

Abstract
Despite the widespread study of genetic variation in admixed human populations, such as African-Americans, there has not been an evaluation of the effects of recent admixture on patterns of polymorphism or inferences about population demography. These issues are particularly relevant because estimates of the timing and magnitude of population growth in Africa have differed among previous studies, some of which examined African-American individuals. Here we use simulations and single-nucleotide polymorphism (SNP) data collected through direct resequencing and genotyping to investigate these issues. We find that when estimating the current population size and magnitude of recent growth in an ancestral population using the site frequency spectrum (SFS), it is possible to obtain reasonably accurate estimates of the parameters when using samples drawn from the admixed population under certain conditions. We also show that methods for demographic inference that use haplotype patterns are more sensitive to recent admixture than are methods based on the SFS. The analysis of human genetic variation data from the Yoruba people of Ibadan, Nigeria and African-Americans supports the predictions from the simulations. Our results have important implications for the evaluation of previous population genetic studies that have considered African-American individuals as a proxy for individuals from West Africa as well as for future population genetic studies of additional admixed populations.
STUDIES of archeological and genetic data show that anatomically modern humans originated in Africa and more recently left Africa to populate the rest of the world (Tishkoff and Williams 2002; Barbujani and Goldstein 2004; Garrigan and Hammer 2006; Reed and Tishkoff 2006; Campbell and Tishkoff 2008; Jakobsson et al. 2008; Li et al. 2008). Given the central role Africa has played in the origin of diverse human populations, understanding patterns of genetic variation and the demographic history of populations within Africa is important for understanding the demographic history of global human populations. The availability of large-scale single-nucleotide polymorphism (SNP) data sets coupled with recent advances in statistical methodology for inferring parameters in population genetic models provides a powerful means of accomplishing these goals (Keinan et al. 2007; Boyko et al. 2008; Lohmueller et al. 2009; Nielsen et al. 2009).
It is important to realize that studies of African demographic history using genetic data have come to qualitatively different conclusions regarding important parameters. Some recent studies have found evidence for ancient (>100,000 years ago) two- to fourfold growth in African populations (Adams and Hudson 2004; Marth et al. 2004; Keinan et al. 2007; Boyko et al. 2008). Other studies have found evidence of very recent growth (Pluzhnikov et al. 2002; Akey et al. 2004; Voight et al. 2005; Cox et al. 2009; Wall et al. 2009) or could not reject a model with a constant population size (Pluzhnikov et al. 2002; Voight et al. 2005). It is unclear why studies found such different parameter estimates. However, these studies all differ from each other in the amount of data considered, the types of data used (e.g., SNP genotypes vs. full resequencing), the genomic regions studied (e.g., noncoding vs. coding SNPs), and the types of demographic models considered (e.g., including migration vs. not including migration postseparation of African and non-African populations).
Another important way in which studies of African demographic history differ from each other is in the populations sampled. Some studies have focused on genetic data from individuals sampled from within Africa (Pluzhnikov et al. 2002; Adams and Hudson 2004; Voight et al. 2005; Keinan et al. 2007; Cox et al. 2009; Wall et al. 2009), while other studies included American individuals with African ancestry (Adams and Hudson 2004; Akey et al. 2004; Marth et al. 2004; Boyko et al. 2008). While there is no clear correspondence between those studies which sampled native African individuals (as opposed to African-Americans) and particular growth scenarios, it is clear from previous studies that African-American populations do differ from African populations in their recent demographic history. In particular, genetic studies suggest that there is wide variation in the degree of European admixture in most African-American individuals in the United States and that they have, on average, ∼80% African ancestry and 20% European ancestry (Parra et al. 1998; Pfaff et al. 2001; Falush et al. 2003; Patterson et al. 2004; Tian et al. 2006; Lind et al. 2007; Reiner et al. 2007; Price et al. 2009; Bryc et al. 2010). Furthermore, both historical records and genetic evidence suggest that the admixture process began quite recently, within the last 20 generations (Pfaff et al. 2001; Patterson et al. 2004; Seldin et al. 2004; Tian et al. 2006). Recent population admixture can alter patterns of genetic variation in a discernible and predictable way. For example, recently admixed populations will exhibit correlation in allele frequencies (i.e., linkage disequilibrium) among markers that differ in frequency between the parental populations. This so-called admixture linkage disequilibrium (LD) (Chakraborty and Weiss 1988) can extend over long physical distances (Lautenberger et al. 2000) and decays exponentially with time the since the admixture process began (i.e., recently admixed populations typically exhibit LD over a longer physical distance than anciently admixed populations).
While it is clear that African-American populations have a different recent demographic history than do African populations from within Africa and that admixture tracts can be identified in admixed individuals (Falush et al. 2003; Patterson et al. 2004; Tang et al. 2006; Sankararaman et al. 2008a,b; Price et al. 2009; Bryc et al. 2010), the effect that admixture has on other patterns of genetic variation remains unclear. For example, Xu et al. (2007) found similar LD decay patterns when comparing African-American and African populations. It is also unclear whether the recent admixture affects our ability to reconstruct ancient demographic events (such as expansions that predate the spread of humans out of Africa) from whole-genome SNP data. Most studies of demographic history have summarized the genome-wide SNP data by allele frequency or haplotype summary statistics. If these summary statistics are not sensitive to the recent European admixture, then the African-American samples may yield estimates of demographic parameters that are close to the true demographic parameters for the ancestral, unsampled, African populations. This would suggest that the differences in growth parameter estimates obtained from African populations cannot be explained by certain studies sampling African-American individuals and others sampling African individuals from within Africa. However, if these statistics are sensitive to recent admixture, then they may give biased estimates of growth parameters.
