

Abstract
Next-generation sequencing technologies are quickly becoming the preferred approach for characterizing and quantifying entire genomes. Even though data produced from these technologies are proving to be the most informative of any thus far, very little attention has been paid to fundamental design aspects of data collection and analysis, namely sampling, randomization, replication, and blocking. We discuss these concepts in an RNA sequencing framework. Using simulations we demonstrate the benefits of collecting replicated RNA sequencing data according to well known statistical designs that partition the sources of biological and technical variation. Examples of these designs and their corresponding models are presented with the goal of testing differential expression.
NEXT-GENERATION sequencing (NGS) has emerged as a revolutionary tool in genetics, genomics, and epigenomics. By increasing throughput and decreasing cost, compared to other sequencing technologies (Hayden 2009), NGS has enabled genome-wide investigations of various phenomena, including single-nucleotide polymorphisms (Craig et al. 2008), epigenetic events (Park 2009), copy number variants (Alkan et al. 2009), differential expression (Bloom et al. 2009), and alternative splicing (Sultan et al. 2008). One application with demonstrated effectiveness over previous technologies [e.g., microarrays and serial analysis of gene expression (SAGE)] is called RNA sequencing (RNA-Seq) (Cloonan et al. 2009). RNA-Seq uses NGS technology to sequence, map, and quantify a population of transcripts (Mortazavi et al. 2008; Morozova et al. 2009). While RNA-Seq is a relatively new method, it has already provided unprecedented insights into the transcriptional complexities of a variety of organisms, including yeast (Nagalakshmi et al. 2008), mice (Mortazavi et al. 2008), Arabidopsis (Eveland et al. 2008), and humans (Sultan et al. 2008).
At present, there are three widely accepted commercially available NGS devices [Illumina's Genome Analyzer, Applied Biosystems' (Foster City, CA) SOLiD, and the 454 Genome Sequencer FLX] for RNA-Seq (Cloonan et al. 2008; Eveland et al. 2008; Marioni et al. 2008). Across platforms, the RNA-Seq methodology is approximately the same. Briefly, RNA is isolated from cells, fragmented at random positions, and copied into complementary DNA (cDNA). Fragments meeting a certain size specification (e.g., 200–300 bases long) are retained for amplification using polymerase chain reaction (PCR). After amplification, the cDNA is sequenced using NGS; the resulting reads are aligned to a reference genome, and the number of sequencing reads mapped to each gene in the reference is tabulated. These gene counts, or digital gene expression (DGE) measures, can be transformed and used to test differential expression (see Morozova et al. 2009 for a review of these technologies as applied to RNA-Seq).
Although there are many steps in this experimental process that may introduce errors and biases, RNA-Seq has been hailed as the future of transcriptome research (Shendure 2008) because it potentially generates an unlimited dynamic range, provides greater sensitivity than microarrays, is able to discriminate closely homologous regions, and does not require a priori assumptions about regions of expression (Cloonan et al. 2009; Morozova et al. 2009). As research transitions from microarrays to sequencing-based approaches, it is essential that we revisit many of the same concerns that the statistical community had at the beginning of the microarray era (Kerr and Churchill 2001a).
Soon after the introduction of microarrays (Schena et al. 1995), a series of articles was published elucidating the need for proper experimental design (Kerr et al. 2000; Lee et al. 2000; Kerr and Churchill 2001a,b; Churchill 2002). All of these articles rely heavily on the three fundamental aspects of sound experimental design formalized by R. A. Fisher (Fisher 1935a) 70 years ago, namely replication, randomization, and blocking. These concepts can be understood by considering the following controlled experiment that is designed to test the effectiveness of two different diets. A sound experimental design would include many different subjects (i.e., replication) recruited from multiple weight loss centers (i.e., blocking). Each center would randomly assign its subjects to one of the two diets (i.e., randomization).
Although the principles of good design are straightforward, their proper implementation often requires significant planning and statistical expertise. To date, many NGS applications, specifically RNA-Seq, have neglected good design. While a few RNA-Seq studies have reported highly reproducible results with little technical variation (e.g., Marioni et al. 2008; Mortazavi et al. 2008), in the absence of a proper design, it is essentially impossible to partition biological variation from technical variation. When these two sources of variation are confounded, there is no way of knowing which source is driving the observed results. No amount of statistical sophistication can separate confounded factors after data have been collected.
Generally, for differential expression analyses, researchers are interested in comparisons across treatment groups in the form of contrasts or pairwise comparisons, and the designs for these analyses are usually quite simple. The good news for NGS technologies is that certain properties of the platforms can be leveraged to ensure proper design. One such feature, available in all three NGS devices, is the capacity to bar code. Genomic fragments can be labeled or bar coded with sample-specific sequences that in turn allow multiple samples to be included in the same sequencing reaction (i.e., multiplexing) while maintaining, with high fidelity, sample identities downstream (Craig et al. 2008; Hamady et al. 2008; http://www3.appliedbiosystems.com/). To date, bar coding has been appreciated only as a means to increase the number of samples per sequencing run. Yet here we demonstrate how multiplexing can be used as a quality control feature that offers the flexibility to construct balanced and blocked designs for the purpose of testing differential expression.
We anticipate that the progression from the current unreplicated unblocked designs to more complex designs will be swift once the full offerings of NGS technologies are appreciated. Toward this end, we provide a brief review of some powerful statistical techniques for testing differential expression under a variety of designs. Although the designs that are presented are specific to RNA-Seq using the Illumina (Solexa) platform, the same statistical principles are applicable to the other NGS devices, as well as to other types of comparative genetic and “omic” data.
REPLICATION
Unreplicated data:
Observational studies with no biological replication are common in the RNA-Seq literature (e.g., Marioni et al. 2008). In an observational study, as opposed to a controlled experiment, the assignment of subjects to treatment groups is not decided by the investigator. In many cases, the different treatment groups consist of different tissue types. For example, in Marioni et al. (2008) messenger RNA (mRNA) was isolated from liver and kidney tissues, randomly fragmented, and sequenced using the Illumina Genome Analyzer (GA). The Illumina technology (aka “Solexa”) relies on a flow cell with eight lanes, or channels, and massively parallel sequencing by synthesis to simultaneously sequence millions of short DNA fragments in each of the lanes. Typically, independent samples of mRNA are loaded into different lanes of the flow cell such that sequencing reactions occur independently between samples. For illustration purposes, consider an example with seven subjects and seven treatment groups (T1,…, T7), where each subject is randomly assigned to one treatment group, and mRNA from each subject is loaded into a different lane (Figure 1). Note that there is no biological replication because there is only a single subject in each treatment group.
Figure 1.—

Hypothetical Illumina GA flow cell with mRNA isolated from subjects within seven different treatment groups  and loaded into individual lanes (e.g., the mRNA from the subject within treatment group 1 is sequenced in lane 1). As a control, a
and loaded into individual lanes (e.g., the mRNA from the subject within treatment group 1 is sequenced in lane 1). As a control, a 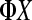 genomic sample is often loaded into lane 5. The bacteriophage
genomic sample is often loaded into lane 5. The bacteriophage  genome is known exactly and can be used to recalibrate the quality scoring of sequencing reads from other lanes (Bentley et al. 2008).
genome is known exactly and can be used to recalibrate the quality scoring of sequencing reads from other lanes (Bentley et al. 2008).
