

Abstract
Light-induced formation of singlet oxygen selectively oxidizes methionines in the heavy chain of IgG2 antibodies. Peptide mapping has indicated the following sensitivities to oxidation: M252 > M428 > M397. Irrespective of the light source, formulating proteins with the free amino acid methionine limits oxidative damage. Conventional peptide mapping cannot distinguish between the S- and R-diastereomers of methionine sulfoxide (Met[O]) formed in the photo-oxidized protein because of their identical polarities and masses. We have developed a method for identification and quantification of these diastereomers by taking advantage of the complementary stereospecificities of the methionine sulfoxide reductase (Msr) enzymes MsrA and MsrB, which promote the selective reduction of S- and R-diastereomers of Met(O), respectively. In addition, an MsrBA fusion protein that contains both Msr enzyme activities permitted the quantitative reduction of all Met(O) diastereomers. Using these Msr enzymes in combination with peptide mapping, we were able to detect and differentiate diastereomers of methionine sulfoxide within the highly conserved heavy chain of an IgG2 that had been photo-oxidized, as well as those in an IgG1 oxidized with peroxide. The rapid identification of the stereospecificity of methionine oxidation by Msr enzymes not only definitively differentiates Met(O) diastereomers, which previously has been indistinguishable using traditional techniques, but also provides an important tool that may contribute to understanding of the mechanisms of protein oxidation and development of new formulation strategies to stabilize protein therapeutics.
Key words: immunoglobulin gamma antibody, methionine sulfoxide, oxidation, photo-oxidation, methionine sulfoxide reductase
Introduction
Protein oxidation, which can occur during bioprocessing and shelf-life storage, is considered one major cause of chemical degradation of biopharmaceuticals. The oxidation of methionine (Met) residues has been well-documented for therapeutic proteins such as alpha1-antitrypsin,1 granulocyte colony-stimulating factor,2,3 coagulation factor VIIa,4 and, in particular, immunoglobulin gamma antibodies (IgGs). Our study focuses on oxidation of Met residues in the Fc region of IgG1 and IgG2.
With a sequence homology that is as high as 84%, the Fc region has up to four Met residues, two of which (M252 and M428) are conserved in all IgGs (Fig. 1).5–8 The presence of M358 in IgG1 is dependent on the allele of the gene;9 the fourth Met residue, M397, is present in IgG2 and IgG3 only. The Fc region of IgGs can interact with Fc gamma receptors, resulting in phagocytosis and cellular cytotoxicity.10,11 The Fc can also interact with neonatal Fc receptors (FcRn), which contributes to the long serum half-life of antibodies.12,13 Because of the critical role of the Fc region, modifications such as Met-oxidation may adversely affect interactions between antibodies and FcRn or Protein A.14,15
Figure 1.

Amino acid sequence homology of the four IgG subclasses.
Oxidation of M252 or M428 (EU numbering scheme16) in IgG1 due to photo-exposure, elevated temperatures or peroxide has been studied previously. Lam et al. reported that a recombinant humanized antibody oxidized primarily at M252, and to a much lesser extent at M428, when exposed to high intensity fluorescent lighting and elevated temperatures, e.g. 30 and 40°C.17 Chumsae et al. indicated M252 and M428 were most susceptible to oxidation after storage for a year at 25°C or following exposure to tert-butylhydroperoxide.18 Results of a separate study indicated that M252 and M428 could be oxidized following exposure to Xenon light, and tert-butylhydroperoxide.19 Liu et al. showed that the melting temperature of the CH2 domain was significantly reduced upon oxidation, and that this change was dependent on the extent of oxidation of both M252 and M428.20 Bertolotti-Ciarlet et al. determined the sensitivity of oxidation to be greater at M252 than M428.15 Compared to IgG1s, there are few reports available to date on Met oxidation in IgG2. Pan et al. determined M252 to be most labile, followed by M428, whereas M358 and M397 both had the slowest oxidation rate.14
Pathways resulting in Met oxidation can be divided into one-electron and two-electron oxidation reactions.21 One-electron oxidation is instigated by radicals that can be formed by metal catalysis (iron II, copper I) or following photo-initiation. Two-electron oxidants are non-radicals such as peroxides, singlet oxygen, and hypochlorous acid. Unless conditions are carefully controlled, protein samples may undergo oxidation as a result of more than one pathway, e.g. a combination of exposure to stray light and contamination with trace amounts of metals or peroxides.
When the free amino acid Met is oxidized by hydrogen peroxide in aqueous solution, it yields two diastereomeric methionine sulfoxide products in equal amounts.22 Here, Met-S-(O) denotes the diastereomer with the (S) configuration at the alpha carbon and (S) configuration at the sulfur, whereas Met-R-(O) indicates the (S) configuration at the alpha carbon and (R) configuration at the sulfur (Fig. 2). Methionine sulfoxide (Met(O)) diastereomers are the only stable protein oxidation products, unlike reversible cysteine and Cys sulfenic acid formation, with known repair mechanisms in vivo.23–25
Figure 2.
