation the association rate constant depends both on translational and on rotational diffusion coefficients. If the diffusion coefficients both obey the Stokes-Einstein relations, i.e. are inversely proportional to solution viscosity, then eq 8 predicts that the association rates will also be inversely proportional to viscosity. A much weaker dependence on viscosity was seen in the experiments.180,198
It is useful to describe here the PEG8000 results, which were particularly clear and striking. When plotted as a function of viscosity, the association times (inverse rates) in PEG8000 solutions were much faster than the SE prediction, and in fact seem to saturate and remain essentially constant above a relative viscosity value of ~5 (Figure 15A, green squares). This result seems to be in accord with the measurement of rotational diffusion times of the proteins in PEG8000 solutions, which also shows a saturating behavior (Figure 12). However, translational diffusion times obey the SE relation well, and when the various diffusion times are inserted into eq. 8 the predicted association times are found to be too large (Figure 15A, red squares). The significant enhancement of association in PEG8000 solutions can be attributed to depletion, i.e. to the effective attraction induced by the polymer solution on the two proteins. Eq 17 can be used to estimate the interaction potential due to the depletion effect as a function of polymer concentration. This results in the depletion-induced interaction potential being linear with concentration (Figure 15B), in line with the AO theory. In other words, the depletion effect on association time is exponential in the polymer concentration. However, calculations using equation 17 show that the effect is significantly weaker than predicted by the AO theory, suggesting that the geometric considerations used to derive the depletion potential in that theory might be oversimplified. This is not surprising, since PEG8000 molecules cannot be treated as spheres, certainly not at the concentrations used in the experiment. On the other hand, these results cannot be explained by the PRISM theory of Schweitzer and coworkers187,188 either, which predicts a linear dependence of the depletion effect on concentration, rather than an exponential dependence. Further theoretical analysis is thus required in order to fully rationalize the experimental findings.
Figure 15.

Effect of PEG8000 on association times of the TEM1-BLIP protein pair. (a) Measured association times (relative to water, green squares) and association times calculated from translational and rotational diffusion coefficients (red squares), as a function of relative viscosity. The black line is the Stokes-Einstein prediction. (b) Depletion energies obtained from the difference between measured and calculated association times are plotted as a function of polymer concentration. Reprinted with permission from Ref. 180. Copyright 2008 American Chemical Society.
The biological implications of the above results are quite interesting. Over a rather broad range of polymer concentrations the rate of association between proteins is likely to be only mildly affected by the large abundance of other macromolecules. This is due to the ‘corrective’ effect of the depletion interaction, which enhances association much beyond the diffusion-limited rate.196
6.3.2 Surprises in concentrated solutions
When the concentration of macromolecules in solution is so large that the polymer solution correlation length becomes comparable to the size of a monomer, an interesting regime is approached. In this regime monomers from one chain interact with monomers on other chains in the same manner as they interact with themselves. This leads to an effective loss of the excluded volume interaction on internal statistics of polymer molecules, and the statistics become those of ideal chains.200 The effect of concentrated macromolecular solutions on association reactions of solute proteins should be similar to the effect in concentrated solutions of the relevant monomers.
Studying the effect of concentrated macromolecular solutions on the kinetics of the TEM1-BLIP association reaction turned out to be problematic for larger polymers, e.g. PEG8000, due to the excessively-high viscosity. Nevertheless, it was possible to study association in concentrated solutions of smaller polymers, e.g. PEG1000.201 It was found that above a certain critical concentration of the polymer a dramatic increase in association times occurred. Quite satisfactorily, the dependence of the change in association times in this phase on polymer concentration was rather similar to the dependence of association times on concentration in solutions of the monomeric ethylene glycol.201 This observation matches the physics of concentrated polymer solutions as described above. It is possible that in cellular environments a continuous change of the crowding effect, induced by changes in concentrations of proteins and other macromolecules, may strongly modulate association kinetics.
7. Conclusions
The association of proteins to form a complex is a multi-step process, which starts by random collisions of the individual proteins. Multiple collisions and rotational diffusion brings the proteins to an orientation that is close to that of the native complex, leading to the formation of a transient complex. This part of the process is diffusion-controlled, and strongly affected by electrostatic interactions. An open question is whether the transient complex develops into the native complex through a transition state, or complex formation is a simple downhill transformation of the transient complex. In Section 1.2 theoretical models are used to describe association as a diffusion-limited process, which is slowed down from the Smoluchowski limit due to orientational constraints satisfied by a stereospecific native complex. On the other hand, in Section 3.2 experimental evidence is presented to show that the transient complex will likely dissociate, and that the transition state for association is related to short-range conformational rearrangement and desolvation, processes that slow the conversion from the transient complex to the native complex. However, structurally, the transient complex and the transition state seem to be similar.