To analyze data from unreplicated designs, the sampling hierarchy must be taken into account. Regardless of the design, we can define three levels of sampling at work in RNA-Seq data: subject sampling, RNA sampling, and fragment sampling. Subjects (e.g., organisms or individuals) are ideally drawn from a larger population to which results of the study may be generalized (unreplicated data consist of a single subject within each treatment group). RNA sampling occurs during the experimental procedure when RNA is isolated from the cell(s). Finally, only certain fragmented RNAs that are sampled from the cells(s) are retained for amplification, and since the sequencing reads do not represent 100% of the fragments loaded into a flow cell, fragment-level sampling is also at play.
Unreplicated data consider only a single subject per treatment group. Typically there is either one subject to which every treatment is applied (e.g., in Marioni et al. 2008, liver and kidney samples were extracted from one human cadaver) or one distinct subject within each treatment group (e.g., Figure 1). In either situation, it is not possible to estimate variability within treatment group, and the analysis must proceed without any information regarding within-group biological variation. As such, in the context of RNA-Seq, statistical methods for finding differences between groups are limited to RNA and fragment-level sampling information.
Since the sampling scheme for RNA-Seq is similar to SAGE (Velculescu et al. 1995), and a sizable statistical literature is already established for analyzing differential DGE measures from unreplicated SAGE data, similar methods can be used for unreplicated RNA-Seq data. See Man et al. (2000), Romualdi et al. (2001), and Ruijter et al. (2002) for reviews and comparisons of techniques and Tino (2009) for further discussion. For both RNA-Seq and SAGE data the analysis usually proceeds on a gene-by-gene basis by organizing the data in a 2 × 2 table (Table 1). Perhaps the most natural test for differential expression in the unreplicated case is Fisher's exact test (Fisher 1935b), which fixes the marginal totals of the 2 × 2 table and tests differential expression using the hypotheses
 |
(1) |
where
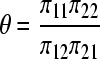 |
and where 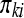 is the true proportion of counts in cell
is the true proportion of counts in cell 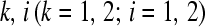 , assuming every transcript was isolated and perfectly sequenced. In Table 1 we can think of having N1. white balls and N2. black balls in an urn. If we draw N.1 balls from the urn, we may ask, “What is the probability of observing an outcome at least as unlikely as
, assuming every transcript was isolated and perfectly sequenced. In Table 1 we can think of having N1. white balls and N2. black balls in an urn. If we draw N.1 balls from the urn, we may ask, “What is the probability of observing an outcome at least as unlikely as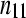 white balls?” If this probability (i.e., the P-value from Fisher's exact test) is small, then the column classification has affected the draw from the urn. In our application, gene A is differentially expressed between Treatment1 and Treatment2. One method of calculating two-sided P-values is to sum the probabilities of all 2 × 2 tables with probabilities less than or equal to that of the observed table where the probability of a 2 × 2 table (e.g., Table 1) is
white balls?” If this probability (i.e., the P-value from Fisher's exact test) is small, then the column classification has affected the draw from the urn. In our application, gene A is differentially expressed between Treatment1 and Treatment2. One method of calculating two-sided P-values is to sum the probabilities of all 2 × 2 tables with probabilities less than or equal to that of the observed table where the probability of a 2 × 2 table (e.g., Table 1) is
 |
(2) |
where  denotes the observed value of
denotes the observed value of 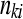 . Note that there are several methods for computing two-sided P-values from Fisher's exact test (Agresti 2002). Figure 2 illustrates the behavior of Fisher's exact test for testing differential expression, between two treatment groups, for every gene in an RNA-Seq data set. It is worth remembering that Fisher's exact test becomes more conservative as expression values decrease to zero, a point concurrent with the fact that genes with small expression values also demonstrate larger variability. This phenomenon as related to RNA-Seq data is discussed in detail in Bloom et al. (2009). The methods used by Kal et al. (1999) (a test of the equality of two binomial proportions) and Audic and Claverie (1997) (a Bayesian model with a Poisson likelihood and a flat prior on the mean) may also be used, although comparisons between these, and other, approaches to Fisher's exact test (Man et al. 2000; Romualdi et al. 2001; Ruijter et al. 2002) show marginal differences in performance.
. Note that there are several methods for computing two-sided P-values from Fisher's exact test (Agresti 2002). Figure 2 illustrates the behavior of Fisher's exact test for testing differential expression, between two treatment groups, for every gene in an RNA-Seq data set. It is worth remembering that Fisher's exact test becomes more conservative as expression values decrease to zero, a point concurrent with the fact that genes with small expression values also demonstrate larger variability. This phenomenon as related to RNA-Seq data is discussed in detail in Bloom et al. (2009). The methods used by Kal et al. (1999) (a test of the equality of two binomial proportions) and Audic and Claverie (1997) (a Bayesian model with a Poisson likelihood and a flat prior on the mean) may also be used, although comparisons between these, and other, approaches to Fisher's exact test (Man et al. 2000; Romualdi et al. 2001; Ruijter et al. 2002) show marginal differences in performance.
TABLE 1.
A 2 × 2 contingency table of (unreplicated) digital gene expression (DGE) measures for testing differential expression between Treatment1 and Treatment2 of gene A
| Treatment1 | Treatment2 | Total | |
|---|---|---|---|
| Gene A | 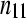 |
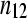 |
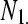 |
| Remaining genes |  |
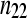 |
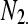 |
| Total |  |
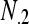 |
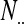 |
The cell counts 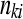 represent the DGE count for gene A (k = 1) or the remaining genes (k = 2) for Treatmenti, I = 1, 2. The
represent the DGE count for gene A (k = 1) or the remaining genes (k = 2) for Treatmenti, I = 1, 2. The 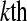 marginal row total is denoted
marginal row total is denoted 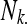 ,
, 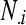 is the marginal total for column i, and
is the marginal total for column i, and 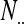 is the grand total.
is the grand total.
Figure 2.—

The log2 fold change, between Treatment1 and Treatment2, of the normalized gene expression is plotted on the y-axis, and the mean log2 expression is plotted on the x-axis. Gene expression counts were normalized by the column totals of the corresponding 2 × 2 table (e.g., Table 1). Blue dots represent significantly differentially expressed genes as established by Fisher's exact test; gray dots represent genes with similar expression. The red horizontal line at zero provides a visual check for symmetry.
Limitations of unreplicated data:
The fundamental problem with generalizing results gathered from unreplicated data is a complete lack of knowledge about biological variation. As Fisher (1935a) noted, without an estimate of variability (i.e., within treatment group), there is no basis for inference (between treatment groups). Although we can test for differential expression between treatment groups from unreplicated data, the results of the analysis only apply to the specific subjects included in the study (i.e., the results cannot be generalized). To better understand the unrealistic conclusions that can be drawn from unreplicated data, suppose that an alien visits Earth and observes only two people, one male named “John” standing 177 cm and one female named “Jane” standing 180 cm. The same reasoning that results in unrealistic conclusions from testing differential expression between treatment groups on the basis of unreplicated data would compel the alien to believe not only that John is shorter than Jane, but also that women are, on average, taller than men.