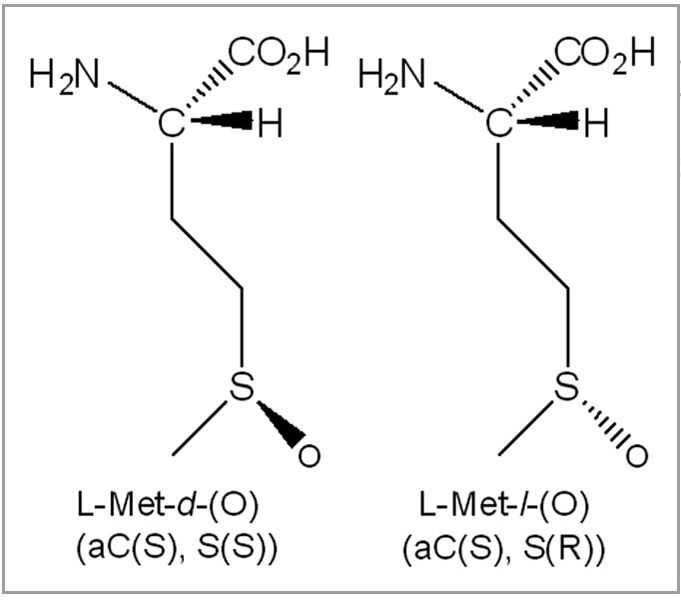
Diastereomeric structure of methionine sulfoxide, Met-S-(O) and Met-R-(O).
The diastereo-selectivity of the oxidation of Met residues in IgGs and other therapeutic proteins is often overlooked in the characterization of their degradation products.3,18 This problem requires further examination because of requirements by regulatory agencies that all degradation products observed in accelerated and long-term stability samples be identified. Under extremely well-optimized, reversed-phase liquid chromatography conditions, peptide fragments containing diastereomers of Met(O) can occasionally be separated,22 but no robust method to assign the S- and R-diastereomers for these peptides has been reported. To address this problem, we developed a method that utilizes peptide mapping in conjunction with a special class of enzymes, methionine sulfoxide reductases (Msr). Two forms of these enzymes exist, MsrA and MsrB, and they are able to reduce Met-S-(O) and Met-R-(O), respectively.26,27 In some genomes, these enzymes are fused into one protein, MsrBA, with the ability to perform dual function.26,28 These Msr enzymes have been demonstrated to be important in protecting cells against oxidative stress.23,29 The upregulation of MsrA in the human epidermis upon exposure to UVA or hydrogen peroxide has been established.30,31
We characterized Met oxidation in an IgG2 exposed to light, and compared the results to those for an IgG1 exposed to peroxide. Using the Msr enzymes, we selectively reduced individual stereo-isoforms of Met(O) and differentiated the S- and R-diastereomers, specifically at Met428. Our method thus extends traditional peptide mapping approaches to enable identification of oxidation products, and provides the first example of Msr enzymes use to characterize Met oxidation in IgGs.
Results
Photo-oxidation of an IgG2.
The tryptic map overlay in Figure 3A provides a global view of changes to the IgG2 antibody following 24 hours of exposure to light from Xe lamps. The loss in native tryptic peptide peaks and corresponding gain in oxidized tryptic peptide peaks are highlighted in B–D of Figure 3. In all three cases, the new peaks present in the Xe-irradiated sample were due to the oxidation of Met to Met(O) as determined by a mass increase of +16 Da of the oxidized M252-, M428- and M397-containing peptides compared with the native ones. The position of the oxidation was determined conclusively by MS/MS data; Figure 4 shows data for the tryptic peptide containing oxidized M428.
Figure 3.

The UV 215 nm overlay of RPHPLC separated tryptic peptides of an IgG2 exposed to 24 hour Xenon (black line) versus control, no exposure to Xenon (gray line). Major modifications are outlined by boxes in (A). Close-up views are shown in (B) (M252), (C) (M428) and (D) (M397).
Figure 4.

Tandem mass spectra of the tryptic peptide containing oxidized M428 from IgG2 irradiated for 24 hrs with Xenon.
Seven consecutive y ions (y16, y17, y18, y19 y20, y21, y22) were mass matched to ions (with varying charge states and loss of ammonia) in the spectrum (Fig. 4). The matching of y11, y12, y13 and b13 ions effectively localizes the +16 Da modification to the Met residue, and discounts modification at Trp or either of the His residues present in H35. Both oxidized peptides containing M428 (Fig. 3C) showed similar MS/MS fragmentation patterns, indicating the presence of two stereomers of Met428. Differentiation of the stereomers by MS/MS was not possible due to their identical masses.
Of the five Met residues in this IgG2, three were determined to be susceptible to photo-oxidation. These oxidation hotspots have been highlighted in B (M252), C (M428) and D (M397) of Figure 3. The bar graphs in Figure 5 show the level of oxidation for each Met residue as a function of exposure time to UV (A) or Xe (B) irradiation. Regardless of the light source, Met 252 showed the highest level of oxidation compared with that of Met397 and Met428. Relative oxidative susceptibility of the three Met residues was as follows: M252 > M428 > M397. For Met252, approximately 14% of the starting material at time zero was already oxidized to Met sulfoxide. This was consistently observed for different manufactured lots of this IgG2.
Figure 5.

(A and B) Bar graphs comparing UV and Xe induced Met modifications in an IgG2. (C) Bar graph comparing the effect of adding free Met (10 mM) to UV and Xe (24 hour exposure) induced Met modifications in IgG2.
A comparison of irradiation by UV to that by Xe indicated that UV irradiation resulted in a gradual increase in Met sulfoxide, whereas Xe irradiation led to a rapid increase in oxidation in all three Met locations (Fig. 5A and B). The level of oxidation in the UV-exposed samples after 48 hours was similar to that in the Xe-exposed samples after 8 hours of exposure. All three Met residues were nearly (93% for M397) or fully oxidized at the 48 hour time point for Xe-exposed samples (Fig. 5B). Met252 was oxidized further to Met sulfone under these conditions. This might be attributed to the intensity of the Xe irradiation being greater than that of the UV, thereby leading to more rapid oxidation in the Xe-exposed samples. Also, as previously reported, Met252 was the most labile of the three Met residues,14 which might facilitate oxidation of this residue from sulfoxide to sulfone.