Biographies
Gideon Schreiber received his Doctorate in Biochemistry at the Hebrew University In Jerusalem in 1992. After a postdoctoral period at the MRC in Cambridge UK at the laboratory of Prof. Alan Fersht, he joined the Weizmann Institute of Science as Senior Scientist. Presently, he is an associate Professor at the department of Biological Chemistry at the same institute. His research interest is in the investigation of protein-protein interactions, from the basic understanding of the mechanism to protein-design. His work spans from bioinformatics and algorithm development, biophysical bench work, protein-design and engineering to applied biology. In addition he is a director of the Israel Structural Proteomic Center, which aim is to provide structures of proteins and protein-complexes to the biological community.

Gilad Haran is currently an associate professor in the department of Chemical Physics of the Weizmann Institute of Science. He did his Ph.D. at the Weizmann Institute with Profs. Ephraim Katchalsky-Katzir and Elisha Haas. He was then a post-doctoral fellow with Prof. Robin Hochstrasser at the University of Pennsylvania. Gilad Haran’s lab is using single-molecule spectroscopy to study molecular dynamics, from protein folding and association to charge transfer dynamics on metal surfaces.

Huan-Xiang Zhou received his Ph.D. from Drexel University in 1988. He did postdoctoral work at the NIH with Attila Szabo. After faculty appointments at HKUST and Drexel, he moved to Florida State University in 2002. His group does theoretical, computational, and experimental research on protein-protein interactions, on crowding and confinement effects of cellular environments, and on biological roles of protein dynamics. He is currently the Editor-in-Chief of PMC Biophysics.

References
- 1.Alsallaq R, Zhou HX. Proteins. 2008;71:320. doi: 10.1002/prot.21679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou HX. Phys Biol. 2005;2:R1. doi: 10.1088/1478-3975/2/3/R01. [DOI] [PubMed] [Google Scholar]
- 3.Terlau H, Shon KJ, Grilley M, Stocker M, Stuhmer W, Baldomero OM. Nature. 1996;381:148. doi: 10.1038/381148a0. [DOI] [PubMed] [Google Scholar]
- 4.Radic Z, Kirchhoff PD, Quinn DM, McCammon JA, Taylor P. J Biol Chem. 1997;272:23265. doi: 10.1074/jbc.272.37.23265. [DOI] [PubMed] [Google Scholar]
- 5.Wallis R, Moore GK, James R, Kleanthous C. Biochemistry. 1995;34:13743. doi: 10.1021/bi00042a004. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber G, Fersht AR. Nat Struct Biol. 1996;3:427. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 7.Schreiber G, Buckle AM, Fersht AR. Structure. 1994;2:945. doi: 10.1016/s0969-2126(94)00096-4. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro R, Ruiz-Gutierrez M, Chen CZ. J Mol Biol. 2000;302:497. doi: 10.1006/jmbi.2000.4075. [DOI] [PubMed] [Google Scholar]
- 9.Johnson RJ, McCoy JG, Bingman CA, Phillips GN, Jr, Raines RT. J Mol Biol. 2007;368:434. doi: 10.1016/j.jmb.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Nature. 2000;404:151. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- 11.Hemsath L, Dvorsky R, Fiegen D, Carlier MF, Ahmadian MR. Mol Cell. 2005;20:313. doi: 10.1016/j.molcel.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 12.Marchand JB, Kaiser DA, Pollard TD, Higgs HN. Nat Cell Biol. 2001;3:76. doi: 10.1038/35050590. [DOI] [PubMed] [Google Scholar]
- 13.Alsallaq R, Zhou HX. Structure. 2007;15:215. doi: 10.1016/j.str.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Kiel C, Selzer T, Shaul Y, Schreiber G, Herrmann C. Proc Natl Acad Sci USA. 2004;101:9223. doi: 10.1073/pnas.0401160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korennykh AV, Piccirilli JA, Correll CC. Nat Struct Mol Biol. 2006;13:436. doi: 10.1038/nsmb1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park C, Raines RT. J Am Chem Soc. 2001;123:11472. doi: 10.1021/ja0164834. [DOI] [PubMed] [Google Scholar]