Replicated data:
Consider extending the example illustrated in Figure 1 (i.e., seven treatment groups with one subject per treatment) to include two more biological subjects within each treatment group (Figure 3). The biological replicates allow for the estimation of within-treatment group (biological) variability, provide information that is necessary for making inferences between treatment groups, and give rise to conclusions that can be generalized.
Figure 3.—

A multiple flow-cell design based on three biological replicates within seven treatment groups. There are three flow cells with eight lanes per flow cell. The control 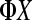 sample is in lane 5 of each flow cell. Tij refers to the
sample is in lane 5 of each flow cell. Tij refers to the 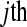 replicate in the
replicate in the 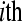 treatment group
treatment group 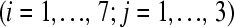 .
.
A simple method for testing differential expression that incorporates within-group (or within-treatment) variability relies on a generalized linear model (GLM) with overdispersion. The model is similar to the one provided by Lu et al. (2005). Consider a per-gene Poisson GLM. If 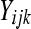 represents the DGE measure for the
represents the DGE measure for the  replicate
replicate  in the
in the 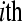 treatment group
treatment group 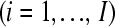 of gene k, and
of gene k, and  represents the overall number of reads from the
represents the overall number of reads from the  replicate in the
replicate in the 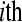 treatment group (e.g.,
treatment group (e.g., 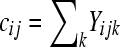 ), then we can model
), then we can model 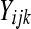 as a
as a 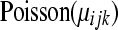 random variable, where
random variable, where 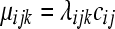 and
and 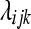 represents the normalized level of expression of gene k in the
represents the normalized level of expression of gene k in the 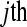 replicate of the ith treatment group. (Marioni et al. 2008). In this example, the inclusion of the
replicate of the ith treatment group. (Marioni et al. 2008). In this example, the inclusion of the 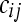 term is equivalent to normalizing by the total number of reads per lane, a common practice in RNA-Seq (Mortazavi et al. 2008). The following model is fit independently to each of k genes,
term is equivalent to normalizing by the total number of reads per lane, a common practice in RNA-Seq (Mortazavi et al. 2008). The following model is fit independently to each of k genes,
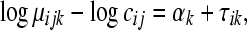 |
(3) |
where 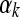 is the mean normalized level of expression across treatments for gene k and
is the mean normalized level of expression across treatments for gene k and 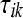 is the effect of the
is the effect of the 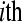 treatment on the overall mean normalized level of expression for gene k. Differential expression of gene k between treatment group
treatment on the overall mean normalized level of expression for gene k. Differential expression of gene k between treatment group 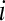 and treatment group
and treatment group 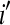 is tested with the following hypotheses:
is tested with the following hypotheses:
 |
(4) |
A simple Poisson GLM assumes that 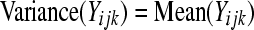 , and when this assumption holds, the hypotheses in (4) can be tested by comparing the likelihood-ratio test statistic (LRT) to a
, and when this assumption holds, the hypotheses in (4) can be tested by comparing the likelihood-ratio test statistic (LRT) to a 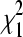 distribution, where the LRT takes the form
distribution, where the LRT takes the form
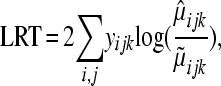 |
(5) |
and  is the maximum-likelihood estimate (MLE) of
is the maximum-likelihood estimate (MLE) of 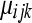 (under the alternative hypothesis) while
(under the alternative hypothesis) while 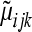 is the MLE of
is the MLE of 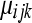 under
under  . If there is any within-treatment-group variability between individuals that is beyond that expected by Poisson sampling, then the required assumption of the Poisson GLM [i.e., that
. If there is any within-treatment-group variability between individuals that is beyond that expected by Poisson sampling, then the required assumption of the Poisson GLM [i.e., that 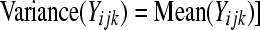 will not hold. In fact, if the LRT in (5) is used to evaluate the hypotheses in (4) when the assumption is violated, it typically results in inflated type I error rates. To maintain the type I error at the appropriate level we need to estimate a dispersion parameter
will not hold. In fact, if the LRT in (5) is used to evaluate the hypotheses in (4) when the assumption is violated, it typically results in inflated type I error rates. To maintain the type I error at the appropriate level we need to estimate a dispersion parameter  , where
, where 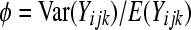 (if
(if 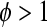 the data are said to be “overdispersed;” in more rare cases,
the data are said to be “overdispersed;” in more rare cases, 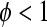 and the data are “underdispersed”). The estimate of
and the data are “underdispersed”). The estimate of 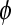 suggested in Faraway (2006) and Tjur (1998) is
suggested in Faraway (2006) and Tjur (1998) is
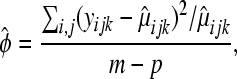 |
(6) |
where m is the total number of observations and p is the number of estimated parameters in model (3) (i.e., in our example with three replicates per treatment, 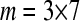 and p=7). Both Faraway (2006) and Tjur (1998) argue that when overdispersion is present, the hypotheses in (4) should be tested by comparing the following test statistic to a
and p=7). Both Faraway (2006) and Tjur (1998) argue that when overdispersion is present, the hypotheses in (4) should be tested by comparing the following test statistic to a 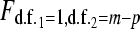 distribution:
distribution:
 |
(7) |
This method is similar to the one described in Baggerly et al. (2004), where an overdispersed logistic regression model is fit to SAGE data to test differential expression. Other methods from the SAGE literature may also be considered; for example, Vêncio et al. (2004) took a Bayesian approach with a Beta-Binomial model accounting for within-class variability. Thygesen and Zwinderman (2006) used a Poisson model with a Gamma Prior in an attempt to model all genes simultaneously. Robinson and Smyth (2007, 2008) incorporated the moderated test statistic approach (Smyth 2004) into a Negative Binomial model to account for both within-class and across-gene variability. Their approach is available in the edgeR package (Robinson et al. 2010) from Bioconductor (Gentleman et al. 2004).
BALANCED BLOCK DESIGNS
Without careful planning an unblocked design faces a fundamental problem with generalizing the results, namely, the potential for confounding. With respect to an RNA-Seq analysis, if the treatment effects are not separable from possible confounding factors, then for any given gene, there is no way of knowing whether the observed difference in abundance between treatment groups is due to the biology or the technology (e.g., amplification or sequencing bias). For example, in Figure 3 all replicates of Treatment1 are sequenced in lane 1 and all replicates of Treatment2 are sequenced in lane 2. Any differences in expression between Treatment1 and Treatment2 are confounded with possible lane effects that may persist across flow cells. In fact, once the data are collected, there is no way of separating the effects due to lane from the effects due to true treatment differences.