The relative area oxidized for samples formulated with 10 mM Met and samples formulated without Met after 24 hours irradiation by UV and Xe is shown in Figure 5C. For both light sources, the presence of 10 mM Met was demonstrated to inhibit oxidation of the IgG2 Met residues by approximately half. This is consistent with previous reports that Met can be added to protein formulations as an anti-oxidant to limit protein oxidation.
Reduction of met sulfoxide diastereomers at M428 in the IgG2.
Since it was not possible to differentiate between the presumed methionine sulfoxide diastereomers by mass spectrometry, Msr enzymes MsrA and MsrB were used for the identification of Met-S-(O) and Met-R-(O), respectively. The diastereomers of oxidized Met containing peptides M397(O) (Fig. 3D) and M428(O) (Fig. 3C) were separated, whereas those from M252(O) were either not resolved or not present in the sample (Fig. 3B).
The effect of MsrBA on M428(O) diastereomers is shown in Figure 6A. Both peaks were completely reduced after 8 hours incubation, confirming the identification of an oxidized methionine residue in the peptide and excluding oxidation of other amino acids such as tryptophan or histidine. The second diastereomeric peak that eluted, designated M428(O)2, was reduced successively with increasing incubation time with MsrA (Fig. 6B). In contrast, chromatographic overlays shown in Figure 6C, revealed the first diastereomeric peak that eluted, designated M428(O)1, was reduced successively with increasing incubation time with MsrB and almost completely reduced after 12 hours of incubation. Concomitantly, this oxidized peak loss was accompanied by a gain in the native M428 peak (Fig. 6D).
Figure 6.

Effect of Msr enzymes on oxidized M428 containing peptides from a 12 hour Xenon irradiated IgG2 sample. The loss of oxidized peaks is shown in (A) (MsrBA), (B) (MsrA) and (C) (MsrB). The increase in the native peak using MsrB is shown in (D).
The percentages that each of these oxidized peaks decreased are depicted in the bar graphs in Figure 7A showing that MsrB incubation resulted in oxidized peak loss predominantly in M428(O)1, while B shows that MsrA incubation led to oxidized peak loss predominantly in M428(O)2. Based on the stereospecificity of the Msr enzymes, we deduced that M428(O)1 and M428(O)2, designated according to elution order, corresponded to Met-R-(O) and Met-S-(O), respectively.
Figure 7.

Effect of MsrB (A) and MsrA (B) incubation on M428(O)1 and M428(O)2 from a 12 hour Xenon irradiated IgG2 sample. MsrBA resulted in >95% loss of M428(O)1 and M428(O)2 in IgG2 after 8 hour incubation (Fig. 6A).
A detailed characterization of M252 and M397 was not possible due to incomplete separation of M252(O) and the low levels of M397(O). Under the conditions used in this study, M397(O) was resolved to two separate peaks. The appearance of two peaks indicated the presence of both stereomers (Fig. 3D). In the case of Met252, separation of the oxidized forms proved to be more difficult (Fig. 3B). The appearance of only one discrete peak of M252(O) suggested that either only one of the two stereomers was present or there was incomplete separation of the two stereomers. The analysis was further complicated by the rapid sulfone formation of Met252(O). These difficulties impeded a more detailed characterization of the diastereomers of M252(O) by MsrA and MsrB.
Characterization of met sulfoxide diastereomers in a peroxide-treated IgG1.
To demonstrate applicability of our method to other IgG subclasses and oxidants, an IgG1 antibody was oxidized using H2O2 and analyzed as described above. Similar to the photo-oxidized IgG2, Met252 in our IgG1 was found to be the most susceptible to oxidation, followed by Met428. The diastereomers of oxidized Met for tryptic peptides containing Met428 were further characterized using the Msr enzymes. The two stereomers of M428(O) containing peptides from this IgG1 were reduced using MsrBA, MsrA and MsrB (Fig. 8). Both stereomers of M428(O) were completely reduced after 8 hours incubation, confirming the identification an oxidized methionine residue in the peptide and excluding oxidation of other amino acids such as tryptophan or histidine (Fig. 8A). Results of the reduction of M428(O)-containing peptides using MsrA and MsrB showed stereospecific reduction of the two methionine stereomers (Fig. 9). Based on the stereospecificity of the Msr enzymes, M428(O)1 and M428(O)2, designated according to elution order, corresponded to Met-R-(O) and Met-S-(O), respectively. Results from this peroxide-treated IgG1 compared well to those from the photo-oxidized IgG2 molecule. In both cases, oxidation resulted in the generation of S- and R-stereomers of Met428 at an approximate ratio of 1:1 (Figs. 6 and 8). As was the case with the photo-oxidized sample, the stereomers of Met252 either could not be resolved with our reversed-phase HPLC method or only one of the two stereomers of M252(O) was present. Using MsrA and MsrB enzymes for the identification of the diastereomers of M252(O) without chromatographic separation was not possible because each enzyme reduced not only the reported substrate Met-S-(O) or Met-R-(O), but the other diastereomer to some degree as well (Fig. 8).
Figure 8.

Effect of Msr enzymes on oxidized M428 containing peptides from a 2 hour 0.3 M H2O2 treated IgG1 sample. The loss of oxidized peaks is shown in (A) (MsrBA), (B) (MsrA), and (C) (MsrB). The increase in the native peak using MsrB is shown in (D).
Figure 9.

Effect of MsrB (A) and MsrA (B) incubation on M428(O)1 and M428(O)2 from a 2 hour 0.3 M H2O2 treated IgG1 sample. MsrBA resulted in >95% loss of M428(O)1 and M428(O)2 in IgG1 after 8 hour incubation (Fig. 8A).