- 17.Perrett S, Zahn R, Stenberg G, Fersht AR. J Mol Biol. 1997;269:892. doi: 10.1006/jmbi.1997.1081. [DOI] [PubMed] [Google Scholar]
- 18.Alsallaq R, Zhou HX. Biophys J. 2007;92:1486. doi: 10.1529/biophysj.106.096024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg OG, von Hippel PH. Annu Rev Biophys Biophys Chem. 1985;14:131. doi: 10.1146/annurev.bb.14.060185.001023. [DOI] [PubMed] [Google Scholar]
- 20.Zhou HX, Wong KY, Vijayakumar M. Proc Natl Acad Sci USA. 1997;94:12372. doi: 10.1073/pnas.94.23.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camacho CJ, Weng Z, Vajda S, DeLisi C. Biophys J. 1999;76:1166. doi: 10.1016/S0006-3495(99)77281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou HX. Biopolymers. 2001;59:427. doi: 10.1002/1097-0282(200111)59:6<427::AID-BIP1047>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Dong F, Zhou HX. J Am Chem Soc. 2005;127:6836. doi: 10.1021/ja042641q. [DOI] [PubMed] [Google Scholar]
- 24.Gabdoulline RR, Wade RC. Biophys J. 1997;72:1917. doi: 10.1016/S0006-3495(97)78838-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eyring H. J Chem Phys. 1932;3:107. [Google Scholar]
- 26.Qin SB, Zhou HX. J Phys Chem B. 2008;112:5955. doi: 10.1021/jp075919k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramers HA. Physica. 1940;7:284. [Google Scholar]
- 28.Northrup SH, Erickson HP. Proc Natl Acad Sci USA. 1992;89:3338. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou HX. Biophys J. 1997;73:2441. doi: 10.1016/S0006-3495(97)78272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camacho CJ, Kimura SR, DeLisi C, Vajda S. Biophys J. 2000;78:1094. doi: 10.1016/S0006-3495(00)76668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlosshauer M, Baker D. Protein Sci. 2004;13:1660. doi: 10.1110/ps.03517304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foote J, Eisen HN. Proc Natl Acad Sci USA. 1995;92:1254. doi: 10.1073/pnas.92.5.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffman TL, LaBranche CC, Zhang W, Canziani G, Robinson J, Chaiken I, Hoxie JA, Doms RW. Proc Natl Acad Sci USA. 1999;96:6359. doi: 10.1073/pnas.96.11.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wassaf D, Kuang G, Kopacz K, Wu QL, Nguyen Q, Toews M, Cosic J, Jacques J, Wiltshire S, Lambert J, Pazmany CC, Hogan S, Ladner RC, Nixon AE, Sexton DJ. Anal Biochem. 2006;351:241. doi: 10.1016/j.ab.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 35.Darling RJ, Kuchibhotla U, Glaesner W, Micanovic R, Witcher DR, Beals JM. Biochemistry. 2002;41:14524. doi: 10.1021/bi0265022. [DOI] [PubMed] [Google Scholar]
- 36.Uter NT, Gruic-Sovulj I, Perona JJ. J Biol Chem. 2005;280:23966. doi: 10.1074/jbc.M414259200. [DOI] [PubMed] [Google Scholar]
- 37.Zhou HX, Szabo A. Phys Rev Lett. 2004;93:178101. doi: 10.1103/PhysRevLett.93.178101. [DOI] [PubMed] [Google Scholar]
- 38.Zhou HX. Biophys J. 2005;88:1608. doi: 10.1529/biophysj.104.052688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luscombe NM, Laskowski RA, Thornton JM. Nucl Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tjong H, Zhou HX. Nucl Acids Res. 2007;35:1465. doi: 10.1093/nar/gkm008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 42.Zhou HX, Wlodek ST, McCammon JA. Proc Natl Acad Sci USA. 1998;95:9280. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdul-Manan N, Aghazadeh B, Liu GA, Majumdar A, Ouerfelli O, Siminovitch KA, Rosen MK. Nature. 1999;399:379. doi: 10.1038/20726. [DOI] [PubMed] [Google Scholar]
- 44.Hage T, Sebald W, Reinemer P. Cell. 1999;97:271. doi: 10.1016/s0092-8674(00)80736-9. [DOI] [PubMed] [Google Scholar]
- 45.Smoluchowski MV. Z Phys Chem. 1917;92:129. [Google Scholar]
- 46.Debye P. Trans Electrochem Soc. 1942;82:265. [Google Scholar]
- 47.Solc K, Stockmayer WH. Int J Chem Kinet. 1973;5:733. [Google Scholar]