In RNA-Seq data, the design is the same for every gene, even though different genes have different variances and are potentially subject to different errors and biases. Of course, there are sources of variation that affect the majority of genes and these should certainly be integrated into the design. However, to ensure a robust analysis across all genes, sources of variation affecting only a minority of genes should be integrated into the design as well (e.g., a PCR-based GC bias may affect only a small proportion of transcript fragments; therefore if it is possible, PCR batch should be integrated into the design). As such, we examine two main sources of variation (beyond the sampling hierarchy explained previously) that may contribute to confounding of effects in RNA-Seq data, namely “batch effects” and “lane effects.” Batch effects include any errors that occur after random fragmentation of the RNA until it is input to the flow cell (e.g., PCR amplification and reverse transcription artifacts). Lane effects include any errors that occur from the point at which the sample is input to the flow cell until data are output from the sequencing machine (e.g., systematically bad sequencing cycles and errors in base calling).
Batch and lane effects have both been observed in previous studies. PCR amplification and reverse transcription artifacts were found to be nonnegligible in both Balwierz et al. (2009) and Chepelev et al. (2009). Chepelev et al. (2009) also observed systematically bad sequencing cycles, and Rougemont et al. (2008) discusses the presence of base-calling errors in the Solexa platform. Although Marioni et al. (2008) found that variation across lanes generally follows a Poisson sampling process, they did observe considerably more variation for a nonnegligible number of genes (on the order of  ).
).
Balanced blocks by multiplexing:
To eliminate confounding caused by batch or lane effects, consider the situation in which all samples of RNA are pooled into the same batch and then sequenced in one lane of a flow cell. This would ensure that any batch effects are the same for all samples, and since the sequencing reaction is contained in one lane, all effects due to lane will be the same for all samples. While indeed this is an idealized situation, it can be accomplished by bar coding the RNA immediately after fragmentation. Once bar codes are attached to the random fragments, the samples can be pooled and processed together through the reverse transcription, size selection, and amplification steps. Typically, each lane is dedicated to sequencing one sample, so the number of samples m is equal to the number of lanes 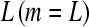 . To not lose sequencing depth compared to the typical layout,
. To not lose sequencing depth compared to the typical layout,  total bar-coded samples can be pooled and processed together through the amplification step. The amplification product can then be divided into
total bar-coded samples can be pooled and processed together through the amplification step. The amplification product can then be divided into 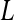 equal parts. Each part is then input to a different lane of the flow cell. By exposing equal portions of every unique (i.e., bar-coded) sample to the same experimental conditions (i.e., same batch in the same lane), “balanced blocks” are formed.
equal parts. Each part is then input to a different lane of the flow cell. By exposing equal portions of every unique (i.e., bar-coded) sample to the same experimental conditions (i.e., same batch in the same lane), “balanced blocks” are formed.
It is worth noting that if L lanes are utilized, there is no loss of sequencing depth compared to running each sample in a lane. Figure 4 shows a comparison of this design to a typical design that confounds sample with batch and lane.
Figure 4.—

Comparison of two designs for testing differential expression between treatments A and B. Treatment A is denoted by red tones and treatment B by blue tones. In the ideal balanced block design (left), six samples 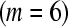 are bar coded, pooled, and processed together. The pool is then divided into six equal portions that are input to six lanes
are bar coded, pooled, and processed together. The pool is then divided into six equal portions that are input to six lanes 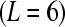 of the flow cell. Bar coding in the balanced block design results in six technical replicates
of the flow cell. Bar coding in the balanced block design results in six technical replicates  of each sample, while balancing batch and lane effects and blocking on lane. The balanced block design also allows partitioning of batch and lane effects from the within-group biological variability. The confounded design (right) represents a typical RNA-Seq experiment and consists of the same six samples, with no bar coding, and does not permit partitioning of batch and lane effects from the estimate of within-group biological variability.
of each sample, while balancing batch and lane effects and blocking on lane. The balanced block design also allows partitioning of batch and lane effects from the within-group biological variability. The confounded design (right) represents a typical RNA-Seq experiment and consists of the same six samples, with no bar coding, and does not permit partitioning of batch and lane effects from the estimate of within-group biological variability.
Balanced incomplete block designs and blocking without multiplexing:
Although the previous balanced block design is convenient for illustration purposes, in reality resources, technical constraints, and the scientific hypotheses under investigation will dictate the number of treatments 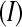 , the number of biological replicates per treatment
, the number of biological replicates per treatment 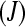 , the number of unique bar codes
, the number of unique bar codes 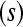 that can be included in a single lane, and the number of lanes available for sequencing
that can be included in a single lane, and the number of lanes available for sequencing 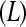 . When the number of unique bar codes in one lane is less than the number of treatments
. When the number of unique bar codes in one lane is less than the number of treatments  , a complete block design (Figure 4) is not be possible. In these cases, we suggest using a balanced incomplete block design (BIBD). If
, a complete block design (Figure 4) is not be possible. In these cases, we suggest using a balanced incomplete block design (BIBD). If 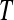 is the total number of possible technical replicates per biological replicate, then a BIBD (according to our scheme of blocking by batch and lane by multiplexing) satisfies
is the total number of possible technical replicates per biological replicate, then a BIBD (according to our scheme of blocking by batch and lane by multiplexing) satisfies 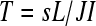 (Oehlert 2000). For a situation where there are three treatments
(Oehlert 2000). For a situation where there are three treatments 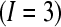 , a single subject within each treatment group
, a single subject within each treatment group 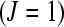 , the ability to include 2 unique bar codes
, the ability to include 2 unique bar codes 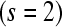 within a lane, and three available lanes for sequencing
within a lane, and three available lanes for sequencing 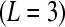 (currently, Illumina advertises 12 different bar codes in a single lane; http://www.illumina.com/documents/products/datasheets/datasheet_sequencing_multiplex.pdf), a BIBD as illustrated in Figure 5 is possible. Extensive lists of other BIBDs are given in Fisher and Yates (1963) and Cochran and Cox (1957).
(currently, Illumina advertises 12 different bar codes in a single lane; http://www.illumina.com/documents/products/datasheets/datasheet_sequencing_multiplex.pdf), a BIBD as illustrated in Figure 5 is possible. Extensive lists of other BIBDs are given in Fisher and Yates (1963) and Cochran and Cox (1957).
Figure 5.—

A balanced incomplete block design (BIBD) for three treatment groups (T1, T2, T3) with one subject per treatment group (T11, T21, T31) and two technical replicates of each (T111, T112, T211, T212, T311, T312). After fragmentation, each of the three samples is bar coded and divided in two (e.g., T11 would be split into T111 and T112) and then pooled and sequenced as illustrated (e.g., T111 is pooled with T212 as input to lane 1).
Clearly multiplexing is useful for generating technical replicates that are effective in blocking on lane and batch effects to reduce confounding. However, it is important to understand that technical replicates are no substitute for independent biological replication and that for a sufficient number of biological replicates certain designs can accommodate lane and/or flow cell as blocking factors. As an illustration of this flexibility, the design in Figure 3 can be rearranged as a balanced complete block design where the blocks are flow cells and a balanced incomplete block design where the blocks are lanes (Figure 6).