Discussion
Photo-induced degradation of proteins is complicated to study due to the ubiquitous presence of photo-sensitizers such as carbonyl groups. Photo initiation of carbonyls can generate radical fragmentation products or an excited triplet state, which is a strong oxidant that can further form singlet oxygen.32 Singlet oxygen is a highly reactive oxidizing species that reacts directly with organic substrates and can form methionine sulfoxides, Met(O), in addition to other products.32 Peroxides and light-sensitive transition metals can undergo photo-induced electron transfer to initiate chain reactions.32 While care is taken to avoid metal and peroxide contamination in all purification processes and all reagents used for analysis, photolytic initiation and subsequent propagation may catalyze normally negligible amounts of metals/peroxides to produce significant oxidation.17 Met(O) can be formed by radical reactions and through hydrogen abstraction in the alpha position to the sulfur.32 Our study examined Met oxidation, although fragmentation of the IgG antibody was also observed by SDS-PAGE (data not shown).
The Met residues most susceptible to oxidation in our study, M252 and M428, are conserved in all four human IgGs and are located in the Fc region. Recent studies have shown that oxidation of analogous M252 and M428 residues in IgGs led to changes in the structure and stability of the Fc region,20,33 which may weaken the binding of intact IgG antibody with two natural protein binding conjugates, Protein A and FcRn.14 A decrease in FcRn binding in particular may translate to a decrease in half-life of the IgG antibody.12,13
To our knowledge, this is the first study that identifies conclusively the doublet product peaks of oxidized Met428 as Met-R-(O) and Met-S-(O). This doublet has often been observed during peptide mapping of oxidized protein degradants for the purpose of formulation development and characterization during long-term and acute stress stability studies,34,35 but the true identity of these peaks had not been positively confirmed until now. This identification was reproducible using an IgG2 exposed to light, and an IgG1 incubated with peroxide. Our method using Msr enzymes, MsrA and MsrB, in combination with peptide mapping is a more direct approach than existing procedures for identification and quantification of methionine sulfoxide diastereomers.
Traditionally, the first step in identifying Met-R-(O) or Met-S-(O) diastereomers at a specific location on a protein is peptide mapping. The peak in the peptide map containing the specific Met is collected and subjected to further hydrolysis. The resulting free amino acids are separated using HPLC with o-phthaldialdehyde derivatization, and the free Met-R-(O) or Met-S-(O) are identified using standards.22,36 The synthesis and confirmation of Met-R-(O) and Met-S-(O) standards can be laborious.27–30 For example, the method described by Lavine37 utilizes the differential solubility of the diastereomeric picrates of Met sulfoxide to fractionate to purity. The final purified Met-R-(O) and Met-S-(O) standards were confirmed by melting point and optical rotation,37 or by 13C NMR analysis38 of signals α and β to the sulfur. Another means to purify the Met(O) standards utilized oxidation of N-phthaloyl derivatives of Met, followed by crystallization, and removal of the protecting group.39 If biocatalytic oxidation by Beauveria caledonica ATCC 64970 was utilized, Met-S-(O) of 90% purity could be obtained.38 Oxidation of the N-phthaloyl derivatives by hydrogen peroxide results in a diastereomeric mixture, which some have countered by using either MsrA or MsrB to consume the lesser diastereomer and achieve acceptable purity.40 The use of the Msr enzymes described here is more efficient than these other approaches, which require collection and hydrolysis of the peak in the peptide map and synthesis and purification of standards that could distinguish Met-R-(O) and Met-S-(O).
Results from our study indicate that, while MsrA and MsrB each exhibited the highest affinity for their reported substrates (Met-S-(O) and Met-R-(O), respectively, each enzyme also reduced the other diastereomer to some degree (Figs. 6 and 8). MsrA has been reported to reduce not only Met-S-(O) but also Met-R-(O) if a sufficiently high concentration of enzyme is used.22 In our case, high concentrations of the Msr enzyme were used to minimize the effects of agents such as residual guanidine HCl and trypsin, both of which were used during sample preparation for peptide mapping, that may have potentially lowered the enzymatic activity. MsrA, MsrB and MsrBA used in our study were purified separately and their activity was not standardized. These factors might explain the difference in the reduction rate of MsrB and MsrA (Figs. 7 and 9).28
Model reactions involving Met radical cations and superoxide have shown a diastereomeric preference in products.41 The diastereo-selectivity of different oxidants such as hydrogen peroxide, alkyl peroxide and singlet oxygen for a given Met residue in the protein calmodulin has been demonstrated, but trending across different Met locations is complicated.36 Most likely, this is because the specific conformational arrangement of the target Met plays a role in diastereoselectivity. In our studies of M428, oxidation by irradiation with Xe lamp, UV light, and hydrogen peroxide showed a slight but consistent preference for the formation of Met-R-(O) over Met-S-(O) (Fig. 2C). While the use of this information to predict the oxidants involved is not possible at this time, the identification of the diastereomers by the methodology demonstrated in this paper will be critical in this endeavor.
Materials and Methods
Materials
The recombinant human monoclonal IgG1 and IgG2 antibodies analyzed in this study were expressed at Amgen and purified using standard manufacturing procedures. Unless otherwise stated, samples were 30 mg/mL in a mildly acidic buffer. All chemicals and reagents used were of analytical grade and purchased from VWR Scientific (West Chester, PA). Amino acids are numbered according to the EU numbering scheme.16
Cloning and expression of Msr isoforms.