- 48.Berg OG. Biophys J. 1985;47:1. doi: 10.1016/S0006-3495(85)83870-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou HX. Biophys J. 1993;64:1711. doi: 10.1016/S0006-3495(93)81543-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shoup D, Lipari G, Szabo A. Biophys J. 1981;36:697. doi: 10.1016/S0006-3495(81)84759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Temkin SI, Yakobson BI. J Phys Chem. 1984;88:2679. [Google Scholar]
- 52.Schlosshauer M, Baker D. J Phys Chem B. 2002;106:12079. [Google Scholar]
- 53.Dafforn TR, Smith CJ. EMBO Rep. 2004;5:1046. doi: 10.1038/sj.embor.7400276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fink AL. Curr Opin Struct Biol. 2005;15:35. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 55.Jemth P, Gianni S. Biochemistry. 2007;46:8701. doi: 10.1021/bi7008618. [DOI] [PubMed] [Google Scholar]
- 56.Levy Y, Cho SS, Onuchic JN, Wolynes PG. J Mol Biol. 2005;346:1121. doi: 10.1016/j.jmb.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 57.Piehler J. Curr Opin Struct Biol. 2005;15:4. doi: 10.1016/j.sbi.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Nieba L. Anal Biochem. 1996;234:155. doi: 10.1006/abio.1996.0067. [DOI] [PubMed] [Google Scholar]
- 59.Piehler J, Schreiber G. J Mol Biol. 1999;289:57. doi: 10.1006/jmbi.1999.2726. [DOI] [PubMed] [Google Scholar]
- 60.Albeck S, Unger R, Schreiber G. J Mol Biol. 2000;298:503. doi: 10.1006/jmbi.2000.3656. [DOI] [PubMed] [Google Scholar]
- 61.Rich RL, Cannon MJ, Jenkins J, Pandian P, Sundaram S, Magyar R, Brockman J, Lambert J, Myszka DG. Anal Biochem. 2008;373:112. doi: 10.1016/j.ab.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 62.Northrup SH, Reynolds JCL, Miller CM, Forrest KJ, Boles JO. J Am Chem Soc. 1986;108:8162. [Google Scholar]
- 63.Elcock AH, Gabdoulline RR, Wade RC, McCammon JA. J Mol Biol. 1999;291:149. doi: 10.1006/jmbi.1999.2919. [DOI] [PubMed] [Google Scholar]
- 64.Altobelli G, Subramaniam S. Biophys J. 2000;79:2954. doi: 10.1016/S0006-3495(00)76532-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fogolari F, Ugolini R, Molinari H, Viglino P, Esposito G. Eur J Biochem. 2000;267:4861. doi: 10.1046/j.1432-1327.2000.01542.x. [DOI] [PubMed] [Google Scholar]
- 66.Gabdoulline RR, Wade RC. J Mol Biol. 2001;306:1139. doi: 10.1006/jmbi.2000.4404. [DOI] [PubMed] [Google Scholar]
- 67.Gabdoulline RR, Kummer U, Olsen LF, Wade RC. Biophys J. 2003;85:1421. doi: 10.1016/S0006-3495(03)74574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zou G, Skeel RD. Biophys J. 2003;85:2147. doi: 10.1016/s0006-3495(03)74641-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin J, Beratan DN. J Phys Chem B. 2005;109:7529. doi: 10.1021/jp045417w. [DOI] [PubMed] [Google Scholar]
- 70.Spaar A, Dammer C, Gabdoulline RR, Wade RC, Helms V. Biophys J. 2006;90:1913. doi: 10.1529/biophysj.105.075507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haddadian EJ, Gross EL. Biophys J. 2006;91:2589. doi: 10.1529/biophysj.106.085936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gabdoulline RR, Wade RC. J Phys Chem. 1996;100:3868. [Google Scholar]
- 73.Northrup SH, Allison SA, McCammon JA. J Chem Phys. 1984;80:1517. [Google Scholar]
- 74.Zhou HX. J Chem Phys. 1990;92:3092. [Google Scholar]
- 75.Zhou HX. J Phys Chem. 1990;94:8794. [Google Scholar]
- 76.Zhou HX, Szabo A. Biophys J. 1996;71:2440. doi: 10.1016/S0006-3495(96)79437-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vijayakumar M, Wong KY, Schreiber G, Fersht AR, Szabo A, Zhou HX. J Mol Biol. 1998;278:1015. doi: 10.1006/jmbi.1998.1747. [DOI] [PubMed] [Google Scholar]
- 78.Zhou HX. J Chem Phys. 1996;105:7235. [Google Scholar]
- 79.Zhou HX, Briggs JM, McCammon JA. J Am Chem Soc. 1996;118:13069. [Google Scholar]