Here, we examine the effect of recent admixture on the estimation of population demography. In particular, we estimate growth parameters from simulated data sets using SNP frequencies as well as a recently developed haplotype summary statistic (Lohmueller et al. 2009). We compare the demographic parameter estimates made from the admixed and nonadmixed populations and find that some parameter estimates are qualitatively similar between the two populations when inferred using allele frequencies. Inferences of growth using haplotype-based approaches appear to be more sensitive to recent admixture than inferences based on SNP frequencies. We discuss implications that our results have for interpreting studies of demography in admixed populations.
METHODS
Demographic model for simulations:
For generating simulated data, we used a demographic model that qualitatively approximates the history of African, European, and African-American human populations. We chose to focus on African-American demography as (1) African-American populations are a significant component of the U.S. population (∼12%, U.S. Census 2000 Summary File 1, http://factfinder.census.gov) and are, therefore, heavily studied by population and medical geneticists in the United States; (2) there is considerable understanding of the historical context surrounding the recent demographic history of African-Americans including the trans-Atlantic slave trade, early American history, and history of African-American migrations within the United States; and (3) the admixture process in other human populations is likely to be more complex.
Figure 1 shows an illustration of the demographic model considered. Essentially, an ancestral population of size NB split tsplit generations ago to form an African population (Pop A) and a European population (Pop E). The African population expanded from its ancestral size (NB) to its current size (NA) tcur generations ago. The European population underwent a bottleneck (using parameters similar to those inferred by Lohmueller et al. 2009). Note that we assumed no gene flow between Pop A and Pop E after the split even though some studies have found evidence for migration between African and European populations (Schaffner et al. 2005; Gutenkunst et al. 2009; Nielsen et al. 2009; Wall et al. 2009). We chose not to include such migration in our models so that our assessments of the effects of recent admixture would not be confounded by other sources of gene flow. Twenty generations ago, the African-American population (Pop AA) was formed and has current size NAA (Pfaff et al. 2001; Patterson et al. 2004; Tian et al. 2006). We assumed 80% of the ancestry of Pop AA comes from Pop A, with the remainder coming from Pop E (Pfaff et al. 2001; Patterson et al. 2004; Tian et al. 2006). Since it is unknown whether there was a founder effect in forming the African-American population, we allowed NAA to vary. All simulations assumed an infinite-sites mutation model and a Wright–Fisher model of reproduction.
Figure 1.—

Demographic model for African (Pop A), African-American (Pop AA), and European (Pop E) populations used to simulate test data sets. For all simulations conducted here, tsplit = 4000 generations, NA = 20,000, NB = 10,000, and Pop E followed the bottleneck model from Lohmueller et al. (2009), except for the ancestral population size (NB), which was set to 10,000. See text for a further description of the parameters.
Inference on simulated data using the site frequency spectrum:
A useful summary of SNP data that potentially contains information regarding the magnitude of recent population growth is the site frequency spectrum (SFS) (Fu 1995; Griffiths and Tavaré 1998; Nielsen 2000; Williamson et al. 2005). Mathematically, the SFS is defined for a set of n sequenced chromosomes across S variable sites (i.e., SNPs) as the random vector (X1, X2, …, Xn−1), where Xi represents the number of SNPs where the n chromosomes are portioned into exactly i copies of the derived allele and n − i copies of the ancestral allele. For example, X1 is the number of singleton SNPs in the data, X2 is the number of SNPs where exactly two chromosomes carry the derived allele, and so on. Note that the sum of the entries in the SFS equals the total number of SNPs in the data set. Informally, one can think of the SFS as a histogram consisting of the number of SNPs at different frequencies in the sample where frequencies are binned at 1/n intervals. To determine the accuracy of parameter estimates for NA, tcur, and NB/NA when using the SFS obtained from Pop AA, for each combination of demographic parameters, we simulated 500 data sets, each consisting of 10,000 unlinked 1-kb regions in n = 24 chromosomes from each population. The size of each data set was meant to roughly mimic the scope of resequencing data sets currently in use, such as the Celera Genomics SNP data set (Bustamante et al. 2005; Lohmueller et al. 2008). We assumed a per-nucleotide mutation rate μ = 10−8 and a per-nucleotide recombination rate, r = 10−8. For each data set, we calculated the SFS for both Pop A and Pop AA, which were then used for inference.
To find the maximum-likelihood estimates (MLEs) for the three growth parameters, we used a Poisson likelihood function (see Nielsen 2000; Williamson et al. 2005; Boyko et al. 2008 for details). Briefly, the observed number of SNPs in each bin, Xi, of the SFS is treated as a Poisson random variable,
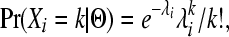 |
(1) |
where the rate parameter of the Poisson distribution λi is the expected number of SNPs in the particular bin of the SFS based on the set Θ = {θ, ν, τ} of mutation rate (θ = 4NAμ) and growth (υ = NB/NA; τ = tcur/2NA) parameters. This expectation has the form where the mutation rate acts as a scaling factor for each data set, so we can rewrite as the product as
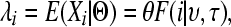 |
(2) |
where F(i | υ, τ) is proportional to the number of SNPs at frequency i/n in the sample and can be found either by coalescent simulations (Nielsen 2000) or via diffusion-based approximations (Williamson et al. 2005). All SNPs and bins of the SFS are treated independently and the final log-likelihood for a given set of growth parameters is the sum of the Poisson log-likelihoods for each bin of the SFS as given below:
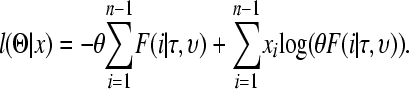 |
(3) |
This is a reasonable approximation to the true log-likelihood and holds when there is ample recombination among SNPs. Since the expected values of the SFS entries are not affected by recombination, when applied to linked data, the above inference scheme can be thought of as a composite-likelihood approach (Zhu and Bustamante 2005; Boyko et al. 2008).