Two putative Msr genes, MsrA (SO2337) and MsrBA (SO2588), identified in the Shewanella oneidensis MR-1 genome42 contained 480- and 903-base pairs, respectively, and their DNA fragments were PCR amplified with MsrA-F (5′-CAC CAR GGC GTT AGC AAC TTT CGG T-3′) and MsrA-R (5′-GTA CTC GAG CTG ACA ACT TGG TAA-3′) primers, and MsrBA-F (5′-CAC CAT GGA CAA ACT GAC TGA TTT TGA A-3′) and MsrBA-R (5′-AAC TTG CAG TTC GGC AAA TAA ATG C-3′) primers. For comparison, the MsrB region of the MsrBA gene containing the first 128 amino acids in the sequence was PCR amplified using the following forward and reverse primers: MSRB#2-F (5′-CAC CAT GAA CAA ACT GAC TGA TTT TGA-3′) and MSRB#2R (5′-AGC ATG TTT TGG GGC AAT AT-3′). This sequence encodes the entire MsrB gene based on homology with other MsrB sequences in a range of microbes, including Bacillus anthracis, Escherichia coli, Mycobacterium tuberculosis, Salmonella typhimurium, Vibrio cholerae, Yersinia pestis. The amplified DNA fragments were cloned into pENTR/SD/D-TOPO vector (Invitrogen, Carlsbad, CA) to contain a V5-His tag sequence (KGG RAD PAF LYK VVI NSK LEG KPI PNP LLG LDS TRT GHH HHH H) engineered at the C-terminus of the expressed proteins and transformed into Escherichia coli strain TOP10 following the manufacturer instruction manual. Positive clones were screened and all DNA sequences were verified by DNA sequencing (Amplicon Express, Pullman, WA).
For overexpression of Msr isoforms in Escherichia coli, individual plasmids were cloned into pETDEST 42 (Invitrogen CAT #12276-010) using LR Clonase (Invitrogen CAT #11791-019) and transformed into Escherichia coli strain BL21Star (DE3). One liter of inoculated culture was grown at 37°C to an optical density of 0.4 at 600 nm. Isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 1.0 mM, and growth was continued overnight at 30°C. Bacteria were harvested by centrifugation, and following resuspension in 10 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 10 mM imidazole, cells were lysed by three passes through a French pressure cell (Glenmills Inc., Clifton, NJ). Lysates were first clarified by centrifugation at 5,000 rpm for 15 min, and the resulting supernatant was then centrifuged at 15,000 rpm for an additional 30 min. The clear supernatant was loaded onto a cobalt column (Talon Metal Affinity Resins; BD Biosciences—Clontech; Palo Alto, CA), and following five volumes of washing buffer [10 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 20 mM imidazole]. MsrBA, MsrA and MsrB recombinant proteins were eluted with 5 ml of elution buffer [10 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 300 mM imidazole]. The N-terminal His-Patch thioredoxin sequence from the MsrB recombinant protein was enzymatically digested using EnterokinaseMax (EKMax; Invitrogen Cat # 190-01). Msr protein concentrations were determined using the Coomassie Plus protein assay (Pierce Inc., Rockford, IL).
Photo-exposure of IgG2 antibody.
Two sources of irradiation were used, UVA light from a Caron 6540 (Caron, Marietta, OH) at a setting of 8 W/m2 and Xenon lamp irradiation (simulating full spectrum sunlight) (Q-SUN, Westlake, OH) calibrated to 0.35 W/m2 at 340 nm. Formulations with 10 mM D/L-Met (30 mg/mL IgG2 in mildly acidic buffer) and without Met were irradiated and aliquots were taken at intervals of 5, 10, 17, 24, 48 hours during UVA light exposure (designated as UV), and 4, 8, 12, 24, 48 hours for Xenon lamp irradiation (designated as Xe). The temperature of the samples during irradiation varied from 20 to 29°C. Samples that were shielded from light (covered using foil) and incubated for the same amount of time in the photo-chambers were used as controls to ensure that identified post-translational modifications were due to photo and not thermal processes.
H2O2 oxidation of IgG1 antibody.
Hydrogen peroxide stock (5 µL) was spiked into a 195 µL solution containing 30 mg/mL IgG1 in a mildly acidic buffer. The final H2O2 concentration was 30 mM, 0.3 mM and 0.0 mM (control). The samples were incubated at 37°C for 2 hours, after which it was quenched with DTT and stored at −70°C until the next sample preparation step (reduction and alkylation).
Sample preparation.
Reduction and alkylation of the intact antibody. Reduction and alkylation was performed on the antibody under denaturing conditions to produce free heavy and light chains for further analytical characterization. The antibody was diluted to a concentration of 5 to 6 mg/mL, using a pH 7.5 buffer composed of 8 M guanidine hydrochloride (Mallinckrodt, Phillipsburg, NJ), 0.25 M Tris-HCl pH 7.5 (Sigma, St. Louis, MO). An aliquot of a 0.5 M stock solution of the reducing agent dithiothreitol (DTT) (Sigma, St. Louis, MO) was added to the protein solution to obtain a final concentration of 10 mM DTT. Subsequently, the overall reaction mixture was incubated at 37°C for 30 minutes, and then cooled to room temperature. Alkylation of the reduced antibody was done by adding an aliquot of a 0.5 M iodoacetamide (IAM) (Sigma, St. Louis, MO) stock solution, making the final concentration of the IAM in the protein solution 20 mM. The protein solution was then stored at room temperature in the dark for an additional 45 minutes. A second addition of DTT (10 mM) was used to quench any unused IAM. To remove the excess DTT and IAM, the protein solution was buffer exchanged with 50 mM Tris HCl, pH 7.5, using a NAP-5 gel filtration column packed with Sephadex G-25 resin (Amersham Pharmacia Biotech, Orsay, France) following the procedure described by the manufacturer. Samples were used immediately for digestion or stored at −70°C until use.