- 80.Zhou HX, Briggs JM, Tara S, McCammon JA. Biopolymers. 1998;45:355. doi: 10.1002/(SICI)1097-0282(19980415)45:5<355::AID-BIP4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 81.Janin J. Proteins. 1997;28:153. doi: 10.1002/(sici)1097-0134(199706)28:2<153::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 82.Krishna MM, Hoang L, Lin Y, Englander SW. Methods. 2004;34:51. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 83.Fersht AR, Matouschek A, Serrano L. J Mol Biol. 1992;224:771. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 84.Nolting B, Golbik R, Neira JL, Soler GA, Schreiber G, Fersht AR. Proc Natl Acad Sci USA. 1997;94:826. doi: 10.1073/pnas.94.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eaton WA, Munoz V, Hagen SJ, Jas GS, Lapidus LJ, Henry ER, Hofrichter J. Annu Rev Biophys Biomol Struct. 2000;29:327. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Haran G. J Phys: Condens Matter. 2003;15:R1291. [Google Scholar]
- 87.Cornish PV, Ha T. ACS Chem Biol. 2007;2:53. doi: 10.1021/cb600342a. [DOI] [PubMed] [Google Scholar]
- 88.Huang F, Sato S, Sharpe TD, Ying L, Fersht AR. Proc Natl Acad Sci USA. 2007;104:123. doi: 10.1073/pnas.0609717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kubelka J, Hofrichter J, Eaton WA. Curr Opin Struct Biol. 2004;14:76. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 90.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. J Mol Biol. 2006;360:865. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 91.Huang X, Zhou HX. Biophys J. 2006;91:2451. doi: 10.1529/biophysj.106.082891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miyashita O, Onuchic JN, Okamura MY. Proc Natl Acad Sci USA. 2004;101:16174. doi: 10.1073/pnas.0405745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Selzer T, Albeck S, Schreiber G. Nat Struct Biol. 2000;7:537. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 94.Selzer T, Schreiber G. Proteins. 2001;45:190. doi: 10.1002/prot.1139. [DOI] [PubMed] [Google Scholar]
- 95.Shoup D, Szabo A. Biophys J. 1982;40:33. doi: 10.1016/S0006-3495(82)84455-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gabdoulline RR, Wade RC. J Mol Recog. 1999;12:226. doi: 10.1002/(SICI)1099-1352(199907/08)12:4<226::AID-JMR462>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 97.Volkov A, Worrall J, Holtzmann E, Ubbink M. Proc Natl Acad Sci USA. 2006;103:18945. doi: 10.1073/pnas.0603551103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang C, Iwahara J, Clore GM. Nature. 2006;444:383. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- 99.Suh JY, Tang C, Clore GM. J Am Chem Soc. 2007;129:12954. doi: 10.1021/ja0760978. [DOI] [PubMed] [Google Scholar]
- 100.Keeble AH, Kleanthous C. J Mol Biol. 2005;352:656. doi: 10.1016/j.jmb.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 101.Estrada S, Pavlova A, Björk I. Biochemistry. 1999;38:7339. doi: 10.1021/bi990003s. [DOI] [PubMed] [Google Scholar]
- 102.Estrada S, Olson ST, Raub-Segall E, Bjork I. Protein Sci. 2000;9:2218. doi: 10.1110/ps.9.11.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fersht AR, Daggett V. Cell. 2002;108:573. doi: 10.1016/s0092-8674(02)00620-7. [DOI] [PubMed] [Google Scholar]
- 104.Mateu MG, Sanchez Del Pino MM, Fersht AR. Nat Struct Biol. 1999;6:191. doi: 10.1038/5880. [DOI] [PubMed] [Google Scholar]
- 105.Taylor MG, Rajpal A, Kirsch JF. Protein Sci. 1998;7:1857. doi: 10.1002/pro.5560070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Nature. 2002;418:552. doi: 10.1038/nature00920. [DOI] [PubMed] [Google Scholar]
- 107.Reichmann D, Cohen M, Abramovich R, Dym O, Lim D, Strynadka NC, Schreiber G. J Mol Biol. 2007;365:663. doi: 10.1016/j.jmb.2006.09.076. [DOI] [PubMed] [Google Scholar]
- 108.Schreiber G, Fersht AR. J Mol Biol. 1995;248:478. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 109.Schreiber G, Fersht AR. Biochemistry. 1993;32:5145. doi: 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- 110.Dong F, Vijayakumar M, Zhou HX. Biophys J. 2003;85:49. doi: 10.1016/S0006-3495(03)74453-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mejean A, Bodenreider C, Schuerer K, Goldberg ME. Biochemistry. 2001;40:8169. doi: 10.1021/bi010611j. [DOI] [PubMed] [Google Scholar]