We used the program PRFREQ (Boyko et al. 2008) to find the expected SFS for a given set of growth parameters using the Poisson, rather than the multinomial, implementation. Here we set μ = 0.1, which was the true value used in the simulations to generate the data (10−8 per nucleotide × 103 nucleotides per region × 104 regions). To optimize the likelihood function, we found the expected SFS for each parameter combination on a three-dimensional grid (NA, tcur, and NB/NA) of parameter values. It should be noted that the grids used are coarser than those used on real data sets.
Inference on simulated data using the haplotype-count number statistic:
Lohmueller et al. (2009) recently suggested a new summary statistic for genome-wide SNP data based on haplotype patterns. Their statistic, termed the haplotype-count number (HCN) statistic, is a two-dimensional histogram containing the joint distribution of the number of haplotypes and count of the most common haplotype in windows across the genome. To determine whether we could accurately estimate NA, tcur, and NB/NA when using the HCN statistic obtained from Pop AA, for each combination of demographic parameters, we simulated 100 data sets, each consisting of 7000 unlinked 250-kb regions in n = 46 chromosomes from each population. Importantly, as in Lohmueller et al. (2009), we used only a random subset of 20 SNPs from each simulated window with minor allele frequency (MAF) >10%. The size of these data sets is meant to mimic the large-scale genotyping surveys currently in use, such as that of Perlegen Sciences, where only a subset of SNPs in the population have been discovered and genotyped (Hinds et al. 2005). We assumed a per-nucleotide mutation rate μ = 10−8 and a per-nucleotide recombination rate r = 10−8. For each data set, we calculated the HCN statistics for Pop A and Pop AA, which were then used for inference.
To find the MLEs of the three demographic parameters from each data set via the HCN statistic, we used the approach of Lohmueller et al. (2009). Briefly, we used a multinomial approximate-likelihood function where the observed number of windows with i haplotypes and where the most common haplotype is at count j come from a multinomial distribution whose parameters are determined by the recombination rate and demographic parameters. We used coalescent simulations to find the parameters for the multinomial distribution for a given set of demographic parameters and recombination rates. Importantly, we fixed the recombination rate in these simulations equal to the true value used to generate the test data sets. The likelihood function was optimized using a grid search.
Analysis of National Institute of Environmental Health Sciences data:
We fitted a growth model to the SFS of the Yoruba and African-American samples from the National Institute of Environmental Health Sciences (NIEHS) data (Livingston et al. 2004). The Yoruba sample contained n = 12 individuals from Ibadan, Nigeria and the African-American sample contained 15 individuals sampled from the United States. We used only noncoding SNPs and excluded SNPs that had genotypes for <10 individuals in at least one of the two populations. Since some SNPs did not have a genotype at every individual, we used the hypergeometric distribution to find the expected SFS for a sample size of 20 chromosomes (Nielsen et al. 2004). In total our analysis included 13,588.4 SNPs in the African-American population and 13,487.9 SNPs in the Yoruba population after projection to a sample size of 20 chromosomes. The projection and subsequent analyses were done on the African-American and Yoruba samples separately. For the analysis of the NIEHS data, we used the folded SFS. The folded SFS tabulates the frequency of the minor allele, rather than the derived allele. The folded SFS still contains substantial information regarding demography without having to accurately infer the ancestral/derived states of SNPs (Adams and Hudson 2004).
We estimated the growth parameters for the Yoruba and African-American data sets using the PRFREQ program (Boyko et al. 2008). As we did for the analysis of the simulated data sets, we used the Poisson likelihood function. This procedure required an accurate estimate of the per-nucleotide mutation rate, μ. To estimate μ, we used the level of human–chimp divergence at the regions sequenced and the relationship K = 2Tμ, where K is the number of human–chimp differences (per nucleotide) and T is the human–chimp divergence time in units of generations. There were 51,770 differences in the 4,644,887 nucleotides sequenced in the builds of the human (hg18) and chimpanzee (pantro2) genomes, giving K = 0.01114559 per nucleotide. Then, assuming a human–chimp divergence time of 6 million years, and 25 years/generation, μ = (0.01114559 × 25)/(2 × 6 × 106) = 2.32 × 10−8 per nucleotide/generation. Since before the hypergeometric projection of the SNP frequencies, 7.83% of SNPs were excluded because they contained genotypes for <20 chromosomes in either one population or both populations, we decreased the total number of nucleotides sequenced by the same amount. This led to 4,281,191 total nucleotides that were used for analysis. We again used a grid search to optimize the likelihood function. Profile log-likelihood curves were used to calculate 95% confidence intervals (C.I.'s) for each parameter. We note that fixing of μ to a particular value does not alter the coverage properties for scaled parameters due to the invariance principle of maximum-likelihood inference (Pawitan 2001).
Analysis of the Perlegen data:
We fitted a growth model to the African-American sample analyzed by Perlegen Sciences (Hinds et al. 2005) using the HCN statistic. We chose to use the Perlegen data rather than other data sets, such as HapMap, because Perlegen genotyped all SNPs that they discovered, without regard to LD status, making subsequent analyses simpler (Lohmueller et al. 2009) and relatively free of the ascertainment biases in HapMap.
We divided the genome into nonoverlapping 0.25-cM windows and selected 20 SNPs from each window to construct the HCN statistic. Note that we selected only SNPs with MAF >10% in both the African-American and European-American data sets. Additionally, SNPs that were discovered using fewer than eight chromosomes were not included in the analysis. In total, the HCN statistic contained 8174 windows. As in Lohmueller et al. (2009), we used Clark's phasing algorithm (Clark 1990) to infer haplotype phase of the SNP data.
In the coalescent simulations used to generate the expected HCN statistic for a given demographic model, we also phased the simulated data using Clark's phasing algorithm and drew the recombination rate for each simulated region from a gamma distribution to allow for errors in the estimated genetic map (Lohmueller et al. 2009). Finally, we used the Schaffner recombination hotspot model (Schaffner et al. 2005) as implemented in Lohmueller et al. (2009).