Figure 6.—

A design based on three biological replicates within seven treatment groups. For each of the three flow cells there are eight lanes per flow cell and a control ( ) sample in lane 5. Tij refers to the
) sample in lane 5. Tij refers to the 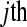 replicate in the
replicate in the 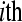 treatment group
treatment group  . In this design the flow cells form balanced complete blocks, and the lanes form balanced incomplete blocks.
. In this design the flow cells form balanced complete blocks, and the lanes form balanced incomplete blocks.
Analyzing a balanced block design:
The generalized linear model in (3) can be expanded to include known blocking factors. Consider Figure 6 where both lane and flow cell form blocks. The model for this design is
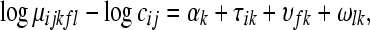 |
(8) |
where 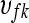 is the effect of the
is the effect of the  flow cell on the mean normalized level of expression for gene k, and similarly
flow cell on the mean normalized level of expression for gene k, and similarly  is the effect of the
is the effect of the 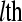 lane on the mean normalized level of expression for gene k. This model can be fit on a gene-by-gene basis implicitly assuming gene-by-block interactions [if one is unwilling to make this assumption, model (8) can easily be modified to fit all genes simultaneously allowing for the estimation of global blocking factors that can be used as offsets in a per-gene model (3)]. Note that model (8) separates the lane and flow-cell effects (i.e., technological variation) from the within-group biological variability. The hypotheses for testing differential expression between Treatment
lane on the mean normalized level of expression for gene k. This model can be fit on a gene-by-gene basis implicitly assuming gene-by-block interactions [if one is unwilling to make this assumption, model (8) can easily be modified to fit all genes simultaneously allowing for the estimation of global blocking factors that can be used as offsets in a per-gene model (3)]. Note that model (8) separates the lane and flow-cell effects (i.e., technological variation) from the within-group biological variability. The hypotheses for testing differential expression between Treatment 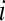 and Treatment
and Treatment 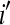 are as in (4), and the dispersion parameter
are as in (4), and the dispersion parameter 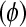 is estimated as
is estimated as
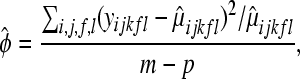 |
(9) |
where m = 21 and p = 15, and the LRT is
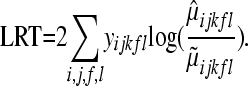 |
(10) |
The F-statistic for testing differential expression is simply the LRT in (10) divided by 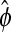 as estimated in (9). Under the null hypothesis of no differential expression, this F-statistic is approximately distributed as an
as estimated in (9). Under the null hypothesis of no differential expression, this F-statistic is approximately distributed as an 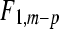 random variable.
random variable.
Analyzing a balanced block design with technical replicates:
Consider the balanced block design in Figure 4. Let 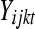 represent the DGE measure for the
represent the DGE measure for the 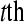 technical replicate
technical replicate 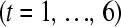 of the
of the 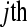 biological replicate
biological replicate 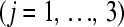 in the
in the 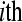 treatment group
treatment group  of gene k. Then
of gene k. Then 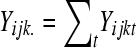 and
and 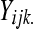 can be modeled as before with a
can be modeled as before with a 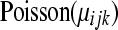 random variable, where
random variable, where 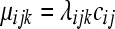 and
and  represents the normalized level of expression of gene
represents the normalized level of expression of gene 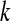 in the
in the  replicate of the
replicate of the 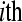 treatment group (the offset term
treatment group (the offset term 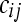 no longer represents the total number of reads per lane, but the total number of reads in the
no longer represents the total number of reads per lane, but the total number of reads in the 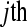 replicate of the
replicate of the  treatment group summed over technical replicates,
treatment group summed over technical replicates,  ). The model, hypotheses, estimation, and testing procedures are the same as in (3–7) with
). The model, hypotheses, estimation, and testing procedures are the same as in (3–7) with 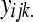 replacing
replacing 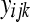 . This analysis strategy does not include lane as a blocking factor; therefore lane effects are not partitioned from estimates of within-treatment group variability (since only one batch is used, batch-to-batch variation is removed from residual error and batch effects need not be included in the model). However, since lane effects are balanced across treatment groups, the potential for confounding on lane effects is eliminated. To accurately partition the lane effects from estimates of within-treatment-group variability a repeated measures GLM (see Faraway 2006) with overdispersion is necessary. To our knowledge, hypothesis testing in this paradigm is currently problematic and a point of future research.
. This analysis strategy does not include lane as a blocking factor; therefore lane effects are not partitioned from estimates of within-treatment group variability (since only one batch is used, batch-to-batch variation is removed from residual error and batch effects need not be included in the model). However, since lane effects are balanced across treatment groups, the potential for confounding on lane effects is eliminated. To accurately partition the lane effects from estimates of within-treatment-group variability a repeated measures GLM (see Faraway 2006) with overdispersion is necessary. To our knowledge, hypothesis testing in this paradigm is currently problematic and a point of future research.
SIMULATIONS
To evaluate the effectiveness of the proposed multiplexed designs, we compare a biologically unreplicated unblocked design (Figure 7A), a biologically unreplicated balanced block design with technical replicates (i.e., multiplexing; Figure 7B), a biologically replicated (triplicate) unblocked design (Figure 7C), and a biologically replicated (triplicate) balanced block design with technical replicates by multiplexing (Figure 7D) in an experimental setting testing differential expression between Treatment 1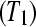 and Treatment 2
and Treatment 2 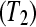 . Gene counts were simulated across treatment groups, and we compare the false positive rate (1 − specificity) and the true positive rate (sensitivity) for each design.
. Gene counts were simulated across treatment groups, and we compare the false positive rate (1 − specificity) and the true positive rate (sensitivity) for each design.
Figure 7.—

Four designs (A–D) are compared in the simulation study for treatments  and
and 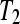 . Design A is a biologically unreplicated unblocked design with one subject for treatment group
. Design A is a biologically unreplicated unblocked design with one subject for treatment group  and one subject for treatment group
and one subject for treatment group 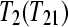 . Design B is a biologically unreplicated balanced block design with
. Design B is a biologically unreplicated balanced block design with 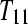 split (bar coded) into two technical replicates
split (bar coded) into two technical replicates 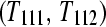 and
and 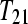 split into two technical replicates
split into two technical replicates 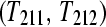 and input to lanes 1 and 2. Design C is a biologically replicated unblocked design with three subjects from treatment group
and input to lanes 1 and 2. Design C is a biologically replicated unblocked design with three subjects from treatment group 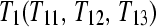 and three subjects from treatment group
and three subjects from treatment group  . Design D is a biologically replicated balanced block design with each subject (e.g.,
. Design D is a biologically replicated balanced block design with each subject (e.g., 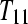 ) split (bar coded) into six technical replicates (e.g.,
) split (bar coded) into six technical replicates (e.g., 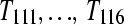 ) and input to six lanes.
) and input to six lanes.