- 112.Carter PJ, Winter G, Wilkinson AJ, Fersht AR. Cell. 1984;38:835. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- 113.Horovitz A. J Mol Biol. 1987;196:733. doi: 10.1016/0022-2836(87)90045-3. [DOI] [PubMed] [Google Scholar]
- 114.Horovitz A, Fersht AR. J Mol Biol. 1990;214:613. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 115.Horovitz A. Fold Des. 1996;1:R121. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 116.Serrano L, Horovitz A, Avron B, Bycroft M, Fersht AR. Biochemistry. 1990;29:9343. doi: 10.1021/bi00492a006. [DOI] [PubMed] [Google Scholar]
- 117.Tetreault M, Cusanovich M, Meyer T, Axelrod H, Okamura MY. Biochemistry. 2002;41:5807. doi: 10.1021/bi012053e. [DOI] [PubMed] [Google Scholar]
- 118.Harel M, Cohen M, Schreiber G. J Mol Biol. 2007;371:180. doi: 10.1016/j.jmb.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 119.Frisch C, Fersht AR, Schreiber G. J Mol Biol. 2001;308:69. doi: 10.1006/jmbi.2001.4577. [DOI] [PubMed] [Google Scholar]
- 120.Miyashita O, Onuchic JN, Okamura MY. Biochemistry. 2003;42:11651. doi: 10.1021/bi0350250. [DOI] [PubMed] [Google Scholar]
- 121.Sheinerman FB, Norel R, Honig B. Curr Opin Struct Biol. 2000;10:153. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 122.Zhou HX. Protein Sci. 2003;12:2379. doi: 10.1110/ps.03216203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miller C. Biochemistry. 1990;29:5320. doi: 10.1021/bi00474a016. [DOI] [PubMed] [Google Scholar]
- 124.Candia S, Garcia ML, Latorre R. Biophys J. 1992;63:583. doi: 10.1016/S0006-3495(92)81630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Escobar L, Root MJ, MacKinnon R. Biochemistry. 1993;32:6982. doi: 10.1021/bi00078a024. [DOI] [PubMed] [Google Scholar]
- 126.Goldstein SAN, Miller C. Biophys J. 1993;65:1613. doi: 10.1016/S0006-3495(93)81200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murrell-Lagnado RD, Aldrich RW. J Gen Physiol. 1993;102:977. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shen BJ, Hage T, Sebald W. Eur J Biochem. 1996;240:252. doi: 10.1111/j.1432-1033.1996.0252h.x. [DOI] [PubMed] [Google Scholar]
- 129.Wendt H, Leder L, Harma H, Jelesarov I, Baici A, Bosshard HR. Biochemistry. 1997;36:204. doi: 10.1021/bi961672y. [DOI] [PubMed] [Google Scholar]
- 130.Baerga-Ortiz A, Rezaie AR, Komives EA. J Mol Biol. 2000;296:651. doi: 10.1006/jmbi.1999.3447. [DOI] [PubMed] [Google Scholar]
- 131.Walker D, Moore GR, James R, Kleanthous C. Biochemistry. 2003;42:4161. doi: 10.1021/bi0273720. [DOI] [PubMed] [Google Scholar]
- 132.Stewart RC, Van Bruggen R. J Mol Biol. 2004;336:287. doi: 10.1016/j.jmb.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 133.Gianni S, Engstrom A, Larsson M, Calosci N, Malatesta F, Eklund L, Ngang CC, Travaglini-Allocatelli C, Jemth P. J Biol Chem. 2005;280:34805. doi: 10.1074/jbc.M506017200. [DOI] [PubMed] [Google Scholar]
- 134.Selzer T, Schreiber G. J Mol Biol. 1999;287:409. doi: 10.1006/jmbi.1999.2615. [DOI] [PubMed] [Google Scholar]
- 135.Piehler J, Schreiber G. J Mol Biol. 1999;294:223. doi: 10.1006/jmbi.1999.3230. [DOI] [PubMed] [Google Scholar]
- 136.Schreiber G, Frisch C, Fersht AR. J Mol Biol. 1997;270:111. doi: 10.1006/jmbi.1997.1080. [DOI] [PubMed] [Google Scholar]
- 137.Clackson T, Wells JA. Science. 1995;267:383. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 138.Horn JR, Kraybill B, Petro EJ, Coales SJ, Morrow JA, Hamuro Y, Kossiakoff AA. Biochemistry. 2006;45:8488. doi: 10.1021/bi0604328. [DOI] [PubMed] [Google Scholar]
- 139.Kouadio JL, Horn JR, Pal G, Kossiakoff AA. J Biol Chem. 2005;280:25524. doi: 10.1074/jbc.M502167200. [DOI] [PubMed] [Google Scholar]