RESULTS
Effect of admixture on patterns of polymorphism:
Figure 2 shows the expected SFS for Pop A with twofold growth as well as for Pop AA under two different values of NAA. First, we note there is an excess of low-frequency SNPs in Pop A compared to the neutral prediction. This result is expected since an excess of low-frequency SNPs is a signature of the population expansion (Tajima 1989a; Slatkin and Hudson 1991). For Pop AA, when NAA = NA, there is an even more pronounced excess of singleton and doubleton SNPs over what is seen in Pop A. However, the remaining bins of the SFS are similar for Pop A and Pop AA. When NAA = 0.1NA, we observe a decrease in the number of singleton SNPs compared to when NAA = NA. However, the number of singleton SNPs with NAA = 0.1NA is still slightly greater than that for Pop A alone. We also examined the SFS when Pop AA was formed 7 generations ago (Price et al. 2009) instead of 20 generations ago, corresponding to more recent admixture (supporting information, Figure S1). When NAA = NA, the SFS for the two different admixture times are indistinguishable. When NAA = 0.1NA, more recent admixture results in a slight increase in the number of singletons, presumably since the more recent founding of Pop AA results in less drift in Pop AA. However, the number of singletons here is still lower than that seen in Pop AA when NAA = NA. Thus, for the parameter combinations investigated here, the SFS for Pop A and Pop AA all appear to have an excess of low-frequency SNPs, with the excess being more pronounced in Pop AA.
Figure 2.—

Expected SFS in a sample size of 24 chromosomes for Pop A and Pop AA under population growth. Note the excess of low-frequency SNPs relative to the neutral prediction (Fu 1995) in all populations as well as the more pronounced excess of low-frequency SNPs in Pop AA relative to Pop A. NA = 20,000; NB/NA = 0.5; tcur = 2400 generations.
We also investigated the effect of recent admixture on the HCN statistic. Figure 3 shows the HCN statistics for simulated data under the three models described above (Pop A; Pop AA, NAA = NA; and Pop AA, NAA = 0.1NA). For Pop AA, when NAA = NA, there is a slight excess (compared to Pop A) of windows that have few haplotypes and a more pronounced excess (again, compared to Pop AA) of windows having the most common haplotype at higher frequency. This pattern likely stems from the fact that some individuals in the admixed population have haplotypes that recently came from Pop E. Since Pop E underwent a bottleneck, it contains less haplotype diversity than Pop A. Thus, the Pop E haplotypes create a shift toward more windows with fewer haplotypes and where the most common haplotype is at higher frequency in the HCN statistic from Pop AA. When NAA = 0.1NA, the difference between the HCN statistics in Pop A and Pop AA becomes even greater due to the loss of haplotypes during the founding of Pop AA. We also examined the HCN statistic when Pop AA was formed 7 generations ago (Price et al. 2009) instead of 20 generations ago (Figure S2). When NAA = NA, the HCNs for the two different admixture times are nearly identical. When NAA = 0.1NA, overall haplotype diversity is still lower than that seen in Pop AA when NAA = NA; however, the shift is less pronounced than when the admixture and founding occurred 20 generations ago. In summary, the admixture process alters the HCN statistic in a manner that is heavily influenced by the current size of Pop AA and the relative duration of the founder effect forming Pop AA.
Figure 3.—

Expected HCN statistics for Pop A and Pop AA. Each cell in the matrix is colored according to the proportion of simulation replicates (windows) having the particular configuration of the number of haplotypes and count of the most common haplotype. For example, red cells contain <2.5% of windows and medium-blue cells contain 20–22.5% of simulated windows. Note the excess of windows with fewer haplotypes and where the most common haplotype is at higher frequency in Pop AA relative to Pop A. This is most pronounced when NAA = 0.1NA. Note that the simulations assume NA = 20,000, NB/NA = 0.5, and tcur = 2400 generations.
Inference of demography from simulated data:
To determine whether the differences in the SFS and HCN statistics from Pop A and Pop AA (Figures 2 and 3 and Figure S1 and Figure S2) are meaningful, we estimated the parameters for a population growth model using the SFS and HCN statistics from Pop A and Pop AA (see methods). The purpose of this analysis was to see if, using data from Pop AA, we could accurately estimate the current population size of Pop A (NA), the time of population growth in Pop A (tcur), and the magnitude of population growth (NB/NA).
Figure 4A shows the distribution of MLEs inferred using the SFS for the three growth parameters when tcur = 2400 generations. For Pop A, the MLEs for all three parameters are clustered at the true parameter values. For Pop AA, when NAA = NA, NA is slightly overestimated and NB/NA is underestimated. The reverse pattern is seen when NAA = 0.1NA. Here, NA is slightly underestimated, but NB/NA is slightly overestimated. Importantly, in both cases, when using the SFS from Pop AA, all the estimates for NA are within 10,000 of the true value and all the estimates for NB/NA are within 0.15 of the true value. The estimates of the timing since the instantaneous growth event (tcur) present a different pattern. For both models of Pop AA, tcur is severely overestimated (see below).
Figure 4.—

Distribution of MLEs for the three growth parameters inferred using (A) the SFS and (B) the HCN method (see text). Solid horizontal bars denote the true parameter values (NA = 20,000; NB/NA = 0.5; tcur = 2400 generations).
Figure 4B shows the distribution of the MLEs inferred using the HCN method. Again, the MLEs from Pop A are clustered around the true parameter values. Unlike the estimates made using the SFS, the MLEs from Pop AA are now quite far from the true parameter values. For example, when NAA = NA, NA is severely overestimated, and NB/NA is underestimated. When NAA = 0.1NA, haplotype diversity is lost, leading to the underestimation of NA when using individuals sampled from Pop AA. Interestingly, tcur is underestimated in both cases.