Data simulation:
We fixed the total number of reads at c = 3,000,000 and the mean sampling rate for treatment group  at four different values
at four different values 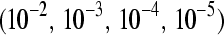 , which correspond to gene counts on the order of
, which correspond to gene counts on the order of 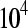 ,
, 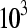 ,
, 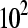 , and
, and  , respectively
, respectively 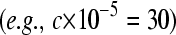 . We varied the mean
. We varied the mean 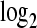 fold change (LFC) between
fold change (LFC) between 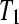 and
and 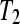 from −3 to 3 in increments of 0.25. The treatment group
from −3 to 3 in increments of 0.25. The treatment group 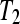 sampling rate was calculated as
sampling rate was calculated as
 |
(11) |
Gene counts  and
and 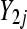 were sampled according to Poisson
were sampled according to Poisson  and Poisson
and Poisson 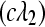 distributions, respectively
distributions, respectively 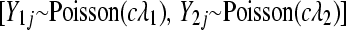 . To evaluate the effectiveness of modeling with an overdispersed Poisson GLM, we added Gaussian noise to each gene count, rounding to the nearest integer:
. To evaluate the effectiveness of modeling with an overdispersed Poisson GLM, we added Gaussian noise to each gene count, rounding to the nearest integer:
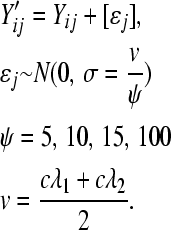 |
Four different simulation settings are considered: batch effect and lane effect [simulation (S)1], batch effect and no lane effect (S2), no batch effect and lane effect (S3), and no batch effect and no lane effect (S4). Batch effects were simulated by adding Gaussian noise to each noisy gene count 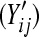 and rounding to the nearest integer:
and rounding to the nearest integer:
 |
No noise was considered in settings S3 and S4 (i.e., for settings S3 and S4, 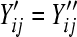 ). Lane effects were simulated by Poisson sampling from
). Lane effects were simulated by Poisson sampling from 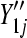 and
and 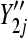 at different rates varying between lanes:
at different rates varying between lanes:
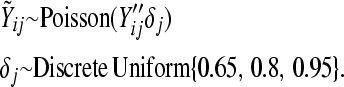 |
For settings with no lane effect (S2 and S4), the Poisson sampling rates were held constant ( ). Since designs B and D (Figure 7) included technical replicates, we distributed the respective sampling rates with no loss of depth for the gene counts in each biological replicate.
). Since designs B and D (Figure 7) included technical replicates, we distributed the respective sampling rates with no loss of depth for the gene counts in each biological replicate.
For design A (Figure 7) we tested for differential expression using Fisher's exact test (1), setting both column totals of the 2 × 2 table to 3,000,000. For design B (Figure 7) we fit a balanced block design with technical replicates model. We set the offset term to c, and since this design does not have any biological replicates (i.e., J = 1), we did not estimate dispersion. The likelihood ratio was compared to a 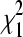 distribution. For design C we fit model (3) with c as the offset, estimated dispersion as shown in (6), and used the F-test described in (7). For design D we fit a balanced block design that acknowledged the technical replicates with c as the offset.
distribution. For design C we fit model (3) with c as the offset, estimated dispersion as shown in (6), and used the F-test described in (7). For design D we fit a balanced block design that acknowledged the technical replicates with c as the offset.
We ran 10,000 simulations under each setting (S1–S4), varying  and
and 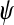 . The false positive rate (i.e., type I error rate) was calculated as the proportion of times a gene was declared to be differentially expressed when the LFC was zero. The true positive rate (i.e., statistical power) was calculated as the proportion of times a gene was determined to be differentially expressed in the correct direction when the LFC was not zero.
. The false positive rate (i.e., type I error rate) was calculated as the proportion of times a gene was declared to be differentially expressed when the LFC was zero. The true positive rate (i.e., statistical power) was calculated as the proportion of times a gene was determined to be differentially expressed in the correct direction when the LFC was not zero.
Results:
Receiver operating characteristic (ROC) curves offer a useful way of comparing false positive rates with true positive rates. Using ROC curves the true positive rate is typically plotted on the vertical axis, and the false positive rate is plotted on the horizontal axis. The resulting curves can be compared by fixing a false positive rate (type I error rate) and contrasting the corresponding true positive rates (statistical power). If one ROC curve is always above another, this indicates its superiority in classifying genes as differentially expressed. The diagonal identity line indicates the performance of classifying a gene as differentially expressed, using a completely random guess (e.g., guessing differential expression 90% of the time yields a 90% true positive and false positive rate).
The designs featuring independent replication (designs C and D) demonstrate remarkably better performance than the unreplicated designs (designs A and B) whenever there is nonnegligible within-treatment-group biological variability (Figure 8; supporting information, Figure S1 and Figure S2) across simulation settings (S1–S4). Even when there is very small within-treatment-group biological variability (Figure S3), the replicated designs still outperform the unreplicated designs.
Figure 8.—

ROC curves for the within-group variability setting  . The x-axis represents the false positive rate and the y-axis represents the true positive rate. The four panels of the graph show results for each of the four simulation settings. The ROC curve for the unblocked unreplicated design (A) is in solid red, for the blocked unreplicated design (B) is in dotted red, for the unblocked replicated design (C) is in solid blue, and for the blocked replicated design (D) is in dotted blue. The replicated designs always outperform the unreplicated designs, and whenever there is a batch effect or a lane effect, the blocked designs outperform their unblocked counterparts.
. The x-axis represents the false positive rate and the y-axis represents the true positive rate. The four panels of the graph show results for each of the four simulation settings. The ROC curve for the unblocked unreplicated design (A) is in solid red, for the blocked unreplicated design (B) is in dotted red, for the unblocked replicated design (C) is in solid blue, and for the blocked replicated design (D) is in dotted blue. The replicated designs always outperform the unreplicated designs, and whenever there is a batch effect or a lane effect, the blocked designs outperform their unblocked counterparts.
To evaluate which designs upheld the typical 0.05 false positive rate, for each of the four different sampling rates 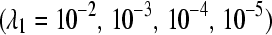 , we calculated the proportion of times a gene was declared differentially expressed (using a P-value cutoff of 0.05) when the LFC was zero (Table 2;
, we calculated the proportion of times a gene was declared differentially expressed (using a P-value cutoff of 0.05) when the LFC was zero (Table 2; 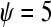 ). The replicated designs (C and D) maintained the nominal significance level (0.05) across the simulation settings, demonstrating that designs featuring independent replication coupled with analyses that estimate within-group variability are robust to batch effects, lane effects, and extra-Poisson variability. The estimated false positive rates for the unreplicated designs (A and B) suffered under all simulation settings, especially for genes with larger expression values. Although the false positive rates tended to decrease slightly as batch and lane effects were removed, in the absence of a batch or lane effect, the type I error rates were still four to five times larger than the nominal significance level for genes with the smallest level of expression (
). The replicated designs (C and D) maintained the nominal significance level (0.05) across the simulation settings, demonstrating that designs featuring independent replication coupled with analyses that estimate within-group variability are robust to batch effects, lane effects, and extra-Poisson variability. The estimated false positive rates for the unreplicated designs (A and B) suffered under all simulation settings, especially for genes with larger expression values. Although the false positive rates tended to decrease slightly as batch and lane effects were removed, in the absence of a batch or lane effect, the type I error rates were still four to five times larger than the nominal significance level for genes with the smallest level of expression (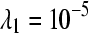 ).
).
TABLE 2.