- 140.Kraich M, Klein M, Patino E, Harrer H, Nickel J, Sebald W, Mueller TD. BMC Biol. 2006;4:13. doi: 10.1186/1741-7007-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Piehler J, Roisman LC, Schreiber G. J Biol Chem. 2000;275:40425. doi: 10.1074/jbc.M006854200. [DOI] [PubMed] [Google Scholar]
- 142.Marvin JS, Lowman HB. Biochemistry. 2003;42:7077. doi: 10.1021/bi026947q. [DOI] [PubMed] [Google Scholar]
- 143.Shaul Y, Schreiber G. Proteins. 2005;60:341. doi: 10.1002/prot.20489. [DOI] [PubMed] [Google Scholar]
- 144.Slutzki M, Jaitin DA, Yehezkel TB, Schreiber G. J Mol Biol. 2006;360:1019. doi: 10.1016/j.jmb.2006.05.069. [DOI] [PubMed] [Google Scholar]
- 145.Lengyel CS, Willis LJ, Mann P, Baker D, Kortemme T, Strong RK, McFarland BJ. J Biol Chem. 2007;282:30658. doi: 10.1074/jbc.M704513200. [DOI] [PubMed] [Google Scholar]
- 146.Sugase K, Dyson HJ, Wright PE. Nature. 2007;447:1021. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 147.Turjanski AG, Gutkind JS, Best RB, Hummer G. PLoS Comput Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bui JM, Radic Z, Taylor P, McCammon JA. Biophys J. 2006;90:3280. doi: 10.1529/biophysj.105.075564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bui JM, McCammon JA. Proc Natl Acad Sci USA. 2006;103:15451. doi: 10.1073/pnas.0605355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Law MJ, Linde ME, Chambers EJ, Oubridge C, Katsamba PS, Nilsson L, Haworth IS, Laird-Offringa IA. Nucl Acids Res. 2006;34:275. doi: 10.1093/nar/gkj436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Katsamba PS, Myszka DG, Laird-Offringa IA. J Biol Chem. 2001;276:21476. doi: 10.1074/jbc.M101624200. [DOI] [PubMed] [Google Scholar]
- 152.Milev S, Bosshard HR, Jelesarov I. Biochemistry. 2005;44:285. doi: 10.1021/bi048907n. [DOI] [PubMed] [Google Scholar]
- 153.Auweter SD, Fasan R, Reymond L, Underwood JG, Black DL, Pitsch S, Allain FH. EMBO J. 2006;25:163. doi: 10.1038/sj.emboj.7600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vijayakumar M, Zhou HX. J Phys Chem B. 2001;105:7334. [Google Scholar]
- 155.Dong F, Zhou HX. Biophys J. 2002;83:1341. doi: 10.1016/S0006-3495(02)73904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang T, Tomic S, Gabdoulline RR, Wade RC. Biophys J. 2004;87:1618. doi: 10.1529/biophysj.104.040964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dong F, Zhou HX. Proteins. 2006;65:87. doi: 10.1002/prot.21070. [DOI] [PubMed] [Google Scholar]
- 158.Qin SB, Zhou HX. Biopolymers. 2007;86:112. doi: 10.1002/bip.20708. [DOI] [PubMed] [Google Scholar]
- 159.Adam G, Delbruck M. In: Structural Chemistry and Molecular Biology. Rich A, Davidson N, editors. Freeman and Company; San Francisco: 1968. [Google Scholar]
- 160.Axelrod D, Wang MD. Biophys J. 1994;66:588. doi: 10.1016/s0006-3495(94)80834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wiegel FW, DeLisi C. Am J Physiol. 1982;243:R475. doi: 10.1152/ajpregu.1982.243.5.R475. [DOI] [PubMed] [Google Scholar]
- 162.Whitty A, Raskin N, Olson DL, Borysenko CW, Ambrose CM, Benjamin CD, Burkly LC. Proc Natl Acad Sci USA. 1998;95:13165. doi: 10.1073/pnas.95.22.13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gavutis M, Lata S, Lamken P, Muller P, Piehler J. Biophys J. 2005;88:4289. doi: 10.1529/biophysj.104.055855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lamken P, Gavutis M, Peters I, Van der Heyden J, Uze G, Piehler J. J Mol Biol. 2005;350:476. doi: 10.1016/j.jmb.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 165.Gavutis M, Jaks E, Lamken P, Piehler J. Biophys J. 2006;90:3345. doi: 10.1529/biophysj.105.072546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sako Y, Minoghchi S, Yanagida T. Nat Cell Biol. 2000;2:168. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 167.Zhou HX, Rivas G, Minton AP. Annu Rev Biophys. 2008;37:375. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hall D, Minton AP. Biochim Biophys Acta. 2003;1649:127. doi: 10.1016/s1570-9639(03)00167-5. [DOI] [PubMed] [Google Scholar]