We also analyzed additional simulated data sets where tcur = 4000 generations. Figure 5 shows the distribution of the MLEs inferred using the SFS (Figure 5A) and the HCN statistic (Figure 5B). Note that when estimating parameters using the SFS, the estimates made from Pop AA again approximate the true parameter values for Pop A. In particular, tcur is not as severely overestimated compared to the case where tcur = 2400 generations. This is especially noticeable when NAA = NA. Thus, part of the explanation for the overestimate of tcur using Pop AA when tcur = 2400 generations is that the estimate was heavily influenced by the population “expansion” that occurred at tsplit, when Pop E and Pop A split (Stadler et al. 2009). When tcur = tsplit, this overestimate is less pronounced, although it is still present.
Figure 5.—

Distribution of MLEs for the three growth parameters inferred using (A) the SFS and (B) the HCN method (see text). Solid horizontal bars denote the true parameter values (NA = 20,000; NB /NA = 0.5; tcur = 4000 generations).
Figure 5B shows that the MLEs inferred using the HCN statistic on individuals from Pop AA are again very far from the true growth parameter values in Pop A. Interestingly, the MLEs of the growth parameters inferred from Pop AA when tcur = 4000 are very similar to those inferred when tcur = 2400 (compare Figure 4B to Figure 5B). This suggests that the admixture process has such a profound influence on the haplotype patterns that changes in the timing of growth in the parental population cannot be detected using individuals from Pop AA. The current size of Pop AA (NAA), on the other hand, has a large impact on the haplotype patterns (compare Pop AA, NAA = NA, to Pop AA, NAA = 0.1NA in Figure 4B). These results, taken together, indicate that recent admixture affects haplotype summary statistics more than it affects the SFS.
Frequently when researchers fit a demographic model to the observed SFS, they will also perform a goodness-of-fit (GOF) test to determine if the best-fitting model can explain the observed SFS (see, for example, Adams and Hudson 2004; Caicedo et al. 2007; Boyko et al. 2008; Nielsen et al. 2009). Given that the simple growth model is the wrong model for Pop AA (the true model involves growth and admixture), we assessed how well the MLEs of the growth parameters generated SFS that fit the observed SFS. Put another way, if a researcher were to fit a growth model to the SFS from an admixed population, how likely is it that the researcher would reject the simple growth model as an explanation for the observed SFS? We performed a simple chi-square GOF test for a particular demographic model where we compared the observed SFS in each of the 500 simulated data sets to the expected SFS at the MLEs. Figure S3 shows a quantile–quantile (Q-Q) plot comparing the GOF P-values from Pop AA to those for Pop A. When tcur = 2400, there is a shift toward smaller P-values in Pop AA compared to Pop A (Figure S3). This effect is less pronounced when tcur = 4000 (Figure S4). We find that when tcur = 2400, 5% of the simulated data sets in Pop A have a P-value <0.014. Note that the fraction of data sets with P < 0.014 is >1.4% due to the fact that some SNPs are linked, thus reducing the effective number of SNPs. Thus, we use 0.014 as an ∼5% rejection region for the GOF test. Using this calibration, we find that 8.8 and 5.8% of data sets for Pop AA have a P-value <0.014, for NAA = NA and NAA = 0.1NA, respectively. When tcur = 4000, 5% of the simulated data sets from Pop A have a P-value <0.0117 compared to 5.6 and 4.8% for NAA = NA and NAA = 0.1NA, respectively. These results suggest that there is a slightly worse GOF for the admixed population (Pop AA) than for the nonadmixed population, but we cannot exclude the possibility that some of this pattern may be due to differences in how accurately we optimized the likelihood function across different models. Nevertheless, for the data sets simulated here (containing ∼17,000 SNPs), the vast majority (91–95%) of data sets from Pop AA will be unable to reject the pure growth model.
Inference of demography from human data:
We estimated the three growth model parameters (tcur, NA, NB/NA) for the African-American and Yoruba populations using the SFS generated from the NIEHS resequencing data set (Livingston et al. 2004). We chose to use this data set since it was generated by complete resequencing of the same genomic regions in both African-American and Yoruba individuals. As such, we can directly measure the effect that admixture has on estimates of the growth model parameters estimated from the SFS by comparing the parameter estimates from the two populations. Importantly, since the same regions were studied and the resequencing was done by the same laboratory for the two populations, any differences in the estimates should not be attributable to differences in selective pressure or laboratory errors.
As expected on the basis of the analysis of simulated data described above, the folded SFS of the African-American and Yoruba samples are fairly similar to each other (P = 0.09; χ2 = 15.02; 9 d.f.; Pearson's chi-square test), but the African-American SFS has slightly more low-frequency SNPs and more SNPs overall (see also Figure S5). Using the SNPs in the folded SFS, Wattersons's θ = 8.88 × 10−4 per nucleotide in the Yoruba sample and 8.95 × 10−4 per nucleotide in the African-American sample. The average number of pairwise differences (π) per nucleotide is 7.99 × 10−4 in the Yoruba sample and 7.93 × 10−4 in the African-American sample. We then estimated the three demographic parameters for the African-American and Yoruba data sets (see methods). Figure 6 shows the profile-likelihood curves for the three parameters. The estimate of NA is slightly higher in the African American population (15,732) compared to the Yoruba population (14,647). However, NB/NA is slightly lower in the African-American (0.46) than in the Yoruba (0.5) sample. The profile-likelihood curves overlap substantially for tcur, with a MLE of 5208 generations in the African-American sample and 5425 generations in the Yoruba sample. Importantly, for all three parameters, the ∼95% C.I.'s from the profile-likelihood curves (<1.92 log-likelihood units) overlap between the African-American and Yorbua estimates, suggesting that the parameter estimates from the two populations are not significantly different from each other.