Tryptic digestion. For peptide mapping, the antibody was first reduced and alkylated as described above. Sequencing grade modified trypsin (Promega, Madison, WI) was prepared by dissolving 20 µg with 40 µL water and immediately added to the reduced and alkylated antibody sample at a 1:20 (w:w) ratio. Digestion was performed for 1 hour at 37°C after which a second aliquot of trypsin was added to make the final enzyme: protein ratio 1:10 (w:w). The total digestion time was 5 hours and digests were frozen at −20°C or immediately analyzed by RP HPLC coupled to ion trap MS.
Methionine sulfoxide reductase (Msr) treatment of met sulfoxide containing tryptic peptides.
The IgG2 sample exposed to Xe light for 12 hours and the IgG1 sample incubated with 30 mM H2O2 were chosen for treatment by Msr because both contained roughly equal amounts of M428 and M428(O) to enable easy tracking of those peaks. These samples were prepared by reduction and alkylation, followed by tryptic digestion as outlined in the steps above. The samples were then incubated with a fresh aliquot of 20 mM DTT and either (1) 1 µM MsrA, (2) 1 µM MsrB (3) 1 µM MsrBA or (4) Control (no enzymes). The IgG concentration was approximately 0.5 g/L and buffered in 50 mM Tris pH 7.5. Temperature of incubation was 37°C and duration was 4 hours after which an aliquot was removed for analysis. The remaining samples were subjected to another cycle of incubation with addition of another 20 mM of DTT and enzymes as outlined above. This was repeated twice for a total of 12 hours. Aliquots removed for sampling were immediately analyzed using RP HPLC coupled to ion trap MS.
Reversed-phase HPLC of tryptic peptides.
Tryptic peptides were separated by RP HPLC using a Polaris Ether column that was 250 × 2 mm and packed with a C18 resin of 3 µm particle size and 300 Å pore size (Varian, Torrance, CA). The mobile phase was 0.1% (v/v) trifluoroacetic acid (TFA) in water in solvent A, and 90:9.915:0.085% (v/v) of acetonitrile: water: TFA in solvent B. Peptides were eluted using a linear gradient from 0% to 50% B over 205 minutes. The column flow rate was 200 µL/min, and its temperature was 50°C. Per analysis, 20–30 µg of protein digest was injected onto the column. The eluate was analyzed by an UV detector, and then directed to an on-line ion trap mass spectrometer.
Ion trap mass spectrometry of tryptic peptides.
A Thermo Finnigan ion trap mass spectrometer LCQ DECA (Thermo Electron, San Jose, CA) was used on-line with the RP HPLC system to identify the digested products. Masses of the peptides and their fragments were obtained using a triple play method, which included full scan, followed by zoom and MS/MS scans. A standard off-axis ESI source was used as the atmospheric vacuum interface. The instrument was tuned with the doubly charged ions from a synthetic peptide at m/z 842 Da. The sequence algorithm from the BioWorks 3.1 software (Thermo Electron, San Jose, CA) and a mass analyzer software developed in house43,44 were used for peptide identifications.
Analysis of met sulfoxide and met sulfone containing tryptic peptides.
Following the identification of native and oxidized tryptic peptide peaks by MS/MS analysis, the peak areas were obtained by reconstructed ion chromatogram using Qual Browser software (Thermo Electron, San Jose, CA). Mass spectrometry data and not UV data was used for quantification because co-eluting peptides interfered with UV quantification. The relative area of oxidized M252, M428 and M397 was calculated using the following equations:
Relative Area, oxidized M252 = {[AM252(O) + AM252(O2)]/[Anative + AM252(O) + AM252(O2)]} × 100
Where, AM252(O)= Peak area of M252 sulfoxide (diastereomers not resolved),
AM252(O2) = Peak area of M252 sulfone,
Anative = Peak area of native M252.
Relative Area, oxidized M428 = {[AM428(O)1 + AM428(O)2]/[Anative + AM428(O)1 + AM428(O)2] × 100
Where, AM428(O)1 = Peak area of M428 sulfoxide, first to elute,
AM428(O)2 = Peak area of M428 sulfoxide, second to elute,
Anative = Peak area of native M428.
Relative Area, oxidized M397 = {[AM397(O)1 + AM397(O)2]/[Anative + AM397(O)1 + AM397(O)2]} × 100
Where, AM397(O)1 = Peak area of M397 sulfoxide, first to elute,
AM397(O)2 = Peak area M397 sulfoxide, second to elute,
Anative = Peak area of native M252.
The data calculated using the above equations are summarized in Figure 4A–C.
Analysis of the effect of methionine sulfoxide reductase (Msr) on met 428 sulfoxide.
Following the identification of M428 native and its two sulfoxide peaks by MS/MS analysis, the peak areas were obtained by UV 215 nm detection using Chemstation for LC B.03.01 software (Agilent, Santa Clara, CA). The first sulfoxide peak to elute was assigned M428(O)1 and the second sulfoxide peak to elute was assigned M428(O)2. The relative peak area was calculated using the following equation:
Relative Area, M428(O)1 = {[AM428(O)1]/[AM428(O)1 + AM428(O)2 + Anative]} × 100
Where, AM428(O)1 = Peak area of M428 sulfoxide, first to elute,
AM428(O)2 = Peak area of M428 sulfoxide, second to elute,
Anative = Peak area of native M428.
Percent oxidized peak loss was calculated for M428(O)1 and M428(O)2 using the following equation for each time point of Msr treatment:
% Oxidized Peak Loss, at time t = [(Rm − Rc)/Rc] × 100
Where, Rm = Relative peak area at time t after Msr treatment,
Rc = Relative peak area at time t, control, no Msr added.
The data calculated using the above equations are summarized in Figures 6 and 8.
Acknowledgements
We thank Dr. Pavel Bondarenko for valuable comments and suggestions during the preparation of the manuscript. This research was supported by Amgen Inc., and the Genomic Science Program at the U.S. Department of Energy.