- 169.Uversky VN. Protein Sci. 2002;11:739. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Swaminathan R, Hoang CP, Verkman AS. Biophys J. 1997;72:1900. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lavalette D, Hink MA, Tourbez M, Tetreau C, Visser AJ. Eur Biophys J. 2006;35:517. doi: 10.1007/s00249-006-0060-z. [DOI] [PubMed] [Google Scholar]
- 172.Doi M, Edwards SF. The Theory of Polymer Dynamics. Clarendon Press; 1988. [Google Scholar]
- 173.Pullarkat PA, Fernandez PA, Ott A. Physics Rep. 2007;449:29. [Google Scholar]
- 174.Dix JA, Verkman AS. Annu Rev Biophys. 2008;37:247. doi: 10.1146/annurev.biophys.37.032807.125824. [DOI] [PubMed] [Google Scholar]
- 175.Haggie PM, Verkman AS. J Biol Chem. 2002;277:40782. doi: 10.1074/jbc.M207456200. [DOI] [PubMed] [Google Scholar]
- 176.Golding I, Cox EC. Phys Rev Lett. 2006;96:098102. doi: 10.1103/PhysRevLett.96.098102. [DOI] [PubMed] [Google Scholar]
- 177.Weiss M, Elsner M, Kartberg F, Nilsson T. Biophys J. 2004;87:3518. doi: 10.1529/biophysj.104.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Odijk T. Biophys J. 2000;79:2314. doi: 10.1016/S0006-3495(00)76477-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Banks DS, Fradin C. Biophys J. 2005;89:2960. doi: 10.1529/biophysj.104.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kuttner YY, Kozer N, Segal E, Schreiber G, Haran G. J Am Chem Soc. 2005;127:15138. doi: 10.1021/ja053681c. [DOI] [PubMed] [Google Scholar]
- 181.Lavalette D, Tetreau C, Tourbez M, Blouquit Y. Biophys J. 1999;76:2744. doi: 10.1016/S0006-3495(99)77427-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.McMillan WG, Mayer JE. J Chem Phys. 1945;13:276. [Google Scholar]
- 183.Minton AP. Methods Enzymol. 1998;295:127. doi: 10.1016/s0076-6879(98)95038-8. [DOI] [PubMed] [Google Scholar]
- 184.Asakura S, Oosawa F. J Chem Phys. 1954;22:1255. [Google Scholar]
- 185.Vrij A. Pure Appl Chem. 1976;48:471. [Google Scholar]
- 186.Verma R, Crocker JC, Lubensky TC, Yodh AG. Phys Rev Lett. 1998;81:4004. [Google Scholar]
- 187.Chatterjee AP, Schweizer KS. J Chem Phys. 1998;109:10464. [Google Scholar]
- 188.Fuchs M, Schweizer KS. J Phys: Condens Matter. 2002;14:R239. [Google Scholar]
- 189.Atha DH, Ingham KC. J Biol Chem. 1981;256:12108. [PubMed] [Google Scholar]
- 190.Rivas G, Minton AP. J Mol Recog. 2004;17:362. doi: 10.1002/jmr.708. [DOI] [PubMed] [Google Scholar]
- 191.Rivas G, Fernandez JA, Minton AP. Proc Natl Acad Sci USA. 2001;98:3150. doi: 10.1073/pnas.051634398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Farnum M, Zukoski C. Biophys J. 1999;76:2716. doi: 10.1016/S0006-3495(99)77424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kulkarni AM, Chatterjee AP, Schweizer KS, Zukoski CF. J Chem Phys. 2000;113:9863. [Google Scholar]
- 194.Liu W, Bratko D, Prausnitz JM, Blanch HW. Biophys Chem. 2004;107:289. doi: 10.1016/j.bpc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 195.Vivares D, Belloni L, Tardieu A, Bonnete F. Eur Phys J E. 2002;9:15. doi: 10.1140/epje/i2002-10047-7. [DOI] [PubMed] [Google Scholar]
- 196.Zhou HX. J Mol Recog. 2004;17:368. doi: 10.1002/jmr.711. [DOI] [PubMed] [Google Scholar]
- 197.Ladurner AG, Fersht AR. Nat Struct Biol. 1999;6:28. doi: 10.1038/4899. [DOI] [PubMed] [Google Scholar]
- 198.Kozer N, Schreiber G. J Mol Biol. 2004;336:763. doi: 10.1016/j.jmb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 199.Schlarb-Ridley BG, Mi H, Teale WD, Meyer VS, Howe CJ, Bendall DS. Biochemistry. 2005;44:6232. doi: 10.1021/bi047322q. [DOI] [PubMed] [Google Scholar]
- 200.de Gennes P-G. Scaling Concepts in Polymer Physics. Cornell university press; Ithaca: 1979. [Google Scholar]
- 201.Kozer N, Kuttner YY, Haran G, Schreiber G. Biophys J. 2007;92:2139. doi: 10.1529/biophysj.106.097717. [DOI] [PMC free article] [PubMed] [Google Scholar]