Figure 6.—

Profile log-likelihood curves for the three growth model parameters estimated using the SFS from the NIEHS resequencing data for the African-American (AA) and Yoruba (YRI) samples and the HCN statistic from the Perlegen SNP genotype data for the African-American (AA) sample. Note that the two estimates based on the SFS (solid curves) are very similar to each other. The horizontal line in each part denotes the approximate asymptotic 95% C.I.
We then estimated the growth parameters for the Perlegen African-American data set (Hinds et al. 2005), using the HCN approach (see methods). Figure 6 also shows the profile-likelihood curves for the three parameters. The estimate of NA (12,500) is slightly smaller than the estimates obtained from the SFS-based analysis. However, the estimates of the other two parameters using the HCN method are quite discordant with the estimates found using the SFS. NB/NA is much larger (0.94) when estimated using the HCN than the SFS (0.46). The timing of growth, tcur is also estimated to be much more recent when using the HCN compared to the SFS. Thus, as predicted by the analysis of simulated data sets described above, the HCN method gives different growth parameter estimates than the SFS method when the population has an admixed demographic history.
DISCUSSION
We have examined how recent admixture affects estimates of population growth when using the SFS and the HCN statistic for inference. For certain parameter combinations, we find that growth parameter estimates made using the SFS in the admixed population are qualitatively similar to the true growth parameters from the unadmixed population. This pattern holds more often for the current population size (NA) and magnitude of growth parameter (NB/NA) than for the timing since the growth parameter (tcur) and seems to be little affected by whether or not the admixed population experienced a reduction in size during its founding. If growth occurs at the time the ancestral populations split from each other (e.g., tcur = tsplit in Figure 1), then estimates of tcur from the admixed population exhibit smaller bias. The HCN approach, on the other hand, is severely affected by recent admixture for all parameters investigated.
Our simulations provide some intuition as to why the SFS and the HCN statistic are differently affected by recent admixture. This difference stems from the manner in which the 20% of ancestry from Pop E affects the SFS and the HCN statistic. The SFS from the admixed population (Pop AA) contains more SNPs and more low-frequency SNPs than does the SFS from the nonadmixed population (Pop A). This is due to the fact that Pop E contains some population-specific SNPs not present in Pop A that are then brought into Pop AA during the admixture process. On the basis of the model assumed here, as well as the analysis of the NIEHS resequencing data, the extra SNPs brought into the admixed population from Pop E do not substantially alter estimates of the population growth parameters. Conversely, the HCN statistic from the admixed population (Pop AA) is shifted toward a higher proportion of windows with fewer haplotypes and where the most common haplotype is at higher frequency than in the nonadmixed Pop A. Essentially, this suggests that there is less haplotype diversity in Pop AA than in Pop A. This pattern arises because ∼20% of chromosomes in Pop AA are from Pop E, rather than from Pop A. Since Pop E has undergone a population bottleneck, it has less haplotype diversity than Pop A does. Consequently, Pop AA has lower haplotype diversity than Pop A simply because it contains ∼20% of its chromosomes from the populations with lower haplotype diversity (Pop E) while Pop A contains 0% of its chromosomes from the population with lower diversity. Put another way, a single chromosome sampled from Pop A is more likely to represent a new haplotype in a sample from Pop A than a single chromosome sampled from Pop E would. This is the opposite of what was seen for single SNPs, where sampling chromosomes from a mixture of Pop A and Pop E results in an increase in the number of SNPs compared to sampling only Pop A (Ptak and Przeworski 2002; Stadler et al. 2009). This difference indicates that these two summaries of SNP data capture different and complementary aspects of ancestral history.
Due to the sensitivity of haplotype-based approaches to the admixture process, these methods may be more informative than SFS-based methods for inferring the extent of recent admixture and detecting founder effects associated with admixture. However, when using haplotype-based approaches, researchers need to explicitly model the admixture process, rather than fitting a simplified growth model. Fitting a simple growth model to an admixed population using haplotype patterns will likely give erroneous results. This conclusion does not imply that one method of inference is superior to the other. Instead, the two methods are complementary and the best approach to be used depends then on what one wishes to learn from the data.
Another interpretation for the lack of sensitivity of the SFS-based method to recent admixture is that the SFS-based approach does not have sufficient power to detect when data were drawn from a more complex model. Our results suggest that LD and haplotype patterns as summarized by the HCN statistic are especially sensitive to recent admixture and may provide better diagnostics for detecting ill-fitting models than the SFS alone. Pairwise LD patterns have recently been used by Hernandez et al. (2007) and Gutenkunst et al. (2009) on large-scale macaque and human resequence data sets to assess the fit of the demographic models estimated from the SFS. A similar approach may also provide a means of rigorously assessing model fit for demographic inference from admixed populations.
Since a plethora of genome-wide resequencing data will soon be available from a variety of human populations, we explored the performance of the SFS to estimate growth parameters using a larger data set from Pop AA. We found that increasing the number of chromosomes sampled from 24 to 100 in Pop AA still gave parameter estimates qualitatively similar to those from the smaller data sets (Figure S6), albeit with lower variance. Applying GOF tests to these data, 21% of data sets rejected GOF for the best-fitting model when we thinned the number of SNPs to match that in the smaller data set and 33% rejected GOF when we did not thin the number of SNPs. Thus, not surprisingly, the SFS generated from larger samples increases the power to reject GOF for an incorrect model. However, our results caution that as data sets grow, many may reject GOF due to minor model misspecifications, while still providing qualitatively reasonable estimates of important parameters. Since all models are simplifications of reality, and the sizes of genetic variation data sets are rapidly expanding, this may be an important practical problem in the near future. GOF tests based on the SFS from large data sets should be carefully interpreted while keeping this fact in mind.