Abbreviations
- IgG
immunoglobulin gamma
- Met(O)
methionine sulfoxide
- Met (O2)
methionine sulfone
- Met-S-(O)
methionine sulfoxide diastereomer, (S) configuration at the alpha carbon and (S) configuration at the sulfur
- Met-R-(O)
methionine sulfoxide diastereomer, (S) configuration at the alpha carbon and (R) configuration at the sulfur
- M428(O)1
M428 sulfoxide containing peak, first to elute
- M428(O)2
M428 sulfoxide containing peak, second to elute
- Msr
methionine sulfoxide reductase
- MsrA
methionine sulfoxide reductase specific to Met-S-(O)
- MsrB
methionine sulfoxide reductase specific to Met-R-(O)
- MsrBA
methionine sulfoxide reductase specific to both Met(O) diastereomers
- RP HPLC
reversed phase high performance liquid chromatography
- MS/MS
tandem mass spectrometry
- TFA
trifluoroacetic acid
- UV
ultraviolet
- Xe
xenon
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- IPTG
isopropyl-β-D-thiogalactopyranoside
- DTT
dithiothreitol
- IAM
iodoacetamide
- FcRn
neonatal Fc receptor
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/11755
References
- 1.Griffiths SW, Cooney CL. Relationship between protein structure and methionine oxidation in recombinant human alpha1-antitrypsin. Biochemistry. 2002;41:6245–6252. doi: 10.1021/bi025599p. [DOI] [PubMed] [Google Scholar]
- 2.Chu JW, Brooks BR, Trout BL. Oxidation of methionine residues in aqueous solutions: free methionine and methinonine in granulocyte colony-stimulation factor. J Am Chem Soc. 2004;126:16601–16607. doi: 10.1021/ja0467059. [DOI] [PubMed] [Google Scholar]
- 3.Chu JW, Yin J, Brooks BR, Wang DIC, Speed Ricci M, Brems DN, Trout BL. A comprehensive picture of non-site specific oxidation of methionine residues by peroxides in protein pharmaceuticals. J Pharm Sci. 2004;93:3096–3102. doi: 10.1002/jps.20207. [DOI] [PubMed] [Google Scholar]
- 4.Kornfelt T, Persson E, Palm L. Oxidation of methionine residues in coagulation factor VIIa. Arch Biochem Biophys. 1999;363:43–54. doi: 10.1006/abbi.1998.1071. [DOI] [PubMed] [Google Scholar]
- 5.Rutishauser U, Cunningham BA, Bennett C, Konisberg WH, Edekman GM. Amino acid sequence of the Fc region of a human gamma G-immunoglobulin. Proc Natl Acad Sci USA. 1968;61:1414–1424. doi: 10.1073/pnas.61.4.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang A, Tung E, Fudenberg HH. The primary sequence of a human IgG2 heavy chain: genetic, evolutionary and functional implications. J Immunol. 1980;125:1048–1054. [PubMed] [Google Scholar]
- 7.Huck S, Fort P, Crawford DH, Lefranc M-P, Lefranc G. Sequence of a human immunoglobulin gamma 3 heavy chain constant region gene: comparison with the other human Cgamma genes. Nucleic Acids Res. 1986;14:1779–1789. doi: 10.1093/nar/14.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellison JW, Buxbaum JN, Hood LE. Nucleotide sequence of a human immunoglobulin Cgamma 4 gene. DNA. 1981;1:11–18. doi: 10.1089/dna.1.1981.1.11. [DOI] [PubMed] [Google Scholar]
- 9.van Loghem E, editor. Allotypic Markers. Basel, Switzerland: Karger; 1986. [PubMed] [Google Scholar]
- 10.Koolwijk P, Spierenburg GT, Frasa HBJ, van de Winkel JG, Bast BJ. Interaction between hybrid mouse monoclonal antibodies and the human high-affinity IgG FcR, huFc gamma RI, on U937. Involvement of only one of the mIgG heavy chains in receptor binding. J Immunol. 1989;143:1656–1662. [PubMed] [Google Scholar]
- 11.Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fcgamma receptor classes on human leukocytes. J Exp Med. 1990;171:1333–1345. doi: 10.1084/jem.171.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Low SC, Mezo AR. Inhibitors of the FcRn:IgG Protein-Protein Interaction. AAPS J. 2009;11:432–434. doi: 10.1208/s12248-009-9120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol. 2000;18:739–766. doi: 10.1146/annurev.immunol.18.1.739. [DOI] [PubMed] [Google Scholar]
- 14.Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to Protein A and FcRn. Protein Sci. 2009;18:424–433. doi: 10.1002/pro.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertolotti-Ciarlet A, Wang W, Lownes R, Pristatsky P, Fang Y, McKelvey T, et al. Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fcγ receptors. Molecular Immunology. 2009;46:1878–1882. doi: 10.1016/j.molimm.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci USA. 1969;63:78–85. doi: 10.1073/pnas.63.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam XM, Yang JY, Cleland JL. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. J Pharm Sci. 1997;86:1250–1255. doi: 10.1021/js970143s. [DOI] [PubMed] [Google Scholar]
- 18.Chumsae C, Gaza-Bulseco G, Sun J, Liu H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibofy. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;850:285–294. doi: 10.1016/j.jchromb.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Gaza-Bulseco G, Zhou L. Mass spectrometry analysis of photo-induced methionine oxidation of a recombinant human monoclonal antibody. J Am Soc Mass Spectrom. 2009;20:525–528. doi: 10.1016/j.jasms.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, Ren D, Huang H, Dankberg J, Rosendfeld R, Cocco MJ, et al. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47:5088–5100. doi: 10.1021/bi702238b. [DOI] [PubMed] [Google Scholar]