The false positive rates (at the 0.05 nominal level) for designs A–D considering four settings of λ1(10−2, 10−3, 10−4, 10−5) and four simulation settings [batch effect and lane effect (S1), batch effect and no lane effect (S2), no batch effect and lane effect (S3), and no batch effect and no lane effect (S4)]
| Simulation setting |
|||||
|---|---|---|---|---|---|
Sampling rate 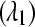
|
Design | S1 | S2 | S3 | S4 |
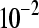 |
A | 0.9655 | 0.9548 | 0.9630 | 0.9508 |
| B | 0.9524 | 0.9521 | 0.9496 | 0.9498 | |
| C | 0.0494 | 0.0499 | 0.0485 | 0.0480 | |
| D | 0.0476 | 0.0463 | 0.0482 | 0.0487 | |
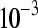 |
A | 0.8789 | 0.8595 | 0.8744 | 0.8434 |
| B | 0.8456 | 0.8479 | 0.8431 | 0.8481 | |
| C | 0.0477 | 0.0480 | 0.0532 | 0.0499 | |
| D | 0.0506 | 0.0491 | 0.0472 | 0.0489 | |
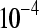 |
A | 0.6521 | 0.5873 | 0.6325 | 0.5527 |
| B | 0.5551 | 0.5622 | 0.5583 | 0.5677 | |
| C | 0.0522 | 0.0505 | 0.0527 | 0.0516 | |
| D | 0.0538 | 0.0529 | 0.0522 | 0.0532 | |
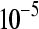 |
A | 0.2662 | 0.2299 | 0.2491 | 0.2111 |
| B | 0.2407 | 0.2452 | 0.2458 | 0.2411 | |
| C | 0.0482 | 0.0524 | 0.0503 | 0.0461 | |
| D | 0.0488 | 0.0460 | 0.0494 | 0.0477 | |
Interestingly, the blocked designs did not outperform the unblocked designs in terms of the false positive rates (Table 2). However, across simulation settings, whenever a batch or lane effect is present, the blocked designs demonstrate distinguishably higher true positive rates than the unblocked designs. We speculate that there are two reasons for this result. First, the blocked designs are all included in the same batch such that batch-to-batch variation is removed from residual error. Second, even though we did not use lane as a blocking factor, lane effects were balanced across every biological replicate in the two treatment groups, thereby reducing the chance that a lane effect would overly influence one treatment group and produce a misleading result (i.e., confounding). Partitioning the variation due to lane through a statistical model that included blocks on lanes may further enhance the performance of the blocked designs by reducing the residual error.
DISCUSSION
Fisher (1935a) was right. Replication, randomization, and blocking are essential components of any well planned and properly analyzed design. RNA-Seq designs and analyses are no exception. Luckily, the current format and properties of the NGS platforms lend themselves nicely to the concepts of randomization and blocking. However, the decision to biologically replicate remains in the hands of the scientist.
Our purpose in writing this article is to demonstrate that variability (which is dependent on the organism, laboratory techniques, and the biological factors under investigation) may be larger than expected and, if not estimated properly, will negatively affect the results of any study. This variability is positively correlated with the magnitude of the overall variability between biological subjects and can be dealt with by employing statistical models that not only accommodate estimation of within-treatment-group biological variability, but also remain faithful to a nominal significance level. Indisputably, the best way to ensure reproducibility and accuracy of results is to include independent biological replicates (technical replicates are no substitute) and to acknowledge anticipated nuisance factors (e.g., lane, batch, and flow-cell effects) in the design.
For both replicated and unreplicated scenarios the proposed balanced block designs benefit from both the NGS platform design and multiplexing. These designs are as good as, if not better than, their unblocked counterparts in terms of power and type I error and are considerably better when batch and/or lane effects are present. Realizing of course that it is not possible to determine whether batch and/or lane effects are present a priori, we recommend the use of block designs to protect against observed differences that are attributable to these potential sources of variation. Since we understand both the expense associated with block designs and the concern of multiplexing, we offer some alternatives. Certainly, it is possible to avoid multiplexing if there are enough biological replicates and sequencing lanes that allow for designs that block on lane and/or flow cell (see Figure 6). However, if resources are limited (i.e., one flow cell), multiplexing offers an alternative that at the very least eliminates the potential for confounding of effects. Multiplexing and blocking aside, the bottom line remains the same: results from unreplicated data cannot be generalized beyond the sample tested (here, differential expression).
Even though the benefits of good designs far outweigh any drawbacks, we anticipate objections to the multiplexed designs related to cost, loss of sequencing depth, and bar-code bias. To mitigate these concerns we provide some reassurances. First, although increased cost may be a concern, the added cost and time to multiplex are negligible when compared to the overall time and resources required for RNA sequencing. Second, there may be additional concerns that multiplexing will result in an overall loss of sequencing depth. This will be problematic only if enough bar codes are incorrectly identified such that the read count for each gene is affected. Recently, Philippe et al. (2009) estimated that, on the Illumina platform, the probability that a 20-base transcript tag contains one or more sequencing errors is 0.0048. Using this as an upper bound on the probability that the bar code will contain one or more sequencing errors (the bar code is 6 bases long), in a lane with 10 million usable sequencing reads and 10 different transcripts, miscalled bar codes would result in an average loss of at most 4.8 reads from the read count per transcript. Third, while there may be technical problems with multiplexing, such as uneven coverage of the samples or bias in the base calls adjacent to the bar code, as long as the problem with uneven coverage is consistent within each sample, normalization schemes that take into account lane- and sample-specific coverage can correct for this (e.g., dividing each gene count by the coverage over sample or the coverage over lane). Read bias associated with bar coding is not problematic if it affects all samples the same way. Specifically, a proper normalization scheme will be robust to bias as long as the problem is consistent within sample.
The design principles that are presented here are applicable to a variety of applications involving quantitative comparisons across samples and can be put into practice on every NGS platform as applied to RNA-Seq. However, for other applications (e.g., ChIP-Seq and copy number variant studies, etc.) a clearly defined statistical model that fully accounts for sources of variation must be developed before specific details of the design can be described.
Acknowledgments
We thank the anonymous reviewers for their thorough and thought-provoking review. We are also grateful to Andrea Rau for helpful comments on the manuscript and to Scott Jackson, Rob Martienssen, and their respective labs for a wealth of sequencing data and biological information. This work is supported by a National Science Foundation Plant Genome grant (DBI-0733857) in part to R.W.D.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.114983/DC1.
References
- Agresti, A., 2002. Categorical Data Analysis, Ed. 2. Wiley, Hoboken, NJ.