While we examined complex demographic models involving population splits, bottlenecks, growth, and admixture, our models are still an oversimplification of the true demography of African and African-American populations. Due to the many parameters in our model, we examined only a few illustrative examples and did not evaluate systematically the effect of changing different parameters. For example, we assumed that the admixture event occurred 20 generations ago and that all Pop AA individuals have, on average, 80% of their ancestry from Pop A. In reality, both these parameters vary among individuals (Pfaff et al. 2001; Patterson et al. 2004). Thus, our models should be taken as illustrative examples of the effect of admixture on inference of ancient growth. It is unclear if the general trends seen from our simulations will hold under more complex models of demography. However, a more recent admixture time (7 generations instead of 20 generations) gave SFS that were qualitatively similar to those found when admixture occurred 20 generations ago (compare Figure 2 to Figure S1), suggesting that our conclusions may apply even if some of the true parameters differ slightly from those used in our models.
Nevertheless, due to these inherent complexities in trying to jointly model African, African-American, and European population history, we also analyzed empirical data from African and African-American populations. The analyses of the NIEHS and Perlegen data allow us to test whether the predictions made from data generated under our simplified demographic models hold for data generated under the true demographic model of these populations. The fact that the growth parameters estimated using the SFS from the NIEHS data in the Yoruba and African-American populations were similar to each other, as predicted by our simulations, suggests that our simple models provide a reasonable guide to reality. Furthermore, the finding that the estimates of growth parameters using the HCN statistic from African-American data significantly differ from those estimated using the SFS is again consistent with the observations from our simulations.
These findings have implications for reconciling differences in estimates of population growth parameters made using African and African-American populations. Our finding from the NIEHS data of similar growth parameters for both the African-American and Yoruba individuals suggests that using African-American individuals as opposed to West African individuals should not lead to large differences in parameter estimates. Instead, we propose that the differences in estimates of growth parameters in different studies are likely to be due to differences in the amounts of natural selection in different data sets, systematic differences in laboratory protocols leading to different SFS among data sets, or differences in modeling methods (e.g., whether or not migration is included) across studies. Consistent with this hypothesis, Wall et al. (2008) found that a summary of the SFS, Tajima's D (Tajima 1989b), significantly differed among different data sets consisting of West African populations. Further studies using more extensive resequencing data with similar laboratory protocols and more advanced demographic models will help obtain more consistent parameter estimates.
Acknowledgments
We thank A. Boyko, A. Auton, and J. Degenhardt for helpful discussions and suggestions throughout the project and J. Degenhardt, B. Lazzaro, A. Di Rienzo, and two anonymous reviewers for comments on an earlier version of the manuscript. We also thank R. Gutenkunst and R. Hernandez for providing the human–chimp divergence estimates for the National Institute of Environmental Health Sciences data. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Human Genome Research Institute for the National Institutes of Health. This work was funded by a National Institutes of Health grant (R01 HG003229 to A.G.C. and C.D.B.), a National Science Foundation Graduate Research Fellowship (to K.E.L.), and a Ruth Kirschstein National Research Service Award from the National Human Genome Research Institute (F32HG005308 to K.E.L.).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.113761/DC1.
References
- Adams, A. M., and R. R. Hudson, 2004. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics 168 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey, J. M., M. A. Eberle, M. J. Rieder, C. S. Carlson, M. D. Shriver et al., 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2 e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbujani, G., and D. B. Goldstein, 2004. Africans and Asians abroad: Genetic diversity in Europe. Annu. Rev. Genomics Hum. Genet. 5 119–150. [DOI] [PubMed] [Google Scholar]
- Boyko, A. R., S. H. Williamson, A. R. Indap, J. D. Degenhardt, R. D. Hernandez et al., 2008. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 4 e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryc, K., A. Auton, M. B. Nelson, J. R. Oksenberg, S. L. Hauser et al., 2010. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl. Acad. Sci. USA 107 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante, C. D., A. Fledel-Alon, S. Williamson, R. Nielsen, M. T. Hubisz et al., 2005. Natural selection on protein-coding genes in the human genome. Nature 437 1153–1157. [DOI] [PubMed] [Google Scholar]
- Caicedo, A. L., S. H. Williamson, R. D. Hernandez, A. Boyko, A. Fledel-Alon et al., 2007. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet. 3 1745–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M. C., and S. A. Tishkoff, 2008. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genomics Hum. Genet. 9 403–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, R., and K. M. Weiss, 1988. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA 85 9119–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A. G., 1990. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol. Biol. Evol. 7 111–122. [DOI] [PubMed] [Google Scholar]
- Cox, M. P., D. A. Morales, A. E. Woerner, J. Sozanski, J. D. Wall et al., 2009. Autosomal resequence data reveal late stone age signals of population expansion in sub-Saharan African foraging and farming populations. PLoS One 4 e6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., 1995. Statistical properties of segregating sites. Theor. Popul. Biol. 48 172–197. [DOI] [PubMed] [Google Scholar]
- Garrigan, D., and M. F. Hammer, 2006. Reconstructing human origins in the genomic era. Nat. Rev. Genet. 7 669–680. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1998. The age of a mutation in a general coalescent tree. Stoch. Models 14 273–295. [Google Scholar]
- Gutenkunst, R. N., R. D. Hernandez, S. H. Williamson and C. D. Bustamante, 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5 e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, R. D., M. J. Hubisz, D. A. Wheeler, D. G. Smith, B. Ferguson et al., 2007. Demographic histories and patterns of linkage disequilibrium in Chinese and Indian rhesus macaques. Science 316 240–243. [DOI] [PubMed] [Google Scholar]