- 21.Schöneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer’s disease. Biochim Biophys Acta. 2005;1703:11129. doi: 10.1016/j.bbapap.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Sharov VS, Ferrington DA, Squier TC, Schöneich C. Diastereoselective reduction of protein-bound methionine sulfoxide by methionine sulfoxide reductase. FEBS Letters. 1999;455:247–250. doi: 10.1016/s0014-5793(99)00888-1. [DOI] [PubMed] [Google Scholar]
- 23.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Brot N, Weissbach H. Biochemistry and physiological role of methionine sulfoxide reductase in proteins. Archives of Biochemistry and Biophysics. 1983;223:271–281. doi: 10.1016/0003-9861(83)90592-1. [DOI] [PubMed] [Google Scholar]
- 25.Brot N, Weissbach H. Peptide methionine sulfoxide reductase: Biochemistry and Physiological role. Biopolymers. 2000;55:288–296. doi: 10.1002/1097-0282(2000)55:4<288::AID-BIP1002>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 26.Grimaud R, Ezraty B, Mitchell JK, Lafitte D, Briand C, Derrick PJ, et al. Repair of oxidized proteins: identification of a new methionine sulfoxide reductase. J Biol Chem. 2001;276:48915–48920. doi: 10.1074/jbc.M105509200. [DOI] [PubMed] [Google Scholar]
- 27.Hansel A, Heinemann SH, Hoshi T. Heterogeneity and function of mammalian MSRs: enzymes for repair, protection and regulation. Biochim Biophys Acta. 2005;1703:239–247. doi: 10.1016/j.bbapap.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Chen B, Markillie LM, Xiong Y, Mayer MU, Squier TC. Increased catalytic efficiency following gene fusion of bifunctional methionine sulfoxide reductase enzymes from Shewanella oneidensis. Biochemistry. 2007;46:14153–14161. doi: 10.1021/bi701151t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (Msr) is a regulator of antioxodant defense and lifespan in mammals. Proc Natl Acad Sci USA. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogawa F, Sander CS, Hansel A, Oehrl W, Kasperczyk H, Elsner P, et al. The repair enzyme peptide methionine-S-sulfoxide reductase is expressed in human epidermis and upregulated by UVA radiation. J Invest Dermatol. 2006;126:1128–1134. doi: 10.1038/sj.jid.5700116. [DOI] [PubMed] [Google Scholar]
- 31.Schallreuter KU. Functioning methionine-S-sulfoxide reductases A and B are present in human skin. J Invest Dermatol. 2006;126:947–949. doi: 10.1038/sj.jid.5700086. [DOI] [PubMed] [Google Scholar]
- 32.Hovorka SW, Schöneich C. Oxidative degradation of pharmaceuticals: theory, mechanism and inhibition. J Pharm Sci. 2000;90:253–269. doi: 10.1002/1520-6017(200103)90:3<253::aid-jps1>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 33.Liu H, Gaza-Bulseco G, Xiang T, Chumsae C. Structural effect of deglycosylation and methionine oxidation on a recombinant monoclonal antibody. Mol Immunol. 2008;45:701–708. doi: 10.1016/j.molimm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Keck RG. The use of t-butyl hydroperoxide as a probe for methionine oxidation in proteins. Anal Biochem. 1996;236:56–62. doi: 10.1006/abio.1996.0131. [DOI] [PubMed] [Google Scholar]
- 35.Bongers J, Cummings JJ, Ebert MB, Federici MM, Gledhill L, Gulati D, et al. Validation of a peptide mapping method for a therapeutic monoclonal antibody: what could we possibly learn about a method we have run 100 times? J Pharm Biomed Anal. 2000;21:1099–1128. doi: 10.1016/s0731-7085(99)00181-8. [DOI] [PubMed] [Google Scholar]
- 36.Sharov VS, Schöneich C. Diastereoselective protein methionine oxidation by reactive oxygen species and diastereoselective repair by methionine sulfoxide reductase. Free Radic Biol Med. 2000;29:986–994. doi: 10.1016/s0891-5849(00)00400-7. [DOI] [PubMed] [Google Scholar]
- 37.Lavine TF. The formation, resolution and optical properties of the diastreomeric sulfoxides derived from L-methionine. J Biol Chem. 1947;169:477–491. [PubMed] [Google Scholar]
- 38.Holland HL, Andreana PR, Brown FM. Biocatalytic and chemical routes to all the stereoisomers of methionine and ethionine sulfoxides. Tetrahedron: Asymmetry. 1999;10:2833–2843. [Google Scholar]
- 39.Bose AK. Organic Synthesis. 1960;40:82–85. [Google Scholar]
- 40.Olry A, Boschi-Muller S, Marraud M, Sanglier-Cianferani S, Dorsselear A, Branlant G. Characterization of the methinine sulfoxide reductase activities of PILB, a probable virulence factor from Neisseria meningitidis. J Biol Chem. 2002;227:12016–12022. doi: 10.1074/jbc.M112350200. [DOI] [PubMed] [Google Scholar]
- 41.Miller BL, Kuczera K, Schöneich C. One-electron photooxidation of N-methionyl peptides: mechanism of sulfoxide and azasulfonium diastereomer formation through reaction of sulfide radical cation complexes with oxygen or superoxide. J Am Chem Soc. 1997;120:3345–3356. [Google Scholar]
- 42.Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, et al. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat Biotechnol. 2002;20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z. De Novo Peptide sequencing based on a divide-and-conquer algorithm and peptide tandem spectrum simulation. Anal Chem. 2004;76:6374–6383. doi: 10.1021/ac0491206. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z. Prediction of low-energy collision-induced dissociation spectra of peptides. Anal Chem. 2004;76:3908–3922. doi: 10.1021/ac049951b. [DOI] [PubMed] [Google Scholar]