- Alkan, C., J. M. Kidd, T. Marques-Bonet, G. Aksay, F. Antonacci et al., 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic, S., and J. Claverie, 1997. The significance of digital gene expression profiles. Genome Res. 7 986–995. [DOI] [PubMed] [Google Scholar]
- Baggerly, K. A., L. Deng, J. S. Morris and C. M. Aldaz, 2004. Overdispersed logistic regression for SAGE: modelling multiple groups and covariates. BMC Bioinformatics 5 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balwierz, P. J., P. Carninci, C.O. Daub, J. Kawai, Y. Hayashizaki et al., 2009. Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data. Genome Biol. 10 R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, D. R., S. Balasubramanian, H. P. Swerdlow, G. P. Smith, J. Milton et al., 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, J. S., Z. Khan, L. Kruglyak, M. Singh and A. A. Caudy, 2009. Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays. BMC Genomics 10 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepelev, I., G. Wei, Q. Tang and K. Zhao, 2009. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic Acids Res. 37 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., 2002. Fundamentals of experimental design for cDNA microarrays. Nat. Genet. 32 490–495. [DOI] [PubMed] [Google Scholar]
- Cloonan, N., A. R. R. Forrest, G. Kolle, B. B. A. Gardiner, G. J. Faulkner et al., 2008. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5 613–619. [DOI] [PubMed] [Google Scholar]
- Cloonan, N., Q. Xu, G. J. Faulkner, D. F. Taylor, T. P. Tang et al., 2009. RNA-MATE: a recursive mapping strategy for high-throughput RNA-sequencing data. Bioinformatics 25 2615–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran, W. G., and G. M. Cox, 1957. Experimental Designs. Wiley, New York.
- Craig, D. W., J. V. Pearson, S. Szelinger, A. Sekar, M. Redman et al., 2008. Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods 5 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveland, A. L., D. R. McCarty and K. E. Koch, 2008. Transcript profiling by 3′-untranslated region sequencing resolves expression of gene families. Plant Physiol. 146 32–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraway, J. J., 2006. Extending the Linear Model With R. Chapman & Hall, Boca Raton, FL.
- Fisher, R. A., 1935. a The Design of Experiments. Ed. 2. Oliver & Boyd, Edinburgh.
- Fisher, R. A., 1935. b The logic of inductive inference. J. R. Stat. Soc. 98 39–82. [Google Scholar]
- Fisher, R. A., and F. Yates, 1963. Statistical Tables for Biological, Agricultural, and Medical Research, Ed. 6. Oliver & Boyd, Edinburgh.
- Gentleman, R. C., V. J. Carey, D. M. Bates, B. Bolstad, M. Dettling et al., 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology 5 R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady, M., J. J. Walker, J. K. Harris, N. J. Gold and R. Knight, 2008. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5 235–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden, E. C., 2009. Genome sequencing: the third generation. Nature 457 769. [DOI] [PubMed] [Google Scholar]
- Kal, A. J., A. J. van Zonneveld, V. Benes, M. van den Berg, M. G. Koerkamp et al., 1999. Dynamics of gene expression revealed by comparison of serial analysis of gene expression transcript profiles from yeast grown on two different carbon sources. Mol. Biol. Cell 10 1859–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, M. K., and G. A. Churchill, 2001. a Statistical design and the analysis of gene expression microarray data. Genet. Res. 77 123–128. [DOI] [PubMed] [Google Scholar]
- Kerr, M. K., and G. A. Churchill, 2001. b Experimental design for gene expression microarrays. Biostatistics 2 183–201. [DOI] [PubMed] [Google Scholar]
- Kerr, M. K., M. Martin and G. A. Churchill, 2000. Analysis of variance for gene expression microarray data. J. Comput. Biol. 7 819–837. [DOI] [PubMed] [Google Scholar]
- Lee, M. T., F. C. Kuo, G. A. Whitmore and J. Sklar, 2000. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc. Natl. Acad. Sci. USA 97 9834–9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J., J. K. Tomfohr and T. B. Kepler, 2005. Identifying differential expression in multiple SAGE libraries: an overdispersed log-linear model approach. BMC Bioinformatics 6 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man, M. Z., X. Wang and Y. Wang, 2000. POWER_SAGE: comparing statistical tests for SAGE experiments. Bioinformatics 16 953–959. [DOI] [PubMed] [Google Scholar]
- Marioni, J. C., C. E. Mason, S. M. Mane, M. Stephens and Y. Gilad, 2008. RNA-Seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova, O., M. Hirst and M. A. Marra, 2009. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 10 135–151. [DOI] [PubMed] [Google Scholar]
- Mortazavi, A., B. A. Williams, K. McCue, L. Schaeffer and B. Wold, 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5 621–628. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi, U., Z. Wang, K. Waern, C. Shou, D. Raha et al., 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlert, G. W., 2000. A First Course in Design and Analysis of Experiments. W. H. Freeman, New York.
- Park, P. J., 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe, N., A. Boureux, L. Brehelin, J. Tarhio, T. Commes et al., 2009. Using reads to annotate the genome: influence of length, background distribution, and sequence errors on prediction capacity. Nucleic Acids Res. 37 e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M. D., and G. K. Smyth, 2007. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23 2881–2887. [DOI] [PubMed] [Google Scholar]
- Robinson, M. D., and G. K. Smyth, 2008. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9 321–332. [DOI] [PubMed] [Google Scholar]
- Robinson, M. D., D. J. McCarthy and G. K. Smyth, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romualdi, C., S. Bortoluzzi and G. Danieli, 2001. Detecting differentially expressed genes in multiple tag sampling experiments: comparative evaluation of statistical tests. Hum. Mol. Genet. 10 2133–2141. [DOI] [PubMed] [Google Scholar]
- Rougemont, J., A. Amzallag, C. Iseli, L. Farinelli, I. Xenarios et al., 2008. Probabilistic base calling of Solexa sequencing data. BMC Bioinformatics 9 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter, J. M., A. H. C. V. Kampen and F. Baas, 2002. Statistical evaluation of SAGE libraries: consequences for experimental design. Physiol. Genomics 11 37–44. [DOI] [PubMed] [Google Scholar]
- Schena, M., D. Shalon, R. W. Davis and P. O. Brown, 1995. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270 467–470. [DOI] [PubMed] [Google Scholar]
- Shendure, J., 2008. The beginning of the end for microarrays? Nat. Methods 5 585–587. [DOI] [PubMed] [Google Scholar]
- Smyth, G. K., 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3 3. [DOI] [PubMed] [Google Scholar]
- Sultan, M., M. H. Schulz, H. Richard, A. Magen, A. Klingenhoff et al., 2008. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321 956–960. [DOI] [PubMed] [Google Scholar]
- Thygesen, H. H., and A. H. Zwinderman, 2006. Modeling Sage data with a truncated gamma-Poisson model. BMC Bioinformatics 7 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tino, P., 2009. Basic properties and information theory of Audic-Claverie statistic for analyzing cDNA arrays. BMC Bioinformatics 10 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjur, T., 1998. Nonlinear regression, quasi likelihood, and overdispersion in generalized linear models. Am. Stat. 52 222–227. [Google Scholar]
- Velculescu, V. E., L. Zhang, B. Vogelstein and K. W. Kinzler, 1995. Serial analysis of gene expression. Science 270 484–487. [DOI] [PubMed] [Google Scholar]
- Vêncio, R. Z., H. Brentani, D. F. Patrão and C. A. Pereira, 2004. Bayesian model accounting for within-class biological variability in serial analysis of gene expression (SAGE). BMC Bioinformatics 5 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]