- Hinds, D. A., L. L. Stuve, G. B. Nilsen, E. Halperin, E. Eskin et al., 2005. Whole-genome patterns of common DNA variation in three human populations. Science 307 1072–1079. [DOI] [PubMed] [Google Scholar]
- Jakobsson, M., S. W. Scholz, P. Scheet, J. R. Gibbs, J. M. VanLiere et al., 2008. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451 998–1003. [DOI] [PubMed] [Google Scholar]
- Keinan, A., J. C. Mullikin, N. Patterson and D. Reich, 2007. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 39 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenberger, J. A., J. C. Stephens, S. J. O'Brien and M. W. Smith, 2000. Significant admixture linkage disequilibrium across 30 cM around the FY locus in African Americans. Am. J. Hum. Genet. 66 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. Z., D. M. Absher, H. Tang, A. M. Southwick, A. M. Casto et al., 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319 1100–1104. [DOI] [PubMed] [Google Scholar]
- Lind, J. M., H. B. Hutcheson-Dilks, S. M. Williams, J. H. Moore, M. Essex et al., 2007. Elevated male European and female African contributions to the genomes of African American individuals. Hum. Genet. 120 713–722. [DOI] [PubMed] [Google Scholar]
- Livingston, R. J., A. von Niederhausern, A. G. Jegga, D. C. Crawford, C. S. Carlson et al., 2004. Pattern of sequence variation across 213 environmental response genes. Genome Res. 14 1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller, K. E., A. R. Indap, S. Schmidt, A. R. Boyko, R. D. Hernandez et al., 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451 994–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller, K. E., C. D. Bustamante and A. G. Clark, 2009. Methods for human demographic inference using haplotype patterns from genomewide single-nucleotide polymorphism data. Genetics 182 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth, G. T., E. Czabarka, J. Murvai and S. T. Sherry, 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 2000. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., M. J. Hubisz and A. G. Clark, 2004. Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics 168 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., M. J. Hubisz, I. Hellmann, D. Torgerson, A. M. Andres et al., 2009. Darwinian and demographic forces affecting human protein coding genes. Genome Res. 19 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, E. J., A. Marcini, J. Akey, J. Martinson, M. A. Batzer et al., 1998. Estimating African American admixture proportions by use of population-specific alleles. Am. J. Hum. Genet. 63 1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, N., N. Hattangadi, B. Lane, K. E. Lohmueller, D. A. Hafler et al., 2004. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 74 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawitan, Y., 2001. In All Likelihood: Statistical Modeling and Inference Using Likelihood. Oxford University Press, Oxford.
- Pfaff, C. L., E. J. Parra, C. Bonilla, K. Hiester, P. M. McKeigue et al., 2001. Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am. J. Hum. Genet. 68 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov, A., A. Di Rienzo and R. R. Hudson, 2002. Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics 161 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A. L., A. Tandon, N. Patterson, K. C. Barnes, N. Rafaels et al., 2009. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 5 e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak, S. E., and M. Przeworski, 2002. Evidence for population growth in humans is confounded by fine-scale population structure. Trends Genet. 18 559–563. [DOI] [PubMed] [Google Scholar]
- Reed, F. A., and S. A. Tishkoff, 2006. African human diversity, origins and migrations. Curr. Opin. Genet. Dev. 16 597–605. [DOI] [PubMed] [Google Scholar]
- Reiner, A. P., C. S. Carlson, E. Ziv, C. Iribarren, C. E. Jaquish et al., 2007. Genetic ancestry, population sub-structure, and cardiovascular disease-related traits among African-American participants in the CARDIA study. Hum. Genet. 121 565–575. [DOI] [PubMed] [Google Scholar]
- Sankararaman, S., G. Kimmel, E. Halperin and M. I. Jordan, 2008. a On the inference of ancestries in admixed populations. Genome Res. 18 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman, S., S. Sridhar, G. Kimmel and E. Halperin, 2008. b Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner, S. F., C. Foo, S. Gabriel, D. Reich, M. J. Daly et al., 2005. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 15 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seldin, M. F., T. Morii, H. E. Collins-Schramm, B. Chima, R. Kittles et al., 2004. Putative ancestral origins of chromosomal segments in individual African Americans: implications for admixture mapping. Genome Res. 14 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., and R. R. Hudson, 1991. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler, T., B. Haubold, C. Merino, W. Stephan and P. Pfaffelhuber, 2009. The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics 182 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. a The effect of change in population size on DNA polymorphism. Genetics 123 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. b Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H., M. Coram, P. Wang, X. Zhu and N. Risch, 2006. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 79 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, C., D. A. Hinds, R. Shigeta, R. Kittles, D. G. Ballinger et al., 2006. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am. J. Hum. Genet. 79 640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff, S. A., and S. M. Williams, 2002. Genetic analysis of African populations: human evolution and complex disease. Nat. Rev. Genet. 3 611–621. [DOI] [PubMed] [Google Scholar]
- Voight, B. F., A. M. Adams, L. A. Frisse, Y. Qian, R. R. Hudson et al., 2005. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc. Natl. Acad. Sci. USA 102 18508–18513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J. D., M. P. Cox, F. L. Mendez, A. Woerner, T. Severson et al., 2008. A novel DNA sequence database for analyzing human demographic history. Genome Res. 18 1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J. D., K. E. Lohmueller and V. Plagnol, 2009. Detecting ancient admixture and estimating demographic parameters in multiple human populations. Mol. Biol. Evol. 26 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, S. H., R. Hernandez, A. Fledel-Alon, L. Zhu, R. Nielsen et al., 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., W. Huang, H. Wang, Y. He, Y. Wang et al., 2007. Dissecting linkage disequilibrium in African-American genomes: roles of markers and individuals. Mol. Biol. Evol. 24 2049–2058. [DOI] [PubMed] [Google Scholar]
- Zhu, L., and C. D. Bustamante, 2005. A composite-likelihood approach for detecting directional selection from DNA sequence data. Genetics 170 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]